

Abstract
STAT3-STAT3 dimerization, which involves reciprocal binding of the STAT3-SH2 domain to phosphorylated tyrosine-705 (Y-705), is required for STAT3 nuclear translocation, DNA binding and transcriptional regulation of downstream target genes. Here we describe a small molecule S3I-1757 capable of disrupting STAT3-STAT3 dimerization, activation and malignant transforming activity. Fluorescence polarization assays and molecular modeling suggest that S3I-1757 interacts with the Y-705 binding site in the SH2 domain and displaces fluorescein-labelled GpYLPQTV phosphotyrosine peptide from binding to STAT3. We generated HA-tagged STAT3 and FLAG-tagged STAT3 and showed using co-immunoprecipitation and co-localization studies that S3I-1757 inhibits STAT3 dimerization and STAT3-EGF receptor binding in intact cells. Treatment of human cancer cells with S3I-1757 (but not a closely related analogue, S3I-1756, that does not inhibit STAT3 dimerization), inhibits selectively the phosphorylation of STAT3 over AKT1 and ERK1/2 (MAPK3/1), nuclear accumulation of P-Y705-STAT3, STAT3-DNA binding and transcriptional activation and suppresses the expression levels of STAT3 target genes such as Bcl-xL (BCL2L1), survivin (BIRC5), cyclin D1 (CCND1) and MMP9. Furthermore, S3I-1757 but not S3I-1756 inhibits anchorage-dependent and -independent growth, migration and invasion of human cancer cells which depend on STAT3. Finally, STAT3-C, a genetically engineered mutant of STAT3 that forms a constitutively dimerized STAT3, rescues cells from the effects of S3I-1757 inhibition. Thus, we have developed S3I-1757 as a STAT3-STAT3 dimerization inhibitor capable of blocking hyper activated STAT3 and suppressing malignant transformation in human cancer cells that depend on STAT3.
Keywords: STAT3, dimerization, tyrosine phosphorylation, migration, invasion
Introduction
The Signal Transducer and Activator of Transcription 3 (STAT3) is an important regulator of many biological processes including proliferation, survival, inflammation and immune responses (1, 2). STAT3 mediates these processes by responding to ligands such as growth factors (i.e. EGF, PDGF) and cytokines (i.e. IL-6, IFN-gamma) which activate STAT3 to translocate to the nucleus and regulate the expression of a number of genes (1, 2). For example, the binding of EGF to its receptor results in tyrosine phosphorylation of the EGF receptor and subsequent recruitment of STAT3 through the binding of the STAT3-SH2 domain to phospho-tyrosines 1068 and 1086 on the receptors (3). Similarly, non-receptor tyrosine kinases such as JAK2 and Src which are part of non-tyrosine kinase receptor complexes (i.e. IL-6 receptor complex) phosphorylate a specific tyrosine on STAT3 which in turn induces STAT3-STAT3 dimerization through two reciprocal phosphotyrosine-SH2 binding interactions (4). The activated STAT3 dimers translocate to the nucleus where they bind to specific DNA sequences on the promoters of the genes regulated by STAT3 (1, 2). Under physiological normal conditions, this STAT3 activation is rapid (within 2 minutes of ligand stimulation) and transient (lost within a few hours due to dephosphorylation). In contrast, STAT3 is found persistently tyrosine phosphorylated and constitutively activated in the majority of cancers including pancreatic, breast, lung, prostrate, ovarian, colon, gastric and head and neck cancers as well as melanoma, leukemia, multiple myeloma and lymphoma (4). Constitutively-activated STAT3 is believed to contribute to malignant transformation at several levels (5). These include uncontrolled proliferation through activation of several cell cycle regulators such as cyclin D1 and c-Myc as well as evasion of apoptosis by inducing the expression of several anti-apoptotic proteins such as Bcl-xL, Bcl-2, Mcl-1 and survivin. STAT3 also activates the expression of proteins involved in other hallmarks of cancer such as invasion and metastasis (i.e. expression of MMPs) and angiogenesis (i.e. expression of VEGF) (2, 4). The fact that STAT3 regulates the expression of a number of genes involved in oncogenesis makes it an attractive and promising target for cancer therapy (6, 7). Validation of STAT3 as a target for cancer drug discovery comes from several lines of evidence. For example, a genetically engineered mutant of STAT3 (STAT3-C) that forms a constitutively dimerized STAT3 through disulfide binds is oncogenic (8). On the other hand, a dominant-negative variant of STAT3, STAT3β, blocks tumor growth by inhibiting STAT3 in tumors where STAT3 is constitutively activated (9, 10). Several approaches have been proposed to suppress constitutive activation of STAT3. These include those inhibiting STAT3 tyrosine phosphorylation (i.e. inhibition of JAK2 or Src), STAT3 recruitment to the receptor and dimerization (i.e. phosphotyrosine peptide mimics that binds the SH2 domain of STAT3), STAT3 nuclear translocation and STAT3-DNA binding and transcriptional activation (6, 7). We have focused our efforts on identifying small molecules capable of disrupting the phosphotyrosine-SH2 binding interactions of STAT3 as potential dimerization inhibitors. Here we report on a novel STAT3-STAT3 dimerization inhibitor that selectively inactivates STAT3 and suppresses STAT3-dependent malignant transformation.
Materials and Methods
Cells and reagents
Human breast cancer (MDA-MB-468, MDA-MB-231, MDA-MB-453), lung cancer (A549, H358, H460) cells, human non-tumorigenic epithelial cells (MCF10A) and human embryonic kidney cells (HEK293) were obtained from ATCC (the American Type Culture Collection, Manassas, VA, USA). These cell lines have not been authenticated. HEK 293 cell lines with stable transfection of HA-STAT3 and FLAG-STAT3 were generated as described below. Cells were grown in Dulbecco's modified Eagle's medium (DMEM), RPMI 1640, and DMEM/F-12 containing 10% heat-inactivated fetal bovine serum. MCF10A was cultured in DMEM/F12, supplemented with 5% horse serum (Invitrogen, CA, USA), hydrocortisone (0.5 μg/ml), mouse epidermal growth factor (EGF; 20 ng/ml), insulin (10 μg/ml), cholera toxin (100 ng/ml, Sigma, MO, USA). Primary antibodies against pY705STAT3, pAKT, AKT, pErk1/2, Erk, MMP9, and Cyclin D1 were purchased from Cell Signaling Technology (Danvers, MA). Primary antibodies against STAT3, Bcl-xL, Survivin, HA (anti-mouse), and HA (anti-rabbit) were purchased from Santa Cruz Biotech (Santa Cruz, CA). Primary antibody against FLAG was purchased from Sigma (St. Louis, MO, USA).
Generation of HA-STAT3 and FLAG-STAT3 constructs and generation of HEK293 cells stably expressing HA-STAT3 and FLAG-STAT3
FLAG-Stat3 plasmid was obtained from Addgene (Cambridge, MA, USA). HA-Stat3 DNA was amplified from FLAG-Stat3 using PCR with HA-pcDNA3 as a vector as described for RhoB by our lab (11). The primers used for PCR were STAT3F-BamH1 CGCGGATCCGCCACCATGGCTCAGTGGAACCAGCTG and STAT3R-EcoR1 CCGGAATTCTCACATGGGGGAGGTAGCACA. The PCR product was digested with BamH1 and EcoR1, and cloned into HA-pcDNA3 vector, further confirmed by sequencing. Ratio (1:1) of pFLAG-STAT3 and pHA-STAT3 plasmid DNAs were co-transfected into HEK-293 cells, and stable G418-resistant (800ug/ml) clones were selected. Transfections were carried out with LipofectAmine Plus (Invitrogen, Carlsbad, CA), according to the manufacturer's protocol.
