

Abstract
Ring chromosomes are structural aberrations commonly associated with birth defects, mental disabilities, and growth retardation1,2. Rings form upon fusion of the long and short arms of a chromosome, sometimes associated with large terminal deletions2. Due to the severity of these large-scale aberrations affecting multiple contiguous genes, no possible therapeutic strategies for ring chromosome disorders have so far been proposed. During cell division ring chromosomes can exhibit unstable behavior, leading to continuous production of aneuploid progeny with low viability and high cellular death rate3-9. The overall consequences of this chromosomal instability have been largely unexplored in experimental model systems. Here we generated human induced pluripotent stem cells (iPSCs)10-12 from patient fibroblasts containing ring chromosomes with large deletions and found that reprogrammed cells lost the abnormal chromosome and duplicated the wild type homologue via the compensatory uniparental disomy (UPD) mechanism. The karyotypically normal iPSCs with isodisomy for the corrected chromosome outcompeted co-existing aneuploid populations, allowing rapid and efficient isolation of patient-derived iPSCs devoid of the original chromosomal aberration. Our results suggest a fundamentally different function of cellular reprogramming as a means of “chromosome therapy”13 to reverse combined loss-of-function across many genes in cells with large-scale aberrations involving ring structures. In addition, our work provides an experimentally tractable human cellular model system for studying mechanisms of chromosomal number control, which is of critical relevance to human development and disease.
We obtained fibroblasts from a Miller Dieker Syndrome (MDS) patient with ring chromosome 17, subsequently referred to as ring(17). MDS is caused by heterozygous deletions of human band 17p13.314,15 (Fig. 1a). This deletion alone leads to craniofacial dysmorphisms, defective neuronal migration, abnormal cortical layering and nearly absent cortical folding with devastating neurological consequences such as mental retardation and intractable epilepsy14, 16. However, in this case the 17p13.3 deletion was in a ring chromosome, and the patient had a typical MDS phenotype14. To separate the effects of ring(17) from the 17p13.3 deletion, we obtained fibroblasts from two additional MDS patients with similar deletions but without ring(17) (Fig. 1b).
Figure 1. Reprogramming from fibroblasts with ring(17) produces multiple iPSC clones that do not have the ring chromosome.
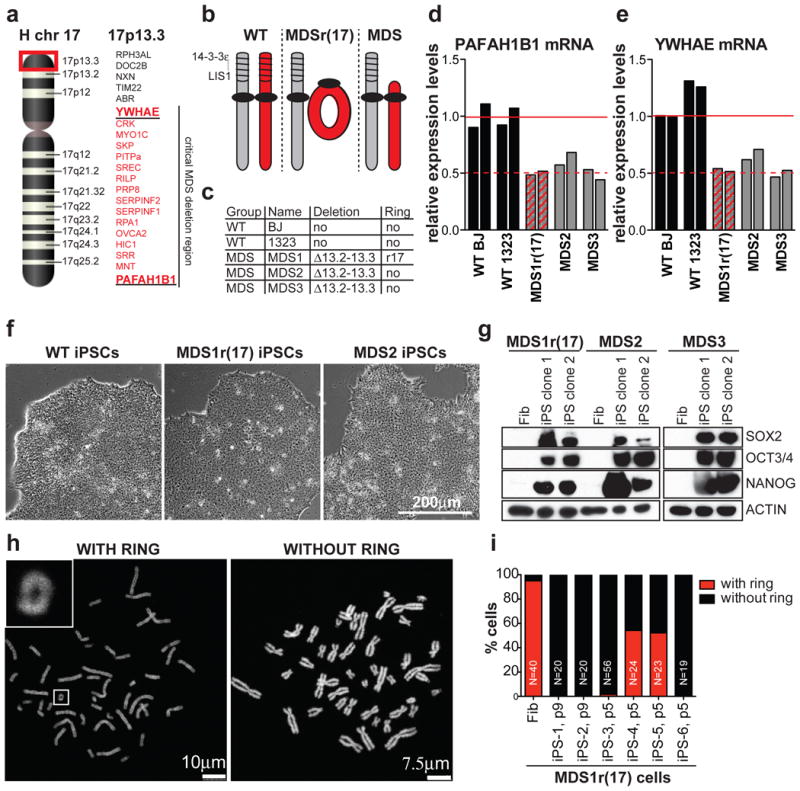
a, Schematic of human chromosome 17, highlighting band 17p13.3, which is deleted in MDS. Encoded genes, including minimal critical MDS deletion are listed on the right. b, Schematic of chr17 status in wild type (WT), MDS patients. c, List of fibroblast lines used in this study. d, e, RT-qPCR for PAFAH1B1 (d) and YWHAE (e) mRNA levels in fibroblasts. f, Images of WT, MDS1r(17) and MDS2 iPSCs. g, Western blot for pluripotency markers in fibroblasts and two iPSC clones. h, Examples of metaphase spreads with or without the ring chromosome. i, Percentage of mitotic cells with or without the ring chromosome in fibroblasts or MDS1r(17) iPSC clones.
Two critical genes deleted in MDS are PAFAH1B1 (encoding LIS1) and Ywhae (encoding 14-3-3ε) (Fig. 1a)15. Accordingly, MDS fibroblasts (MDS1r(17), MDS2 and MDS3, (Fig. 1c)), expressed reduced PAFAH1B1 and YWHAE mRNA compared to control fibroblasts (Fig. 1d, 1e). MDS1r(17) fibroblasts had a 46,XY,r(17) karyotype in 95% of the cells (Fig. 1i, 2a and Supplemental Fig. 1), with the remaining 5% exhibiting ring loss or secondary ring derivatives (Fig. 2b).
Figure 2. Karyotypically normal cells predominate in early passage iPSC clones derived from MDS1r(17) fibroblasts.
