

Abstract
(2,6-Dichloro-4-methoxyphenyl)(2,4-dichlorophenyl) methyl trichloroacetimidate (3) and its polymer-supported reagent 4 can be successfully applied to a one-pot protection-glycosylation reaction to form the disaccharide derivative 7d for the synthesis of lipid II analogues. The temporary protecting group or linker at the C-6 position and N-Troc protecting group of 7d can be cleaved simoultaneousely via a reductive condition. Overall yield of syntheses of lipid II, 1 and neryl-lipid II-Nε-dansylthiourea are significantly improved via the described methods.
Lipid II, 1 is a membrane-anchored cell-wall precursor that is essential for the growth and replication of both Gram-positive and Gram-negative bacteria. The effectiveness of targeting the enzymes associated with lipid II or lipid II itself as an antibacterial strategy is highlighted by the fact that it is the target for at least four different classes of antibiotics, including the clinically important glycopeptide antibiotics.[1] The reactions necessary for the biogenesis of peptidoglycan (PG) have been known for decades, and the biosynthesis of peptidoglycan of E. coli has been discussed extensively in reviews by van Heijenoort.[2] Lipid II is an important biochemical tool for studying MurG, flippase that translocates lipid II across the cytoplasmic membrane, penicillin binding proteins (PBPs), and the mode of action of glycopeptide antibiotics.[3] Lipid II displays very poor water solubility, and thus, is not trivial to apply the natural form of lipid II to their biological investigations or to assay for screening inhibitor molecules. To date, several lipid II analogs have been synthesized and applied to the biochemical studies.[4] We have recently identified that neryl lipid II-Nε-dansyl analogue, 2-Nε-dansylthiourea, could be recognized by mycobacterial transglycosylase (TGase) to form polymerized products with significant enhancement of visible-light absorption at 400 nm. Because the dansyl group itself does not have absorption in visible-light region, bathochromic shift of the polymerized-2-Nε-dansylthiourea is an unusual physicochemical observation. Thus, the polymerized-2-Nε-dansylthiourea via Mtb TGase can be distinguished from unreacted 2-Nε-dansylthiourea in the reaction mixture via visible-light without separation (see, supporting information). The observed physicochemical property of polymerized-2-Nε-dansylthiourea has been applied as a convenient assay for lead discovery of new TGase inhibitor molecules in our laboratory. In order to perform high-throughput screening against TGase, it is indispensable to establish a convenient synthetic route to access neryl-lipid II, 2. To date, total chemical and biochemical syntheses of lipid II and its derivatives have been accomplished by a few research groups.[4] We have accomplished chemoenzymatic and total chemical synthesis of UDP-MurNAc-pentapeptide (Park’s nucleotide) and prenyl-MurNAc-pentapeptide (lipid I).[5] However, to the best of our knowlege, total chemical synthesis is more feasible than other methods for generation of enough lipid II analogues for high-throughput screening (HTS). Herein, we report an improved chemical synthesis of lipid II, 1 and neryl-lipid II analogue, 2-Nε-dansylthiourea via one-pot protection and glycosylation reactions.
Total chemical synthesis of lipid II reported previously revealed that limited combinations of the protecting groups of glycosyl acceptors and donors (i and ii in Figure 2) can be applied to successful synthesis of the lipid II disaccharide (iii and iv); the glycosyl donors such as N-phthaloyl 3,4,6-O-triacetyl-2-deoxyl-2-amino-D-glucopyranosyl 1-bromide (P2 = phthaloyl, L = Br in i), N-2,2,2-trichloroethoxycarbonyl 3,4,6-O-triacetyl-2-deoxyl-2-amino-D-glucopyranosyl 1-bromide (P2 = Troc, L = Br in i), or N-phthaloyl 2-deoxy-2-amino-3,4,6-O-triacetate-D-glucopyranosyl 1-(2,2,2-trichloroacetoimidate) (P2 = phthaloyl, L = O-imidate in i) have been utilized in the synthesis of the disaccharide iv with the C6-protected donor ii (R3 = Ac or Bn).[4] In general, the glycosyl acceptors ii, whose C6-position was protected with acyl groups showed slow reaction rate and low conversion.[4f]
Figure 2.