Nuclear Extract Preparation and STAT3 Filter Plate Assay
Nuclear extract preparation was carried out as previously described (12).The STAT3-DNA binding filter plate assay was performed following the manual of the filter plate assay kit (Signosis, Sunnyvale, CA), as described previously for NFkB (13). The TF Binding buffer was mixed with the STAT3 probe (biotin labeled STAT3 DNA binding sequence) and nuclear extract and incubated at 16°C for 30 minutes to form the STAT3-DNA complex. The STAT3-DNA complex was then separated from free probe by using a filter plate. After several steps of binding and washing, bound STAT3 probe is retained on the filter in the corresponding well of Filer Plate and the free DNA probe is removed. The bound pre-labeled STAT3 probe was then eluted from the filter plate by centrifuge with elution buffer. Eluted probes were then hybridized into 96-well Hybridization Plate for quantitative analysis. The captured STAT3 probe was further detected by conjugation with streptavidin-HRP. The chemiluminescence of each well was detected by 2104 EnVision® Multilabel Reader of PerkinElmer (Pekin Elmer, Waltham, Massachusetts, USA) within 5 minutes after mixture with substrates.
Fluorescence Polarization Assay
Fluorescence polarization (FP) assay was conducted based on fluorescence signal differences between free and STAT3-bound fluorescently labeled peptide as described by Schust and Berg (14, 15). Briefly, all reactions contained 10 nM of the fluorescent peptide 5-FAM –G(pTyr) LPQTV-CONH2 (Genscript, Piscataway, NJ, USA) and 100 nM GST-tagged, full-length human STAT3 protein (SignalChem, Richmond, BC,Canada) in 96-well half-area black plates (Corning, Tewksbury, MA, USA). For evaluating compounds, STAT3 protein was incubated with various concentrations of S3I-1757 or S3I-1756 at room temperature for 60 min in the assay buffer (50mM NaCl, 10mM Hepes, pH 7.5, 1mM EDTA, 0.01% Triton-X100, 2mM dithiothreitol). Fluorescent peptide was added at a final concentration of 10 nM and incubated for 30 min at room temperature following which the FP measurements were examined by 2104 EnVision® Multilabel Reader (Pekin Elmer, Waltham, Massachusetts, USA) using FITC FP Dual module with excitation filter of FITC FP 480 and emission filter of FITC FP P-pol535 and S-pol535. The Z′ value was derived per the equation Z′=1 − (3SDbound+3SDfree)/(mPbound−mPfree), where SD is the standard deviation and mP is the average of fluorescence polarization.
STAT3 Transcriptional Activity
MDA-MB-468 cells were plated into 12-well plate with 4×105 cells per well. The cells were transiently transfected with pLucSRE, pLucTKS3 or STAT3-C with β-gal and then were treated with vehicle, S3I-1756 or S3I-1757 for 48 hours. Then cytosolic extracts of equal total protein were prepared from S3I-1757-treated or -untreated and analyzed for luciferase activity using a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA, USA) described by us previously (16).
Co-localization
HEK-293/FLAG-Stat3/HA-Stat3 cells were cultured (4000 cells per well) with G418 400ug/ml in 8 well chamber slide. The cells were treated with S3I-1757 or S3I-1756 for 1 hour, 2 hours, or 4 hours. The cells were then rinsed with phosphate buffered saline (PBS), fixed with ice-cold methanol for 15 minutes, washed 3 times in PBS, permeabilized with 0.2% Triton X- 100 for 15 minutes, and further washed 3–4 times with PBS. Cells were then blocked in 1% bovine serum albumin (BSA) for 30 min and incubated with anti-HA (Santa Cruz, Santa Cruz, CA, USA) or anti-FLAG (Sigma, St. Louis, MO, USA) primary antibody at 1:100 dilution at 4 °C overnight. Subsequently, cells were rinsed 4–5 times in PBS, incubated with Alexa fluor secondary antibody (Invitrogen, Carlsbad, CA, USA) for 1 hour at room temperature in the dark. Cells were then washed 5 times with PBS, covered with cover slides with VECTASHIELD mounting medium containing DAPI (Vector Lab, Inc., Burlingame, CA), and examined immediately under Zeiss Upright Fluorescence Microscope (Zeiss, Thornwood, NY, USA).
STAT3 Nuclear accumulation
MDA-MB-468 cells were plated at 4000 cells/well in 8-chamber slide. The cells were treated the following day with Vehicle, S3I-1756 or S3I-1757 for 2 hours, 4 hours, or 18 hours. Cells were fixed, washed, and permeabilized as describe above. Specimens were then blocked in 1% bovine serum albumin (BSA) for 30 min and incubated with anti-pSTAT3 (Cell Signaling), antibody at 1:50 dilution at 4 °C overnight. Subsequently, cells were rinsed 4–5 times in PBS, incubated with Alexa Fluor secondary antibody for1 hour at room temperature in the dark. Specimens were then washed 5 times with PBS, covered with cover slides with VECTASHIELD mounting medium containing DAPI (Vector Lab, Inc., Burlingame, CA), and examined immediately under Zeiss Upright Fluorescence Microscope (Zeiss, Thornwood, NY, USA).
MTT Assay
MTT assay was performed exactly as described by our lab (17) to determine the effects of S3I-1757 on cell proliferation. Briefly, cells were plated in a 96 well tissue culture plate (2000 cells per well) and incubated for 12 hours. After incubation the cells were treated with vehicle S3I-1757 or S3I-1756 for 48 hours. After incubation, freshly prepared MTT (3mg/ml) in 1XPBS was added to each well and incubated for 3 hours and the plate was read at 570 nm.
Colony Survival Assay
Cells were cultured at 500 cells per well in 12-well plate with regular growth medium. Cells were treated by vehicle, and 1757 at 50 μM, 100 μM and 200 μM on the following day. And Cells were allowed to grow for 2-3 weeks until the colonies were visible. 3mg/ml MTT in PBS buffer (Sigma, St. Louis, MO, USA) was used to stain the colonies for 4 hours.
Wound Healing Assay
A549, MDA-MB-231 and H460 cells were seeded at 6 × 105 cells per dish into 60mm plate and allowed to grow overnight. Wounds were made the following day by scratching the cells with pipette tips (1-10μL). Cells were treated with vehicle, S3I-1756 or S3I-1757 and allowed to migrate into the scratched area for 16 hours in regular growth medium. The migration of cells was visualized at 10X magnification using a Leica Microscope at time 0 (right before the drug was added) and 16 hours after vehicle, S3I-1756, or S3I-1757 treatments.
Invasion Assay
Invasion assay was performed in BD BioCoat™ Matrigel™ Invasion Chamber in 24-well plates. A549, MDA-MB-468, MDA-MB-231 and H460 cells were seeded at 25,000 cells/ insert in the top chamber over the Matrigel. The bottom chamber contains 20% FBS as the “chemoattractant”. Vehicle, S3I-1756, or S3I-1757 were added the following day. The cells were incubated for 48 hours, after which the cells in the top chamber were carefully removed and the filter membranes containing the invaded cells on the outside of the filter were fixed with methanol, stained with crystal violet and photographed.
Anchorage Independent Growth by Soft Agar Assay
Soft agar colony formation assays were performed in 12-well plate as described previously by us (17). Briefly, cells were seeded at 2000 cells per well in regular growth media containing 0.3% agar (Sigma) and S3I-1757 was added the following day. Colonies were allowed to grow for 3-4 weeks, and quantified by staining with 1mg/mL MTT (Sigma, St. Louis, MO, USA) overnight.
Molecular docking and Modeling
Briefly, the GLIDE docking software (available from Schrödinger, Inc.) was employed to dock small molecule 3D structures from NCI Plated Set to the ApY*LK site derived from the X-ray crystal structure of the STAT3 dimer bound to DNA (18). Schrödinger's Maestro 9.1 was used as the primary graphical user interface. Schrödinger's LigPrep 2.41 was used to prepare molecules for docking and Schrödinger's Protein Preparation workflow was used in the preparation of the protein structure. Schrödinger's GLIDE 5.6 was used for the generation of grid files and docking. Initially, structures were subjected to docking with GLIDE SP and then the structures from each SP job were subjected to GLIDE XP docking. Generally at least three poses were saved for each run for visual inspection. PyMol (Schrodinger, Inc.) was used for graphical presentation of the results in the figures.