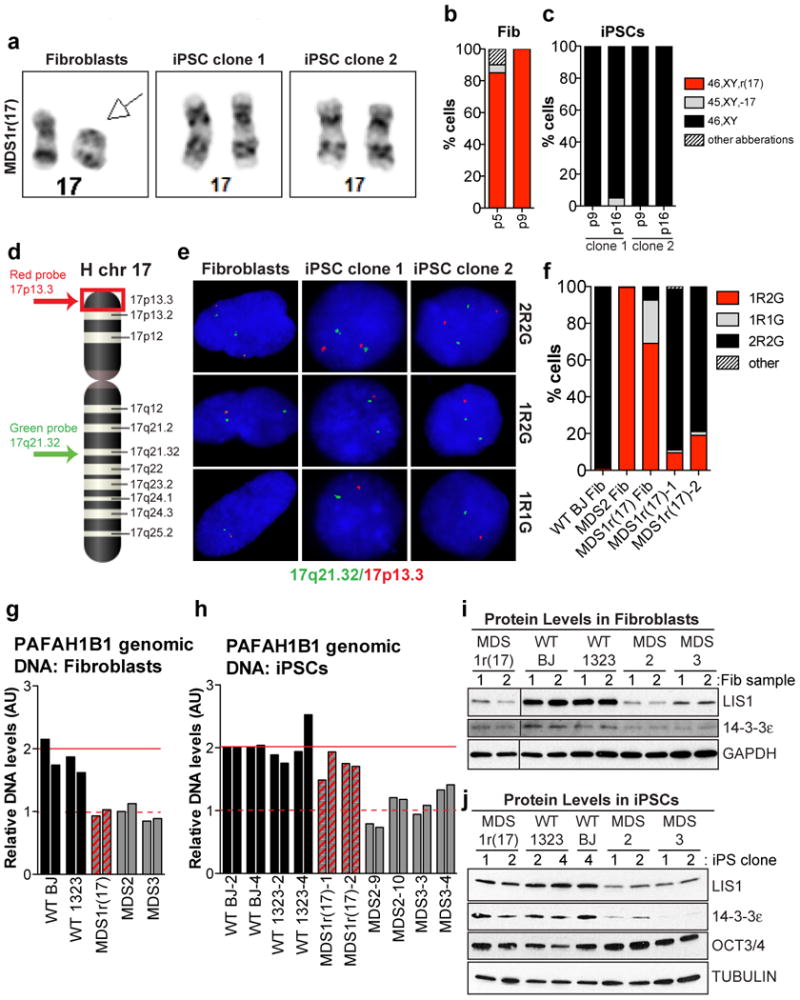
a, Images of chr17 pairs in MDS1r(17) fibroblasts and iPSC clones. b, c, Proportion of MDS1r(17) fibroblasts (b) and iPSC clones 1 & 2 (c) with various karyotypes (n=20 each). d, Approximate position of FISH probes on chr17. e, Signal patterns obtained with FISH probes in (d). f, Proportions of cells with various signal patterns (n=200 each). g, h, qPCR for genomic PAFAH1B1 DNA in fibroblasts (g) and iPSCs (h). i,j, Western blot for LIS1 and 14-3-3ε in fibroblasts (i) (2 samples/fibroblast line collected on different days) and iPSCs (2 clones/line) (j).
To investigate the behavior of ring chromosomes in actively proliferating cells, we generated iPSCs using non-integrating episomal vectors17. All MDS iPSCs were morphologically indistinguishable from wild type (Fig. 1f) and expressed stem cell markers (Fig. 1g and Supplemental Fig. 2a-2d). We confirmed that MDS iPSCs were free of exogenous factor integration (Supplemental Fig. 3a, 3b) and were functionally pluripotent, producing cell types of the three germ layers (Supplemental Fig. 4, 5).
We then analyzed six early passage MDS1r(17) iPSC clones for the presence of the ring and, remarkably, found that four out of six clones grew well, had proper morphology and did not have any detectible ring chromosomes (Fig. 1h, 1i). The two clones with rings differentiated or stopped growing upon subsequent passaging (Supplemental Fig. 6a, 6b). Analysis of chromosome composition revealed that stable clones had 46 chromosomes and no ring in 85-100% of cells, in contrast to <15% of cells in unstable clones (Fig. 1i and Supplemental Fig. 6). These results suggested that ring(17) was incompatible with reprogramming and/or stem cell maintenance using our methods.
Further cytogenetic analysis of the first two MDS1r(17) iPSC clones demonstrated a normal 46,XY karyotype without ring(17) (Fig. 2a-2c). In addition, the deletion, which was readily detectible by G-banding in MDS2 and MDS3 iPSCs, was not apparent in MDS1r(17) iPSCs (Fig. 2a and Supplemental Fig. 1). These findings could be explained by either clonal expansion of rare cells with a normal karyotype from mosaic fibroblasts; or repair or replacement of the ring chromosome during or after reprogramming. We reasoned that presence of a small fraction of cells with the deleted ring(17) in predominantly corrected iPSC clones would solidify their origin from an abnormal fibroblast. To test this, we analyzed interphase nuclei by fluorescence in situ hybridization (FISH) using a red probe to detect the subtelomeric band 17p13.3 together with a green probe to detect the long-arm band 17q21.32 (Fig. 2d, 2e). Consistent with karyotype data, ≥80% of cells in iPSC clones 1 and 2 demonstrated a normal signal pattern (2R2G) (Fig. 2f), suggesting that they have two intact copies of chromosome 17. However, 10-20% of interphase iPSCs had a 1R2G signal pattern, indicative of ring(17) with p13.3 deletion (Fig. 2f). These results revealed persistent mosaicism in MDS1r(17) iPSCs, confirming their origin from ring-containing fibroblasts in support of the second hypothesis above. Of note, despite several attempts (>120 cells analyzed in multiple experiments), no evidence of ring(17) was found in metaphase iPSCs, suggesting that in the pluripotent state such cells may be terminal and non-dividing. The expectation is that remaining interphase cells with ring(17) would be gradually depleted.
Cytogenetic and FISH results suggested that the MDS-associated deletion was rescued in MDS1r(17) iPSC clones 1 and 2. Consistent with that rescue, PAFAH1B1 genomic DNA levels were restored from 50% of wild type in the original fibroblasts (Fig. 2g) to wild type levels in iPSCs (Fig. 2h). On the other hand, in MDS2 and MDS3 cells without ring(17), PAFAH1B1 genomic DNA levels were consistently reduced before and after reprogramming (Fig. 2g and 2h). Similar results demonstrating rescue were obtained by western blot analysis for LIS1 and 14-3-3ε proteins (Fig. 2i, 2j). To investigate the status of the deleted locus before and after reprogramming in more detail, we performed single nucleotide polymorphism (SNP) microarrays. Analysis of total copy number for SNPs on chromosome 17 showed a clear decrease between positions 1 and 5700000 in MDS1r(17) fibroblasts, which was not observed in iPSCs (Fig. 3a, 3b). In contrast, large terminal deletions were apparent in iPSCs derived from MDS2 and MDS3 fibroblasts (Fig. 3c, 3d). Of note, the extent of the deletion in MDS3 cells was exactly the same as in MDS1r(17) fibroblasts, making it an ideal control for the effect of the ring. Therefore, 17p13.3 deletion is recapitulated in iPSCs, and does not severely compromise pluripotent stem cell growth, viability or function. In contrast, ring(17) appears to compromise in vitro stem cell maintenance and proliferation, and leads to preferential survival of iPSCs that lost or repaired the ring chromosome.
Figure 3. Rescue of MDS-associated deletion in iPSCs derived from ring(17) fibroblasts through compensatory uniparental isodisomy (UPD).
