

Abstract
Inhibitor-resistant class A β-lactamases of the TEM and SHV families that arise by single amino acid substitutions are a significant threat to the efficacy of β-lactam/β-lactamase inhibitor combinations. To better understand the basis of the inhibitor-resistant phenotype in SHV, we performed mutagenesis to examine the role of a second-shell residue, Asn276. Of the 19 variants expressed in Escherichia coli, only the Asn276Asp enzyme demonstrated reduced susceptibility to ampicillin/clavulanate (MIC increased from 50/2 → 50/8 μg/mL) while maintaining high-level resistance to ampicillin (MIC = 8192 μg/mL). Steady-state kinetic analyses of Asn276Asp revealed slightly diminished kcat/Km for all substrates tested. In contrast, we observed a 5-fold increase in Ki for clavulanate (7.4 ± 0.9 μM for Asn276Asp vs 1.4 ± 0.2 μM for SHV-1) and a 40% reduction in kinact/KI (0.013 ± 0.002 μM−1 s−1 for Asn276Asp vs 0.021 ± 0.004 μM−1 s−1 for SHV-1). Timed electrospray ionization mass spectrometry of clavulanate-inhibited SHV-1 and SHV Asn276Asp showed nearly identical mass adducts, arguing for a similar pathway of inactivation. Molecular modeling shows that novel electrostatic interactions are formed between Arg244Nη2 and both 276AspOδ1 and Oδ2; these new forces restrict the spatial position of Arg244, a residue important in the recognition of the C3/C4 carboxylate of β-lactam substrates and inhibitors. Testing the functional consequences of this interaction, we noted considerable free energy costs (+ΔΔG) for substrates and inhibitors. A rigid carbapenem (meropenem) was most affected by the Asn276Asp substitution (46-fold increase in Ki vs SHV-1). We conclude that residue 276 is an important second-shell residue in class A β-lactamase-mediated resistance to substrates and inhibitors, and only Asn is able to precisely modulate the conformational flexibility of Arg244 required for successful evolution in nature.
The development of β-lactam antibiotics (penicillins, cephalosporins, and carbapenems, Figure 1) remains one of the most significant advances in modern medicine. β-Lactam antibiotics manifest their bactericidal effects by inhibiting enzymes involved in cell wall synthesis. The broad spectrum of activity and low toxicity of β-lactams make them the “antimicrobial agents of choice” in the treatment of many infections. Unfortunately, the efficacy of these life-saving antibiotics is seriously threatened by the evolution of β-lactamases (1). β-Lactamases (E.C. 3.5.2.6) are periplasmic bacterial enzymes (hydrolases) that are frequently found in Enterobacteriaceae and are responsible for increasing resistance to penicillins, cephalosporins, and carbapenems. There are four classes of β-lactamases: classes A, C, and D possess Ser as the active site nucleophile, while class B uses a metal ion (Zn2+) to inactivate β-lactams (2).
FIGURE 1.

Chemical structures of compounds tested in this study. The structures of cephalothin and clavulanate are labeled with the accepted ring numbering system.
The most prevalent β-lactamases in Enterobacteriaceae (e.g., Escherichia coli and Klebsiella pneumoniae) are the class A enzymes (e.g., TEM and SHV)1 (1). In order to overcome class A β-lactamase-mediated resistance and preserve the efficacy of these antibiotics, β-lactamase inhibitors (clavulanate, sulbactam, and tazobactam, Figure 1) were coupled with β-lactam antibiotics. These mechanism-based inhibitors greatly enhance the utility of their partner β-lactams (amoxicillin, ampicillin, piperacillin, cefoperazone, and ticarcillin) in the treatment of serious infections caused by Gram-negative bacteria in the hospital, skilled nursing facilities, and community settings (3).
Regrettably, selective pressure from excess antibiotic use accelerated the emergence of resistance to β-lactam/β-lactamase inhibitor combinations. Single amino-acid substitutions in the TEM and SHV families of class A β-lactamases resulted in enzymes with reduced affinity for β-lactamase inhibitors (2, 4). At the time of this writing, 25 inhibitor-resistant TEMs (IRTs) and five inhibitor-resistant SHVs have been described (http://www.lahey.org/studies/webt.asp). “Inhibitor-resistant” refers to resistance to amoxicillin/clavulanate as defined by antimicrobial susceptibility testing and may, but does not necessarily, include resistance to sulbactam and tazobactam combinations (5). Because resistance to β-lactam/β-lactamase inhibitor combinations challenges our ability to successfully treat serious infections, attention has focused on designing and discovering novel inhibitors which can help overcome IR enzymes (6–9). A thorough understanding of the molecular basis of IR β-lactamases is essential to inform inhibitor development and anticipate future resistance determinants.
Currently, two primary structural changes explain resistance to clavulanate in TEM and SHV enzymes: (i) alterations in the geometry or shape of the “oxyanion hole” or electrophilic center; and (ii) changes in the position of Ser130 or Arg244 (we use the Ambler numbering system for class A β-lactamases) (10–15). The substitutions most frequently isolated in the clinic and producing the most pronounced inhibitor resistance in TEM include variants at Ambler positions Met69, Ser130, Arg244, Asn275, and Asn276 (16). Met69 and Ser130 substitutions are seen in clinical IR SHVs, but Arg244, Asn275, and Asn276 substitutions have not yet been detected. Despite 68% sequence identity between SHV and TEM enzymes, these closely related β-lactamases demonstrate differences in the kinetic details which confer resistance to clavulanate and sulbactam (13, 17).
Previous studies indicated that Asn276 in TEM-1 plays a critical role in inhibitor inactivation by way of its interaction with Arg244 (18, 19). Arg244 is positioned between the binding pocket and Asn276, and contributes directly to substrate and inhibitor affinity in TEM and SHV through hydrogen bonding with the conserved β-lactam carboxylate (5, 13, 20, 21). The Asn276Asp substitution in TEM results in an amoxicillin/clavulanate-resistant phenotype, reduced kinact, and increased Ki values (18, 19, 22, 23). The X-ray crystal structure of TEM Asn276Asp highlighted the importance of the electrostatic environment surrounding residues 244 and 276 (19). The TEM Asn276Asp substitution created a new interaction with Arg244 (276AspOδ2:Arg244Nη2); surprisingly, movement or repositioning of Arg244 was not observed. In the case of the wild-type (WT) TEM-1 enzyme crystal structure, there is only a hydrogen bond between Asn276 and Arg244; the TEM Asn276Asp variant has both a hydrogen bond and a salt bridge between these residues (24).
Based upon these reported findings, we were compelled to address the role of Asn276 in the SHV family of β-lactamases and discern its contribution to both substrate hydrolysis and clavulanate inhibition. Accounting for the different kinetic parameters of inhibition between TEM and SHV enzymes, we specifically inquired whether Asn276, located remotely from the active site (a second-shell residue), would have a major impact on the ability of SHV β-lactamase to discriminate between various β-lactams and clavulanate. To this end, we performed susceptibility testing on a full panel of amino acid variants at residue 276 constructed by site-saturation and site-directed mutagenesis and completed steady-state kinetic characterization of the clavulanate-resistant Asn276Asp variant. To compare the nature of the intermediates in the inactivation process, we inhibited SHV-1 and SHV Asn276Asp with clavulanate and resolved the resulting adducts by timed electrospray ionization mass spectrometry (ESI-MS). In addition, novel boronic acid transition state inhibitors (BATSIs), a methylidene penem, and meropenem (Figure 1) were tested as probes of the functional consequences of the Asn276Asp substitution on β-lactam carboxylate recognition and affinity. Our data leads us to conclude that in both SHV and TEM, perturbations of the electrostatic relationships between Asp276 and Arg244 create unique structural/conformational changes that have significant consequences on the turnover of substrates and inhibitors. However, the Asn276Asp substitution in SHV “enjoys” clavulanate resistance at a lower cost to catalytic efficiencies than most other IR SHVs and IRTs.