Effects of inhibitors on the levels of P-STAT3, P-Erk, P-Akt, Bcl-xL, Survivin, MMP9 and Cyclin D1 by immunoblotting
MDA-MB-468 cells were treated with vehicle (DMSO), S3I-1757 at 50 μM, 100 μM and 200 μM, and S3I-1756 at 200 μM for 18 hours. Cells were then lysed with RIPA buffer (20mM Tris-HCl (pH 7.4), 5mM EDTA, 10mM Na4P2O7, 100mM NaF, 2mM Na3VO4, 1% NP-40,1mM PMSF and 10mg/ml aprotinin). The cell lysates were denatured by boiling with 5× SDS-PAGE sample buffer for 5 minutes and run on SDS-PAGE gel. The proteins were then transferred to membranes that were blocked with 5% non-fat milk in Tris Buffer Saline with 0.1% Tween-20 (TBST) buffer for 30 minutes at room temperature, and incubated with primary antibodies (pY705STAT3, pAKT, AKT,pERK1/2,MMP9,and CyclinD1) 4°C overnight at dilution of 1:1000 in 3% BSA, followed by washing and incubation with secondary antibody at dilution of 1:1000 in 5% non-milk Tris Buffer Saline with 0.1% Tween-20 (TBST) buffer for 1 hours at room temperature. The membranes were then washed with 1XPBS buffer for 10 minutes for 3 times and developed with ECL kit (PerkinElmer, Waltham, MA, USA).
Immunoprecipitation and immunoblotting
HEK-293/FLAG-STAT3/HA-STAT3 cells were treated for 4 h with vehicle, S3I-1757, S3I-1756, Ac-G{pTYR}LPQTV-AAVLLPVLLAAP-NH2 or Ac-GYLPQTV-AAVLLPVLLAAP-NH2 and then lysed in 20mM Tris-HCl (pH 7.4), 5mM EDTA, 10mM Na4P2O7, 100mM NaF, 2mM Na3VO4, 1% NP-40,1mM PMSF and 10mg/ml aprotinin. For EGF stimulation group, HEK-293/FLAG-STAT3/HA-STAT3 cells were treated by 100ng/ml EGF for 30 minutes before making cell lysate. Protein A or G agarose (EMD Millipore, Billerica, MA, USA) was washed twice with PBS and restore to 50% slurry with PBS. 500 μg of lysate were pre-cleared by mixture of protein A and protein G -agarose for 1 h at 4°C and then remove protein A and G-agarose by centrifuge at 1300rpm for 3minutes. And then 500 μg of lysate was immunoprecipitated with 50 ng of HA antibody overnight at 4°C on shaker and then capture the immunocomplex by adding 100 μl Protein A and G agarose/sepharose bead slurry for 1 hour at 4°C. Samples were washed five times with lysis buffer and then boiled in 5× SDS-PAGE sample buffer and run on SDS-PAGE gel. Protein was transferred to nitrocellulose membrane and then blotted as described above for HA, FLAG, pSTAT3, EGFR, and STAT3.
Results
S3I-1757 inhibits the binding of fluorescein-labelled GpYLPQTV phosphotyrosine peptide to STAT3 much more potently than its closely related analogue S3I-1756
In an effort to identify STAT3-STAT3 dimerization disruptors, we have undertaken a major chemistry effort based on structure activity relationship (SAR) studies using our previously published S3I-201 compound (16) as a starting point. These SAR studies identified several STAT3 inhibitors (19). In the present manuscript, we focused on S3I-1757 and its closely related analogue S3I-1756 (Figure 1A). We first determined the potency of these molecules to disrupt the binding of STAT3 to fluorescein-labelled GpYLPQTV phosphotyrosine peptide by fluorescence polarization assays as described under Methods (the GpYLPQTV phosphotyrosine peptide corresponds to amino acids 903-909 from the gp-130 subunit of the IL-6 receptor and is known to bind the STAT3-SH2 domain (20, 21)). Figure 1A shows that S3I-1757 inhibits the binding of STAT3 to the phosphotyrosine peptide in a concentration-dependent manner with an IC50 value of 13.5 ± 0.5 μM. In contrast, the closely related analogue S3I-1756 had little effects with concentrations as high as 400 μM (Figure 1A). This data shows that replacing the cyclohexyl group in S3I-1757 with a methoxy group as in S3I-1756 resulted in great (over 26-fold) loss of potency to disrupt STAT3 phosphotyrosine-peptide binding.
Figure 1. Chemical structures of S3I-1757 and S3I-1756, fluorescence polarization (FP) assay and molecular modeling.
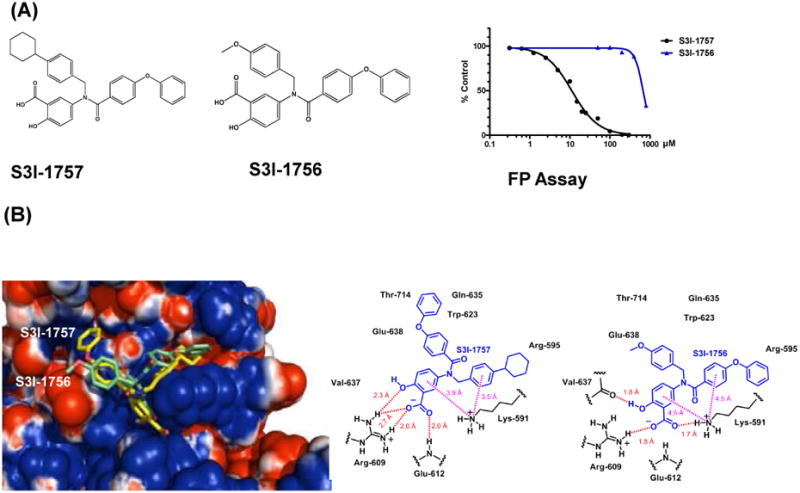
(A) S3I-1757 and S3I-1756 structures are shown. The effects of S3I-1757 and S3I-1756 on the binding of STAT3 to fluorescein-labelled GpYLPQTV phophotyrosine peptide was determined by FP assays as described under Methods. (B) Solvent accessible surface (probe radius of 0.75 Å) of the STAT3 SH2 domain color coded by electrostatic potential calculated using the APBS plugin in PyMol. Red represents regions of negative electrostatic potential and blue positive regions. S3I-1756 and S3I-1757 are shown using a tube rendering with carbon atoms colored green for S3I-1756 and yellow for S3I-1757.
Molecular modeling suggests that S3I-1757 binds the STAT3-SH2 domain in the same binding site where the native phosphotyrosine peptide binds
To determine the potential mode of binding of S3I-1757 to the STAT3-SH2 domain, we carried out docking studies as described under Methods. Figure 1B shows a surface rendering of the SH2 domain of STAT3 with the active analog, S3I-1757 (yellow), and the inactive analog, S3I-1756 (green), bound based upon GLIDE XP docking. A ligand-protein interaction diagram is also shown in Figure 1B. Figure1B shows that S3I-1757 occupies the phosphotyrosine binding site of STAT3. Furthermore, the salicylic acid group of S3I-1757 interacts with SH2 domain amino acids (i.e. Arg-609 and Lys-591) known to interact with P-Y-705 of STAT3 (18). The experimentally observed greater potency of S3I-1757 compared to S3I-1756 is most likely due to a number of factors as illustrated in Figure 1B: (a) S3I-1757 forms 4 hydrogen bonds (H-bonds between Arg-609 and Glu-612) in the phospho-tyrosine binding pocket, whereas S3I-1756 only forms 3 (H-bonds between Val-637, Arg-609 and Lys 591); (b) S3I-1757 forms 2 excellent cation-π interactions with Lys 591 (with an N to phenyl ring centroid distance of 3.9 Å for the salicylate phenyl group and an N to phenyl ring centroid distance of 3.5 Å for the phenyl group to which the cyclohexyl substituent is attached); (c) S3I-1756 forms 2 weaker cation-π interactions with Lys 591 (with an N to phenyl ring centroid distance of 4.6 Å for the salicylate phenyl group and an N to phenyl ring centroid 4.5 Å for the phenyl group to which the phenoxy group is attached); (d) the surface rendering of the protein in Figure 1B shows that the cyclohexyl substituent of S3I-1757 is buried in a pocket with a rather negative electrostatic potential whereas the phenoxy substituent of S3I-1756 is similarly buried, although not as deeply and the πelectron system of the phenoxy substituent would not interact favorably with this negatively charged environment; (e) the methoxy-phenyl substituent of S3I-1756 is mostly solvent exposed and forms almost no interactions with the protein.
