

Abstract
Actin remodeling is an area of interest in biology in which correlative microscopy can bring a new way to analyze protein complexes at the nanoscale. Advances in EM, X-ray diffraction, fluorescence, and single molecule techniques have provided a wealth of information about the modulation of the F-actin structure and its regulation by actin binding proteins (ABPs). Yet, there are technological limitations of these approaches to achieving quantitative molecular level information on the structural and biophysical changes resulting from ABPs interaction with F-actin. Fundamental questions about the actin structure and dynamics and how these determine the function of ABPs remain unanswered. Specifically, how local and long-range structural and conformational changes result in ABPs induced remodeling of F-actin needs to be addressed at the single filament level. Advanced, sensitive and accurate experimental tools for detailed understanding of ABP–actin interactions are much needed. This article discusses the current understanding of nanoscale structural and mechanical modulation of F-actin by ABPs at the single filament level using several correlative microscopic techniques, focusing mainly on results obtained by Atomic Force Microscopy (AFM) analysis of ABP–actin complexes.
Background and significance
To understand biological processes at the molecular level it is essential to identify the involved proteins and their assemblies, to characterize their structure and function, and to unravel their interplay with other proteins and molecules.1 Actin remodeling is an area in biology where AFM can bring a new way to analyze protein complexes at the nanoscale. Actin2 forms filamentous protein complexes (F-actin) (Fig. 1) found in almost all eukaryotic cells.3 Actin cytoskeleton is an important, exciting and fascinating area of research in cell biology: important, because actin and the dynamic remodeling of its various assembly forms are the central players in cell movement and shape,3 cytokinesis, intracellular transport, and other biological processes;4–6 exciting, because the fast progress in this field has increased greatly over the last few years our knowledge of the proteins and factors that affect and/or regulate different stages of actin filament nucleation and elongation,7–9 bundling and cross-linking,10 and severing and depolymerization;11–13 and fascinating, because many aspects of the remodeling of actin filaments are yet to be clarified.
Fig. 1.

Monomeric (G-actin) and filamentous (F-actin) forms of actin. (A) G-actin (PDB code: 1j6z)2 with highlighted ADP and divalent cation. (B) Monomers assemble to form long filaments.
Current high-resolution techniques to study F-actin–ABP complexes
In different cells actin cytoskeleton is regulated by more than 150 ABPs that enable its important physiological functions.14 Many of those proteins bind to/interact with two or more actin protomers within the filament creating conditions and natural pathways for cooperative spreading of changes in F-actin. Techniques like X-ray crystallography,15–19 electron microscopy20–23 and nuclear magnetic resonance spectroscopy23,24 have contributed greatly to elucidate the structure and properties of F-actin–ABP complexes. Some of the major advantages and limitations of these techniques are summarized in Table 1.
Table 1.
F-actin–ABP structural determination techniques
| Technique | Advantages | Limitations |
|---|---|---|
| X-ray crystallography |
|
|
| NMR |
|
|
| Electron crystallography |
|
|
| Electron microscopy |
|
|
| Cryo-EM |
|
|
| Atomic force microscopy |
|
|
| Light microscopy/FRET/TIRF |
|
|
Electron microscopy (EM) is the principle technique in common use for high-resolution studies of F-actin structures (Fig. 2). Based on images obtained by transmission electron microscopy (TEM), actin filaments have been shown to exhibit a double strand helical structure, with a helical pitch of 36–37 nm and length of a few microns. More recently, reconstruction of filaments from many cryo-electron microscopy (cryo-EM) images has proven to be more powerful in elucidating the F-actin architecture,25–28 and is capable of revealing details at resolutions in the range of 4 to 5 Å. 29,30 Cryo-EM, however, has certain inherent limitations. It is useful mainly for analyzing regular F-actin structures having uniform helical symmetry. It yields the “average” structure for the entire F-actin filament population or sub-populations and does not elucidate heterogeneity of individual helix changes. More advanced applications of cryo-EM25,31 provide information on structures lacking helical symmetry. For actin, the common assumption is that all filaments have identical helical parameters. EM images have so far been difficult to analyze (Fig. 2) when the helical structures are polymorphic due to incomplete saturation of binding sites.
Fig. 2.
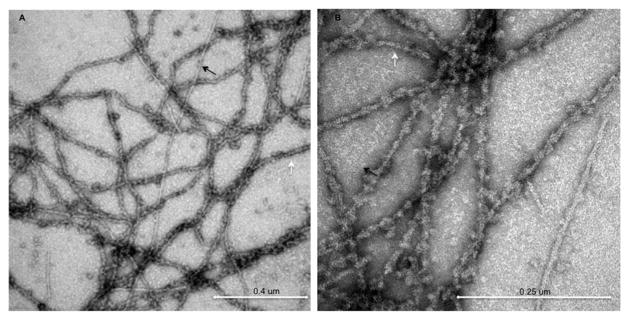
TEM images of negatively stained drebrin–F-actin. Undecorated and drebrin decorated filaments are marked by black and white arrows respectively.
Applications of AFM to high-resolution studies of proteins form and function
AFM32 is a powerful technique for biophysical nanoscale characterization of biological structures.33–38 It offers unique capability for direct 3D imaging of single actin filaments without electron dense staining, fixation, or extreme temperatures, and with imaging resolution comparable to that of electron microscopes. Additionally, AFM is a versatile technique that also brings information on structures, mechanics, dynamics and specific bio-molecular binding interactions under physiological solvent conditions.37,39–41 The atomic force microscope is a member of the scanning probe microscopy techniques, which utilize a probing tip that scans the surface of a sample. Soft cantilevers with spring constants of about 0.01–0.1 N m−1 can sense forces as low as few piconewtons, and piezoelectric scanners can translate the sample or tip in x, y and z-directions with sub-nanometer resolution. This combined functionality enables the atomic force microscope to render three-dimensional images of the sample with atomic resolution.42 Since the development of AFM as an imaging technique in structural biology, a number of selected proteins and protein conformational changes have been investigated at sub-molecular resolution. As a contact microscope, AFM does not suffer from diffraction limits, unlike optical microscopes, enabling it to reach atomic resolution. In particular, tapping mode imaging43 involves minimal physical contact with the samples, which helps in preventing tip-induced damage to soft biological molecules. Consequently, AFM’s exceptionally high signal-to-noise sub-nanometer lateral resolution and vertical resolution of 1 Å can be achieved in selected cases. 36,44–46
Several examples from the literature show the potential of AFM to study both the “form” and “function” of proteins, thereby resolving questions in proteomics and structural biology quasi-simultaneously. Among biomolecules, membrane proteins in their native state have been imaged most extensively.36,46 The surface topography of bacteriorhodopsin was resolved down to 1 nm,45 and vertical resolution of up to ~1 Å was attained for the native complex in photosynthetic membranes.44,47 Besides isolated viruses and phages,48,49 specific substructures like viral capsomeres, bacteriophage connectors and tails50,51 have been imaged at a resolution comparable to EM. Nevertheless, native-like 2D arrays or structurally oriented (but not necessarily homogeneous) conformations (e.g., membranes) or linear biomolecules such as DNA,34 RNA,52 collagen,53 or amyloid,54 are better suited for high resolution AFM imaging than single isolated protein molecules. This technique was successfully used for imaging ‘bare’ actin filaments.56
AFM imaging for mapping structural changes and remodeling of F-actin induced by ABPs
To date only a few examples of ABPs changing the helical twist of F-actin have been reported. The most striking case is that of cofilin, which exerts its severing function by capturing and stabilizing an ‘over-twisted’ actin conformation and consequently, leading to filaments severing. Quantitative single filament level information on changes in helical twist, increase in filament volume and change in mechanical properties such as persistence length is needed to advance our understanding of filament stabilization or disassembly by cofilin and other ABPs. AFM imaging can be effectively used to provide detailed biophysical nanoscale characterization by determining ABP-induced changes in individual helix of unstained, unfixed actin filaments at high resolution, without averaging.