MATERIALS AND METHODS
Mutagenesis and Sequencing
Using the template blaSHV-1 gene in the phagemid vector pBC SK(−) (Stratagene, La Jolla, CA), we employed Stratagene’s QuikChange Mutagenesis kit with degenerate oligonucleotides at Ambler position 276 to make the full complement of amino acid substitutions. After PCR mutagenesis, we electroporated the resultant plasmids into E. coli ElectroMAX DH10B cells (Invitrogen, Carlsbad, CA). These plasmids were isolated and DNA sequencing of the blaSHV genes was performed. Variants not acquired after sequencing of 100 clones were created with specific mutagenic oligonucleotides as previously described (25).
Immunoblotting
E. coli DH10B cells were grown to OD600 = 0.8 and lysed by 10 min incubation at 100 °C in SDS loading dye buffer. Immunoblots were performed with an anti-SHV polyclonal antibody to confirm production of the SHV β-lactamases from all 19 constructs as previously described (26).
Antimicrobial Susceptibility Tests
MICs for E.coli DH10B cells expressing the blaSHV-1 or mutant blaSHV Asn276Xxx genes cloned in phagemid pBC SK(−) were obtained by lysogeny broth agar dilution. The MICs for various antibiotics were determined using a Steers Replicator that delivered 10 μL of a diluted overnight culture containing 104 colony forming units. Ampicillin, piperacillin, cephalothin, and cefotaxime were purchased from Sigma (St. Louis, MO); meropenem was purchased from AstraZeneca Pharmaceuticals (Wilmington, DE). Lithium clavulanate (GlaxoSmithKline, Surrey, United Kingdom), sulbactam (Pfizer, La Jolla, CA), and tazobactam (Wyeth Pharmaceuticals, Pearl River, NY) were kind gifts and were tested in combination with 50 μg/mL ampicillin.
β-Lactamase Expression and Purification
β-Lactamase enzymes were expressed and purified as described previously (27). Briefly, E. coli DH10B cells containing the blaSHV-1 or blaSHV Asn276Asp genes in pBC SK(−) were grown overnight in SOB medium, harvested by centrifugation at 4 °C, and frozen at −20 °C. After thawing, β-lactamase was liberated using stringent periplasmic fractionation with 40 μg/mL lysozyme (Sigma) and 1 mM EDTA, pH 7.8. Preparative isoelectric focusing was performed with the lysate in a Sephadex granulated gel (GE Healthcare, Piscataway, NJ) using ampholines in the pH range 3.5–10 and running the gel overnight at a constant power of 8 W on a Multiphor II isoelectric focusing apparatus (GE Healthcare). Purity was assessed by 5% stacking, 12% resolving sodium dodecyl sulfate–polyacrylamide gel electrophoresis. β-Lactamase concentration was determined using the Bio-Rad (Hercules, CA) Bradford protein assay with bovine serum albumin standards. If purity was <90%, a second purification step was performed using size-exclusion chromatography with a Pharmacia ÄKTA Purifier system (fast protein liquid chromatography instrument) (Uppsala, Sweden) (28).
Kinetic Measurements
Steady-state kinetics were performed using continuous assays at room temperature in an Agilent 8453 diode array spectrophotometer (Agilent, Palo Alto, CA). Each assay was performed in 10 mM phosphate-buffered saline (PBS) at pH 7.4. Measurements were obtained using nitrocefin (NCF) (BD Biosciences, San Jose, CA) (Δε482 = 17400 M−1 cm−1), ampicillin (Δε235 = −900 M−1 cm−1), piperacillin (Δε235 = −820 M−1 cm−1), and cephalothin (Δε262 = −7660 M−1 cm−1). In order to specifically investigate the role of the C3/C4 carboxylate in substrate recognition, we used (i) an achiral and chiral cephalothin BATSI (13), (ii) a methylidene penem inhibitor bearing a dihydropyrazolo[1,5-c][1,3]thiazole moiety (Wyeth Pharmaceuticals) (29), and (iii) the carbapenem meropenem.
The kinetic parameters, Vmax and Km, were obtained with nonlinear least-squares fit of the data (Henri Michaelis–Menten equation) using Origin 7.5 (OriginLab, Northampton, MA)
| (1) |
The dissociation constants of the preacylation complex, Kis, were determined by direct competition assays. The initial velocity was measured in the presence of a constant concentration of enzyme (10 nM) with increasing concentrations of inhibitor against the indicator substrate, NCF. Care was taken to initiate the reaction with the addition of both NCF and the inhibitor and to limit the initial velocity determination to the first 5 s.
Ki values for the BATSIs were determined as described above. However, because of an incubation effect, SHV β-lactamase and the chiral boronic acid compound were preincubated for 5 min in PBS before initiating the reaction with the addition of substrate (NCF), as has been described previously (9, 13).
The first-order rate constant for enzyme and inhibitor complex inactivation, kinact, was obtained by monitoring the reaction time courses in the presence of inhibitor. Fixed concentrations of enzyme (10 nM) and NCF (150 μM) and increasing concentrations of inhibitor were used in each assay. The kobs was determined using a nonlinear least-squares fit of the data using Origin 7.5:
| (2) |
Here, A is absorbance, v0 (expressed in variation of absorbance per unit time) is initial velocity, vf is final velocity, and t is time. Each kobs was plotted versus I and fit to determine kinact. The value of kinact was used to determine KI (equivalent to Km) according to the following equation using Origin 7.5:
| (3) |
Both the Ki and KI data were corrected to account for the affinity of NCF for SHV-1 and Asn276Asp according to the following equations (30):
| (4a) |
| (4b) |
The partitioning of the initial enzyme inhibitor complex between hydrolysis and inactivation, that is, the partition ratio (kcat/kinact or tn), was calculated by plotting the relative β-lactamase activity against the inhibitor/enzyme ratio as previously described by Bush et al. (31). Briefly, increasing amounts of inhibitor were incubated with a fixed concentration of β-lactamase (10 nM). After 24 h, an aliquot was removed from the mixture and the initial velocity was measured and compared with a control sample with no inhibitor added. The proportion of inhibitor to β-lactamase that resulted in 90% inactivation after 24 h was defined here as the partition ratio.
We calculated the difference in binding energies between SHV-1 and Asn276Asp β-lactamases and the substrates and inhibitors (32, 33). For the substrates, the interaction energies of the acyl-enzyme of SHV-1 relative to Asn276Asp (ΔΔG) were determined from the following equation where R is the universal gas constant and T is in degrees Kelvin (33):
| (5) |
For the inhibitors, Ki values were used to obtain the differential energies needed to form the preacylation complex with SHV-1 or Asn276Asp.
Mass Spectrometry
For intact protein mass spectrometry to determine intermediates and products of inactivation, we incubated 40 μM of SHV-1 or Asn276Asp with and without addition of 40 mM clavulanate for 15 min. Each reaction was terminated by the addition of 0.1% trifluoroacetic acid and immediately desalted and concentrated using a C18 ZipTip (Millipore, Bedford, MA) according to the manufacturer’s protocol. Samples were then placed on ice and analyzed within 1 h.
Spectra of the intact SHV-1 and Asn276Asp proteins were generated on an Q-STAR XL quadrupole-time-of-flight mass spectrometer (Applied Biosystems, Framingham, MA) equipped with a nanospray source. Experiments were performed by diluting the protein sample with 50% acetonitrile/0.1% trifluoroacetic acid to a concentration of 10 μM. This protein solution was then infused at a rate of 0.5 μL/min and the data were collected for 2 min. Spectra were deconvoluted using the Analyst program (Applied Biosystems). All measurements have an error of ±3 Da.