Co-immunoprecipitation and co-localization experiments reveal that S3I-1757 but not S3I-1756 disrupts intracellular STAT3-STAT3 dimerization and STAT3-EGFR binding
The data from the FP assays of Figure 1A coupled with the molecular modeling results of Figure 1B suggest that S3I-1757 inhibits STAT3 dimerization. However, the FP assay only measured the ability of S3I-1757 to displace the 7 amino acid fluorescein-labelled GpYLPQTV phosphotyrosine peptide from the SH2 domain of STAT3 protein in vitro. Therefore, we next determined if the intracellular reciprocal binding of 2 full-length STAT3 proteins is inhibited since the ultimate goal is to develop S3I-1757 as a small molecule capable of disrupting STAT3 dimerization and subsequently inactivating constitutively activated STAT3. To this end, we developed an assay that measures directly STAT3-STAT3 dimerization in intact cells by cloning HA-tagged STAT3 and FLAG-tagged STAT3, generating HEK293 cells that stably co-express HA-STAT3 and FLAG-STAT3 and using these cells for co-immunoprecipitation and co-localization experiments as described under Methods. Figure 2A shows that FLAG-STAT3 co-immunoprecipitated with HA-STAT3 in HEK293 cells that co-express FLAG-STAT3 and HA-STAT3 but not in empty vector-transfected HEK293 cells (Vector). We validated this assay by treating the FLAG-STAT3/HA-STAT3 HEK293 cells with tyrosine phosphorylated or non-phosphorylated GYLPQTV peptide fused to a membrane-translocating sequence (MTS) to allow cell uptake. Figure 2A shows that, in vehicle-treated cells (V), FLAG-STAT3 co-immunoprecipitated with HA-STAT3, and that treatment with phosphorylated GpYLPQTV (P-Pep) but not non-phosphorylated GYLPQTV (Pep) inhibited FLAG-STAT3 from co-immunoprecipitating with HA-STAT3. To determine if the compounds can inhibit the binding of HA-STAT3 to FLAG-STAT3, HA-STAT3/FLAG-STAT3/ HEK293 cells were treated with vehicle (V), S3I-1757 or S3I-1756 and the cells were processed for immunoprecipitation with HA antibodies and immunoblotted with FLAG antibodies as described under Methods. Figure 2A shows that in vehicle- treated cells, FLAG-STAT3 co-immunoprecipitated with HA-STAT3, and that treatment with S3I-1757 inhibited the binding of FLAG-STAT3 to HA-STAT3 in a concentration-dependent manner. In contrast, S3I-1756 did not inhibit this binding with concentrations as high as 200 uM (Figure 2A). We next determined if S3I-1757 could disrupt the binding of HA-STAT3 to the EGF receptor since the same STAT3-SH2 domain that is used for STAT3-STAT3 dimerization is also used to bind EGFR on p-Tyr-1068 and p-Tyr-1086 on EGFR (see Introduction). Figure 2B shows that stimulation of HA-STAT3/FLAG-STAT3/HEK293 cells with EGF increased the levels of EGFR and P-STAT3 that co-immunoprecipitated with HA-STAT3, and that treatment of these cells with S3I-1757 inhibited these interactions. These results suggest that S3I-1757 is capable of inhibiting STAT3-STAT3 dimerization and STAT3 binding to EGFR in intact cells. Next, HA-STAT3/FLAG-STAT3/HEK 293 cells were treated with vehicle, S3I-1757 or S3I-1756 and the cells were processed for immunofluorescence staining for FLAG-STAT3 (red) and HA-STAT3 (green) and analyzed by confocal microscopy as described under Methods. As shown in Figure 2C, cells treated with vehicle harbored strong yellow color suggesting co-localization of FLAG-STAT3 (red) and HA-STAT3 (green). In contrast, cells treated with S3I-1757, but not S3I-1756, demonstrated progressively less yellow color over time, suggesting that the dimerization of FLAG-STAT3 and HA-STAT3 was disrupted. Therefore, using methods that investigated STAT3 intracellular dimerization with tagged STAT3 proteins, we demonstrated that STAT3-STAT3 protein-protein binding in intact cells was disrupted with a small molecule designed to disrupt phosphotyrosine binding to STAT3-SH2 domain.
Figure 2. S3I-1757 but not S3I-1756 inhibits STAT3-STAT3 dimerization and STAT3-EGFR binding in intact cells.
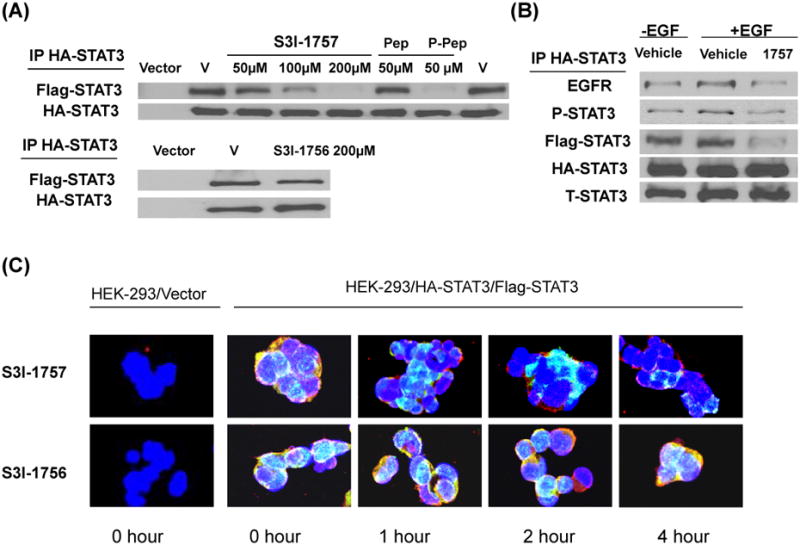
(A) Co-immuno-precipitation. HEK293 cells stably co-expressing FLAG- and HA-tagged STAT3 were treated with either vehicle, Ac-G{pTYR}LPQTV-AAVLLPVLLAAP-NH2 (phospho-peptide with MTS), Ac-GYLPQTV-AAVLLPVLLAAP-NH2 (non-phospho-peptide with MTS), S3I-1757 or S3I-1756 at the indicated concentration, processed for immuno-precipitation with HA antibody and immuno-blotting with FLAG antibody as described in Methods. FLAG-STAT3 co-immuno-precipitated with HA-STAT3 in HEK293 cells that co-express HA-STAT3 and FLAG-STAT3 but not in vector transfected HEK293 cells. Ac-G{pTYR}LPQTV-AAVLLPVLLAAP-NH2 (phospho-peptide) but not Ac-GYLPQTV-AAVLLPVLLAAP-NH2 inhibited the binding of FLAG-STAT3 to HA-STAT3. S3I-1757 but not S3I-1756 inhibited the binding of FLAG-STAT3 to HA-STAT3. (B) HEK293 cells stably co-expressing FLAG- and HA-tagged STAT3 were treated as described in (A) except that prior to treating with S3I-1757, they were first treated either with vehicle or EGF as described in Methods. The cells were then immuno-precipitated with HA antibody and blotted with antibodies to EGFR, P-Y-705-STAT3, FLAG, HA or total STAT3 as described in Methods. Results are representative of 3 independent experiments. (C) Co-localization. HEK293 cells stably co-expressing FLAG- and HA-tagged STAT3 were plated on cover slides over night and then treated with either vehicle, S3I-1757 or S3I-1756 for 0, 1, 2 or 4 hours and processed for co-localization studies with HA-STAT3 (Green) and FLAG-STAT3 (Red) as described under Methods. DAPI nuclear staining is shown in blue. Data are representative of 3 independent experiments.