Our recent work reported images of bare actin filaments (Fig. 3A) obtained using tapping mode AFM imaging.56 The measured helical pitch (36 nm) (Fig. 3C) and persistence length of actin filaments obtained from AFM images agreed well with the EM57 and thermal fluctuation58 observations, respectively. This validates the use of the AFM technique for quantitative in vitro studies of structural and mechanical remodeling of actin filaments. In a novel application of AFM, we imaged the remodeling of F-actin by drebrin A (a neuronal ABP) and showed that it involves changes of helical twist (Fig. 3D) and actin filament stiffness (55% persistence length increase and higher elastic modulus).56 To further test the resolution and validate the analysis of AFM actin filament images, we examined cofilin bound actin filaments. Cofilin is an extensively studied ABP, which is known to change the F-actin structure and mechanics based on EM studies.22 Cofilin-decorated F-actin had a significantly reduced helical pitch (~28.7 nm), in good agreement with previously reported results.22 Structural differences between unbound- and drebrin and cofilin bound-F-actin, as measured by AFM, demonstrate that this method can determine how ABPs change the structure of the actin filaments.56 Thus, AFM can be added to the list of techniques suitable for obtaining molecular level structural details of F-actin complexes at nanoscale resolution. AFM imaging analysis can easily provide individual helix measurements of ABP–F-actin complexes that are difficult to obtain using other imaging methods, such as EM or cryo-EM. The proof-of-concept demonstration of AFM based F-actin remodeling by drebrin and cofilin paves the way for similar measurements on other ABPs. It is likely that the use of AFM would expand, making it a routine and complementary technique to EM and cryo-EM to study ABP–F-actin complexes, and to analyze the structural and mechanical modulation of F-actin by these proteins.
Fig. 3.

AFM tapping mode images of (A) bare F-actin and (B) drebrin–F-actin. (C) and (D) show helix pitch from line profiles along bare F-actin and drebrin–F-actin. Adapted from Sharma S. et al.56 with permission from American Chemical Society.
Cooperativity of ABP-binding and F-actin remodeling
Most molecular models of F-actin assume precisely defined filaments with a homogeneous rigid rod-like structure. However, there is abundant evidence revealing polymorphic states of F-actin and variations in the helical twist of the filament.59–61 First, actin filaments can bend and flex along their long axis with a persistence length (a mechanical parameter that quantifies stiffness) of about 17 μm (in a complex with phalloidin).62 Second, actin subunits in a filament can also twist and tilt,63 leading to long-range rearrangements of actin subunits within filaments along their long axis. Lastly, several actin subdomains, such as the DNase-I-binding loop, are mobile. Thus, actin filaments can adopt multiple conformations. This is the case not only for actin filaments in different nucleotide states (ATP, ADP-Pi or ADP),64 but also for actin filaments with identical bound nucleotides.29,65 Moreover, different structures of the filament have been reported for its complexes with ABPs. Many ABPs exhibit binding preference for specific actin filament conformations in vitro.66 ABPs might stabilize specific actin filament conformations with long-range effects, or selectively bind to preferred actin filament populations. ADF/cofilin has been shown to interact with and stabilize a specific conformational state of ADP–F-actin, and to modify the mechanical properties of the bound filaments.11,67,68 Structural modifications of F-actin have also been reported for myosins,69 gelsolin70,71 and other proteins.
The different conformations of an actin filament regulate its binding properties (F-actin binding sites are non-equivalent and non-identical). Actin filaments are not decorated with similar ratios of all the ABPs present in the cell. Most likely, F-actin remodeling mechanisms allow for dynamic synergy or cooperativity of binding or dissociation of specific ABPs. It is proposed that the properties of F-actin are modified upon binding with ABP, with long-range effects that subsequently affect the binding of other ABPs.72 Allosteric interactions can have long-range effects on individual actin filaments, maintaining the filaments in a specific conformation over biologically relevant distances,66,70 although not necessarily over the entire length of the actin filaments.61 Thus, binding a single ABP may be sufficient to modify the structure of the filament, explaining why additional ABPs would bind cooperatively to such filaments to form regions saturated with this ABP e.g. binding of ADF/cofilin.
Indirect evidence for such effects comes from the structure and mechanical properties of actin filaments at the molecular scale. Direct visualization of how ABPs bind to F-actin and influence the structure and conformation of the decorated actin segment, as well as the probability of ABPs binding and structural effects at the neighboring F-actin sites, can reveal fundamental aspects of ABP interactions and ABPs induced F-actin remodeling. Cooperativity is an important property in the regulation and function of several ABPs such as cofilin, tropomyosin73 and others. Cooperativity of protein–protein binding is defined as positive if the binding of one protein molecule strengthens the binding of subsequent molecules, and as negative if the binding of one molecule weakens the binding of subsequent molecules. Quantitative measurements of cooperativity often rely on fitting the Hill equation74 to protein–protein binding data, considering actin as an infinitely cooperative lattice with n sites that exist in only two forms, fully bound or unbound states. The binding density ν (protein molecules bound per actin subunit) is given by n = Ka[L]nH/1 + Ka[L] nH where [L] is the concentration of free protein at equilibrium, Ka is the association equilibrium constant, and nH is the Hill coefficient. In the case of all-or-none binding, the Hill cooperativity coefficient n indicates the binding stoichiometry, but when cooperativity occurs and the stoichiometry is known from other data, then n differs from the exact stoichiometry and provides information about intra-molecular communication.75,76 Recently the nearest-neighbor cooperativity model was applied successfully to the binding of cofilin to actin filaments.73 In bulk studies, individual macromolecules within an ensemble have been traditionally assumed to be identical. However, recent single-molecule methods reveal that the presence of molecular heterogeneity results in lower cooperativity values in bulk measurements.77
Heterogeneity in the F-actin substructure and the binding cooperativity of ABPs are important in modeling of actin interactions
Conventional solution experiments are unable to detect single filament level changes in helical periodicity or conformations of F-actin upon drebrin or other ABP binding. Imaging of dynamic structural changes spreading across the filament in real time would require temporal resolution on the microsecond scale with sub-nm spatial resolution. Fluorescence microscopy, using FRET/fluorescence correlation spectroscopy, provides information on labeled molecules on a microsecond timescale, but its limited spatial resolution (~300 nm) precludes tracking of individual molecules. Evanescent wave fluorescence can detect individual fluorescent molecules, but is unable to resolve closely associated molecules like actin protomers within the filament.78 Single molecule FRET can directly measure dynamic changes in F-actin, but provides no structurally resolved information on changes such as in the helical parameters of F-actin.79 The binding characteristics of ABPs are frequently derived from dynamic fluorescence measurements such as TIRF (Total Internal Reflection Fluorescence) microscopy,80 allowing for preliminary conclusions regarding the effect of these proteins on filaments size distribution, assembly or severing.81–83 For example, drebrin1–300 binding to actin filaments was imaged in a recent study by two-color TIRF microscopy (Fig. 4).