One-pot protection-glycosylation to synthesize GlcNAc-MurNAc-peptide iv for lipid II.
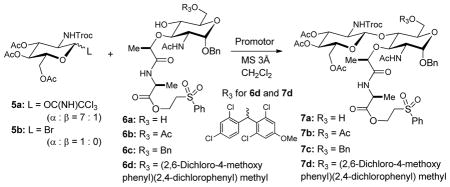
In our studies, glycosylation of the C6-acetylated MurNAc derivative 6a[5a] with the glycosyl imidate 5a did not yield the desired product 7a (e.g. Entry 1 in Table 1). Because the N-Troc-protected imidate 5a is one of the convenient GlcNAc sources in chemical glycosylations, we have sought an appropriate C6-ether-protected MurNAc to efficiently synthesize the lipid II disaccharide iv (Figure 2).[6] The C6-benzyl ether-protected MurNAc 6b was introduced for Königs-Knorr type glycosylation of 5b by Saha et al.[7] The AgOTf-catalysed glycosylation of 6c with 5b was performed in our laboratory; however, these reactions resulted in the formation of the desired β-linked disaccharide 7c in moderate yield (Entry 5 in Table 1). The same glycosyl acceptor 6c was applicable to the glycosylation with the imidate 5a; TMSOTf-catalyzed glycosylation of 6c with 5a furnished 7c in a moderate yield of 50% (Entry 2 in Table 1).[8] Although synthesis of the C6-benzyl-protected glycosyl acceptor 6b from the corresponding diol ii (R3 = H in Figure 2) and selective debenzylation of the primary benzyl ether 7b after the glycosylation were reported, in our hands, each step for these transformations provided the undesired diastereomers(s) that require time-consuming chromatography, and/or the yield of each step was 45–65%.[7] We have previously reported that primary alcohols can be protected selectively with (2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methyl trichloroacetimidates [3, monomethoxydiphenylmethyl acetimidate (MDPM-imidate)] to afford the corresponding ethers in good to excellent yields at controlled temperatures.[9] The MDPM-ether protecting group of 6d was stable under Schmidt glycosylation conditions for glycosyl trichloroacetimidate; TMSOTf-catalyzed glycosylation of 6d with 5a gave rise to the β-glycoside 7d as a mixture of two inseparable diastereomers in 65% yield in 3h (Entry 3 in Table 1). The isolation yield of the same glycosylation reaction of 6d with 5a was increased by 20% using BF3•OEt2 (Entry 4 in Table 1).
Table 1.
Synthesis of lipid II disaccharide 7.[a]
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Promoter | Temp (°C) | Time (h) | Product | Yield (%) |
| 1 | 5a | 6b | TMSOTf | 0 | 12 | 7b | 0 |
| 2 | 5a | 6c | TMSOTf | 0 | 3 | 7c | 50 |
| 3 | 5a | 6d | TMSOTf | 0 | 3 | 7d | 65 |
| 4 | 5a | 6d | BF3 • OEt2 | 0 | 3 | 7d | 85 |
| 5 | 5a | 6c | AgOTf | 0 | 3 | 7c | 45[b] |
2.5 Equivalents of glycosyl donor was used.
The reported isolation yield is 74%.