S3I-1757 but not S3I-1756 decreases phosphotyrosine STAT3 levels in the nucleus of human MDA-MB-468 breast cancer cells
For STAT3 to regulate the expression of its target genes it needs to translocate from the cytosol to the nucleus, a process that requires STAT3 tyrosine phosphorylation and subsequent STAT3-STAT3 dimerization (1, 2). The fact that S3I-1757 was able to inhibit EGFR-STAT3 binding and STAT3-STAT3 protein-protein binding (Figure 2) suggests that it would also inhibit the levels of tyrosine phosphorylated STAT3 and its nuclear accumulation. To determine if this is the case, MDA-MB-468 breast cancer cells, which harbor persistently Y-705 phosphorylated STAT3, were treated with either vehicle, S3I-1757 or S3I-1756 for 2 hours and 4 hours and then was subjected to immunofluorescence staining by a specific p-Tyr-705-STAT3 primary antibody and Alexa Fluor 594 secondary antibody in medium containing DAPI to stain the nuclei as described in Methods. Figure 3A shows that, in vehicle treated cells, pSTAT3 was localized predominantly in the nucleus. In contrast, the levels of p-STAT3 in the nucleus were dramatically decreased in S3I-1757 treated cells particularly after 4 hours of treatment. To determine whether this inhibition is sustained over a longer period of time, we treated MDA-MB-468 cells with various concentrations of S3I-1757 for 18 hours. Figure 3B shows that S3I-1757-inhibited P-STAT3 nuclear accumulation in a concentration-dependent manner starting at 50 μM. In contrast, S3I-1756 had little effect with concentrations as high as 200 μM.
Figure 3. S3I-1757 but not S3I-1756 induces nuclear accumulation of P-STAT3 and inhibits STAT3-DNA binding.
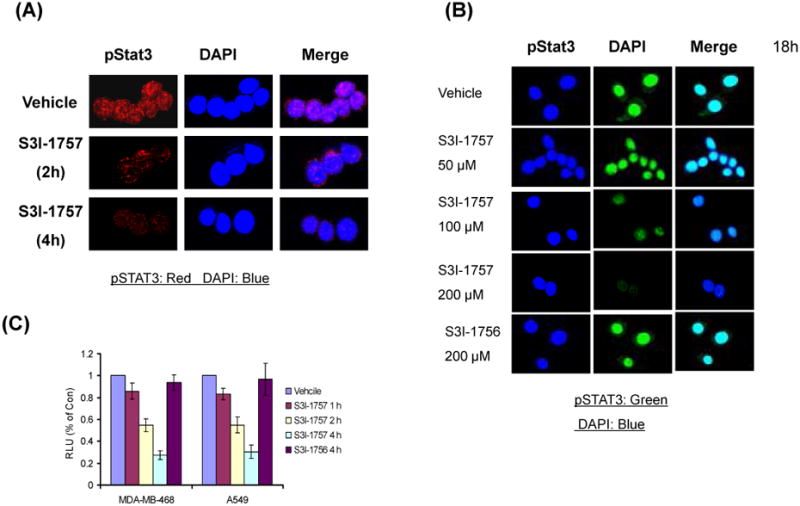
(A and B) MDA-MB-468 cells were plated on cover slides over night and then treated with either vehicle, S3I-1757 or S3I-1756 at the indicated concentration for either 2 or 4 hours (A) or 18 hours (B) and processed for P-STAT3 immuno-fluorescence as described in Methods. (C) MDA-MB-468 and A-549 cells were treated with either vehicle, S3I-1757 or S3I-1756 for the indicated interval of time and the nuclear extract isolated from the treated cells as described in the Methods. The nuclear extracts were then incubated with a biotin labeled STAT3 DNA binding probe and the complexes isolated by a STAT3-DNA binding assay described under Methods. Results are representative of 4 independent experiments.
Treatment of human breast (MDA-MB-468) and lung (A-549) cancer cells with S3I-1757, but not S3I-1756, decreases the amount of activated STAT3 capable of binding to DNA
The ability of S3I-1757 to inhibit STAT3-EGFR binding, tyrosine phosphorylation, STAT3-STAT3 dimer formation and nuclear accumulation would be predicted to result in blocking STAT3-DNA binding activity. To evaluate this possibility, we treated MDA-MB-468 and A-549 cells with vehicle, S3I-1757 or S3I-1756 for 1, 2 or 4 hours and collected nuclear extracts for STAT3-DNA binding activity using a STAT3 filter plate assay as described under Methods. Figure 3C shows that nuclear extracts from vehicle treated cells contained activated STAT3 capable of binding the biotin labeled STAT3 DNA binding probe. In contrast, the nuclear extracts from S3I-1757 treated cells contained less activated STAT3 capable of binding the STAT3 DNA binding probe with the least amount found after 4 hours of treatment. Figure 3C also shows that S3I-1756 did not decrease the amount of STAT3 capable of binding DNA.
S3I-1757, but not S3I-1756, inhibits STAT3- but not SRE-dependent transcriptional activation: STAT3-C rescues this inhibition
We next evaluated the ability of S3I-1757 to inhibit STAT3-dependent transcriptional activation using a luciferase reporter assays. To this end, MDA-MB-468 cells were transiently co-transfected with a STAT3-responsive promoter-firefly luciferase reporter (pLucTKS3) and β-gal reporter used to normalize the transfection efficacy. To determine the selectivity of S3I-1757 to suppress STAT3-dependent over STAT3-independent transcriptional activation, MDA-MB-468 cells were also co-transfected with SRE promoter-renilla luciferase reporter (pLucSRE) and β-gal reporter. Figure 4A shows that, compared to mock transfected cells, cells transfected with STAT3-responsive reporter (pLucTKS3) had increased luciferase activity in the absence of drug treatment. In contrast, less luciferase activity was observed when the cells were treated with S3I-1757 but not S3I-1756. S3I-1757 inhibited STAT3-dependent but not STAT3-independent transcriptional activity as demonstrated by the minimal effect it had on SRE-driven luciferase activity (Figure 4A). We next used a constitutively-dimerized mutant form of STAT3, STAT3-C, to further demonstrate the STAT3-dependence of the inhibition with S3I-1757. STAT3-C spontaneously dimerizes via disulfide bonds in the absence of tyrosine phosphorylation (see Introduction section), and is therefore not predicted to be inhibited by a small molecule that is designed to mimic phosphotyrosine binding. Figure 4A shows that transfection of MDA-MB-468 cells with STAT3-C increased the transcriptional activity of the STAT3- but not the SRE-responsive promoter, and rescued from the S3I-1757 inhibition.
Figure 4. S3I-1757, but not S3I-1756, inhibits STAT3-dependent transcriptional activity and decreases the expression of STAT3-regulated genes.
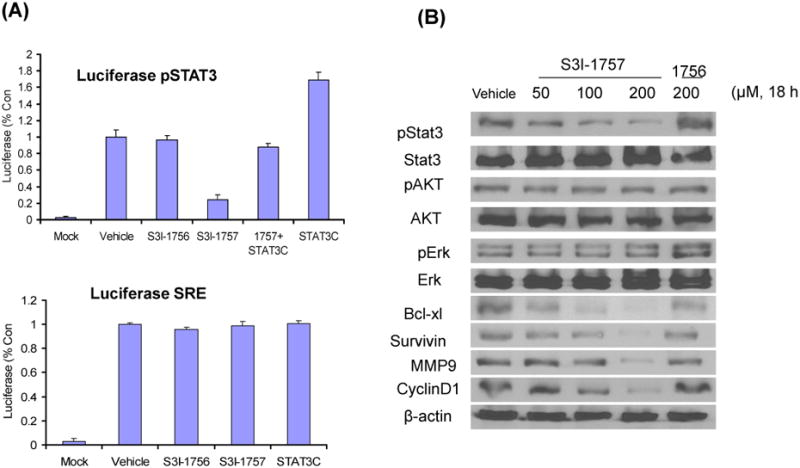
(A) MDA-MB-468 cells were transiently transfected with pLucSRE, pLucTKS3 or STAT3C along with β-gal and then were treated with vehicle, S3I-1756 or S3I-1757 as described in Methods. The cytosolic extracts were prepared and analyzed for luciferase activity as described in Methods. The results are representative of 2 independent experiments. (B) MDA-MB-468 cells were treated for 18 hours with the indicated concentrations of S3I-1757 or S3I-1756 and processed for western immuno-blotting with the indicated antibodies as described in Methods. The results are representative of 3 independent experiments.