Fig. 4.

(A) TIRF imaging revealed a cluster-like attachment of drebrin (green) to actin filaments (red), providing the first indications of cooperative interaction between drebrin1–300 and F-actin. (B) AFM images of F-actin decorated with drebrinA1–300 at sub-saturating ratios reveal their cooperative binding (binding clusters are marked by yellow arrows along individual filaments). (C) and (D) show drebrin clusters along the actin filament at higher resolution. (E) Identification of the segments of drebrin free F-actin; 1st, 2nd and 3rd neighbor relative to the drebrin-bound helix in the AFM image of partially decorated Drb1–300–F-actin. Adapted from Sharma S. et al.84 with permission from Cell Press.
Resolution achieved by optical techniques limits the measuring/mapping of nanoscale protein binding and cluster assembly on individual actin filaments. These current techniques employ bulky probes, which may affect the protein and have insufficient resolution to measure the size of the bound protein cluster (<sub diffraction limit i.e. 300 nm) and the resulting structural changes in F-actin. Clearly, high-resolution images of actin–ABP complexes are required to enable measurement of local changes in F-actin induced by ABPs. Although such a resolution is achieved in EM analysis of these complexes, the averaging of structural information inherent in EM analysis precludes the examination of individual F-actin helical structures to distinguish between localized (directly bound) and transmitted (unbound regions) effects of ABPs.
In contrast to ensemble measurements and single molecule spectroscopy techniques, one can easily measure the ABP binding assembly (cluster size) and F-actin modulation at the single filament level, at the nanoscale, via AFM imaging. AFM allows imaging of individual actin filaments that are partially saturated with ABPs. It yields high-resolution quantitative 3D structural information on the end-to-end contiguous versus non-contiguous protein binding sites under such conditions. Moreover, it enables the analysis of changes in helical periodicity of ABP-bound and unbound F-actin regions.
We have shown AFM measurements for drebrin (under-twist) (Fig. 3) and cofilin (over-twist)56 induced changes in the actin helix (in opposite directions). The same approach can be used to analyze the helical changes in actin filaments for other ABPs. We also measured the ABP binding assembly (nanoscale cluster size) and F-actin modulation at the single filament level via AFM imaging to test if the protein binds in a cooperative manner to F-actin. F-actin filaments can be partially decorated with ABPs (at under-saturating conditions) to study the binding patterns, cooperative effects, and the influence on F-actin modulation/stabilization by ABP using AFM imaging. If ABP binding has positive co-operativity, clusters of protein will form along the actin filament. Though AFM resolution limits the imaging of individual protein molecules, the fact that ABP may bind in nano-scale clusters along the actin filament makes AFM an excellent tool to directly visualize the overall ABP binding at the single actin filament level. Both non-contiguous ABP bound (decorated) regions of actin filaments and regions free of ABP (undecorated) can be analyzed. The size of the ABP clusters over F-actin would depend on the density of binding and the extent of its cooperativity.
Additionally, one can exploit the nanoscale resolution of ABP–actin complexes in AFM to measure directly the periodicity for individual filaments, and thus to determine the helical pitch of the filament both in its bare and ABP bound regions. Our recent study measured the changes in helical periodicity of F-actin upon drebrin binding and the propagation of its binding effects within the filament.84 AFM showed that drebrin binding induces helical changes in F-actin in both the directly occupied and the unbound regions, i.e., it revealed the propagation of the drebrin-induced structural changes to the adjacent, undecorated regions on the filaments. This work provides the first un-averaged experimental information on the remodeling of ABP occupied and unoccupied regions of F-actin at the single-filament level.
Mechanics of single molecule F-actin–ABP complexes
The mechanical properties of actin filaments are tightly correlated with the helical structure of the polymer, and it is expected that factors affecting the structure of the filament also affect its flexibility. There are several ways in which ABPs are known to modulate actin mechano-structure and function. ABPs have been shown to alter filament rigidity by cross-linking them, or to create bends and promote cooperativity in actin,22 which may facilitate regulation or remodeling of the actin cytoskeleton. Mechanical properties of actin have been investigated thus far using a number of techniques including EM, fluorescence and thermal fluctuation measurements.57,58 The resolution achieved with AFM imaging allows determining the local flexibility of specific sites within the filament similar to other techniques, but at a higher resolution (helix level). Flexibility of actin filaments can be obtained from a comparison of the contour length and the mean-square distance between the ends or other sites in the molecule.85,86 For semi-flexible polymers like actin (Lp ~L), the Worm-Like Chain (WLC) 2D model can be used (with the assumption of equilibration) to determine the persistence length of actin filaments from AFM topography images (Fig. 5). Persistence length (p) is a measure of length-scale for molecule stiffness i.e., the distance over which the direction of a polymer segment persists, in the time or ensemble average, owing to limited flexibility of the polymer.
Fig. 5.

Representative spline (medial axis) profile along drebrin–F actin. (A) Points chosen along the filament length are shown as red markers. (B) Spline fit along the markers is shown in yellow. (C) Profile along chosen points on the filament.
If R is defined as the end-to-end distance of the overall polymer and L is the contour length of the polymer, the relationship between them and the persistence length, p, of the polymer is87
where s, and s′ are two positions on the polymer chain; Ū (s) and Ū (s′) are unit vector tangents to the polymer chain at positions s and s′. Another method to estimate the persistence length of actin filaments is by measuring the local filament angle distribution.88 By tracing the actin contours, we can calculate the local bend angle, θ, of two consecutive segments at different step size l (vector length). Using the probability distribution function for the WLC model in two dimensions it can be shown that
Volume analysis of ABP–F-actin complexes
The binding ratio of ABP to actin can be determined from flattened and plane-fitted AFM images by measuring the overall increase in the volume of the actin–ABP complex compared to bare actin filaments (Fig. 6). The surface height is measured using the histogram function and subtracted from the measured height of actin or ABP bound actin for volume determination. The filaments (helical repeats = 37 nm) are then detected individually using the grain analysis function. The density slice selects the pixels above the surface that represent filament sections to be analyzed. The image analysis function scans the image and detects all highlighted segments within the density slice. It generates height and area information, which permits calculation of the net volume of actin filaments. The increase in the overall volume of actin filaments upon ABP binding, compared to bare F-actin, can be used to assess the number of ABP molecules bound per actin helical repeat.
Fig. 6.