Molecular Modeling
The crystal structure coordinates of the SHV-1 β-lactamase from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB entry 1SHV) were used to generate a representation of SHV Asn276Asp β-lactamase inactivated by clavulanate. Hydrogen atoms were added (pH = 7.5), and the structure minimized at a constant dielectric of 1 with the conjugate gradient method and a Constant Valence Force Field (Accelrys Insight II, Discover_3, San Diego, CA). The energy minimization of the PDB file was followed by an Asn276Asp substitution. The model for clavulanate was constructed using the Builder Module of Insight II. In order to represent the acyl-enzyme intermediate, a bond was created between C7 of clavulanate and Ser70. The enzyme–inhibitor complex was energy minimized to a convergence of 0.02Å (10 000 iterations).
RESULTS
Construction of Variants at Ambler Position 276; Mutagenesis and Immunoblotting
Using degenerate oligonucleotides, we performed site-saturation mutagenesis on the blaSHV-1 gene at Ambler position 276. Seventeen of 19 amino acid substitutions were obtained in the initial sequencing screen (100 blaSHV genes selected and sequenced). blaSHV Asn276Phe and blaSHV Asn276Thr were constructed by site-directed mutagenesis. Expression of β-lactamase proteins was confirmed by immunoblotting. With the possible exception of the SHV Asn276Pro variant, all enzymes were expressed at steady state (0.8 OD) (Supporting Information, Figure 1).
Antimicrobial Susceptibility Tests
To test the impact of the single amino acid substitutions at Asn276 on β-lactam susceptibility, MICs against all 19 variants were determined in a uniform E. coli DH10B background. Results are summarized in Table 1.
Table 1.
MIC values (μg/mL) of E. coli DH10B Expressing SHV-1 and Asn276 Variantsa
| ampicillin | piperacillin | cephalothin | ampicillin/clavulanate | ampicillin/sulbactam | ampicillin/tazobactam | |
|---|---|---|---|---|---|---|
| DH10B | 1 | 1 | 4 | 0.06 | 0.06 | 0.06 |
| SHV-1 | > 16384 | 2048 | 64–128 | 2 | 512 | 64 |
| Asn276Asp | 8192 | 512 | 8–16 | 8 | 256 | 64 |
| Asn276Gly | 4096 | 256 | 16 | 2 | 64 | 16 |
| Asn276Ser | 4096 | 512 | 16 | 1 | 32–64 | 16 |
| Asn276Val | 2048 | 512 | 16 | 0.5 | 256 | 32 |
| Asn276Thr | 1024–2048 | 128–256 | 8–16 | 1 | 32 | 8 |
| Asn276Gln, -Glu, -Cys | 1024–2048 | 64–128 | 8–16 | 0.5 | 64–128 | 8 |
| Asn276His | 1024 | 256 | 16 | 1 | 128 | 16 |
| Asn276Ala, -Leu, -Ile | 256–512 | 16–32 | 4–8 | 0.25–0.5 | 8–16 | 2 |
| Asn276Lys, -Arg, -Tyr, -Phe | 64 | 8–16 | 4-8 | 0.125–0.25 | 1–4 | 0.5–1 |
| Asn276Met | 32 | 4 | 4 | 0.06 | 0.06 | 0.06 |
| Asn276Pro, -Trp | 8–16 | 2–4 | 4–8 | 0.06 | 0.06 | 0.06 |
Inhibitors were evaluated in the presence of 50 μg/mL ampicillin.
All Asn276 variants possessed lower MICs for the penicillins and cephalothin as compared to SHV-1. The ampicillin and piperacillin MICs (8192 and 512 μg/mL, respectively) were relatively preserved for the Asn276Asp variant compared to the remaining Asn276 variants, suggesting that the Asn276Asp variant retains greater ability to hydrolyze penicillins. It is particularly noteworthy that there is a very wide range of MICs (2–8192 μg/mL) and that the pattern seen is not based upon size of the R group, charge, polarity, or hydrophobicity. Each of the variants at Asn276 demonstrated reduced susceptibility to cephalothin (range 4–16 μg/mL). MICs against the extended-spectrum cephalosporin cefotaxime and meropenem were also performed and all Asn276 variants had values comparable to SHV-1 (Supporting Information, Table 1).
With regard to β-lactam/β-lactamase inhibitor combinations, only the Asn276Asp enzyme resulted in a higher MIC value for the ampicillin/inhibitor combinations: increased resistance to ampicillin/clavulanate and a value comparable to SHV-1 for ampicillin/sulbactam and ampicillin/tazobactam.
Kinetic Behavior of Asn276Asp with β-Lactam Substrates
Because of its phenotype and potential clinical relevance, we purified the Asn276Asp variant enzyme for kinetic analysis. Except for ampicillin, we observed that the Asn276Asp variant has a higher Km against all substrates (Table 2). In contrast, the kcat value is lowered save for piperacillin, which has a value similar to SHV-1. Overall, the catalytic efficiencies, kcat/Km, were slightly reduced for the Asn276Asp variant.
Table 2.
Kinetic Properties of SHV-1 and Asn276Asp for Ampicillin, Piperacillin, Nitrocefin, and Cephalothin
| SHV-1 | Asn276Asp | |
|---|---|---|
| Ampicillin | ||
| Km(μM) | 183 ± 34 | 134 ± 13 |
| kcat (s−1) | 3100 ± 300 | 989 ± 99 |
| kcat/Km (μM−1 s−1) | 17 ± 4 | 7 ± 1 |
| Piperacillin | ||
| Km(μM) | 77 ± 7 | 105 ± 18 |
| kcat (s−1) | 898 ± 90 | 976 ± 98 |
| kcat/Km (μM−1 s−1) | 12 ± 1 | 9 ± 1 |
| Nitrocefin | ||
| Km(μM) | 21 ± 3 | 28 ± 3 |
| kcat (s−1) | 237 ± 24 | 137 ± 14 |
| kcat/KM (μM−1 s−1) | 11 ± 2 | 5 ± 1 |
| Cephalothin | ||
| Km(μM) | 29 ± 1 | 83 ± 11 |
| kcat (s−1) | 14 ± 1 | 10 ± 1 |
| kcat/Km (μM−1 s−1) | 0.47 ± 0.03 | 0.12 ± 0.02 |
Kinetic Behavior of Asn276Asp with Clavulanate
Table 3 summarizes the kinetic data for SHV-1 and the Asn276Asp variant inactivated by clavulanate. Both the clavulanate Ki and KI values are increased for Asn276Asp as compared to SHV-1 (5.4- and 1.6-fold, respectively). Unlike studies of SHV Arg244Ser, we did not find a difference in clavulanate kinact for Asn276Asp and SHV-1 (13). We measured the partition ratio for both SHV-1 and Asn276Asp and observed only a slight increase in the number of clavulanate molecules hydrolyzed by the variant enzyme before inactivation was achieved (40 vs 50). Similarly, the kcat calculations indicate a small increase in the rate of inhibitor hydrolysis for the Asn276Asp variant.
Table 3.