S3I-1757 but not S3I-1756 decreases the tyrosine phosphorylation of STAT3 selectively over the phosphorylation of Akt and Erk1/2, and decreases the expression of genes that are transcriptionally-regulated by STAT3
Figures 1, 2, 3 and 4A demonstrated that S3I-1757 inhibits STAT3 dimerization, accumulation of nuclear P-Y705-STAT3, STAT3-DNA binding and transcriptional activity. We next determined if the ability of STAT3 to regulate the expression of its target genes is affected by S3I-1757. First, we confirmed that S3I-1757 inhibits the phosphorylation of STAT3-Y705 by western blotting and determined whether this is selective. To this end, MDA-MB-468 cells were treated with vehicle, S3I-1756 or increasing concentration of S3I-1757 and processed for western blotting as described under Methods. Figure 4B shows that S3I-1757, but not S3I-1756, inhibited the phosphorylation of STAT3-Y705 in a concentration-dependent manner starting at 50 μM. This inhibition was selective for STAT3 over Akt and Erk1/2 phosphorylation. Figure 4B also shows that S3I-1757, but not S3I-1756, inhibited the expression of STAT3 target genes such as the anti-apoptotic proteins Bcl-xL and survivin, the cell cycle protein cyclin D1 and the pro-metastatic protein MMP9.
S3I-1757 but not S3I-1756 inhibits anchorage-dependent and -independent growth, migration and invasion selectively in cancer cells harboring constitutively active STAT3 over those that do not
The data presented so far in Figures 1, 2, 3 and 4 demonstrated that S3I-1757 effectively blocks STAT3 dimerization and constitutive activation and suppresses its ability to persistently up-regulate the expression of genes known to mediate hallmarks of malignant transformation. We, therefore, next investigated whether S3I-1757 can suppress uncontrolled anchorage-dependent and –independent tumor cell growth, migration and invasion. To this end, we used several cell lines, some with persistently Y705 phosphorylated STAT3 and others without (Figure 5A). Figures 5A shows that S3I-1757, but not S3I-1756, inhibited in a dose dependent manner anchorage-dependent proliferation by MTT only in cells that harbor (MDA-MB-468, MDA-MB-231, H358, A549) but not in those that do not harbor (H460, MDA-MB-453, HEK293 and MCF10A) persistently activated STAT3. Similar results were obtained with colony formation on plastic (Figure 5B). The effects of S3I-1757 on cancer cell anchorage-independent growth was next determined by soft-agar assay as described under Methods. The results show that S3I-1757 significantly inhibited the anchorage independent growth of cancer cells with constitutively active STAT3 such as MDA-MB-468, but had little effect on anchorage-independent growth of H460 cells which has low activated STAT3 level (Figure 6).
Figure 5. S3I-1757 inhibits anchorage-dependent proliferation selectively in human cancers that harbor high levels of P-STAT3.
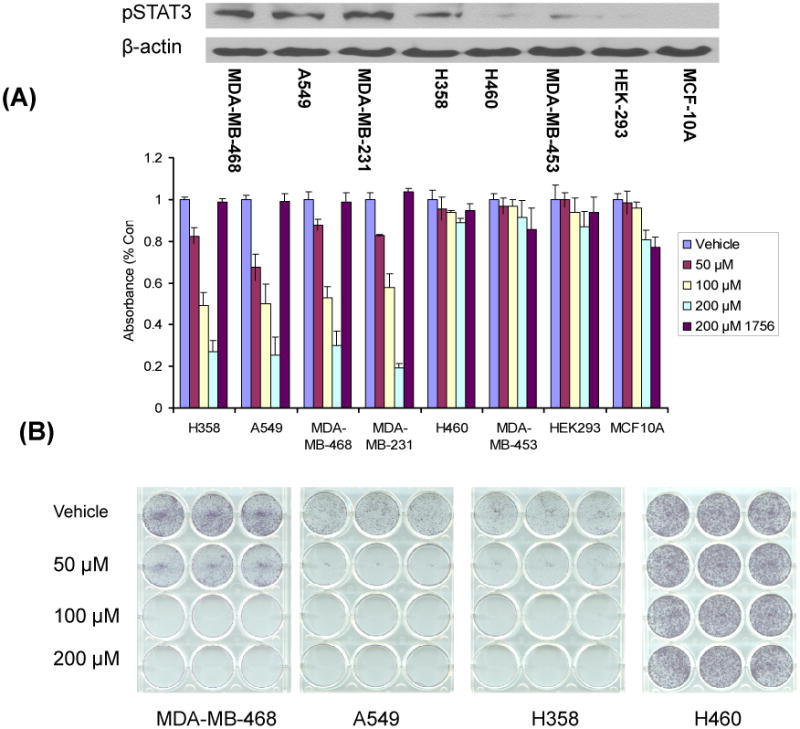
(A) H358, A549, MDA-MB-468 and MDA-MB-231 cells (high P-STAT3) and H460, MDA-MB-453, HEK293, and MCF-10A cells (lowP-STAT3) were plated in 96-well plates and treated with the indicated concentrations of S3I-1757 or S3I-1756 for 48 hours and processed for MTT assays as described in Methods. The levels of P-STAT3 were analyzed by western blotting with a P-Y-705-STAT3 antibody as described in Methods. (B) Cells were treated exactly as described for (A) except that the cells were plated in 12-well plates and treated for 21 days. The results are representative of 2 independent experiments.
Figure 6. S3I-1757 inhibits anchorage-independent proliferation selectively in human cancers that harbor high levels of P-STAT3.
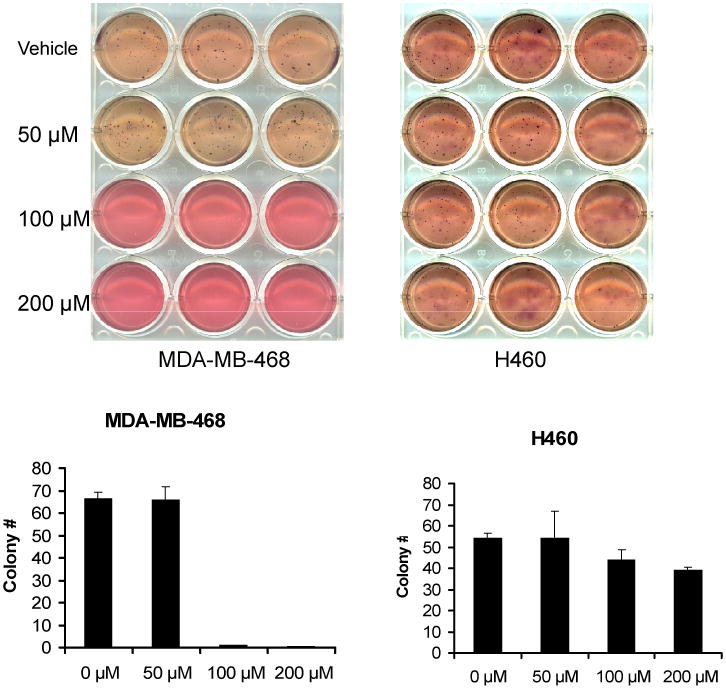
The cells were treated exactly as described for Figure 5B except that they were first seeded at 2000 cells per well in regular growth media containing 0.3% agar (Sigma) and S3I-1757 was added the following day, and colonies were allowed to grow for 3-4 weeks as described in Methods. The experiment was performed once in triplicates.
The ability of S3I-1757 to inhibit selectively the migration of cancer cells that depend on STAT3 over those that do not was next evaluated by a wound healing assay. The cancer cells were cultured with serum-starved medium for 8 hours before scratching the cells with a pipette tip and treating with increasing concentrations of S3I-1757 for 24 hours. Figure 7A shows that in the absence of drug, the cells migrated within 24 hours to fill the scratched area. S3I-1757, but not S3I-1756, treatment prevented this migration in cells with persistently activated STAT3 (MDA-MB-231 and A-549). In contrast, the migration of H460 (with low levels of P-STAT3) was minimally affected by the same treatment condition. Finally, the ability of S3I-1757 to inhibit selectively invasion was determined as described under Methods. Figure 7B shows that S3I-1757 but not S3I-1756, inhibited invasion in MDA-MB-468, MDA-MB-231 and A-549 but not in H-460 cells.