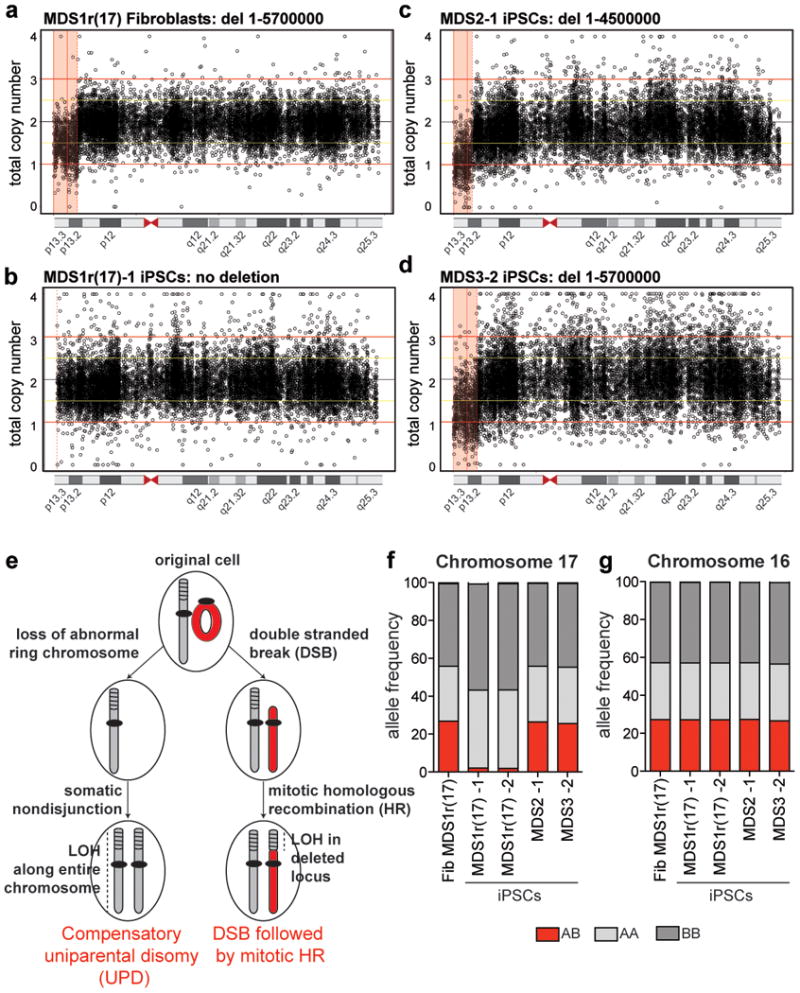
a-d, Total copy number of SNPs across chr17 in MDS1r(17) fibroblasts (a), MDS1r(17) iPSC clone 1 (b), MDS2 iPSC clone 1 (c), and MDS3 iPSC clone 2 (d). The pink shaded areas represent the deletions. e, Proposed mechanisms for how cells with ring(17) end up with two intact chromosomes 17 after reprogramming. f, g, Frequency of heterozygous (red) or homozygous (shades of gray) SNPs on chr17 (f) or chr16 (g).
We hypothesized two possible mechanisms that could lead to loss of ring(17) and rescue of the deleted locus. The first mechanism, called compensatory uniparental disomy (UPD), also referred to as monosomy correction, is known to occur during human development, and involves replacement of an abnormal chromosome with a copy of its normal homologue18. This is a two-step process that could proceed through loss of ring(17) due to anaphase lag, followed by duplication of the wild type chromosome 17 by nondisjunction (Fig. 3e). The second mechanism could occur through a ring opening due to a double strand break19, followed by homology based repair or somatic recombination20,21 with the wild type homologue (Fig. 3e). In both cases selection is likely to favor cells with 46 normal chromosomes. However, in the first scenario we would expect both chromosomes 17 in iPSCs to be isodisomic—i.e. completely homozygous, whereas in the second scenario we would expect partial loss of heterozygosity (LOH) at the repaired locus with the rest of chromosome 17 being heterozygous (Fig. 3e). To distinguish between these possibilities, we used data from SNP arrays to determine heterozygosity on chromosome 17 in fibroblasts and iPSCs. Consistent with compensatory UPD, the two homologues of chromosome 17 were entirely homozygous in both iPSC clones derived from MDS1r(17) cells, whereas the original fibroblasts as well as MDS2 and MDS3 iPSCs exhibited 27% heterozygosity (Fig. 3f). This was a specific effect for the corrected chromosome 17 (Fig. 3g).
Nonrandom gain of a third chromosome 17 occurs frequently in long-term culture of human pluripotent cells due to a proliferative growth advantage 22-25. To test whether chromosomes not typically gained or implicated in advantageous stem cell growth can replace the corresponding ring chromosome in iPSCs, we reprogrammed fibroblasts from two different cases involving ring(13) with large deletions (Fig 4a, 4f) associated with multiple congenital anomalies. We confirmed the presence of ring(13) in ∼80-100% of the fibroblasts, with the remaining cells exhibiting 45 chromosomes minus ring(13) or non-clonal aberrations involving the ring structure (Fig. 4b, 4d and Supplemental Fig. 7a, 7d). Initial examination of iPSCs at passage six revealed 46,XY,r(13) karyotypes, although a fraction of cells had lost the ring chromosome (Fig. 4c, 4e). However, six out of nine clones that were examined after passage eight had a normal karyotype (Fig. 4a, 4c, 4e, and Supplemental Fig. 7). In addition, GM00285 iPSC clone 4 transitioned from predominantly 46,XY,r(13) at passage six to 46,XY at passage twelve (Fig. 4c). These results demonstrate dynamic mosaicism between clonally related cells, leading to preferential survival of karyotypically normal iPSCs in a matter of six passages, which did not occur in the fibroblasts (Fig. 4b. 4d). In agreement, FISH studies revealed normal signal patterns in the corrected iPSC clones (Fig. 4f-4h). Finally, SNP microarray analysis confirmed rescue of the deleted regions (Supplemental Fig. 8a-8e) and showed that corrected iPSCs are completely homozygous for chromosome 13 (Supplemental Fig. 8f, 8g). The repaired iPSCs expressed stem cell markers (Supplemental Fig. 2e-2j), differentiated into three germ layers (Supplemental Fig. 9), and were free of exogenous factor integration (Supplemental Fig. 3c, 3d).
Figure 4. Derivation of iPSC clones with a normal karyotype from fibroblasts with ring(13).
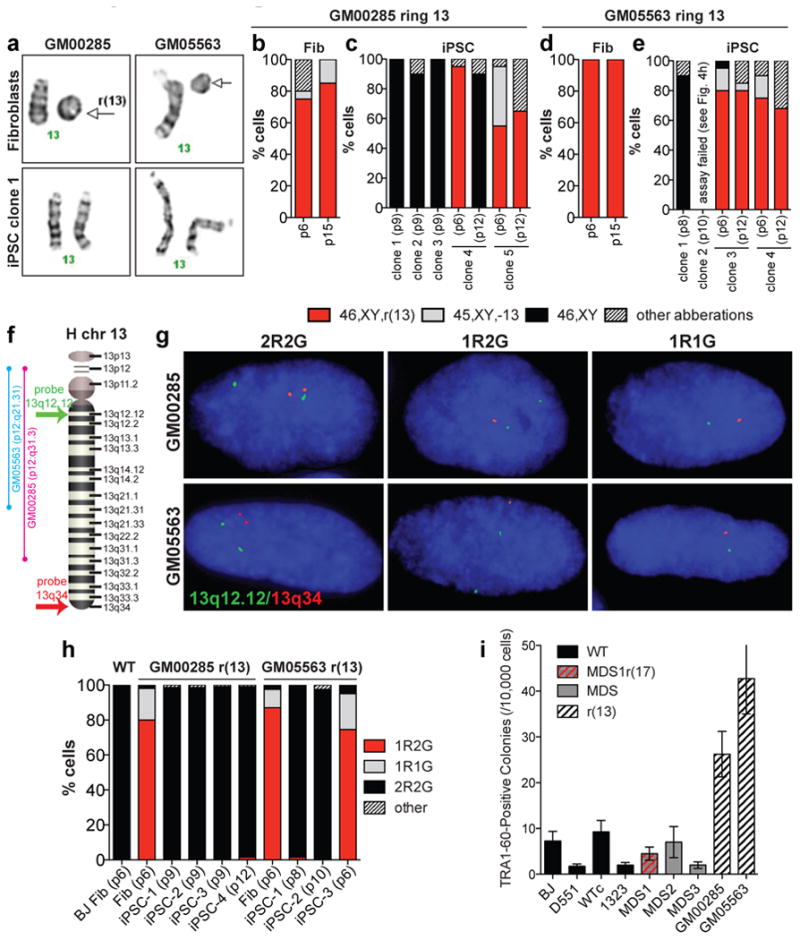
a, Images of chr13 pairs in fibroblasts and repaired iPSCs from two individuals with ring(13). b-e, Proportions of fibroblasts and iPSCs with various karyotypes (n=20 each). f, Schematic of chr13 showing approximate position of FISH probes used in (g). g, Examples of signal patterns obtained with FISH probes. h, Proportions of cells with various signal patterns (n=200 each). i, Number of TRA-1-60-positive colonies on day 25 per 10,000 electroporated cells. Results are mean±s.e.m., (N = 4).