MDPM-imidate 3 reacted against only the primary alcohol of 6a even at room temperatures with near quantitative yield, and the by-product, 2,2,2-trichloroacetamide is an innocent species in Schmidt glycosylations. Thus, a one-pot protection-glycosylation protocol was envisioned for the synthesis of 7d directly from the diol 6a (Figure 2). As expected, the desired lipid II disaccharide derivative 7d could be synthesized from 6a in 75–85% yields in a one-pot two steps strategy (Scheme 1). It is worth mentioning that (2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methyl ether protecting group possesses a characteristic UV absorption, and isolation of the disaccharide 7d from the crude reaction mixture via chromatography was relatively simple compared to that of 7c (Entry 2 in Table 1).
Scheme 1.

One-pot protection-glycosylation to synthesize GlcNAc-MurNAc-peptide 7.
In order to further facilitate the synthesis of the key intermediate iv in Figure 2, (2,6-dichloro-4-alkoxyphenyl)(2,4-dichlorophenyl)methyl trichloroacetimidate linker resin 4 was applied to a one-pot two steps strategy for the synthesis of iii (Figure 2).[10] Loading of the diol 6a onto the linker resin 4 was completed with BF3•OEt2 (5 equivalents) at room temperature in 1h; in this step, progress of the reaction was monitored by consumption of 6a via LC-MS. Once the loading step was completed, the imidate 5a was added into the reaction mixture to afford the desired β-glycoside resin 7e in 65–80% yield which was determined based on the isolated 7a (Table 1) after the cleavage of 7e with 30% TFA in CH2Cl2 for 1h. Accordingly, convenient synthetic procedures for 7d and 7e for the syntheses of lipid II analogues were accomplished. The other convenient feature of (2,6-dichloro-4-alkoxyphenyl)(2,4-dichlorophenyl)methyl ether-protecting group and -linker is that they can be deptrotected or cleaved simultaneously when the N-Troc group is removed under a reductive condition. The N-Troc and C6-ether protecting group or -linker of 7d or 7e were deprotected under the condition of Zn in 30% TFA-AcOH to furnish the amino-alcohol 8 (Scheme 2). Acetylations of the free amine and alcohol of 8 afforded 9 in 85–90% yield from 7d or 7e. α-Phosphorylation and diphosphate ester formation of 9 were carried out via the established protocols with minor modifications. Deprotection of the anomeric Bn protecting group was performed by Pd-C catalysed hydrogenation reaction to afford a mixture of α/β-anomers, which were subjected to α-selective phosphite formation using dibenzyl N,N-diisopropylphosphoramidite and 5-(ethylthio)-1H-tetrazole. The generated α-phosphite intermediate was oxidized with tBuO2H to afford the α-phosphate 10 in 70% overall yield for three steps. Deprotection of the 2-(phenylsulfonyl)ethanol protecting group of 10 was achieved by the treatment with DBU to furnish the α-phosphoryl GlcNAc-MurNAc-monopeptide derivative. The tetrapeptide, HCl•H-L-Ala-γD-Glu(OMe)-L-Lys(COCF3)-D-Ala-D-Ala-OMe (11) was synthesized in water media via water-soluble reagents (glyceroacetonide-Oxyma (12), EDCI, and NaHCO3) (see supporting information).[11] Coupling of the free carboxylic acid, α-phosphoryl GlcNAc-MurNAc-monopeptide with the tetrapeptide 11 under a mild condition (12, EDCI, and NaHCO3) in H2O yielded the α-phosphoryl GlcNAc-MurNAc-pentapeptide 13 in over 90% overall yield from 10. Conveniently, all reagents and excess tetrapeptide used in this step could be removed via a basic water work-up. Hydrogenolytic debenzylations of 10 followed by the treatment with Et3N resulted in the corresponding monotriethylammonium phosphate 14 in quantitative yield, whose structure was established by 1H-NMR analysis. Triethylammonium phosphate 14 was then applied to a carbonyldiimidazole (CDI) promoted diphosphate-formation reaction.