

Abstract
Antisense oligonucleotides with iron binding hydroxamate linkages are designed to act as sequence-selective cleaving agents of complementary nucleic acids through Fenton chemistry. Oligothymidylate analogs with hydroxamate linkages were efficiently synthesized from coupling of nucleoside intermediates, activated as p-nitrophenyl carbonates, with hydroxylamine derivatized nucleosides. Iron binding studies showed that hydroxamate linked oligonucleotides are effective iron chelators when there are three nonadjacent internucleosidic hydroxamate linkages available in the same oligonucleotide molecule. However, analysis of the CD spectra of an oligothymidylate 16mer, which contained complete substitution of all phosphates with hydroxamates, indicated that the hydroxamate linkage was too rigid to allow the analog to base pair with the complementary DNA d(A16). Syntheses of mix-linked thymidine oligomers with up to three hydroxamate linkages incorporated in the center of the sequence are also reported. Iron binding of the thymidine oligomer with hydroxamate linkages was confirmed by matrix assisted laser desorption mass spectrometry analysis. Nuclease stability assays showed that the modified oligonucleotides have enhanced resistance toward nuclease S1 (endonuclease) compared to natural oligonucleotides. A thymidine 16mer with three hydroxamate linkages incorporated in the center of the sequence was shown to be able to bind with both iron and its complementary polyA strand. A small destablizing effect was observed when the phosphodiester linkage was changed to the hydroxamate linkage. Under Fenton chemistry conditions, this novel iron binding oligothymidylate analog cleaved the complementary DNA strand sequence-selectively.
Keywords: Hydroxamic acid, Iron binding, Fenton chemistry, Antisense oligonucleotides, Chemical nucleases
Introduction
The therapeutic potential of antisense oligonucleotides continues to attract attention in medicinal chemistry (Mansoor and Melendez 2008; Aboul-Fadl 2005; Manoharan 2002; Lebedeva and Stein 2001; Wickstrom 1991; Crooke and Lebleu 1993; Agrawal 1996; Chu and Baker 1993; Sanghvi and Cook 1994; Uhlmann and Peyman 1990). The sequence-specific inhibition mechanism of antisense oligos potentially can reduce the toxicity problems encountered with current antiviral and anticancer therapies. However, in recent years antisense research has suffered from many technical problems, most notably inadequate sequence specificity and insufficient cellular uptake (Agrawal 1999; Stein 1999). In the usual antisense approach, oligonucleotides are designed to bind to a specific mRNA to inhibit the translation process via duplex formation and/or RNase H-induced degradation of the mRNA (Minshull and Hunt 1986). Previously, we proposed that novel antisense oligonucleotides with hydroxamate linkages (Fig. 1; Li and Miller 1999, 2000) would have the potential to cleave the corresponding natural sense strands by a new cleavage pathway. Since it is well known that hydroxamic acids effectively chelate Fe(III), among other metals (Hashimoto and Nakamur 1995; Roosenberg et al. 2000), and the bound Fe(III) can undergo Fenton chemistry to generate hydroxyl radicals (Ghosh et al. 1995), antisense oligonucleotides with hydroxamate linkages could potentially cleave the target mRNA through radical reactions. Moreover, hydroxamate linked oligonucleotides, when chelated with Fe(III), might have the potential to assist cellular uptake of antisense oligonucleotides through active iron uptake systems (Roosenberg et al.2000). Antisense oligonucleotides chelating other metals such as indium-111 or technicium-99 could also potentially be used in MRI-assisted detection of gene expression in tissues, as has been done with labeled peptides (Arano et al. 1996; Fischman et al. 1993). Herein, we report the syntheses of thymidine oligomers with hydroxamate linkages, binding studies, nuclease stability studies and the sequence-selective cleavage of a complementary DNA strand through Fenton chemistry. As proof of concept, complementary DNA strands were used for the binding and sequence-selective cleavage studies.
Fig. 1.

Proposed novel antisense oligonucleotide
Materials and methods
General synthetic methods and procedures
Melting points (mp) were performed on a Thomas-Hoover Capillary Melting Point Apparatus and are uncorrected. NMR spectra were recorded on a Varian 300 instrument at 300 MHz (1H), 75 MHz (13C), and 121 MHz (31P), unless otherwise noted. 13C and 31P NMR spectra were fully proton decoupled. The chemical shifts are reported in parts per million (ppm) relative to TMS or residual solvent and relative to external 85% phosphoric acid (0 ppm) in 31P NMR. Coupling constants are reported in Hz. IR spectra were obtained on a Perkin Elmer Paragon 1000 FT-IR. UV spectra were measured on a Beckman DU-40 spectro-photometer. HPLC analysis was performed on a Waters-Millipore instrument. TLC was performed utilizing Silica gel 60 F254. Visualization was performed utilizing UV light and PMA stain. Silica gel flash column chromatography was performed using Silica Gel 60 (30–70 μm irregular particles). Reverse phase chromatography was performed on Whatman LRP-2 C-18 silica gel (37–53 μm particles). Sephadex Microspin G-25 columns were purchased from Amersham Pharmacia Biotech. Super RX X-ray film was purchased from Fuji.