Finally, to further rule out the possibility that repaired iPSC clones originated from rare 46,XY fibroblasts, we measured reprogramming rates for the lines used in this study. If there is a bias against reprogramming from cells with ring chromosomes, then we would expect reduced reprogramming efficiency from ring-containing fibroblast populations. However, we found no significant difference between wild type and MDS cells, while reprogramming efficiency from ring(13) fibroblasts was increased compared to wild type (Fig. 4i). Therefore, ring chromosomes do not interfere with reprogramming using this method. More likely, reprogramming increases cell divisions, increasing the probability that random nondisjunction compensates for the loss of the corresponding ring. Nondisjunction leading to chromosomal trisomy is the most commonly reported karyotypic change in cultured human ES/iPS cells23, because mitotic checkpoints that ensure proper alignment and disentanglement of sister chromatids before separation are less efficient than in differentiated cells 23,26-28. In this case, isodisomic cells have a growth advantage over cells with ring chromosomes including large deletions; however, they are not exactly the same as wild type. UPD could lead to undesirable clinical consequences through homozygosity for a recessive mutation or disruption of imprinting18. Thus, expression of imprinted genes and presence of recessive mutations would have to be carefully monitored and/or corrected for any therapeutic applications.
Compensatory UPD and dynamic mosaicism occur in actively proliferating cells in vivo, including cases involving ring(21), where patients' lymphocytes transitioned from predominantly 46,XX,r(21) to 46,XX (with UPD21) several years later 29,30. These observations further suggest that taking advantage of this intrinsic property of iPSCs may be a feasible approach to correct combined loss-of-function and structural aberrations associated with additional ring chromosomes. Given that repair of large deletions involving multiple genes is not possible with current genome editing techniques, our results may have broad implications for regenerative medicine and advance the newly emerging concept of chromosome therapy13. More broadly, our findings underscore the importance of studying chromosome repair in stem cells and establish a novel platform for investigating the mechanisms that regulate chromosome number during early human development.
Methods (Online Version)
Cell Culture
hiPSCs were generated from these HDFs within six passages after the receipt. The study involved use of existing specimens that are publicly available and provided without any identifiers linked to the subjects. All procedures were approved by the UCSF Committee on Human Research and the Germ and Embryonic Stem Cell Research Committee. Human fibroblasts and SNL feeder cells (Health Protection Agency Culture Collection) were cultured in DMEM containing 10% FBS, 1% GlutaMAX, 0.1 mM MEM nonessential amino acids (Invitrogen), 1 mM sodium pyruvate, and penicillin-streptomycin (all from Invitrogen). For passaging, fibroblasts were washed once with Dulbecco's PBS (DPBS; calcium and magnesium free) and incubated with TrypLE Express (Invitrogen) at 37°C. When cells were dissociated, the TrypLE Express was removed, and the cells were washed with culture medium described above. The cells were collected into a 15-ml conical tube and spun down. The contents were transferred to a new dish. The split ratio was routinely 1:3 to 1:20. Established hiPSC lines were cultured in mTeSR1 medium (Stem Cell Technologies) on Matrigel (BD) or Synthemax (Corning) -coated dishes. For passaging, hiPSCs were washed once with DPBS and incubated with Accutase (Millipore) at 37°C. When colonies at the edge of the dish started dissociating from the bottom, Accutase was removed, and mTeSR1 medium was added. Cells were scraped and replated into new dishes in mTeSR1 with 10 μM ROCK inhibitor (Y-27632: Millipore). The split ratio was routinely 1:3 to 1:20. Next day, the media was replaced with mTeSR1 without ROCK inhibitor.
Generation of Human iPSCs with Episomal Vectors
Human iPSCs were generated with episomal plasmids as described17 with some modifications. Three micrograms of expression plasmid mixtures (epiY4) were electroporated into 3 × 105 fibroblasts with a Neon Electroporation Device (Invitrogen) with a 100-μl kit, according to the manufacturer's instructions. Conditions for electroporations were 1,650 V, 10 ms, and three pulses. Cells were detached within 1 week after electroporation and seeded at 1-3× 105 cells per 10-cm dish onto irradiated or mitomycin-C treated SNL feeder cells. The culture medium was replaced the next day with primate ESC medium (Reprocell) containing bFGF. Colonies were counted 25 days after electroporation, and those colonies similar to hESCs were selected for further cultivation and evaluation. For calculating reprogramming efficiency, 1 × 104 electroporated cells were seeded per well of 6-well plate, and the number of TRA-1-60 positive colonies were counted on day 25. The generation and characterization of WT iPSC clones from BJ and HDF-1323 was previously described 31,32. These lines were free of episomal integration, maintained pluripotency, self-renewal and normal karyotype 31,32.
In Vitro Differentiation
For EB formation from hiPSCs, the cells were harvested by treating with Accutase. The clumps of the cells were transferred to ultra-low-attachment dishes (Corning) in DMEM/F12 containing 20% knockout serum replacement (KSR, Invitrogen), 2 mM L-glutamine, 100 mM nonessential amino acids, 100 mM 2-mercaptoethanol (Invitrogen), and penicillin-streptomycin. The medium was changed every other day. After 8 days as a floating culture, EBs were transferred to gelatin-coated plate and cultured in the same medium for another 8 days.
Teratoma Formation
hiPSCs cells were dissociated by Accutase treatment. Cell pellets were resuspended in mTeSR1 complete medium with 10 μM Rock inhibitor, Y-27632. One confluent well of cells from a 6-well-plate was used for each injection into the testis of 8-10 week old CB17 SCID male mice (Charles River). Tumor samples were collected 6.5-11 weeks after injection, fixed with PBS containing 4% paraformaldehyde, and processed for paraffin-embedded sections by the Gladstone Histology and Microscopy Core. All mouse studies were approved by the UCSF Institutional Animal Care and Use and Committee.