Kinetic Properties of SHV-1 and Asn276Asp for Clavulanate, Cephalothin BATSIs, Methylidene Penem, and Meropenem
| SHV-1 | Asn276Asp | |
|---|---|---|
| Clavulanate | ||
| Ki(μM) | 1.37 ± 0.23 | 7.44 ± 0.84 |
| KI(μM) | 0.72 ± 0.11 | 1.12 ± 0.11 |
| kinact (s−1) | 0.015 ± 0.002 | 0.015 ± 0.002 |
| kinact/KI (μM−1 s−1) | 0.021 ± 0.004 | 0.013 ± 0.002 |
| kcat/kinact (tn) | 40 ± 10 | 50 ± 10 |
| kcat (s−1) | 0.60 ± 0.17 | 0.75 ± 0.18 |
| Achiral Cephalothin BATSI | ||
| Ki(μM) | 42 ± 4 | 36 ± 4 |
| Chiral Cephalothin BATSI | ||
| Ki(μM) | 0.68 ± 0.07 | 3.8 ± 0.4 |
| Methylidene Penem | ||
| Ki(μM) | 0.057 ± 0.006 | 0.102 ± 0.001 |
| Ki(μM) | 0.026 ± 0.004 | 0.039 ± 0.002 |
| kinact (s−1) | 0.17 ± 0.01 | 0.24 ± 0.02 |
| kinact/KI (μM−1 s−1) | 6.5 ± 1.0 | 6.2 ± 0.6 |
| kcat/kinact (tn) | 2 ± 1 | 2 ± 1 |
| kcat (s−1) | 0.34 ± 0.17 | 0.48 ± 0.24 |
| Meropenem | ||
| Ki(μM) | 31 ± 3 | 1411 ± 14 |
Kinetic Behavior of Asn276Asp with β-Lactamase Inhibitors: Probes of the Active Site
In order to investigate the functional interactions responsible for the SHV Asn276Asp IR phenotype, we assayed two BATSIs, an investigational methylidene penem, and meropenem for their activities against SHV-1 and Asn276Asp enzymes (Table 3). The BATSIs tested contain the R1 side chain of cephalothin and differ only by the addition of a meta-carboxyphenyl group on the chiral compound. The meta-carboxyphenyl moiety contains a carboxylate that is linked to the sp2-hybridized carbon atom of a phenyl ring, and is intended to imitate the interactions with the C3/C4 carboxylate of penicillins and cephalosporins. These boronic acid compounds allow us to assess the extent to which the Asn276Asp substitution contributes to affinity for the substrate and/or inhibitor carboxylate by its interaction with Arg244 (13).
The chiral BATSI with the meta-carboxyphenyl group demonstrated 62-fold greater affinity for SHV-1 than the achiral compound (42 μM vs 0.68 μM, respectively), suggesting that interactions with the C3/C4 carboxylate are very important for increasing affinity for the enzyme’s active site. Testing the BATSIs with Asn276Asp also showed that the presence of the meta-carboxyphenyl on the chiral compound increased affinity, but the improvement in Ki of binding the chiral compound over the achiral was reduced to only 9-fold (36 μM vs 3.8 μM, respectively).
We next studied a methylidene penem that contains an sp2-hybridized C3 bearing the carboxylate and a bicyclic R1 side chain. The Ki and KI values of the penem for SHV-1 and SHV Asn276Asp were in the nM range, with modest increases in both constants for the Asn276Asp variant (1.7- and 1.5-fold for Ki and KI, respectively). The kinact rate was also slightly higher for the Asn276Asp variant, but the overall inhibitor efficiency (kinact/KI) and partition ratios were very similar between SHV-1 and Asn276Asp enzymes.
Meropenem resists hydrolysis by SHV β-lactamases and forms a stable acyl-enzyme (34). Here, the affinity of meropenem for Asn276Asp was assessed by a competition reaction with NCF to obtain the dissociation constant, Ki. The affinity loss of the Asn276Asp enzyme for the carbapenem was greatest, a 46-fold increased Ki as compared to SHV-1.
In Table 4, we summarize our calculations of differences in binding energies between SHV-1 and Asn276Asp (see eq 5). With the exceptions of ampicillin and the achiral cephalothin BATSI, where energy differences are very small, the Asn276Asp enzyme required more energy to bind substrates and inhibitors than did SHV-1.
Table 4.
Differences in Substrate and Inhibitor Binding Energies for SHV-1 and Asn276Asp
| ΔΔG (kcal/mol) | |
|---|---|
| β-Lactam Substrates | |
| ampicillin | −0.18 |
| piperacillin | + 0.18 |
| cephalothin | + 0.62 |
| Inhibitors | |
| clavulanate | + 1.00 |
| achiral cephalothin BATSI | −0.09 |
| chiral cephalothin BATSI | + 1.02 |
| methylidene penem | + 0.34 |
| meropenem | + 2.26 |
Determining the Nature of the Intermediates: ESI-MS of SHV-1 and Asn276Asp with Clavulanate
Timed ESI-MS was performed with SHV-1 and Asn276Asp β-lactamase inactivated by clavulanate using an inhibitor/enzyme ratio of 1000:1 to ensure detection of the covalent intermediates in the inactivation pathway. As shown in Figure 2, when SHV-1 and Asn276Asp were incubated with clavulanate for 15 min, nearly identical covalent intermediates were formed. This included adducts at Δ +51, Δ +70, Δ +88, Δ +140, and Δ +158 which were observed previously with TEM-2, SHV-1, and the IR SHV Arg244Ser (all measurements have an error of ± 3 Da, see Supporting Information, Figure 2, for proposed reaction intermediates corresponding to adducts) (13, 35, 36). These similarities in the spectra of inactivation species argue strongly for a common pathway and reaction mechanism.
FIGURE 2.

Deconvoluted ESI-MS spectra of (A) SHV-1 and (B) SHV Asn276Asp before and after 15 min inactivation with clavulanate at a inhibitor/enzyme ratio of 1000:1. All measurements have an error of ± 3 Da. Eight distinct mass shifts were identified with both enzymes inactivated with clavulanate. This includes adducts postulated to represent: Δ + 51, terminally inactivated cross-linked or propynoyl enzyme species; Δ + 70, aldehyde; Δ + 88, hydrated aldehyde; Δ + 158, decarboxylated imine; and Δ + 198, acyl-enzyme, imine, cis- or trans-enamine (see Supporting Information, Figure 2 for structures of proposed reaction intermediates) (13, 35).
DISCUSSION
We undertook an in-depth study of Ambler position 276 in SHV to enhance our understanding of the IR phenotype in class A β-lactamase enzymes, and to explore potentially relevant differences between IR SHVs and IRTs. Our goal was to elucidate the role of this second-shell residue on the catalytic profile of SHV enzymes.
The Impact of Substitutions at Ambler Position Asn276 on Phenotype
Susceptibility testing of the full repertoire of SHV Asn276 variants showed that only the Asn276Asp enzyme was uniquely able to reduce susceptibility to ampicillin/clavulanate. Furthermore, this variant displayed another distinct feature that contrasts what is typically observed with IR enzymes: the Asn276Asp variant retained a high level of resistance to ampicillin and piperacillin (Table 1). It is noteworthy that all the other variants at position 276 were uniformly less resistant to ampicillin/sulbactam and ampicillin/tazobactam than SHV-1 expressed in E. coli DH10B. This pattern was observed by Thomson et al. in studies exploring the role of Arg244 in SHV (5, 13). On the basis of the Asn276Asp enzyme’s ability to balance these “desirable” catalytic properties (i.e., avoiding inhibition by clavulanate and retaining hydrolytic activity for penicillins) and its potential clinical relevance, we selected Asn276Asp for detailed study.
Steady-State Kinetics
Steady-state kinetic analyses were performed to explain the biochemical correlates of the Asn276Asp phenotype for substrate and inhibitor catalysis. Overall, we found that the Asn276Asp β-lactamase is moderately impaired when compared to SHV-1 (kcat/Km ratios are 41–75% of WT) (Table 2). Of the parameters measured, the Km and catalytic efficiency (kcat/Km) for the penicillins were least affected by the 276 substitution. NCF and cephalothin demonstrated slightly greater reductions in affinities (higher Km) and turnover (lower kcat). The most striking difference compared to SHV-1 was that Asn276Asp showed a 46-fold change in Ki for meropenem. Based upon previous analysis and work in SHV performed by Nukaga et al. and Thomson et al., we attribute this pattern (loss of penicillin > cephalothin > meropenem affinities) to the interaction of the Asn276Asp β-lactamase with the C3/C4 carboxylate of these substrates (13, 34). Carboxylates linked to C3 or C4 sp2-hybridized carbons (meropenem and cephalothin, respectively) have reduced rotational freedom due to conjugation as compared to carboxylates linked to a C3 sp3-hybridized carbon (i.e., penicillins). Thomson et al. suggested that the carboxylate linked to a C3/C4 sp2-configured carbon is brought in closer approximation to Arg244 of SHV in the Henri-Michaelis complex, likely leading to increased hydrogen bonds with this residue (13). This contrasts with the notion that Arg244 may make only one relatively weak hydrogen bond with the penicillin carboxylate linked to a C3 sp3-hybridized carbon; the anchoring interactions come from other residues in the binding pocket, such as Thr235 (20). Thus, by examining substrates with different stereochemistry at the C3/C4 position, we infer that the substitutions at residue 276 in SHV affect the affinity and kinetics of how Arg244 binds to substrates.