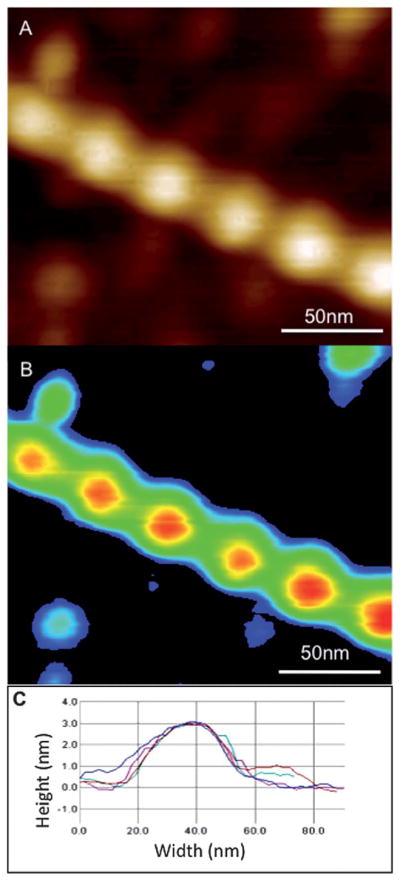
Volume determination of ABP–F-actin in AFM 3D images. (A) Unprocessed AFM tapping mode height image. (B) Zero order flattened and background subtracted image. (C) Multiple line profiles taken along the filament cross-section to detect height and width of the filaments. Adapted from Sharma S. et al.56 with permission from American Chemical Society.
Imaging F-actin–ABP complexes under physiological conditions
A major advantage of AFM is the capability of high-resolution imaging of biomolecules in their native environment. However, to date AFM studies on actin–ABP complexes have been done mostly in ambient air and therefore under non-physiological conditions. AFM has been used previously to study complex processes such as the dynamics of proteins89 and DNA–protein interactions.90 AFM imaging of F-actin in solution is difficult since the protein–substrate interactions need to be optimized, in order to withstand tip–sample interaction forces during imaging. Here we present the sub-nanometer AFM images of rabbit skeletal F-actin obtained in buffer conditions (Fig. 7). Data obtained in air and in solution have been compared and found to yield the same helix pitch (36 nm) for bare F-actin. AFM imaging under physiological conditions in aqueous solution was also used previously to show distinct variations in monomer stacking and an increase in pitch of the helix for malaria parasite actin compared to skeletal actin filaments.91
Fig. 7.
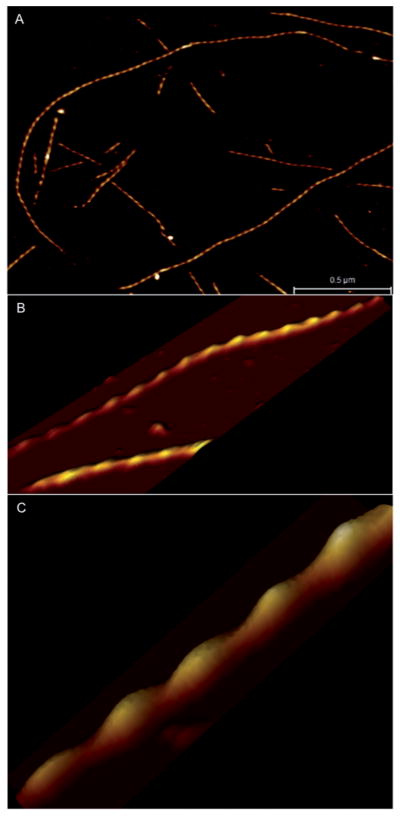
AFM imaging of F-actin under physiological conditions. (A) Image of several actin filaments shows stable attachment to a silane modified mica surface. (B) Higher resolution image of F-actin and (C) 3D view of actin filament section showing well resolved helical periodicity.
Additionally, the development of high-speed atomic force microscopy (HS-AFM)92,93 has enabled studying the structure and dynamics of functioning biomolecules simultaneously. Kodera et al.94 used HS-AFM to observe with unprecedented time resolution myosin V moving along actin filaments. Their work provided visual evidence supporting the ‘swinging lever-arm’ model for myosin “walking” along the filament.
Frequency-modulation AFM (FM-AFM)95 is another imaging mode where the tip–sample interaction is probed via minute changes in the cantilever resonance frequency (in contrast to amplitude in tapping mode). Recently, FM-AFM has been successfully applied to single biomolecules to visualize different structural conformations of two-polynucleotide strands in the DNA double helix.96
Probing nanomechanical changes in ABP–F-actin complexes
Stress and strain are important factors modulating cells’ reaction to their environment. Since actin filaments are associated with a large number of actin-binding proteins, including nucleators, capping proteins and cross-linking proteins, it is important to understand how these other proteins modulate and regulate the response of an actin filament to mechanical stress. Stress induced structural modifications in actin filaments have been studied biochemically or spectroscopically.97–99 Tension on an actin filament can regulate its disassembly by ABPs.100 Hayakawa et al.101 show that actin filament binding and severing by ADF/cofilin are inhibited when the filament is under tension. They also show that in cells ADF/cofilin localizes preferentially to relaxed rather than stretched actin stress fibers.101 Thus, mechanical stress and contractility may regulate the dynamics of actin filaments in cells, perhaps by altering filament twist to inhibit ADF/cofilin binding. Yet, it is still not fully understood how forces affect the molecular interactions, what role ABPs play in regulating the mechanical behavior and remodeling of F-actin, and how strong a force is required to pull the associated proteins apart in the absence or presence of other ABPs. These are some of the basic questions that can be potentially addressed using AFM based force spectroscopy measurements. One can obtain combined topography and rupture force maps of bare F-actin, and fully and partially decorated F-actin–ABP complexes. Furthermore, AFM tips can also be functionalized102–104 to measure and map the unbinding of ABPs as the molecules are pulled away from F-actin.
Thus, AFM imaging yields information on: (1) nanoscale 3D morphology of single filaments, helix parameters, and variations in helical twist, binding cooperativity, filament bending modulus and persistence length; (2) ABPs induced change in helical twists of individual filaments in the regions directly bound to the protein; (3) propagation of changes, if any, and the extent of the propagation of helical twist change in neighboring, unbound regions of F-actins; and (4) direct measurement of the bound protein cluster size and assembly, for determination of the binding cooperativity of the protein at a single filament level.
Higher-order ABP–F-actin structures
Within the cells, actin filaments rarely exist as isolated single filaments but instead associate into higher-order structures or networks, together with many different ABPs. Such higher-order cellular organization of actin is crucial to both the form and function of cells. Modest changes in the concentration of ABPs can significantly modify the structure of the network and can lead to concomitant changes in the structure, and mechanical properties of the cell. The changes in the structure occur over a wide range of length scales, ranging from a few nanometers to several micrometers.
Current AFM technology is powerful in visualizing nanoscale structural and dynamic (HS-AFM) events in isolated and purified biological molecules such as F-actin, but limited in the dynamic observation of the outer surface and the interior of live cells (i.e. in situ imaging).105–107 The extension of high-speed AFM to imaging live cells, which allows direct in situ observation of dynamic processes of biomolecules, remains an exciting challenge for the near future. Emerging super-resolution light microscopy techniques, such as stimulated emission depletion (STED),108 allow sub-diffraction limit visualization of cellular structures109 and are beginning to provide detailed information about the higher-order actin structures within the cells (Fig. 8). It will be interesting to see how combining the capabilities of STED and AFM could improve our understanding of actin filaments and networks regulated by various ABPs in the cellular environment.