Immunocytochemistry
For counting the TRA-1-60-positive colonies in hiPSC generation experiments, cells were fixed with PBS containing 4% paraformaldehyde for 10 minutes at room temperature and reacted with ImmunoPure Peroxidase Suppressor (Thermo Scientific). The cells were washed with PBS and treated with 1% BSA for blocking. Primary antibodies used were biotin-conjugated anti-TRA-1-60 antibody (1 μg/ml, eBiosciences). Immunostaining was performed using HRP-conjugated streptavidin (1 μg/ml, Thermo Scientific) and ImmunoHisto Peroxidase Detection Kit (Thermo Scientific), according to the manufacturer's instructions. For immunocytochemistry of pluripotency and differentiation markers, cells were fixed with PBS containing 4% paraformaldehyde for 10 minutes at room temperature, permeabilized with PBS containing 0.1% Triton X-100 for 10 minutes at room temperature and then washed with PBS and treated with 1% BSA for blocking. The following primary antibodies were used: SSEA3 (0.5 μg/ml, eBiosciences), SSEA4 (0.5 μg/ml, eBiosciences), TRA-1-60 (0.5 μg/ml, eBiosciences), TRA-1-81 (0.5 μg/ml, eBiosciences; 1:250, MAB4381, Millipore), NANOG (2 μg/ml, AF1997, R&D Systems; 1:250, Ab21624, Abcam), SOX2 (1:500, Ab59776, Abcam), OCT3/4 (1:250, sc-5279, Santa Cruz), OCT4 (1:250, 09-0023, Stemgent), KLF4 (1:100, Ab75486, Abcam), βIII-tubulin, aka TUJ1 (1:100, MAB1637, Millipore; 1:1,000, MMS-435P, Covance), α-smooth muscle actin, aka SMA (pre-diluted, N1584, DAKO; 1:200, A2547, Sigma), α-fetoprotein aka AFP (2 μg/ml, MAB1368, R&D Systems; 1:200, A8452, Sigma), and MAP2 (1:200, MAB3418, Millipore). Secondary antibodies used were Alexafluor488 or 555-conjugated goat anti-mouse IgG (1:200, Invitrogen), Alexa488 or 555-conjugated goat anti-rabbit IgG (1:200, Invitrogen), and Alexa488 or 555-conjugated donkey anti-goat IgG (1:200, Invitrogen). Nuclei were stained with DAPI contained in the Vectashield set (Vector Laboratories) or DAPI in ProLong gold antifade reagent (Invitrogen).
Western blot
Cells were lysed with RIPA buffer (150mM sodium chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50mM Tris, pH 8.0) supplemented with protease and phosphotase inhibitor cocktails (Roche). Protein concentration was determined with BCA assay (Biorad) according to manufacturer's instructions, and samples were then diluted to the same concentration with lysis buffer. Prior to loading, samples were denatured with Laemmli 6× buffer and boiled for 5min. Equal amounts of total protein (typical range 15-30μg) were loaded onto 4-20% TGX gradient gels (Biorad) and separated by SDS-PAGE. Proteins were transferred onto nitrocellulose membranes (Biorad), blocked for 1hr with TBS-T buffer containing 5% nonfat dry milk, incubated with primary antibodies in blocking buffer overnight at 4°C, rinsed with TBS-T, and incubated with secondary antibodies in blocking buffer for 1hr at room temp. The following primary antibodies were used: SOX2 (1:1,000, Ab59776, Abcam), OCT3/4 (1:250, sc-5279, Santa Cruz), NANOG (1:250, Ab21624, Abcam), ACTIN (1:10,000, A2228, Sigma), LIS1 (1:1,000, kind gift of Dr. Shinji Hirotsune, Osaka, Japan), 14-3-3ε (1:250, sc-1020, Santa Cruz), GAPDH (1:5,000, ACR001P, Acris), α-TUBULIN (1:2,000, T6074, Sigma). HRP-conjugated secondary antibodies (Jackson immunoresearch laboratories) were used at 1:10,000. Development was done with ECL detection kits (Pierce) according to manufacturer's instructions.
Quantitative PCR (qPCR)
Genomic DNA was isolated with QIAmp DNA micro kit (Qiagen). To isolate RNA, cells were lysed with TRIzol reagent. Following chloroform extraction, aqueous phase containing RNA was mixed with 70% ethanol, transferred onto mini spin column, isolated with RNeasy mini kit (Qiagen), and digested with DNase I (Qiagen). DNA/RNA quality and concentration were assessed using a nanodrop. cDNA was prepared with iScript cDNA synthesis kit (Biorad). qPCR was done with SsoFast EvaGreen Supermix (Biorad) using a Vii7 instrument (Applied Biosystems). To check exogenous reprogramming factor integration, we performed qPCR on genomic DNA isolated from iPSCs, using published primers17. To measure endogenous genomic DNA levels by qPCR, the following primers sets were used: PAFAH1B1 (set1 Forward: 5′-ACCCTTCAGGGCTTTCATTT; Reverse: 5′-CCATTGGGCATAGGTCTCAT; set4 Forward: 5′-TTCTCGCCTGAACCATCTTT; Reverse: 5′-GTGCTGCCTGTAGTGTGGAA) normalized to ACTA1 (Forward: 5′-CAGAGAGGATGGGCACTAGC; Reverse: 5′-CGAGACCCCACTGAGAAGTC) and B2M (Forward: 5′-TGCTGTCTCCATGTTTGATGTATCT; Reverse: 5′-TCTCTGCTCCCCACCTCTAAGT). For qPCR from cDNA, the following primers were used: PAFAH1B1 (Forward: 5′-TGGGTACGTGGAGTTCTGTT; Reverse: 5′-TGTGGAAATCCAAGGAGGTA), YWHAE (Forward: 5′-ATGGATGTGGAGCTGACAGT; Reverse: 5′- CCAGTGTTAGCTGCTGGAAT) normalized to TBP (Forward: 5′-TATAATCCCAAGCGGTTTGC; Reverse: 5′-GCTGGAAAACCCAACTTCTG). We calculated amplification efficiency (E) for all the primers above, and ensured that % efficiency was close to 100% (±5%) in order to detect 2-fold differences. Relative ratios were determined using Pfaffl method33, 34, taking into account primer efficiency.
SNP genotyping and CNV Calling
SNP genotyping to determine CNV presence and UPD status for MDS samples was performed using the Affymetrix Axiom EUR array platform by the Genomics Core Facility (GCF) at UCSF, using standard protocols. The Axiom EUR array assays approximately 675,000 SNPs across the genome, and is optimized for genome-wide, gene-based, and candidate-SNP coverage35. SNP genotype calls were generated using the “apt-probeset-genotype” function in Affymetrix Power Tools. Heterozygosity was determined in R36 by counting the number of individuals with homozygous genotypes and the ones with heterozygous genotypes. This has been done on chromosome 17, the chromosome of interest, and chromosome 16, the control chromosome.