Resistance to clavulanate in SHV Asn276Asp is mediated by decreased affinity. This is a property shared with the IR TEM Asn276Asp enzyme. However, the magnitude of affinity loss is less for the SHV variant than the TEM variant (18, 19). The inactivation efficiencies (kinact/KI) reveal that SHV Asn276Asp retains more susceptibility to clavulanate (19).
Molecular Modeling
On the basis of phenotypic and kinetic study of SHV Asn276Asp, we created a molecular model to complement our observations. Our model reveals that new electrostatic interactions are created by the Asn276Asp substitution in SHV (Figure 3). In SHV Asn276Asp, the distances between atoms Nη2 of Arg244 and both Oδ1 and Oδ2 of 276Asp are decreased as compared to SHV-1, by 0.9 and 0.6 Å, respectively. These observations are reminiscent of the distances observed in the atomic structure of TEM Asn276Asp (Nη2 of Arg244 and Oδ2 of 276Asp are closer by 1.0Å) (19, 24). Yet, why does Asn276Asp maintain catalytic efficiency against penicillins, demonstrate resistance to inactivation by clavulanate, and show such wide changes in Ki when probes are tested? Why are all the other 276 substitutions so ineffective? We posit that the Asp substitution is singular in its ability to create a more “rigid enzyme” by impairing the conformational flexibility of Arg244. Close inspection of the Nε atom of Arg244 in the crystal structures of TEM-1 and TEM Asn276Asp (PDB entries 1BTL and 1CK3, respectively) reveals that this residue exists in multiple conformations. The two new interactions between Arg244 and 276Asp in SHV Asn276Asp restrict facile hydrogen bond formation of the Arg244 guanidinium group with the C3 carboxylate of clavulanate. As Arg244 plays a key role in substrate recognition, we would anticipate that major effects would be seen for substrates as well. Our MIC data show that 18 of the 19 substitutions at Asn276 result in a less robust penicillinase as a result of this rigidity (the other residues at 276 do not permit productive interactions with Arg244). Most importantly, we also see resistance to clavulanate. This latter property is most likely a consequence or benefit of restricted flexibility. This argument is supported by the increased Km, Ki, and ΔG values and changes in MICs.
FIGURE 3.
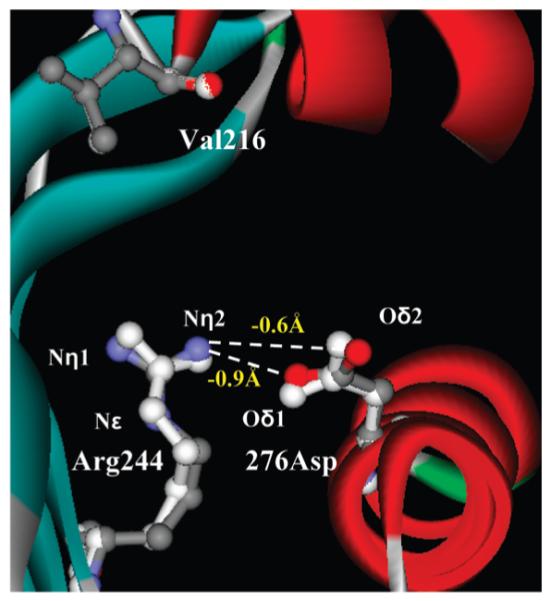
Molecular representation of SHV Asn276Asp based on SHV-1 (PDB entry 1SHV) showing the increased interaction between Arg244 and 276Asp mediated by shortened distances between Arg244 Nη2 and both 276Asp Oδ1 and Oδ2 (decreased by 0.9 and 0.6Å, respectively). Residue 216 is shown for reference. SHV-1 amino acid positions in white and SHV Asn276Asp model in color.
To explore further the functional interactions of the Asn276Asp substitution, we next turned to inhibitors (BATSIs, a methylidene penem, and meropenem) with carboxylates linked to sp2-hybridized atoms. The inhibitors were chosen because they help us answer (i) the extent to which the position and stereochemistry of the carboxylate impacts affinity for the Asn276Asp variant and (ii) if other interactions besides Arg244-Asn276Asp are important for inhibitor binding.
BATSIs were selected because they replace the β-lactam motif with boronic acid and form reversible, dative covalent bonds with the active site serine (37, 38). This leads to formation of an adduct which resembles the geometry of the tetrahedral transition state of the β-lactamase hydrolytic reaction. By modifying these compounds to contain the R1 side chains of natural substrates at the same distance from the electrophilic moiety, affinities in the nanomolar range for both class A TEM-1 and extended-spectrum-type SHV and class C enzymes were reported (39, 40). Here, we tested one achiral BATSI bearing the R1 amide side chain found in cephalothin and a second chiral BATSI that has an additional meta-carboxyphenyl ring on the carbon atom alpha to the boron which resembles the dihydrothiazine ring of cephalosporins. Since the geometry and the distances in the chiral BATSI are the same as in the natural substrate, the carboxylate moiety of the meta-carboxyphenyl group corresponds to the C3/C4 carboxylate found on all β-lactams. Thus, comparing affinity of the achiral and chiral compounds allowed us to probe selectively how the Asn276Asp substitution affects binding of the carboxylate of cephalosporins.
The affinity gain of the carboxylate (i.e., the difference in Ki between the achiral and chiral compounds) for the SHV-1 enzyme was almost seven times increased for the Asn276Asp variant. Because of the selective design of these inhibitors, we attribute this difference to a less favorable interaction between Arg244 and the inhibitor carboxylate in the Asn276Asp variant. Quantitatively, the meta-carboxyphenyl group contributes 1.0 kcal/mol less energy to the affinity of the enzyme interaction in the Asn276Asp variant as compared to SHV-1 (Table 4). In total, these results support our hypothesis that residue 276, despite not making direct bonds with the substrate, plays a significant role in coordinating the carboxylate in the substrate-enzyme complex.
Methylidene penems and their derivatives are effective inactivators of classes A, C, and D β-lactamases, including IR class A enzymes (5, 41–43). Assays of one of these novel penems yielded similar inhibitor efficiencies for SHV-1 and Asn276Asp. As proposed in studies with the SHV Arg244Ser variant, the affinity losses at the C3 carboxylate of the penem may be compensated by favorable interactions with the inhibitor’s bicyclic R1 side chain (5). Previous molecular models and structure determinations have shown that the R1 side chain may be involved in π–π interactions with Tyr105 (42). Despite the sp2-hybridized carboxylate, the preserved efficiency of this penem against the clavulanate-resistant Asn276Asp variant stresses that more than one significant (compensatory) interaction is possible in the active site (43).
In contrast to stabilizing interactions of the R1 side chain of methylidene penem inhibitors, carbapenems contain a hydroxyethyl group at the R1 position and a large R2 side chain, both of which have destabilizing effects in crystal structures of carbapenem/class A β-lactamase complexes (34, 44, 45). Thus, that SHV Asn276Asp experiences the greatest affinity loss for meropenem, as compared to all other substrates and inhibitors tested, is consistent with the notion that the interactions between the variant enzyme and the ligand carboxylate are impaired. Meropenem, with its limited flexibility and rigid carboxylate on the sp2 carbon atom, requires an additional 2.3 kcal/mol for formation of the preacylation complex with Asn276Asp vs SHV-1 (Table 4). Previous studies of the SHV Arg244Ser variant revealed a cost of 3.6 kcal/mol for the same process; perturbations of the inherent flexibility of residue 244 impair carboxylate recognition (13).