[4e,4f] Triethylammonium α-phosphoryl GlcNAc-MurNAc-pentapeptide 14 was first activated with CDI and the excess CDI was quenched with MeOH to afford 1H-imidazole-1-carboxylic (phosphoric) anhydride and methyl 1H-imidazole-1-carboxylate, which was not reactive against the phosphate nucleophiles. All volatiles were extensively removed and the remaining mixture was subjected to the cross-coupling reaction with the ammonium undecaprenyl phosphate (15) or neryl phosphate (16). Progress in the coupling reactions was monitored via reverse-phase HPLC (0.05 M NH4HCO3 : MeOH to MeOH for lipid II). The reaction mixture was lyophilized and the fully-protected product was subjected to global deprotection reactions with aq. LiOH. Lipid II, 1 was synthesized in 45% overall yield from 13 after purification via reverse-phase HPLC. The structure of 1 was confirmed by 1H-NMR, negative ESI-TOF-MS spectroscopy, and retention time in HPLC analysis.[4e] Similary, neryl-lipid II analogue, 2 could be synthesized in 75% overall yield by using excess ammonium neryl phosphate (16). The dansyl group was conjugated to the Nε-lysine moiety of 2 with 4-(dansylamino)phenyl isothiocyanate to furnish neryl-lipid II-Nε-dansylthiourea, 2-Nε-dansylthiourea, in greater than 90% yield.
Scheme 2.

Synthesis of lipid II and neryl lipid II analogues.
In conclusion, chemical syntheses of lipid II and neryl-lipid II analogues were accomplished via one-pot protection-glycosylation protocols. (2,6-Dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methyl trichloroacetimidate (3) and its linker-resin 4 were demonstrated to be useful temporary protecting groups for the primary alcohol of the diol 6a that could be compatible with Schmidt glycosylation reactions. In addition, (2,6-dichloro-4-alkoxyphenyl)(2,4-dichlorophenyl)methyl ether linkage could be deprotected simoutaneousely during the deprotection of N-Troc groups. Accodingly, the synthetic intermediate 10 for lipid II was efficiently synthesized from 6a in 45–54% overall yield with minimum number of chemical steps. Gram-quantity of the tetrapeptide building block 11 can readily be synthesized in water media, and the peptide-forming reagents used for the synthesis of 13 could be removed via simple water work-ups. Detailed experimental procedures for improved syntheses of lipid II and its neryl analogues are summarized in Supporting Information. As mentioned in the introduction, neryl lipid II-Nε-dansylthiourea, 2-Nε-dansylthiourea is a very useful biochemical tool for studying transglycosylase (TGase). Characterization of polymerized 2-Nε-dansylthiourea by TGase and development of high-throughput screening (HTS) against TGase will be reported elsewhere.
Experimental Section
One-pot protection and glycosylation reaction of 6a
To a stirred suspension of 6a (0.20 g, 0.32 mmol), (2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methyl trichloroacetimidate (3, 0.29 g, 0.57 mmol), and MS 3Å (0.5 g) in CH2Cl2 (2.5 mL) was added BF3•OEt2 (0.5 mmol) at 0 °C. After 1h, the imidate 5a (0.34 g, 0.54 mmol) in dichloromethane (1.0 mL) was added. The reaction mixture was stirred for 3 h at 0 °C, and quenched with sat. aq. NaHCO3 solution. The resulting suspension was filtered through celite. The aqueous layer was extracted with ethyl acetate and the combined organic layer was dried over Na2SO4, filtered, and evaporated. Purification by silica gel column chromatography (hexanes/ethyl acetate = 20/80) afforded 7d (0.39 g, 85%).
(2,6-Dichloro-4-alkoxyphenyl)(2,4-dichlorophenyl)methyl trichloroacetimidate linker resin 4
(2,6-Dichloro-4-alkoxyphenyl)(2,4-dichlorophenyl)methanol linker resin was prepared according to the procedure reported previously. The resin (5g, active surface: ~1.0 mmol/g) was suspended in CH2Cl2 (10 mL) and CCl3CN (5 mmol) and DBU (1 mmol). The suspension was gently stirred for 3h. The polymer resins were washed with THF-iPrOH (3/1), THF, and THF-CH2Cl2 (2/1), and dried under high vacuum to furnish 4 (5.72g, quantitative yield).