For CNV estimation and visualization, we used a similar procedure to previous successful methods, as available CNV calling tools such as PennCNV, QuantiSNP and Birdsuite are not able to handle Axiom data. First, quantile normalization (QN) transforms the quantiles for each array to have the same distribution (Bolstad, 2001). This was performed using the “apt-probeset-genotype” function in Affymetrix Power Tools on a complete plate of arrays (N=95). Second, to estimate integer copy number, we use the COPy number Polymorphism Evaluation Routine (COPPER), previously developed to call CNVs from SNP array data37. In brief, COPPER uses genotype calls to set the median for 0, 1, and 2 copies of each allele and transform the intensity axes appropriately to derive scaled intensity estimates. This procedure was implemented in R. After QN and COPPER, we sum up the intensities on each axis, which represents a total copy number estimate at each locus for each individual.
After the first iteration of this procedure, genome-wide variability in each individual's copy number graph is manually assessed and individuals with excess variability are excluded. Typical individuals have variance ∼1, so individuals with variance across entire chromosomes ≫1 are excluded, leaving 87 samples in this case for the second run of QN and COPPER to perform CNV calling. To take into account the non-independence of consecutive SNPs, we utilize a 5-SNP median, as the Copy Number (CN) of a locus is estimated more accurately by including information from SNP probes on each side. We then apply a HMM to make the CNV calls, similar to QuantiSNP 38, implemented in Python.
SNP genotyping to determine CNV presence and UPD status for ring(13) samples was performed using the Agilent SurePrint G3 Human CGH+SNP 4×180K array platform by Cell Line Genetics, using standard protocols. Labeling reactions were prepared using the Agilent Enzymatic Labeling Protocol for aCGH (Version 7.1) with 1 μg total DNA input. The protocol consists of two parts: Labeling of the DNA and hybridization. First, 1 μg of DNA is fragmented with restriction enzymes and the test DNA labeled with Cyanine 5dUTP and reference DNA with Cyanine 3-dUTP by ExoKlenow fragment. Labeled DNA is then purified using Millipore Amicon Ultracel-30 filters and the labeling efficiency determined by the NanoDrop spectrophotometer. Next, the labeled DNA is prepared for hybridization with human Cot1 DNA and placed on the Sureprint G3 human CGH + SNP 4×180k microarray and hybridized at 65°C for ∼ 24hours. Finally, the arrays are washed and scanned at 3μM resolution on an Agilent G2565CA High Resolution Scanner. Data was processed through Agilent's Feature Extraction software version 11.0.1.1 using the protocol CGH_1100_Jul11 and the 029830_D_F_20100916 grid file. Analyses of CNV estimation and visualization were performed using Agilent genomic Workbench 7.0 software.
The microarray data reported in this paper have been deposited to NCBI GEO with the accession numbers GSE52585 and GSE52691.
Karyotype and FISH Analysis
Non-banded chromosome number and ring status analyses were performed as described previously39. DNA was stained with DAPI in ProLong gold antifade reagent (Invitrogen) and mitotic chromosomes were visualized using a 100× objective on a Leica TCS SP5 confocal microscope. Additional chromosomal analyses were performed on twenty G-banded metaphase cells from each sample at Cell Line Genetics or the UCSF cytogenetics laboratory. FISH analyses were performed on two hundred interphase nuclei from each sample at Cell Line Genetics as described previously39. A green probe to detect the pericentromeric long-arm band 13q12.12 together with a red probe to detect the subtelomeric band 13q34 were used for chromosome 13. A green probe to detect the subtelomeric long-arm band 17q21.32 together with a red probe to detect the subtelomeric short-arm band 17p13.3 were used for chromosome 17.
DNA fingerprinting
The short tandem repeat (STR) profile assays were performed at Cell Line Genetics. They used the Powerplex 16 (Promega) kit and followed the manufacturer's recommendation to obtain the results. These samples were run in duplicate and blinded to the interpreter. All the established iPSC lines in this study were confirmed to be identical with their original fibroblasts by DNA fingerprinting (Supplemental Table 1).
Supplementary Material
Supplemental Table 1. Summary of DNA fingerprinting for cells used in this study. The origin of all the iPSC lines derived from fibroblasts with ring chromosomes was confirmed to be from the expected fibroblast source. All the patterns in these samples did not match the cell lines published in the ATCC, NIH, or DSMZ website.
{kind=link}
Supplemental Figure 9. EB differentiation from ring(13)-derived iPSCs. a-c, Immunocito-chemistry showing expression of endoderm-derived cells positive for AFP (a), mesoderm-derived cells positive for SMA (b), and ectoderm-derived cells positive for MAP2 (c) generated in vitro using embryoid body method with corrected iPSC clones GM00285-1, GM00285-3, and GM05563-1.
{kind=link}
Supplemental Figure 1. Karyotypes of MDS cells used in this study. a, b, Representative karyotypes of MDS1r(17) fibroblasts (a) and two independent iPSC clones from each of the three MDS patients (b). Magnifications of chromosome 17 pairs (dotted squares) are shown in Fig. 2a for MDS1r(17) cells (boxes 1-3) and to the right of panel (a) for MDS2 and MDS3 iPSCs (boxes 4-7). c, Summary of karyotype data from G-banding analyses of MDS fibroblasts and iPSC clones 1 and 2 (n=20 each).
{kind=link}
Supplemental Figure 2. Expression of pluripotency markers in iPSCs. Immunocytochemistry of MDS (a-d) and ring(13)-derived (e-j) iPSCs with the indicating antibodies.
Supplemental Figure 3. No evidence of episomal factor integration in MDS iPSCs. qPCR from genomic DNA using primers specific for exogenous KLF4, SOX2, OCT4, L-MYC, and LIN28, as described previously17. a, b, Relative levels of episomal factors in MDS fibroblasts on day 15 after electroporation with epiY4 mixture17 (positive control), WT and MDS iPSCs (passages 20-25), as well as WT human embryonic stem cell line HSF-1 (negative control) (N=1). c, d, Episomal factor copy number per cell in ring(13) fibroblasts electroporated with epiY4 mixture (positive control) and iPSCs (N=1)
{kind=link}
Supplemental Figure 4. EB differentiation from MDS iPSCs. a-c, Immunocytochemistry showing expression of endoderm-derived cells positive for AFP (a), mesoderm-derived cells positive for SMA (b), and ectoderm-derived cells positive for TUJ1 (c) generated in vitro using embryoid body method with MDS1r(17), MDS2 and MDS3 iPSCs.
{kind=link}
Supplemental Figure 5. Teratoma formation from MDS iPSCs. a-c, Histological sections from 6.5-11 week teratomas developed in the testis of SCID mice following injection with MDS1r(17), MDS2 and MDS3 iPSCs. Hematoxylin and eosin (H&E) staining reveals characteristic tissues from the mesoderm (a), endoderm (b), and ectoderm (c). d, List of MDS iPSC lines that were injected and teratoma efficiency.
{kind=link}
Supplemental Figure 6. Cell morphology and chromosome distribution of additional MDS1r(17) iPSC clones. a-c, Cell morphology of poorly growing MDS1r(17) clones 4 and 5 at passage 4 on feeders (a) and passage 5 on matrigel (b), compared to well growing clones 3 and 6 at passage 5 on matrigel (c). d, examples of metaphase spreads observed in MDS1r(17) clones. e, Quantification of cell populations in MDS1r(17) clones 3-6 with various chromosome compositions shown in (d).