Fig. 8.
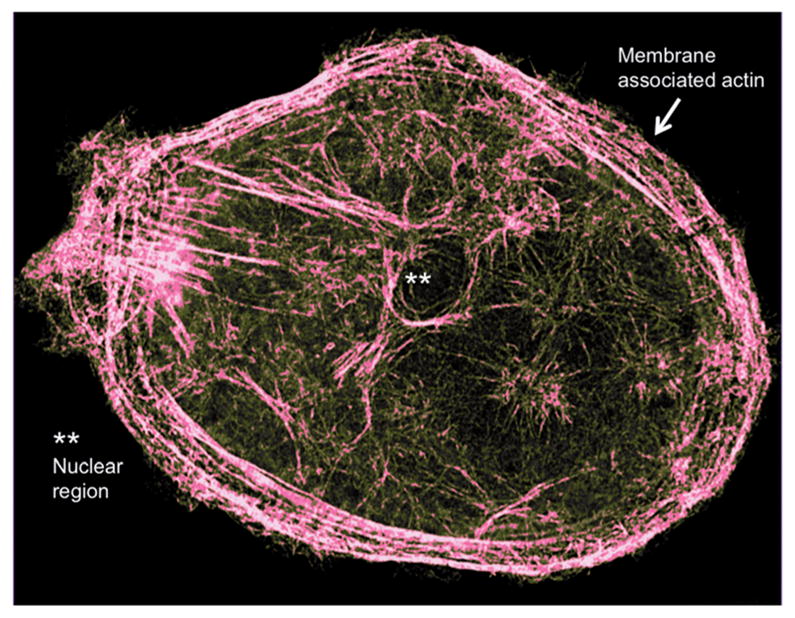
Super-resolution STED image of an ovarian cancer cell showing detailed actin cytoskeleton. Adapted from Sharma S. et al.109 with permission from American Chemical Society.
Summary.
The unique capability of AFM as a biophysical nano-characterization tool in studying F-actin remodeling induced by ABPs, not offered by other biochemical and biophysical techniques, includes sub-nanometer resolution – quantification at the single filament level – of structural (individual helix determination, volume analysis) and mechanical changes (persistence length) induced by the bound ABPs. AFM offers direct visualization of cooperative binding interactions and enables determination of the extent of propagation of these changes within the unbound regions of filaments, as demonstrated in our proof-of-concept studies involving neuronal ABP drebrin. The AFM based approach can be used to study F-actin interactions with cofilin, tropomyosin, coronin, formin, and other ABPs, and to obtain fundamental and quantitative molecular level information on the structural and biophysical F-actin remodeling and its regulation by ABPs at the single filament level.
Acknowledgments
Authors would like to acknowledge support from International Center for Materials Nanoarchitectonics Satellite (MANA), National Institute for Materials Science (NIMS), Tsukuba, Japan (J.K.G.), California Nanosystems Institute, UCLA (S.S.) and USPHS grant R01 GM 077190 (E.R.).
Biographies
 Dr Shivani Sharma is a Project Scientist at the California NanoSystems Institute at the University of California, Los Angeles. She received her PhD from the Centre for Biomedical Engineering, Indian Institute of Technology, Delhi, India. Her research focuses on biological and medical applications of scanning probe microscopy including actin structural remodeling induced by actin binding proteins, nanoscale characterization of exosomes and cellular mechanics for cancer diagnostics. She also serves as a Regional Editor (USA) for International Journal of Bioassays and Editorial Board member for Exosomics and Journal of Oncology.
Dr Shivani Sharma is a Project Scientist at the California NanoSystems Institute at the University of California, Los Angeles. She received her PhD from the Centre for Biomedical Engineering, Indian Institute of Technology, Delhi, India. Her research focuses on biological and medical applications of scanning probe microscopy including actin structural remodeling induced by actin binding proteins, nanoscale characterization of exosomes and cellular mechanics for cancer diagnostics. She also serves as a Regional Editor (USA) for International Journal of Bioassays and Editorial Board member for Exosomics and Journal of Oncology.
 Huanqi Zhu obtained his Bachelor’s degree in physics in 2010 at the University of Science and Technology of China (USTC), China. He is a PhD candidate in Physical Chemistry at the University of California, Los Angeles (UCLA) at Prof. Gimzewski’s lab. His research interests include characterization of the structure of filament molecules and force spectroscopy of cells.
Huanqi Zhu obtained his Bachelor’s degree in physics in 2010 at the University of Science and Technology of China (USTC), China. He is a PhD candidate in Physical Chemistry at the University of California, Los Angeles (UCLA) at Prof. Gimzewski’s lab. His research interests include characterization of the structure of filament molecules and force spectroscopy of cells.
 Dr Grintsevich is a Staff Research Associate in Emil Reisler’s group at the Department of Chemistry and Biochemistry of University of California, Los Angeles. She received her PhD in Chemistry in 2003. Her PhD work involved exploration of the role of human thyroid peroxidase in metabolic activation of carcinogenic compounds. Her current research interests include regulation of the actin cytoskeleton in normal and diseased states of neuronal system.
Dr Grintsevich is a Staff Research Associate in Emil Reisler’s group at the Department of Chemistry and Biochemistry of University of California, Los Angeles. She received her PhD in Chemistry in 2003. Her PhD work involved exploration of the role of human thyroid peroxidase in metabolic activation of carcinogenic compounds. Her current research interests include regulation of the actin cytoskeleton in normal and diseased states of neuronal system.
 Emil Reisler is a Distinguished Professor of Biochemistry in the Department of Chemistry and Biochemistry and the Molecular Biology Institute of University of California, Los Angeles. Prof. Reisler’s group has carried out research focused on structural understanding of motor proteins and of the actin based cytoskeleton system. Over the last decade the focus of the group has been on interactions of actin binding proteins with actin filaments and monomers. In close to 200 publications, reviews and book chapters, his group has contributed in a major way to the present mechanistic understanding of protein interactions and the remodeling of cellular actin assemblies by the bound proteins.
Emil Reisler is a Distinguished Professor of Biochemistry in the Department of Chemistry and Biochemistry and the Molecular Biology Institute of University of California, Los Angeles. Prof. Reisler’s group has carried out research focused on structural understanding of motor proteins and of the actin based cytoskeleton system. Over the last decade the focus of the group has been on interactions of actin binding proteins with actin filaments and monomers. In close to 200 publications, reviews and book chapters, his group has contributed in a major way to the present mechanistic understanding of protein interactions and the remodeling of cellular actin assemblies by the bound proteins.
 Dr James K. Gimzewski is a Distinguished Professor in the Department of Chemistry and Biochemistry and Member of the California NanoSystems Institute (CNSI) at UCLA. Previously, he worked for 20 years at the IBM Corporate Research Labs in Zurich, Switzerland. Dr Gimzewski pioneered research on mechanical and electrical contacts with single atoms and molecules using scanning tunneling microscopy (STM) and was one of the first persons to image molecules with STM. He has received honorary doctorates from the University of Aix-en-Provence and University of Strathclyde. He is a Fellow of the Royal Society, a Fellow of the Royal Academy of Engineering, a Fellow of the Institute of Physics and a Fellow of the World Innovation Foundation. His current interests are in the nanomechanics of biomolecules, cells and bacteria where he collaborates with the UCLA Medical and Dental Schools. He is involved in projects that range from the operation of X-rays, ions and nuclear fusion using pyroelectric crystals, direct deposition of carbon nanotubes and single molecule DNA profiling. His work has received worldwide press coverage.