Figure 7. S3I-1757, but not S3I-1756, inhibits migration and invasion selectively in human cancers that harbor high levels of P-STAT3.
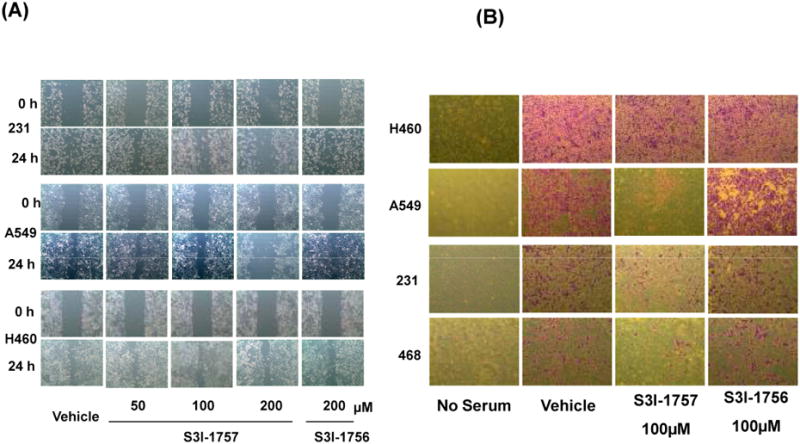
(A) Migration: MDA-MB-231, A549 and H460 cells were seeded and allowed to grow overnight prior to scratching the cells with pipette tips. Cells were then treated with vehicle, S3I-1756 or S3I-1757 and allowed to migrate into the scratched area for 16 hours in regular growth medium as described in Methods. (B) Invasion: H460, A549, MDA-MB-468 and MDA-MB-231 cells were seeded over the Matrigel in the top chamber of insert, and treated with vehicle, S3I-1756, or S3I-1757 the following day. The cells were incubated for 48 hours, and the invaded cells were fixed with methanol, stained with crystal violet and photographed as described under Methods. Experiments were performed in triplicates with identical results.
STAT3-C rescues from S3I-1757 inhibition of gene expression, tumor cell growth, migration and invasion as well as from apoptosis induction
The fact that S3I-1757 but not its inactive analogue S3I-1756 inhibits malignant transformation selectively in cells that harbor hyperactivated STAT3 suggested that S3I-1757 mediates its effects by inhibiting STAT3. To give further support to this suggestion, we determined whether the effects of S3I-1757 can be rescued by STAT3-C, a genetically engineered mutant of STAT3 that forms a constitutively dimerized STAT3 through disulfide bonds in the absence of tyrosine phosphorylation (8). To this end, we transfected MDA-MB-468 cells with STAT3-C, then treated the cells with S3I-1757 and processed the cells for western blotting, cell growth, migration and invasion. Figure S1 A shows that expression of STAT3-C increased the levels of BclxL, cyclin D1 and survivin. In contrast, S3I-1757 inhibited the expression levels of these proteins and induced activation of caspase 3 and PARP cleavage. Furthermore, Figure S1 A also shows that ectopic expression of STAT3-C inhibited the ability of S3I-1757 to down regulate the expression of BclxL, cyclin D1 and survivin and to induce apoptosis. Similarly, Figure S1 B shows that STAT3-C inhibited S3I-1757 from inhibiting the proliferation/survival of MDA-MB-468 cells as determined by MTT assays. We next investigated the effects of STAT3-C on S3I-1757 effects on migration and invasion. Figures S2 A and B show that S3I-1757 inhibited the migration and invasion of MDA-MB-468 cells transfected with vector DNA. In contrast, the ability of S3I-1757 to inhibit migration and invasion was partially rescued in cells transfected with STAT3-C.
Discussion
The first demonstration that STAT3 is involved in malignant transformation (12) was reported in 1995 only a year after its discovery (22). Less than 6 years later peptides and peptide mimics of the phospho-tyrosine peptide PpYLKTK that bind STAT3 SH2 domain were shown to inhibit STAT3 dimerization in vitro and STAT3 activity in intact cells (23). Yet, eleven years later, there are no small molecule STAT3 dimerization inhibitors in clinical trials. One of the major reasons for this is that STAT3-STAT3 dimerization is a protein-protein interaction that involves a large surface area which is difficult to target with drug-like small molecules. The second reason, which is even more challenging, is that the negatively charged phospho-tyrosine which is required for binding to the SH2 domain is difficult to mimic with moieties that can be easily taken up by cells. Nevertheless, because of the critical role of STAT3 in oncogenesis, several groups have put major efforts towards developing STAT3 dimerization inhibitors based on Phospho-peptide mimics as novel anti cancer drugs (6, 7). For example, McMurray and colleagues have succeeded at obtaining cell permeable peptidomimetics of pYLPQ where pY was replaced by phosphocinnamide derivatives to improve peptidase resistance and used the pivaloyloxymethyl prodrug strategy to improve cellular uptake which lead to potent inhibition of STAT3 activity in whole cells (24). Similarly, Wang and colleagues (25) have also succeeded at designing a conformationally constrained pYLPQTV peptidomimetic with a long hydrocarbon chain to improve cell permeability. Although these are outstanding achievements, there still remain physicochemical challenges concerning the use of phospho-tyrosine peptidomimetics in vivo (7). Therefore non-peptidic small molecules capable of disrupting STAT3-STAT3 dimerization is an attractive alternative approach to inhibiting STAT3 directly. We have used structure-based virtual screening and identified S3I-201, a salicylic acid sulfonamide-based compound, which inhibits STAT3 dimerization in vitro and STAT3 activity in whole cells (16). Turkson, Gunning and colleagues have subsequently reported on salicylic acid sulfonamide S3I-201 analogues with improved potency (26, 27). Our recent chemistry efforts resulted in a series of novel non-sulfonamide-containing salicylic acid based compounds (19). In this manuscript, we have demonstrated that one of these, S3I-1757, inhibited STAT3 dimerization in vitro and in whole cells, STAT3 tyrosine phosphorylation, nuclear accumulation, transcriptional activity and expression of STAT3-regulated genes as well as anchorage-dependent and –independent growth, migration and invasion.
The ability of S3I-1757 to displace fluorescein-GpYLPQTV in the FP assay in vitro suggested that S3I-1757 binds the SH2 domain of STAT3 at the phospho-tyrosine-705 binding site. Our molecular modeling studies give further support to this suggestion. In deed our molecular modeling indicated that S3I-1757 makes several contacts with Arg-609 and Lys-591, 2 critical amino acids in the SH2 domain that are known to bind phospho-Tyr-705 of PpYLKTK of STAT3 as well as phospho-Tyr-904 of GpYLPQTV of the gp-130 subunit of the IL-6 receptor. In whole cells, S3I-1757 disrupted the binding of HA-STAT3 to FLAG-STAT3 as demonstrated both by co-immuno-precipitation and co-localization, consistent with the in vitro FP and molecular modeling results. Taken together, these results strongly suggest that S3I-1757 is a STAT3-STAT3 dimerization inhibitor. Furthermore, STAT3 is also known to associate with the EGF receptor (EGFR) through binding of the STAT3-SH2 domain to phospho-tyrosines 1068 and 1086 on EGFR (3), and S3I-1757 inhibited the binding of STAT3 to EGFR. It is, therefore, not surprising that S3I-1757 inhibited STAT3 tyrosine phosphorylation. This suggests that the ability of S3I-1757 to inhibit nuclear translocation, DNA binding and transcriptional activation may be due to its ability to directly disrupt STAT3-STAT3 dimerization as well as inhibition of STAT3-EGFR binding and subsequent suppression of STAT3 tyrosine phosphorylation which would also lead to preventing STAT3 dimerization. The fact that STAT3-C, a genetically engineered mutant of STAT3 that forms a constitutively dimerized STAT3 through disulfide bonds in the absence of tyrosine phosphorylation (8), was able to rescue from S3I-1757 inhibition of transcriptional activity further solidifies the suggestion that S3I-1757 is a STAT3 dimerization inhibitor.