{kind=link}
Supplemental Figure 7. Karyotypes of ring(13) cells and morphology of corrected iPSC clones. a-c, Representative karyotypes of GM00285 fibroblasts (a) and corrected GM00285 iPSC clones 1 and 3 (b, c). d,e, Representative karyotypes of GM05563 fibroblasts (d) and corrected GM05563 iPSC clone 1 (e). f-h, Representative morphologies of corrected iPSC clones derived from ring(13) fibroblasts.
{kind=link}
Supplemental Figure 8. Rescue of ring(13)-associated deletions in corrected iPSC clones through compensatory UPD. a-e, Total copy number of SNPs across chromosome 13 in ring(13) GM00285 fibroblasts (a), GM05563 fibroblasts (b), and karyotypically normal GM00285 iPSC clones 1 (c) and clone 3 (d), and GM05563 iPSC clone 1 (e). The areas shaded in pink represent the deletions. f,g, Frequency of heterozygous (blue) or homozygous (red) SNPs in ring(13) fibroblasts and karyotypically normal iPSC clones for chromosome 13 (f) and chromosome 12 (g). The corrected iPSC clones are completely homozygous for chromosome 13, supporting the compensatory UPD mechanism.
{kind=link}
Acknowledgments
The authors wish to thank Nick Larocque and Susan Fisher for access to the UCSF Human Embryonic Stem Cell Shared Research and Training Facility. We thank Paul Tesar and Zachary Nevin for comments on the manuscript; Deepak Srivastava and Bruce Conklin for advice; Haim Belinson, Alex Pollen, and Tom Nowakowski for helpful discussions; Shinji Hirotsune for LIS1 antibody; Karena Essex for administrative support; and the Gladstone Stem Cell, Histology and Microscopy, and Bioinformatics Cores for technical support. The research was made possible by NIH/NIGMS postdoctoral training grant in medical genetics (Grant Number GM007085-32) and a postdoctoral training fellowship from the California Institute for Regenerative Medicine (Grant Number TG2-01153) to MB; and the Uehara Memorial Foundation and USCF's Program for Breakthrough Biomedical Research to YH; and NHLBI/NIH (UO1HL098179), the Leading Project of MEXT (Japan), the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program) of the JSPS (Japan), Grants-in-Aid for Scientific Research of the JSPS and MEXT (Japan), the Program for Promotion of Fundamental Studies in Health Sciences of NIBIO (Japan), L.K. Whittier Foundation, and the Roddenberry Foundation to SY. Gladstone Institutes received support from a National Center for Research Resources Grant RR18928. SY is a member without salary of the scientific advisory boards of iPierian, iPS Academia Japan, Megakaryon Corporation, and HEALIOS K.K. Japan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of CIRM or any other agency of the State of California.
Footnotes
Author Contributions: MB conceived and designed the study, generated and characterized MDS iPSC lines, performed experiments, analyzed data, created the figures and wrote the manuscript. YH helped generate MDS iPSC lines, generated and characterized WT and ring(13) iPSC lines, designed the study, performed experiments and analyzed data. GD and LW performed SNP array genotyping and CNV calling analyses for MDS samples. EH, SS and KT provided technical support for various experiments. AK provided advice, access to equipment and laboratory space for MB. SY supervised the study, provided advice, laboratory space and financial support. AWB supervised the study, provided advice, helped with design and interpretation, and provided laboratory space and financial support. AWB, YH, SY, AK, EH and LW edited the manuscript. All authors read and approved the final version of the manuscript.
References
- 1.Cote GB, et al. The cytogenetic and clinical implications of a ring chromosome 2. Ann Genet. 1981;24:231–235. [PubMed] [Google Scholar]
- 2.Kosztolanyi G. Does “ring syndrome” exist? An analysis of 207 case reports on patients with a ring autosome. Hum Genet. 1987;75:174–179. doi: 10.1007/BF00591082. [DOI] [PubMed] [Google Scholar]
- 3.McClintock B. The production of homozygous deficient tissues with mutant characteristics by means of the aberrant mitotic behavior of ring-shaped chromosomes. Genetics. 1938;23:315–376. doi: 10.1093/genetics/23.4.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kistenmacher ML, Punnett HH. Comparative behavior of ring chromosomes. Am J Hum Genet. 1970;22:304–318. [PMC free article] [PubMed] [Google Scholar]
- 5.Tommerup N, Lothe R. Constitutional ring chromosomes and tumor suppressor genes. Med Genet. 1992;29:879–882. doi: 10.1136/jmg.29.12.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jobanputra V, et al. Changes in an inherited ring (22) due to meiotic recombination? Implications for genetic counseling. Am J Med Genet Part A. 2009;149A:1310–1314. doi: 10.1002/ajmg.a.32680. [DOI] [PubMed] [Google Scholar]
- 7.Mantzouratau A, et al. Meiotic and mitotic behaviour of a ring/deleted chromosome 22 in human embryos determined by preimplantation genetic diagnosis for a maternal carrier. Mol Cytogenet. 2009;2 doi: 10.1186/1755-8166-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sodre CP, et al. Ring chromosome instability evaluation in six patients with autosomal rings. Genet & Mol Res. 2010;9:134–143. doi: 10.4238/vol9-1gmr707. [DOI] [PubMed] [Google Scholar]
- 9.Zhang HZ, et al. Unique genomic structure and distinct mitotic behavior of ring chromosome 21 in two unrelated cases. Cytogenet & Genome Res. 2012;136:180–187. doi: 10.1159/000336978. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi K, et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 11.Yu J, et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 12.Park IH, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2007;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 13.Jiang J, et al. Translating dosage compensation to trisomy 21. Nature. 2013 doi: 10.1038/nature12394. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobyns WB, et al. Miller-Dieker syndrome: Lissencephaly and monosomy 17p. J Ped. 1983;102:552–558. doi: 10.1016/s0022-3476(83)80183-8. [DOI] [PubMed] [Google Scholar]
- 15.Cardoso C, et al. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet. 2003;72:918–930. doi: 10.1086/374320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wynshaw-Boris A, et al. Lissencephaly: mechanistic insights from animal models and potential therapeutic strategies. Semin Cell Dev Biol. 2010;21:823–830. doi: 10.1016/j.semcdb.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nat Method. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 18.Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays. 2000;22:452–459. doi: 10.1002/(SICI)1521-1878(200005)22:5<452::AID-BIES7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 19.Speevak MD, et al. Molecular characterization of an inherited ring (19) demonstrating ring opening. Am J Med Genet Part A. 2003;121A:141–145. doi: 10.1002/ajmg.a.20184. [DOI] [PubMed] [Google Scholar]
- 20.Hussein SM, et al. Genome damage in induced pluripotent stem cells: assessing the mechanisms and their consequences. Bioessays. 2013;35:152–162. doi: 10.1002/bies.201200114. [DOI] [PubMed] [Google Scholar]