Dr James K. Gimzewski is a Distinguished Professor in the Department of Chemistry and Biochemistry and Member of the California NanoSystems Institute (CNSI) at UCLA. Previously, he worked for 20 years at the IBM Corporate Research Labs in Zurich, Switzerland. Dr Gimzewski pioneered research on mechanical and electrical contacts with single atoms and molecules using scanning tunneling microscopy (STM) and was one of the first persons to image molecules with STM. He has received honorary doctorates from the University of Aix-en-Provence and University of Strathclyde. He is a Fellow of the Royal Society, a Fellow of the Royal Academy of Engineering, a Fellow of the Institute of Physics and a Fellow of the World Innovation Foundation. His current interests are in the nanomechanics of biomolecules, cells and bacteria where he collaborates with the UCLA Medical and Dental Schools. He is involved in projects that range from the operation of X-rays, ions and nuclear fusion using pyroelectric crystals, direct deposition of carbon nanotubes and single molecule DNA profiling. His work has received worldwide press coverage.
Contributor Information
Shivani Sharma, Email: sharmas@ucla.edu.
James K. Gimzewski, Email: gim@chem.ucla.edu.
References
- 1.Alberts B. Cell. 1998;92:291–294. doi: 10.1016/s0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- 2.Otterbein LR, Graceffa P, Dominguez R. Science. 2001;293:708–711. doi: 10.1126/science.1059700. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Bernstein BW, Bamburg JR. Trends Biochem Sci. 2000;25:19–23. doi: 10.1016/s0968-0004(99)01511-x. [DOI] [PubMed] [Google Scholar]
- 4.Engqvist-Goldstein AE, Drubin DG. Annu Rev Cell Dev Biol. 2003;19:287–332. doi: 10.1146/annurev.cellbio.19.111401.093127. [DOI] [PubMed] [Google Scholar]
- 5.Parsons JT, Horwitz AR, Schwartz MA. Nat Rev Mol Cell Biol. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaksonen M, Toret CP, Drubin DG. Nat Rev Mol Cell Biol. 2006;7:404–414. doi: 10.1038/nrm1940. [DOI] [PubMed] [Google Scholar]
- 7.Paul AS, Pollard TD. Cell Motil Cytoskeleton. 2009;66:606–617. doi: 10.1002/cm.20379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rouiller I, Xu XP, Amann KJ, Egile C, Nickell S, Nicastro D, Li R, Pollard TD, Volkmann N, Hanein D. J Cell Biol. 2008;180:887–895. doi: 10.1083/jcb.200709092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominguez R. Crit Rev Biochem Mol Biol. 2009;44:351–366. doi: 10.3109/10409230903277340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmid MF, Sherman MB, Matsudaira P, Chiu W. Nature. 2004;431:104–107. doi: 10.1038/nature02881. [DOI] [PubMed] [Google Scholar]
- 11.McCullough BR, Blanchoin L, Martiel JL, De la Cruz EM. J Mol Biol. 2008;381:550–558. doi: 10.1016/j.jmb.2008.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kueh HY, Charras GT, Mitchison TJ, Brieher WM. J Cell Biol. 2008;182:341–353. doi: 10.1083/jcb.200801027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gandhi M, Goode BL, Chan CSM. Genetics. 2006;174:665–678. doi: 10.1534/genetics.106.058180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chhabra ES, Higgs HN. Nat Cell Biol. 2007;9:1110–1121. doi: 10.1038/ncb1007-1110. [DOI] [PubMed] [Google Scholar]
- 15.Holmes KC, Popp D, Gebhard W, Kabsch W. Nature. 1990;347:44–49. doi: 10.1038/347044a0. [DOI] [PubMed] [Google Scholar]
- 16.Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 17.Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 18.Dominguez R, Freyzon Y, Trybus KM, Cohen C. Cell. 1998;94:559–571. doi: 10.1016/s0092-8674(00)81598-6. [DOI] [PubMed] [Google Scholar]
- 19.Fedorov AA, Lappalainen P, Fedorov EV, Drubin DG, Almo SC. Nat Struct Biol. 1997;4:366–369. doi: 10.1038/nsb0597-366. [DOI] [PubMed] [Google Scholar]
- 20.Volkmann N, Amann KJ, Stoilova-McPhie S, Egile C, Winter DC, Hazelwood L, Heuser JE, Li R, Pollard TD, Hanein D. Science. 2001;293:2456–2459. doi: 10.1126/science.1063025. [DOI] [PubMed] [Google Scholar]
- 21.McLaughlin PJ, Gooch JT, Mannherz HG, Weeds AG. Nature. 1993;364:685–692. doi: 10.1038/364685a0. [DOI] [PubMed] [Google Scholar]
- 22.McGough A, Pope B, Chiu W, Weeds A. J Cell Biol. 1997;138:771–781. doi: 10.1083/jcb.138.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lukoyanova N, VanLoock MS, Orlova A, Galkin VE, Wang K, Egelman EH. Curr Biol. 2002;12:383–388. doi: 10.1016/s0960-9822(02)00678-4. [DOI] [PubMed] [Google Scholar]
- 24.Schutt CE, Myslik JC, Rozycki MD, Goonesekere NC, Lindberg U. Nature. 1993;365:810–816. doi: 10.1038/365810a0. [DOI] [PubMed] [Google Scholar]
- 25.Galkin VE, Orlova A, Cherepanova O, Lebart MC, Egelman EH. Proc Natl Acad Sci U S A. 2008;105:1494–1498. doi: 10.1073/pnas.0708667105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang J, Taylor DW, Taylor KA. J Mol Biol. 2001;310:845–858. doi: 10.1006/jmbi.2001.4789. [DOI] [PubMed] [Google Scholar]
- 27.Volkmann N, DeRosier D, Matsudaira P, Hanein D. J Cell Biol. 2001;153:947–956. doi: 10.1083/jcb.153.5.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galkin VE, Orlova A, VanLoock MS, Rybakova IN, Ervasti JM, Egelman EH. J Cell Biol. 2002;157:243–251. doi: 10.1083/jcb.200111097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oda T, Iwasa M, Aihara T, Maeda Y, Narita A. Nature. 2009;457:441–445. doi: 10.1038/nature07685. [DOI] [PubMed] [Google Scholar]
- 30.Murakami K, Yasunaga T, Noguchi TQ, Gomibuchi Y, Ngo KX, Uyeda TQ, Wakabayashi T. Cell. 2010;143:275–287. doi: 10.1016/j.cell.2010.09.034. [DOI] [PubMed] [Google Scholar]
- 31.Voss NR, Potter CS, Smith R, Carragher B. Methods Enzymol. 2010;482:381–392. doi: 10.1016/S0076-6879(10)82016-6. [DOI] [PubMed] [Google Scholar]