One-pot loading and glycosylation of 6a
To a stirred suspension of 6a (0.20 g, 0.32 mmol) and (2,6-dichloro-4-alkoxyphenyl)(2,4-dichlorophenyl)methyl trichloroacetimidate linker resin 4 (1.6 g, ~1.0 mmol/g), and in CH2Cl2 (5.0 mL) was added BF3•OEt2 (0.64 mmol) at r.t. After 1h, the imidate 5a (0.596 g, 0.96 mmol) in dichloromethane (2.0 mL) was added at 0 °C. The reaction mixture was gently stirred for 3h at 0 °C, and the resins were washed THF-iPrOH (3/1), THF, and THF-CH2Cl2 (2/1), and dried under high vacuum to furnish 7e (0.25 g). In order to determine the overall yield, the disaccharide resin 7e was cleaved with 30% TFA for 1h and the generated 7a was quantitated via LC-MS (65–80% yield).
Deprotections of N-Troc and (2,6-dichloro-4-methoxyphenyl)(2,4-dichlorophenyl)methyl ether groups and acetylations
To a stirred solution of 7d (0.35 g, 0.25 mmol) in 30% TFA-AcOH (3.5 mL) was added zinc powder (0.16 g, 2.5 mmol). After stirring the solution for 6h at r.t, the reaction mixture was filtered and all volatiles were evaporated to afford the crude 8. To a solution of 8 in pyridine (2.0 mL) was added Ac2O (2.0 mL). After 4h at r.t, all volatiles were removed to afford the crude product. Purified by silica gel column chromatography (ethyl acetate/methanol = 95/5) to afford 9 (0.24 g, 95% overall yield). Similarly, the disaccharide resin 7e was converted to 9 (85% overall yield).
Supplementary Material
Figure 1.

Structures of lipid II and neryl-lipid II analogues for studying transglycosylase (TGase).
Acknowledgments
The National Institutes of Health is greatly acknowledged for financial support of this work (AI084411). We also thank University of Tennessee for generous financial support. NMR data were obtained on instruments supported by the NIH Shared Instrumentation Grant.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201xxxxxx.
References
- 1.a) Varneyl KM, Bonvin AMJJ, Pazgier M, Malin J, Yu W, Ateh E, Oashi T, Lu W, Huang J, Diepeveen-de Buin M, Bryant J, Breukink E, MacKerell AD, Jr, de Leeuw EPH. PLOS. 2013;9:e1003732. doi: 10.1371/journal.ppat.1003732. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yuan Y, Fuse S, Ostash B, Sliz P, Kahne D, Walker S. ACS Chem Biol. 2008;3:429–436. doi: 10.1021/cb800078a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Martin NI, Breukink E. Future Micro. 2007;2:513–525. doi: 10.2217/17460913.2.5.513. [DOI] [PubMed] [Google Scholar]; d) Breukink E, de Kruijff B. Nature Rev Drug Disc. 2006;5:321–323. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 2.a) van Heijenoort J. Mol Biol Rev. 2007;71:620–635. doi: 10.1128/MMBR.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Auger G, van Heijenoort J, Mengin-Lecreulx D, Blanot D. FEMS Microbiol Lett. 2003;219:115–119. doi: 10.1016/S0378-1097(02)01203-X. [DOI] [PubMed] [Google Scholar]; c) Bupp K, van Heijenoort J. J Bacteriol. 1993;175:1841–1843. doi: 10.1128/jb.175.6.1841-1843.