- 21.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Draper JS, et al. Recurrent gain of chromosomes17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22:53–54. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- 23.Baker DE, et al. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol. 2007;25:207–215. doi: 10.1038/nbt1285. [DOI] [PubMed] [Google Scholar]
- 24.Spits C, et al. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat Biotechnol. 2008;26:1361–1363. doi: 10.1038/nbt.1510. [DOI] [PubMed] [Google Scholar]
- 25.Azuhata T, et al. The inhibitor of apoptosis protein survivin is associated with high-risk behavior of neuroblastoma. J Pediatr Surg. 2001;36:1785–1791. doi: 10.1053/jpsu.2001.28839. [DOI] [PubMed] [Google Scholar]
- 26.Damelin M, et al. Decatenation checkpoint deficiency in stem and progenitor cells. Cancer Cell. 2005;8:479–484. doi: 10.1016/j.ccr.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Wilton L. Preimplantation genetic diagnosis for aneuploidy screening in early human embryos: a review. Prenat Diagn. 2002;22:512–518. doi: 10.1002/pd.388. [DOI] [PubMed] [Google Scholar]
- 28.Vanneste E, et al. Chromosome instability is common in human cleavage-stage embryos. Nat Med. 2009;15:577–583. doi: 10.1038/nm.1924. [DOI] [PubMed] [Google Scholar]
- 29.Petersen MB, et al. Uniparental isodisomy due to duplication of chromosome 21 occurring in somatic cells monosomic for chromosome 21. Genomics. 1992;13:269–274. doi: 10.1016/0888-7543(92)90242-k. [DOI] [PubMed] [Google Scholar]
- 30.Bartsch O, et al. “Compensatory” uniparental disomy of chromosome 21 in two cases. J Med Genet. 1994;31:534–540. doi: 10.1136/jmg.31.7.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsiao EC, et al. Human Fibrodysplasia Ossificans Progressiva Induced Pluripotent Stem Cells show Increased Mineralization and Cartilage Formation. doi: 10.1186/1750-1172-8-190. In revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayashi Y, et al. Natural Human Mutation in ACVR1 Promotes Reprogramming to Pluripotency by BMP-SMAD-ID Axis by Suppressing p16/INK4A-Dependent Cell Senescence. In revision. [Google Scholar]
- 33.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Real-Time PCR applications guide. Bio-Rad Laboratories. 2006 [Google Scholar]
- 35.Hoffmann TJ, et al. Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics. 2011;90:79–89. doi: 10.1016/j.ygeno.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. URL http://www.R-project.org. [Google Scholar]
- 37.Weiss LA, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 38.Colella S, et al. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meisner LF, Johnson JA. Protocols for cytogenetic studies of human embryonic stem cells. Methods in Stem Cell Research. 2008;45:133–141. doi: 10.1016/j.ymeth.2008.03.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Summary of DNA fingerprinting for cells used in this study. The origin of all the iPSC lines derived from fibroblasts with ring chromosomes was confirmed to be from the expected fibroblast source. All the patterns in these samples did not match the cell lines published in the ATCC, NIH, or DSMZ website.
Supplemental Figure 9. EB differentiation from ring(13)-derived iPSCs. a-c, Immunocito-chemistry showing expression of endoderm-derived cells positive for AFP (a), mesoderm-derived cells positive for SMA (b), and ectoderm-derived cells positive for MAP2 (c) generated in vitro using embryoid body method with corrected iPSC clones GM00285-1, GM00285-3, and GM05563-1.
Supplemental Figure 1. Karyotypes of MDS cells used in this study. a, b, Representative karyotypes of MDS1r(17) fibroblasts (a) and two independent iPSC clones from each of the three MDS patients (b). Magnifications of chromosome 17 pairs (dotted squares) are shown in Fig. 2a for MDS1r(17) cells (boxes 1-3) and to the right of panel (a) for MDS2 and MDS3 iPSCs (boxes 4-7). c, Summary of karyotype data from G-banding analyses of MDS fibroblasts and iPSC clones 1 and 2 (n=20 each).
Supplemental Figure 2. Expression of pluripotency markers in iPSCs. Immunocytochemistry of MDS (a-d) and ring(13)-derived (e-j) iPSCs with the indicating antibodies.
Supplemental Figure 3. No evidence of episomal factor integration in MDS iPSCs. qPCR from genomic DNA using primers specific for exogenous KLF4, SOX2, OCT4, L-MYC, and LIN28, as described previously17. a, b, Relative levels of episomal factors in MDS fibroblasts on day 15 after electroporation with epiY4 mixture17 (positive control), WT and MDS iPSCs (passages 20-25), as well as WT human embryonic stem cell line HSF-1 (negative control) (N=1). c, d, Episomal factor copy number per cell in ring(13) fibroblasts electroporated with epiY4 mixture (positive control) and iPSCs (N=1)
Supplemental Figure 4. EB differentiation from MDS iPSCs. a-c, Immunocytochemistry showing expression of endoderm-derived cells positive for AFP (a), mesoderm-derived cells positive for SMA (b), and ectoderm-derived cells positive for TUJ1 (c) generated in vitro using embryoid body method with MDS1r(17), MDS2 and MDS3 iPSCs.
Supplemental Figure 5. Teratoma formation from MDS iPSCs. a-c, Histological sections from 6.5-11 week teratomas developed in the testis of SCID mice following injection with MDS1r(17), MDS2 and MDS3 iPSCs. Hematoxylin and eosin (H&E) staining reveals characteristic tissues from the mesoderm (a), endoderm (b), and ectoderm (c). d, List of MDS iPSC lines that were injected and teratoma efficiency.
Supplemental Figure 6. Cell morphology and chromosome distribution of additional MDS1r(17) iPSC clones. a-c, Cell morphology of poorly growing MDS1r(17) clones 4 and 5 at passage 4 on feeders (a) and passage 5 on matrigel (b), compared to well growing clones 3 and 6 at passage 5 on matrigel (c). d, examples of metaphase spreads observed in MDS1r(17) clones. e, Quantification of cell populations in MDS1r(17) clones 3-6 with various chromosome compositions shown in (d).
Supplemental Figure 7. Karyotypes of ring(13) cells and morphology of corrected iPSC clones. a-c, Representative karyotypes of GM00285 fibroblasts (a) and corrected GM00285 iPSC clones 1 and 3 (b, c). d,e, Representative karyotypes of GM05563 fibroblasts (d) and corrected GM05563 iPSC clone 1 (e). f-h, Representative morphologies of corrected iPSC clones derived from ring(13) fibroblasts.
Supplemental Figure 8. Rescue of ring(13)-associated deletions in corrected iPSC clones through compensatory UPD. a-e, Total copy number of SNPs across chromosome 13 in ring(13) GM00285 fibroblasts (a), GM05563 fibroblasts (b), and karyotypically normal GM00285 iPSC clones 1 (c) and clone 3 (d), and GM05563 iPSC clone 1 (e). The areas shaded in pink represent the deletions. f,g, Frequency of heterozygous (blue) or homozygous (red) SNPs in ring(13) fibroblasts and karyotypically normal iPSC clones for chromosome 13 (f) and chromosome 12 (g). The corrected iPSC clones are completely homozygous for chromosome 13, supporting the compensatory UPD mechanism.