S3I-1757 inhibited anchorage-dependent proliferation/survival and colony formation as well as anchorage-independent soft agar growth, migration and invasion, consistent with its ability to suppress the expression of genes that are known to drive these hallmarks of cancer such as cyclin D1, BclxL, survivin, and MMP9. The fact that S3I-1757 did not inhibit other signal transduction pathways such as those leading to hyper-activated P-Akt and P-Erk suggest that S3I-1757 induces these effects through inhibition of STAT3. Further support for this suggestion comes from the fact that S3I-1757 inhibited anchorage-dependent and –independent tumor cell growth, migration and invasion selectively in human cancer cells that dependent on STAT3 over those that do not. S3I-1756, a closely related structural analogue of S3I-1757 that does not inhibit STAT3-STAT3 dimerization, STAT3 tyrosine phosphorylation, DNA binding and transcriptional activation, was not able to inhibit anchorage-dependent and –independent tumor cell growth, migration and invasion. Finally, the fact that STAT3-C, was able to rescue from S3I-1757 induction of apoptosis and inhibition of gene expression, tumor cell growth, migration and invasion strongly supports the suggestion that S3I-1757 mediates its effects through inhibition of STAT3.
In summary, we have identified a non-sulfonamide-containing salicylic analogues of S3I-201, S3I-1757, which directly inhibits STAT3-STAT3 dimerization as well as STAT3-EGFR binding leading to inhibition of STAT3 tyrosine phosphorylation and nuclear translocation. Using several approaches we have also demonstrated that S3I-1757 is able to inhibit malignant transformation and that this is due to its ability to inhibit STAT3 function.
Supplementary Material
Acknowledgments
We would like to thank the Moffitt Chemical Biology Core and the Analytic Microscopy Core for their outstanding technical expertise. This work was partially supported by NCI grant CA140681 (SMS and NJL). We would also like to thank Dr Bromberg for providing us with the STAT3-C DNA construct.
Grant Support: This work was partially supported by NCI grant CA140681 (SMS and NJL).
Financial Support: This work was partially supported by NCI grant CA140681 (SMS and NJL).
Footnotes
Conflict of Interests: The authors have no conflict of interests.
References
- 1.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264(5164):1415–21. doi: 10.1126/science.8197455. Epub 1994/06/03. [DOI] [PubMed] [Google Scholar]
- 2.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nature reviews Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. Epub 2009/10/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shao H, Cheng HY, Cook RG, Tweardy DJ. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer research. 2003;63(14):3923–30. Epub 2003/07/23. [PubMed] [Google Scholar]
- 4.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nature reviews Cancer. 2004;4(2):97–105. doi: 10.1038/nrc1275. Epub 2004/02/18. [DOI] [PubMed] [Google Scholar]
- 5.Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F, Sawaya R, et al. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004;23(20):3550–60. doi: 10.1038/sj.onc.1207383. Epub 2004/04/30. [DOI] [PubMed] [Google Scholar]
- 6.Debnath B, Xu S, Neamati N. Small Molecule Inhibitors of Signal Transducer and Activator of Transcription 3 (Stat3) Protein. Journal of medicinal chemistry. 2012 doi: 10.1021/jm300207s. Epub 2012/06/02. [DOI] [PubMed] [Google Scholar]
- 7.Masciocchi D, Gelain A, Villa S, Meneghetti F, Barlocco D. Signal transducer and activator of transcription 3 (STAT3): a promising target for anticancer therapy. Future medicinal chemistry. 2011;3(5):567–97. doi: 10.4155/fmc.11.22. Epub 2011/04/30. [DOI] [PubMed] [Google Scholar]
- 8.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98(3):295–303. doi: 10.1016/s0092-8674(00)81959-5. Epub 1999/08/24. [DOI] [PubMed] [Google Scholar]
- 9.Caldenhoven E, van Dijk TB, Solari R, Armstrong J, Raaijmakers JA, Lammers JW, et al. STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. The Journal of biological chemistry. 1996;271(22):13221–7. doi: 10.1074/jbc.271.22.13221. Epub 1996/05/31. [DOI] [PubMed] [Google Scholar]
- 10.Niu G, Heller R, Catlett-Falcone R, Coppola D, Jaroszeski M, Dalton W, et al. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer research. 1999;59(20):5059–63. Epub 1999/10/28. [PubMed] [Google Scholar]
- 11.Wang DA, Sebti SM. Palmitoylated cysteine 192 is required for RhoB tumor-suppressive and apoptotic activities. The Journal of biological chemistry. 2005;280(19):19243–9. doi: 10.1074/jbc.M411472200. Epub 2005/02/17. [DOI] [PubMed] [Google Scholar]
- 12.Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, et al. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269(5220):81–3. doi: 10.1126/science.7541555. Epub 1995/07/07. [DOI] [PubMed] [Google Scholar]
- 13.Ying WZ, Wang PX, Aaron KJ, Basnayake K, Sanders PW. Immunoglobulin light chains activate nuclear factor-kappaB in renal epithelial cells through a Src-dependent mechanism. Blood. 2011;117(4):1301–7. doi: 10.1182/blood-2010-08-302505. Epub 2010/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schust J, Berg T. A high-throughput fluorescence polarization assay for signal transducer and activator of transcription 3. Analytical biochemistry. 2004;330(1):114–8. doi: 10.1016/j.ab.2004.03.024. Epub 2004/06/09. [DOI] [PubMed] [Google Scholar]
- 15.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chemistry & biology. 2006;13(11):1235–42. doi: 10.1016/j.chembiol.2006.09.018. Epub 2006/11/23. [DOI] [PubMed] [Google Scholar]
- 16.Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(18):7391–6. doi: 10.1073/pnas.0609757104. Epub 2007/04/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balasis ME, Forinash KD, Chen YA, Fulp WJ, Coppola D, Hamilton AD, et al. Combination of farnesyltransferase and Akt inhibitors is synergistic in breast cancer cells and causes significant breast tumor regression in ErbB2 transgenic mice. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(9):2852–62. doi: 10.1158/1078-0432.CCR-10-2544. Epub 2011/05/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394(6689):145–51. doi: 10.1038/28101. Epub 1998/07/22. [DOI] [PubMed] [Google Scholar]
- 19.Urlam MK, Pireddu R, Ge Y, Zhang X, Sun Y, Luo Y, et al. Development of new N-Arylbenzamides as STAT3 Dimerization Inhibitors. Submitted. 2012 doi: 10.1039/C3MD20323A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haan S, Hemmann U, Hassiepen U, Schaper F, Schneider-Mergener J, Wollmer A, et al. Characterization and binding specificity of the monomeric STAT3-SH2 domain. The Journal of biological chemistry. 1999;274(3):1342–8. doi: 10.1074/jbc.274.3.1342. Epub 1999/01/09. [DOI] [PubMed] [Google Scholar]
- 21.Ren Z, Cabell LA, Schaefer TS, McMurray JS. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorganic & medicinal chemistry letters. 2003;13(4):633–6. doi: 10.1016/s0960-894x(02)01050-8. Epub 2003/03/18. [DOI] [PubMed] [Google Scholar]
- 22.Zhong Z, Wen Z, Darnell JE., Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264(5155):95–8. doi: 10.1126/science.8140422. Epub 1994/04/01. [DOI] [PubMed] [Google Scholar]
- 23.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. The Journal of biological chemistry. 2001;276(48):45443–55. doi: 10.1074/jbc.M107527200. Epub 2001/10/02. [DOI] [PubMed] [Google Scholar]
- 24.Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, et al. Potent and selective phosphopeptide mimetic prodrugs targeted to the Src homology 2 (SH2) domain of signal transducer and activator of transcription 3. Journal of medicinal chemistry. 2011;54(10):3549–63. doi: 10.1021/jm2000882. Epub 2011/04/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Bai L, Bernard D, Nikolovska-Coleska Z, Gomez C, Zhang J, et al. Structure-Based Design of Conformationally Constrained, Cell-Permeable STAT3 Inhibitors. ACS medicinal chemistry letters. 2010;1(2):85–9. doi: 10.1021/ml100010j. Epub 2010/07/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT, Turkson J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochemical pharmacology. 2010;79(10):1398–409. doi: 10.1016/j.bcp.2010.01.001. Epub 2010/01/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Yue P, Page BD, Li T, Zhao W, Namanja AT, et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(24):9623–8. doi: 10.1073/pnas.1121606109. Epub 2012/05/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.