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Zervosen A, Sauvage E, Frère JM, Charlier P, Luxen A. Molecules. 2012;17:12478–12505. doi: 10.3390/molecules171112478. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) van Damk V, Sijbrandi R, Kol M, Swiezewska E, de Kruijff B, Breukink E. Mol Micro. 2007;64:1105–1114. doi: 10.1111/j.1365-2958.2007.05722.x. [DOI] [PubMed] [Google Scholar]; c) Zawadzke LE, Wu P, Cook L, Fan L, Casperson M, Kishnani M, Calambur D, Hofstead SJ, Padmanabha R. Anal Biochem. 2003;314:243–252. doi: 10.1016/s0003-2697(02)00622-x. [DOI] [PubMed] [Google Scholar]; d) Hu Y, Chen L, Ha S, Gross B, Falcone B, Walker D, Mokhtarzadeh M, Walker S. Proc Natl Acad Sci USA. 2003;100:845–849. doi: 10.1073/pnas.0235749100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Lebar MD, Lupoli TJ, Tsukamoto H, May JM, Walker S, Kahne D. J Am Chem Soc. 2013;135:4632–4635. doi: 10.1021/ja312510m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen KT, Kuan YC, Fu WC, Liang PH, Cheng TJR, Wong CH, Cheng WG. Chem Eur J. 2013;19:834–838. doi: 10.1002/chem.201203251. [DOI] [PubMed] [Google Scholar]; c) Shih HW, Chen KT, Cheng TJR, Wong CH, Cheng WC. Org Lett. 2011;13:4600–4003. doi: 10.1021/ol201806d. [DOI] [PubMed] [Google Scholar]; d) Yi Zhang Y, Fechter EJ, Wang T-SA, Barrett D, Walker S, Kahne DE. J Am Chem Soc. 2007;129:3080–3081. doi: 10.1021/ja069060g. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Van Niieuwenhze MS, Mauldin SC, Zia-Ebrahimi M, Winger BE, Hornback WJ, Saha SL, Aikins JA, Blaszack LC. J Am Chem Soc. 2002;124:3656–3660. doi: 10.1021/ja017386d. [DOI] [PubMed] [Google Scholar]; f) Schwartz B, Markwalder JA, Wang Y. J Am Chem Soc. 2001;12:11638–11643. doi: 10.1021/ja0166848. [DOI] [PubMed] [Google Scholar]; g) Ha S, Chang E, Lo MC, Park P, Ge M, Walker S. J Am Chem Soc. 1999;121:8415–8426. [Google Scholar]; h) Hitchcock SA, Eid CN, Aikins JA, Zia-Ebrahimi M, Blaszczak LC. J Am Chem Soc. 1998;120:1916–1917. [Google Scholar]
- 5.a) Kurosu M, Li K. Hetrocycles. 2008;76:445–469. [Google Scholar]; b) Kurosu M, Mahapatra S, Narayanasamy P, Crick DC. Tetrahedron Lett. 2007;48:799–803. [Google Scholar]
- 6.Zhang Z, Ollmann I, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 7.Saha SL, Van Nieuwenhze MS, Hornback WJ, Aikins JA, Blaszczak LC. Org Lett. 2001;3:3575–3577. doi: 10.1021/ol016692t. [DOI] [PubMed] [Google Scholar]
- 8.Termin A, Schmidt RR. Liebigs Ann Chem. 1992:527–533. [Google Scholar]
- 9.a) Kurosu M, Li K. Synthesis. 2009;21:3633–3641. [Google Scholar]; b) Kurosu M, Biswas K, Crick DC. Org Lett. 2007;9:1141–1145. doi: 10.1021/ol070150f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ali IAI, El Ashry EH, Schmidt RR. Eur J Org Chem. 2003:4121–4131. [Google Scholar]
- 10.Wang Y, Kurosu M. Tetrahedron. 2012;68:4797–4804. doi: 10.1016/j.tet.2012.03.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Q, Wang Y, Kurosu M. Org Lett. 2012;14:3372–3375. doi: 10.1021/ol3013556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.