Comparison to IR SHV and TEM Enzymes
Can we compare IR SHV enzymes with IRTs and what do we learn from this? Examined together, our analysis of substrate and inhibitor catalysis in SHV-1 and SHV Asn276Asp represents a “fine tuning” of the IR phenotype. In general, IR enzymes must establish a balance between retaining the ability to hydrolyze β-lactam substrates, but decreasing efficacy of the structurally related β-lactam inhibitors (4). The majority of the IR enzymes “accept” a significant decrease in substrate hydrolytic efficiency in exchange for dramatically decreased inhibitory efficiency. This produces an IR phenotype when organisms expressing these enzymes are tested against β-lactam/β-lactamase inhibitor combinations. MICs represent the “sum” of an enzyme’s ability to hydrolyze the combination of both β-lactam (e.g., ampicillin) and the β-lactamase inhibitor (e.g., clavulanate) in a relatively short time frame. Our data shows that in the case of SHV Asn276Asp, the increased ampicillin/clavulanate resistance results from a small decrease in clavulanate inhibition, and is also due to the preserved ability of the enzyme to hydrolyze the ampicillin present in the β-lactam/β-lactamase inhibitor combination. We further illustrate these unique properties of SHV Asn276Asp by comparison with the kinetic data of a collection of IR SHV and IRT variants; first, the relatively high catalytic efficiency for ampicillin; and second, the slightly decreased inhibitory efficiency of clavulanate (see Supporting Information, Tables 2 and 3, for substrate and inhibitor efficiencies of a panel of IR SHV and IRT enzymes). The Asn276Asp alteration of the IR phenotype in the SHV β-lactamase may represent a significant evolutionary advance, as the enzyme maintains a balance of the desired catalytic properties.
For clavulanate-resistant TEM enzymes, substitutions at Asn276 and Arg244 lead to decreased occupancy of a water molecule essential for inactivation by clavulanate (18–20, 46). To support our model for SHV, we propose that the alteration imposed by the Asn276Asp substitution may not affect the (yet to be identified) water molecule crucial to clavulanate inactivation in SHV (Figure 4). Previous examination of SHV and TEM crystal structures suggests that SHV enzymes may not rely on the water molecule coordinated by Arg244 and Val216 for inactivation by clavulanate (13, 17, 24). Consequently, the attenuated IR SHV Asn276Asp phenotype is produced by decreased affinity for the carboxylate of the inhibitors, and not by disruption of the inactivation mechanism from a dislocated water molecule. These important differences in TEM and SHV mechanisms support a divergent evolution of these enzymes viz a viz substrate and inhibitor profiles.
FIGURE 4.
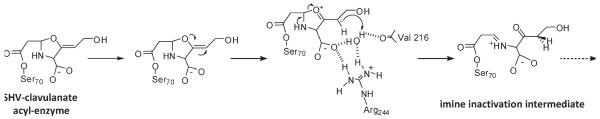
Proposed reaction mechanism for SHV-1 and clavulanate showing the formation of the imine inactivation intermediate. Scheme shows the contribution of Arg244 in the coordination of the water molecule hypothesized to donate a proton for C2 double bond saturation and subsequent secondary ring-opening. In SHV-1, it is postulated that the water molecule is recruited or relocated into the active site with the binding of clavulanate (13). The crystal structure of TEM Asn276Asp (PDB entry 1CK3) reveals that this crucial water is missing, offering an explanation for the IR phenotype as protonation of clavulanate would be impaired, and thus lead to more ready hydrolysis of the inhibitor than in TEM-1, where the water is clearly refined (19).
Thus, we posit that the SHV Asn276Asp variant may exist in clinical isolates of Enterobacteriaceae. However, as a result of the attenuated IR phenotype, this variant has not risen to clinical attention. This is in contrast to the TEM Asn276Asp enzyme, which exhibits a more significant decrease in catalytic activity, but more pronounced IR. In general, the prevalence of IR β-lactamases may be underestimated as identification of IR enzymes requires specific kinetic characterization and susceptibility testing (4, 47). Recognition of existing IR SHV variants, particularly those with a less pronounced phenotype, may have escaped detection.
Conclusion
In summary, we highlight the important role of a second-shell residue in class A β-lactamases and describe the unique properties of Ambler residue Asn276 in SHV. Structure–function studies of proteins often focus on first-shell residues that make direct interactions with active-site ligands. Recent studies of metallo-enzymes, including metallo-β-lactamases, have emphasized that second-shell residues can contribute to metal binding affinities and active site charge distributions (48, 49). Additional investigations show that second- and third-shell residues in nitric oxide synthase enzymes modulate important conformational changes in invariant first-shell residues (50). In these proteins, new binding pockets are revealed upon ligand binding, and selective inhibitors can be rationally designed to take advantage of how the second-shell residues modulate enzyme flexibility. Our data demonstrate that perturbations of the β-lactamase second-shell residue 276 are transferred to the active site by interaction with Arg244 and manifested in decreased affinity for piperacillin, NCF, cephalothin, and clavulanate. This new interpretation and deeper understanding for the tertiary structure and interactions between residues are essential to the success of selective and effective inhibitors of these β-lactamase enzymes. The lessons learned from the “finely tuned” enzyme can focus our efforts to meet the challenges raised by the impressive efficiency of these β-lactamases.
Supplementary Material
ACKNOWLEDGMENT
Special thanks to Ms. Magda Taracila for assistance with molecular modeling, Ms. Andrea Hujer and Dr. Andrea Endimiani for critical review of this manuscript, Dr. Vernon Anderson for guidance on kinetic analyses, and Wyeth Pharmaceuticals for their kind contribution of the penem inhibitor.
Footnotes
SUPPORTING INFORMATION AVAILABLE MIC table of SHV-1 and Asn276 variants for cefotaxime and meropenem, tables of substrate and inhibitor efficiencies for a panel of IR SHV and TEM enzymes, immunoblot of Asn276 variants probed with SHV-1 polyclonal antibody, and proposed reaction intermediates observed in ESI-MS spectra of SHV-1 and SHV Asn276Asp inactivated with clavulanate are available free of charge via the Internet at http://pubs.acs.org.
Financial support for this work was provided by the Veterans Affairs Merit Review Program and the National Institutes of Health (1R01 A1063517-01). R.A.B. is also supported by the Veterans Integrated Service Network 10 Geriatric Research, Education, and Clinical Center. S.M.D. was supported in part by NIH Grants T32 GM07250, T32 HL083823, and the Case Medical Scientist Training Program. F.P. and E.C. gratefully thank Fondazione Cassa di Risparmio di Modena for financial support.
Abbreviations: TEM, class A β-lactamase named after the patient (Temoneira) from whom the first β-lactamase was isolated; SHV, class A β-lactamase named after the property sulfhydryl reagent variable; IR, inhibitor resistant; MIC, minimum inhibitory concentration; BATSI, boronic acid transition state inhibitor; IRT, inhibitor-resistant TEM; ESI-MS, electrospray ionization mass spectrometry; WT, wild-type; NCF, nitrocefin; PBS, phosphate-buffered saline; PDB, Protein Data Bank.