- 32.Binnig G, Quate CF, Gerber C. Phys Rev Lett. 1986;56:930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- 33.Radmacher M, Tillamnn RW, Fritz M, Gaub HE. Science. 1992;257:1900–1905. doi: 10.1126/science.1411505. [DOI] [PubMed] [Google Scholar]
- 34.Bustamante C, Vesenka J, Tang CL, Rees W, Guthold M, Keller R. Biochemistry. 1992;31:22–26. doi: 10.1021/bi00116a005. [DOI] [PubMed] [Google Scholar]
- 35.Hansma HG, Hoh JH. Annu Rev Biophys Biomol Struct. 1994;23:115–139. doi: 10.1146/annurev.bb.23.060194.000555. [DOI] [PubMed] [Google Scholar]
- 36.Muller DJ, Schabert FA, Buldt G, Engel A. Biophys J. 1995;68:1681–1686. doi: 10.1016/S0006-3495(95)80345-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muller DJ, Dufrene YF. Nat Nanotechnol. 2008;3:261–269. doi: 10.1038/nnano.2008.100. [DOI] [PubMed] [Google Scholar]
- 38.Sharma S, Gimzewski J. In: Life at the nanoscale: Atomic Force Microscopy of live cells. Dufrene Y, editor. ch 20. Pan Stanford Publishing; 2011. pp. 421–436. [Google Scholar]
- 39.Hinterdorfer P, Dufrene YF. Nat Methods. 2006;3:347–355. doi: 10.1038/nmeth871. [DOI] [PubMed] [Google Scholar]
- 40.Muller DJ, Helenius J, Alsteens D, Dufrene YF. Nat Chem Biol. 2009;5:383–390. doi: 10.1038/nchembio.181. [DOI] [PubMed] [Google Scholar]
- 41.Florin EL, Moy VT, Gaub HE. Science. 1994;264:415–417. doi: 10.1126/science.8153628. [DOI] [PubMed] [Google Scholar]
- 42.Ohnesorge F, Binnig G. Science. 1993;260:1451–1456. doi: 10.1126/science.260.5113.1451. [DOI] [PubMed] [Google Scholar]
- 43.Hansma HG, Sinsheimer RL, Groppe J, Bruice TC, Elings V, Gurley G, Bezanilla M, Mastrangelo IA, Hough PV, Hansma PK. Scanning. 1993;15:296–299. doi: 10.1002/sca.4950150509. [DOI] [PubMed] [Google Scholar]
- 44.Fotiadis D, Jeno P, Mini T, Wirtz S, Muller SA, Fraysse L, Kjellbom P, Engel A. J Biol Chem. 2001;276:1707–1714. doi: 10.1074/jbc.M009383200. [DOI] [PubMed] [Google Scholar]
- 45.Schabert FA, Henn C, Engel A. Science. 1995;268:92–94. doi: 10.1126/science.7701347. [DOI] [PubMed] [Google Scholar]
- 46.Scheuring S, Ringler P, Borgnia M, Stahlberg H, Muller DJ, Agre P, Engel A. EMBO J. 1999;18:4981–4987. doi: 10.1093/emboj/18.18.4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scheuring S, Seguin J, Marco S, Levy D, Robert B, Rigaud JL. Proc Natl Acad Sci U S A. 2003;100:1690–1693. doi: 10.1073/pnas.0437992100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuznetsov Y, Gershon PD, McPherson A. J Virol. 2008;82:7551–7566. doi: 10.1128/JVI.00016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuznetsov YG, Victoria JG, Robinson WE, Jr, McPherson A. J Virol. 2003;77:11896–11909. doi: 10.1128/JVI.77.22.11896-11909.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ikai A, Yoshimura K, Arisaka F, Ritani A, Imai K. FEBS Lett. 1993;326:39–41. doi: 10.1016/0014-5793(93)81756-p. [DOI] [PubMed] [Google Scholar]
- 51.McPherson A, Kuznetsov YG. Methods Mol Biol. 2011;736:171–195. doi: 10.1007/978-1-61779-105-5_12. [DOI] [PubMed] [Google Scholar]
- 52.Hansma HG, Oroudjev E, Baudrey S, Jaeger L. J Microsc. 2003;212:273–279. doi: 10.1111/j.1365-2818.2003.01276.x. [DOI] [PubMed] [Google Scholar]
- 53.Fujita Y, Kobayashi K, Hoshino T. J Electron Microsc. 1997;46:321–326. doi: 10.1093/oxfordjournals.jmicro.a023526. [DOI] [PubMed] [Google Scholar]
- 54.Adamcik J, Jung JM, Flakowski J, De Los Rios P, Dietler G, Mezzenga R. Nat Nanotechnol. 2010;5:423–428. doi: 10.1038/nnano.2010.59. [DOI] [PubMed] [Google Scholar]
- 55.Shao Z, Shi D, Somlyo AV. Biophys J. 2000;78:950–958. doi: 10.1016/S0006-3495(00)76652-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma S, Grintsevich EE, Phillips ML, Reisler E, Gimzewski JK. Nano Lett. 2011;11:825–827. doi: 10.1021/nl104159v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takebayashi T, Morita Y, Oosawa F. Biochim Biophys Acta. 1977;492:357–363. doi: 10.1016/0005-2795(77)90086-1. [DOI] [PubMed] [Google Scholar]
- 58.Isambert H, Venier P, Maggs AC, Fattoum A, Kassab R, Pantaloni D, Carlier MF. J Biol Chem. 1995;270:11437–11444. doi: 10.1074/jbc.270.19.11437. [DOI] [PubMed] [Google Scholar]
- 59.Reisler E, Egelman EH. J Biol Chem. 2007;282:36133–36137. doi: 10.1074/jbc.R700030200. [DOI] [PubMed] [Google Scholar]
- 60.Trachtenberg S, Stokes D, Bullitt E, DeRosier D. Ann N Y Acad Sci. 1986;483:88–99. doi: 10.1111/j.1749-6632.1986.tb34500.x. [DOI] [PubMed] [Google Scholar]
- 61.Galkin VE, Orlova A, Schroder GF, Egelman EH. Nat Struct Mol Biol. 2010;17:1318–1323. doi: 10.1038/nsmb.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ott A, Libchaber A. Phys Rev E: Stat Phys, Plasmas, Fluids, Relat Interdiscip Top. 1993;48:R1642–R1645. doi: 10.1103/physreve.48.r1642. [DOI] [PubMed] [Google Scholar]
- 63.Egelman EH, Francis N, Derosier DJ. Nature. 1982;298:131–135. doi: 10.1038/298131a0. [DOI] [PubMed] [Google Scholar]
- 64.Belmont LD, Orlova A, Drubin DG, Egelman EH. Proc Natl Acad Sci U S A. 1999;96:29–34. doi: 10.1073/pnas.96.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kueh HY, Mitchison TJ. Science. 2009;325:960–963. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hild G, Bugyi B, Nyitrai M. Cytoskeleton. 2010;67:609–629. doi: 10.1002/cm.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prochniewicz E, Janson N, Thomas DD, De la Cruz EM. J Mol Biol. 2005;353:990–1000. doi: 10.1016/j.jmb.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 68.De La Cruz EM. Biophys Rev. 2009;1:51–59. doi: 10.1007/s12551-009-0008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prochniewicz E, Thomas DD. Biochemistry. 1997;36:12845–12853. doi: 10.1021/bi971201r. [DOI] [PubMed] [Google Scholar]