REFERENCES
- 1.Paterson DL, Bonomo RA. Extended-spectrum β-lactamases: a clinical update. Clin. Microbiol. Rev. 2005;18:657–686. doi: 10.1128/CMR.18.4.657-686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helfand MS, Bonomo RA. Beta-lactamases: a survey of protein diversity. Curr. Drug Targets Infect. Disord. 2003;3:9–23. doi: 10.2174/1568005033342181. [DOI] [PubMed] [Google Scholar]
- 3.Bush K. β-Lactamase inhibitors from laboratory to clinic. Clin. Microbiol. Rev. 1988;1:109–123. doi: 10.1128/cmr.1.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buynak JD. Understanding the longevity of the β-lactam antibiotics and of antibiotic/β-lactamase inhibitor combinations. Biochem. Pharmacol. 2006;71:930–940. doi: 10.1016/j.bcp.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 5.Thomson JM, Distler AM, Bonomo RA. Overcoming resistance to β-lactamase inhibitors: comparing sulbactam to novel inhibitors against clavulanate resistant SHV enzymes with substitutions at Ambler position 244. Biochemistry. 2007;46:11361–11368. doi: 10.1021/bi700792a. [DOI] [PubMed] [Google Scholar]
- 6.Weiss WJ, Petersen PJ, Murphy TM, Tardio L, Yang Y, Bradford PA, Venkatesan AM, Abe T, Isoda T, Mihira A, Ushirogochi H, Takasake T, Projan S, O’Connell J, Mansour TS. In vitro and in vivo activities of novel 6-methylidene penems as β-lactamase inhibitors. Antimicrob. Agents Chemother. 2004;48:4589–4596. doi: 10.1128/AAC.48.12.4589-4596.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helfand MS, Bonomo RA. Current challenges in antimicrobial chemotherapy: the impact of extended-spectrum β-lactamases and metallo-β-lactamases on the treatment of resistant Gram-negative pathogens. Curr. Opin. Pharmacol. 2005;5:452–458. doi: 10.1016/j.coph.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 8.Buynak JD. The discovery and development of modified penicillin- and cephalosporin-derived β-lactamase inhibitors. Curr. Med. Chem. 2004;11:1951–1964. doi: 10.2174/0929867043364847. [DOI] [PubMed] [Google Scholar]
- 9.Morandi F, Caselli E, Morandi S, Focia PJ, Blazquez J, Shoichet BK, Prati F. Nanomolar inhibitors of AmpC β-lactamase. J. Am. Chem. Soc. 2003;125:685–695. doi: 10.1021/ja0288338. [DOI] [PubMed] [Google Scholar]
- 10.Thomas VL, Golemi-Kotra D, Kim C, Vakulenko SB, Mobashery S, Shoichet BK. Structural consequences of the inhibitor-resistant Ser130Gly substitution in TEM β-lactamase. Biochemistry. 2005;44:9330–9338. doi: 10.1021/bi0502700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Totir MA, Padayatti PS, Helfand MS, Carey MP, Bonomo RA, Carey PR, van den Akker F. Effect of the inhibitor-resistant M69V substitution on the structures and populations of trans-enamine β-lactamase intermediates. Biochemistry. 2006;45:11895–11904. doi: 10.1021/bi060990m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delaire M, Labia R, Samama JP, Masson JM. Site-directed mutagenesis at the active site of Escherichia coli TEM-1 β-lactamase. Suicide inhibitor-resistant mutants reveal the role of arginine 244 and methionine 69 in catalysis. J. Biol. Chem. 1992;267:20600–20606. [PubMed] [Google Scholar]
- 13.Thomson JM, Distler AM, Prati F, Bonomo RA. Probing active site chemistry in SHV β-lactamase variants at Ambler position 244. Understanding unique properties of inhibitor resistance. J. Biol. Chem. 2006;281:26734–26744. doi: 10.1074/jbc.M603222200. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Minasov G, Shoichet BK. The structural bases of antibiotic resistance in the clinically derived mutant beta-lactamases TEM-30, TEM-32, and TEM-34. J. Biol. Chem. 2002;277:32149–32156. doi: 10.1074/jbc.M204212200. [DOI] [PubMed] [Google Scholar]
- 15.Ambler RP, Coulson AF, Frere JM, Ghuysen JM, Joris B, Forsman M, Levesque RC, Tiraby G, Waley SG. A standard numbering scheme for the class A β-lactamases. Biochem. J. 1991;276(1):269–270. doi: 10.1042/bj2760269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Rasmussen BA, Shlaes DM. Class A β-lactamases–enzyme-inhibitor interactions and resistance. Pharmacol. Ther. 1999;83:141–151. doi: 10.1016/s0163-7258(99)00027-3. [DOI] [PubMed] [Google Scholar]
- 17.Kuzin AP, Nukaga M, Nukaga Y, Hujer AM, Bonomo RA, Knox JR. Structure of the SHV-1 β-lactamase. Biochemistry. 1999;38:5720–5727. doi: 10.1021/bi990136d. [DOI] [PubMed] [Google Scholar]
- 18.Saves I, Burlet-Schiltz O, Swaren P, Lefevre F, Masson JM, Prome JC, Samama JP. The asparagine to aspartic acid substitution at position 276 of TEM-35 and TEM-36 is involved in the β-lactamase resistance to clavulanic acid. J. Biol. Chem. 1995;270:18240–18245. doi: 10.1074/jbc.270.31.18240. [DOI] [PubMed] [Google Scholar]
- 19.Swaren P, Golemi D, Cabantous S, Bulychev A, Maveyraud L, Mobashery S, Samama JP. X-ray structure of the Asn276Asp variant of the Escherichia coli TEM-1 β-lactamase: direct observation of electrostatic modulation in resistance to inactivation by clavulanic acid. Biochemistry. 1999;38:9570–9576. doi: 10.1021/bi990758z. [DOI] [PubMed] [Google Scholar]
- 20.Imtiaz U, Billings EM, Knox JR, Manavathu EK, Lerner SA, Mobashery S. Inactivation of class A β-lactamases by clavulanic acid: the role of Arginine 244 in a proposed noncerted sequence of events. J. Am. Chem. Soc. 1993;115:4435–4442. [Google Scholar]
- 21.Zafaralla G, Manavathu EK, Lerner SA, Mobashery S. Elucidation of the role of Arginine-244 in the turnover processes of class A β-lactamases. Biochemistry. 1992;31:3847–3852. doi: 10.1021/bi00130a016. [DOI] [PubMed] [Google Scholar]
- 22.Canica MM, Caroff N, Barthelemy M, Labia R, Krishnamoorthy R, Paul G, Dupret JM. Phenotypic study of resistance of β-lactamase-inhibitor-resistant TEM enzymes which differ by naturally occurring variations and by site-directed substitution at Asp276. Antimicrob. Agents Chemother. 1998;42:1323–1328. doi: 10.1128/aac.42.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vakulenko SB, Geryk B, Kotra LP, Mobashery S, Lerner SA. Selection and characterization of β-lactam-β-lactamase inactivator-resistant mutants following PCR mutagenesis of the TEM-1 β-lactamase gene. Antimicrob. Agents Chemother. 1998;42:1542–1548. doi: 10.1128/aac.42.7.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jelsch C, Mourey L, Masson JM, Samama JP. Crystal structure of Escherichia coli TEM1 β-lactamase at 1.8Å resolution. Proteins. 1993;16:364–383. doi: 10.1002/prot.340160406. [DOI] [PubMed] [Google Scholar]
- 25.Hujer AM, Hujer KM, Helfand MS, Anderson VE, Bonomo RA. Amino acid substitutions at Ambler position Gly238 in the SHV-1 β-lactamase: exploring sequence requirements for resistance to penicillins and cephalosporins. Antimicrob. Agents Chemother. 2002;46:3971–3977. doi: 10.1128/AAC.46.12.3971-3977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hujer AM, Bethel CR, Bonomo RA. Antibody mapping of the linear epitopes of CMY-2 and SHV-1 β-lactamases. Antimicrob. Agents Chemother. 2004;48:3980–3988. doi: 10.1128/AAC.48.10.3980-3988.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin S, Thomas M, Shlaes DM, Rudin SD, Knox JR, Anderson V, Bonomo RA. Kinetic analysis of an inhibitor-resistant variant of the OHIO-1 β-lactamase, an SHV-family class A enzyme. Biochem. J. 1998;333(2):395–400. doi: 10.1042/bj3330395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pattanaik P, Bethel CR, Hujer AM, Hujer KM, Distler AM, Taracila M, Anderson VE, Fritsche TR, Jones RN, Pagadala SR, van den Akker F, Buynak JD, Bonomo RA. Strategic design of an effective β-lactamase inhibitor: LN-1–255, A 6-alkylidene-2′-substituted penicillin sulfone. J. Biol. Chem. 2009;284:945–953. doi: 10.1074/jbc.M806833200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venkatesan AM, Agarwal A, Abe T, Ushirogochi H, Yamamura I, Kumagai T, Petersen PJ, Weiss WJ, Lenoy E, Yang Y, Shlaes DM, Ryan JL, Mansour TS. Novel imidazole substituted 6-methylidene-penems as broad-spectrum β-lactamase inhibitors. Bioorg. Med. Chem. 2004;12:5807–5817. doi: 10.1016/j.bmc.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 30.De Meester F, Joris B, Reckinger G, Bellefroid-Bourguignon C, Frere JM, Waley SG. Automated analysis of enzyme inactivation phenomena. Application to β-lactamases and DD-peptidases. Biochem. Pharmacol. 1987;36:2393–2403. doi: 10.1016/0006-2952(87)90609-5. [DOI] [PubMed] [Google Scholar]
- 31.Bush K, Macalintal C, Rasmussen BA, Lee VJ, Yang Y. Kinetic interactions of tazobactam with β-lactamases from all major structural classes. Antimicrob. Agents Chemother. 1993;37:851–858. doi: 10.1128/aac.37.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bethel CR, Hujer AM, Hujer KM, Thomson JM, Ruszczycky MW, Anderson VE, Pusztai-Carey M, Taracila M, Helfand MS, Bonomo RA. Role of Asp104 in the SHV β-lactamase. Antimicrob. Agents Chemother. 2006;50:4124–4131. doi: 10.1128/AAC.00848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fersht AR. Enzyme Structure and Mechanism. 2nd ed W. H. Freeman and Company; New York: 1984. [Google Scholar]
- 34.Nukaga M, Bethel CR, Thomson JM, Hujer AM, Distler A, Anderson VE, Knox JR, Bonomo RA. Inhibition of class A β-lactamases by carbapenems: crystallographic observation of two conformations of Meropenem in SHV-1. J. Am. Chem. Soc. 2008;130:12656–12662. doi: 10.1021/ja7111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown RP, Aplin RT, Schofield CJ. Inhibition of TEM-2 β-lactamase from Escherichia coli by clavulanic acid: observation of intermediates by electrospray ionization mass spectrometry. Biochemistry. 1996;35:12421–12432. doi: 10.1021/bi961044g. [DOI] [PubMed] [Google Scholar]
- 36.Sulton D, Pagan-Rodriguez D, Zhou X, Liu Y, Hujer AM, Bethel CR, Helfand MS, Thomson JM, Anderson VE, Buynak JD, Ng LM, Bonomo RA. Clavulanic acid inactivation of SHV-1 and the inhibitor-resistant S130G SHV-1 β-lactamase. Insights into the mechanism of inhibition. J. Biol. Chem. 2005;280:35528–35536. doi: 10.1074/jbc.M501251200. [DOI] [PubMed] [Google Scholar]
- 37.Chen CC, Rahil J, Pratt RF, Herzberg O. Structure of a phosphonate-inhibited β-lactamase. An analog of the tetrahedral transition state/intermediate of β-lactam hydrolysis. J. Mol. Biol. 1993;234:165–178. doi: 10.1006/jmbi.1993.1571. [DOI] [PubMed] [Google Scholar]
- 38.Crompton IE, Cuthbert BK, Lowe G, Waley SG. β-Lactamase inhibitors. The inhibition of serine β-lactamases by specific boronic acids. Biochem. J. 1988;251:453–459. doi: 10.1042/bj2510453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caselli E, Powers RA, Blasczcak LC, Wu CY, Prati F, Shoichet BK. Energetic, structural, and antimicrobial analyses of β-lactam side chain recognition by β-lactamases. Chem Biol. 2001;8:17–31. doi: 10.1016/s1074-5521(00)00052-1. [DOI] [PubMed] [Google Scholar]
- 40.Thomson JM, Prati F, Bethel CR, Bonomo RA. Use of novel boronic acid transition state inhibitors to probe substrate affinity in SHV-type extended-spectrum β-lactamases. Antimicrob. Agents Chemother. 2007;51:1577–1579. doi: 10.1128/AAC.01293-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venkatesan AM, Agarwal A, Abe T, Ushirogochi H, Yamamura I, Ado M, Tsuyoshi T, Dos Santos O, Gu Y, Sum FW, Li Z, Francisco G, Lin YI, Petersen PJ, Yang Y, Kumagai T, Weiss WJ, Shlaes DM, Knox JR, Mansour TS. Structure-activity relationship of 6-methylidene penems bearing 6,5 bicyclic heterocycles as broad-spectrum β-lactamase inhibitors: evidence for 1,4-thiazepine intermediates with C7 R stereochemistry by computational methods. J. Med. Chem. 2006;49:4623–4637. doi: 10.1021/jm060021p. [DOI] [PubMed] [Google Scholar]
- 42.Nukaga M, Abe T, Venkatesan AM, Mansour TS, Bonomo RA, Knox JR. Inhibition of class A and class C β-lactamases by penems: crystallographic structures of a novel 1,4-thiazepine intermediate. Biochemistry. 2003;42:13152–13159. doi: 10.1021/bi034986b. [DOI] [PubMed] [Google Scholar]
- 43.Bethel CR, Distler AM, Ruszczycky MW, Carey MP, Carey PR, Hujer AM, Taracila M, Helfand MS, Thomson JM, Kalp M, Anderson VE, Leonard DA, Hujer KM, Abe T, Venkatesan AM, Mansour TS, Bonomo RA. Inhibition of OXA-1 β-lactamase by penems. Antimicrob. Agents Chemother. 2008;52:3135–3143. doi: 10.1128/AAC.01677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maveyraud L, Mourey L, Kotra LP, Pedelacq J-D, Guillet V, Mobashery S. Structural basis for clinical longevity of carbapenem antibiotics in the face of challenge by the common class A β-lactamases from the antibiotic-resistant bacteria. J. Am. Chem. Soc. 1998;120:9748–9752. [Google Scholar]
- 45.Wang X, Minasov G, Shoichet BK. Noncovalent interaction energies in covalent complexes: TEM-1 β-lactamase and β-lactams. Proteins. 2002;47:86–96. [PubMed] [Google Scholar]
- 46.Wang X, Minasov G, Shoichet BK. The structural bases of antibiotic resistance in the clinically derived mutant β-lactamases TEM-30, TEM-32, and TEM-34. J. Biol. Chem. 2002;277:32149–32156. doi: 10.1074/jbc.M204212200. [DOI] [PubMed] [Google Scholar]
- 47.Chaibi EB, Farzaneh S, Morand A, Peduzzi J, Barthelemy M, Sirot D, Labia R. Problems encountered in the characterization of IRT β-lactamase-producing clinical Escherichia coli isolates intermediate-resistant to cephalothin. J. Antimicrob. Che-mother. 1996;37:190–191. doi: 10.1093/jac/37.1.190. [DOI] [PubMed] [Google Scholar]
- 48.Ataie NJ, Hoang QQ, Zahniser MP, Tu Y, Milne A, Petsko GA, Ringe D. Zinc coordination geometry and ligand binding affinity: the structural and kinetic analysis of the second-shell serine 228 residue and the methionine 180 residue of the aminopeptidase from Vibrio proteolyticus. Biochemistry. 2008;47:7673–7683. doi: 10.1021/bi702188e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tomatis PE, Rasia RM, Segovia L, Vila AJ. Mimicking natural evolution in metallo-β-lactamases through second-shell ligand mutations. Proc. Natl. Acad. Sci. U. S. A. 2005;102:13761–13766. doi: 10.1073/pnas.0503495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat. Chem. Biol. 2008;4:700–707. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.