- 70.Orlova A, Prochniewicz E, Egelman EH. J Mol Biol. 1995;245:598–607. doi: 10.1006/jmbi.1994.0049. [DOI] [PubMed] [Google Scholar]
- 71.Prochniewicz E, Zhang Q, Janmey PA, Thomas DD. J Mol Biol. 1996;260:756–766. doi: 10.1006/jmbi.1996.0435. [DOI] [PubMed] [Google Scholar]
- 72.Higgs HN. Trends Biochem Sci. 2005;30:342–353. doi: 10.1016/j.tibs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 73.De La Cruz EM. J Mol Biol. 2005;346:557–564. doi: 10.1016/j.jmb.2004.11.065. [DOI] [PubMed] [Google Scholar]
- 74.Hill AV. J Physiol. 1910:40. [Google Scholar]
- 75.Weiss JN. FASEB J. 1997;11:835–841. [PubMed] [Google Scholar]
- 76.Wyman J., Jr Adv Protein Chem. 1964;19:223–286. doi: 10.1016/s0065-3233(08)60190-4. [DOI] [PubMed] [Google Scholar]
- 77.Solomatin SV, Greenfeld M, Herschlag D. Nat Struct Mol Biol. 2011;18:732–734. doi: 10.1038/nsmb.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kovar DR, Harris ES, Mahaffy R, Higgs HN, Pollard TD. Cell. 2006;124:423–435. doi: 10.1016/j.cell.2005.11.038. [DOI] [PubMed] [Google Scholar]
- 79.Kozuka J, Yokota H, Arai Y, Ishii Y, Yanagida T. Nat Chem Biol. 2006;2:83–86. doi: 10.1038/nchembio763. [DOI] [PubMed] [Google Scholar]
- 80.Schneckenburger H. Curr Opin Biotechnol. 2005;16:13–18. doi: 10.1016/j.copbio.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 81.Kuhn JR, Pollard TD. Biophys J. 2005;88:1387–1402. doi: 10.1529/biophysj.104.047399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan C, Beltzner CC, Pollard TD. Curr Biol. 2009;19:537–545. doi: 10.1016/j.cub.2009.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cai L, Makhov AM, Schafer DA, Bear JE. Cell. 2008;134:828–842. doi: 10.1016/j.cell.2008.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sharma S, Grintsevich EE, Hsueh C, Reisler E, Gimzewski JK. Biophys J. 2012;103:275–283. doi: 10.1016/j.bpj.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kratky O, Porod G. Recl Trav Chim Pays-Bas. 1949;68:1106–1122. [Google Scholar]
- 86.Landau LD. Sov Phys. 1958;7:182–184. [Google Scholar]
- 87.Rivetti C, Guthold M, Bustamante C. J Mol Biol. 1996;264:919–932. doi: 10.1006/jmbi.1996.0687. [DOI] [PubMed] [Google Scholar]
- 88.Frontali C, Dore E, Ferrauto A, Gratton E, Bettini A, Pozzan MR, Valdevit E. Biopolymers. 1979;18:1353–1373. doi: 10.1002/bip.1979.360180604. [DOI] [PubMed] [Google Scholar]
- 89.Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 90.Rivetti C, Guthold M, Bustamante C. EMBO J. 1999;18:4464–4475. doi: 10.1093/emboj/18.16.4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schmitz S, Schaap IA, Kleinjung J, Harder S, Grainger M, Calder L, Rosenthal PB, Holder AA, Veigel C. J Biol Chem. 2010;285:36577–36585. doi: 10.1074/jbc.M110.142638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ando T, Uchihashi T, Fukuma T. Prog Surf Sci. 2008;83:337–437. [Google Scholar]
- 93.Hansma PK, Schitter G, Fantner GE, Prater C. Science. 2006;314:601–602. doi: 10.1126/science.1133497. [DOI] [PubMed] [Google Scholar]
- 94.Kodera N, Yamamoto D, Ishikawa R, Ando T. Nature. 2010;468:72–76. doi: 10.1038/nature09450. [DOI] [PubMed] [Google Scholar]
- 95.Fukuma T, Kobayashi K, Matsushige K, Yamada H. Appl Phys Lett. 2005;87:034101/1–034101/3. [Google Scholar]
- 96.Leung C, Bestembayeva A, Thorogate R, Stinson J, Pyne A, Marcovich C, Yang J, Drechsler U, Despont M, Jankowski T, Tschope M, Hoogenboom BW. Nano Lett. 2012;12:3846–3850. doi: 10.1021/nl301857p. [DOI] [PubMed] [Google Scholar]
- 97.Tsuda Y, Yasutake H, Ishijima A, Yanagida T. Proc Natl Acad Sci U S A. 1996;93:12937–12942. doi: 10.1073/pnas.93.23.12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shimozawa T, Ishiwata S. Biophys J. 2009;96:1036–1044. doi: 10.1016/j.bpj.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kojima H, Ishijima A, Yanagida T. Proc Natl Acad Sci U S A. 1994;91:12962–12966. doi: 10.1073/pnas.91.26.12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Galkin VE, Orlova A, Egelman EH. Curr Biol. 2012;22:R96–R101. doi: 10.1016/j.cub.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hayakawa K, Tatsumi H, Sokabe M. J Cell Biol. 2011;195:721–727. doi: 10.1083/jcb.201102039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yakovenko O, Sharma S, Forero M, Tchesnokova V, Aprikian P, Kidd B, Mach A, Vogel V, Sokurenko E, Thomas WE. J Biol Chem. 2008;283:11596–11605. doi: 10.1074/jbc.M707815200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sharma S, Rasool HI, Palanisamy V, Mathisen C, Schmidt M, Wong DT, Gimzewski JK. ACS Nano. 2010;4:1921–1926. doi: 10.1021/nn901824n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sharma S, Gonzalez O, Petzold O, Baker W, Walczak W, Yongsunthon R, Baker D, Gimzewski JK. J Scanning Probe Microsc. 2009;4:7–16. [Google Scholar]
- 105.Rotsch C, Radmacher M. Biophys J. 2000;78:520–535. doi: 10.1016/S0006-3495(00)76614-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Azeloglu EU, Costa KD. Methods Mol Biol. 2011;736:303–329. doi: 10.1007/978-1-61779-105-5_19. [DOI] [PubMed] [Google Scholar]
- 107.Arce FT, Meckes B, Camp SM, Garcia JG, Dudek SM, Lal R. Nanomedicine: Nanotechnology, Biology, and Medicine. 2013 doi: 10.1016/j.nano.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hell SW, Wichmann J. Opt Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- 109.Sharma S, Santiskulvong C, Bentolila LA, Rao J, Dorigo O, Gimzewski JK. Nanomedicine: Nanotechnology, Biology, and Medicine. 2012;8:757–766. doi: 10.1016/j.nano.2011.09.015. [DOI] [PubMed] [Google Scholar]