Unless otherwise noted, most reactions were carried out in septum-capped, oven-dried, and argon-purged flasks. THF was continuously refluxed from sodium benzophenone ketyl and distilled immediately before use. Dichloromethane was distilled from CaH2 under argon. DMF was dried over 3Å molecular sieves. Pyridine was dried over anhydrous KOH pellets. All chemicals and reagents were obtained from Aldrich. T4 polynucleotide kinase and 10× buffer were purchased from New England Biolabs. Nuclease S1 was purchased from Boehringer Mannheim. Distilled water was purified on a Milli-Q system (Millipore). DNA oligomers were obtained from solid phase DNA syntheses at the Bioscience Core facility at the Department of Chemistry and Biochemistry, University of Notre Dame. Their concentrations were determined spectrophotometrically using extinction coefficients E254 of d(A16) (221.76), d(T16) (126.72) and 32mer (377.28) (Eckstein 1991). T4 Kinase was purchased from Epicentre Technology and used with its supplied buffer. Deoxyadenosine 5′-[γ-32P]triphosphate was purchased from ICN.
O4-Benzyl-3′-O-(tert-butyldimethylsilyl)-5′-O-(p,p′-dimethoxytrityl)thymidine (2) Compound 1 (32.9 g, 0.05 mol) was dissolved in anhydrous dichloromethane (200 ml). Then DMAP (3 g, 0.025 mol) and triethylamine (28 ml, 0.2 mol) were added to the solution, followed by p-toluenesulfonyl chloride (14.3 g, 0.075 mol). The reaction was stirred at room temperature (rt) overnight. Then benzyl alcohol (10.35 ml, 0.1 mol) and DBU (14.95 ml, 0.1 mol) were added to the solution. The reaction was stirred another overnight period and then was diluted with dichloromethane (200 ml) and washed with saturated aqueous NaHCO3 (3 × 200 ml). The organic layer was separated, dried over Na2SO4, filtered, and evaporated. The residue was purified by flash column chromatography (hexanes/EtOAc = 8:1, 4:1, 2:1) to give 2 (32.95 g, 88%) as a clear oil. 1H NMR (CDCl3) δ 8.04 (s, 1 H), 7.44-7.20 (m, 14 H), 6.84 (dd, J = 1.5 Hz, 8.7 Hz, 4 H), 6.37 (t, J = 5.7 Hz, 1 H), 5.45 (ABq, J = 12.9 Hz, 2 H), 4.52 (q, J = 5.4 Hz, 1 H), 4.00 (m, 1 H), 3.76 (s, 6 H), 3.56 (dd, J = 2.4 Hz, 10.8 Hz, 1 H), 3.29 (dd, J = 2.7 Hz, 10.8 Hz, 1 H), 2.58–2.50 (m, 1 H), 2.31–2.22 (m, 1 H), 1.54 (s, 3 H), 0.82 (s, 9 H), 0.018 (s, 3 H), −0.049 (s, 3 H); 13C NMR (CDCl3) δ 169.97, 158.52, 155.60, 144.18, 139.69, 135.84, 135.28, 129.91, 128.31, 128.00, 127.92, 127.81, 127.75, 126.90, 113.05, 113.03, 104.33, 86.51, 86.37, 86.17, 70.84, 68.43, 62.14, 55.01, 42.06, 25.54, 17.73, 11.56, −4.80, −5.12; IR (neat): 2,930, 1,671, 1,508, 1,252 cm−1; FABMS found for C44H53N2O7Si (M+H)+: 749.
O4-Benzyl-3′-O-(tert-butyldimethylsilyl)thymidine (3) Compound 2 (25 g, 0.033 mol) was dissolved in dichloromethane (400 ml). Then dichloroacetic acid (40 ml) was added to the reaction solution. The reaction mixture was stirred at rt for 30 min and then quenched by saturated aqueous NaHCO3 (400 ml) at 0°C. The solution was stirred for another 15 min, layers were separated and the aqueous layer was extracted with dichloromethane (2 × 200 ml). The organic layers were combined, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (hexanes/EtOAc = 4:1, 2:1, 1:1) to give 3 (12.95 g, 87%) as a white solid. Mp 109–110°C; 1H NMR (CDCl3) δ 7.91 (d, J = 0.9 Hz, 1 H), 7.42–7.29 (m, 5 H), 6.19 (t, J = 6.3 Hz, 1 H), 5.41 (ABq, J = 12.9 Hz, 2 H), 4.53 (m, 1 H), 3.99–3.95 (m, 2 H), 3.80 (dd, J = 3.3 Hz, 12.3 Hz, 1 H), 2.36 (m, 2 H), 1.95 (d, J = 0.9 Hz, 3 H), 0.89 (s, 9 H), 0.074 (s, 6 H); 13C NMR (CDCl3) δ 170.06, 155.92, 141.34, 135.65, 128.35, 128.02, 127.80, 104.68, 88.16, 88.02, 71.32, 68.54, 61.52, 41.19, 25.59, 17.80, 12.07, −4.85, −5.01; IR (neat): 3,361, 2,930, 1,661, 1,105 cm−1; HRFABMS calcd. for C23H35N2O5Si (M+H)+: 447.2315, found: 447.2295.
O4-Benzyl-3′-O-(tert-butyldimethylsilyl)-5′-deoxy-5′-N-(N-tert-butoxycarbonyl-O-benzyl)hydroxylaminothymidine (4) Compound 3 (13.5 g, 0.03 mol), triphenylphosphine (9.53 g, 0.036 mol) and BocN-HOBn (8.1 g, 0.036 mol) were dissolved in anhydrous THF (200 ml). Then di-tert-butyl azodi-carboxylate (DBAD) (8.36 g, 0.036 mol) was added to the solution at 0°C. The reaction mixture was stirred at rt overnight. TLC indicated complete consumption of starting material. Then THF was removed under reduced pressure. The residue was purified by flash column chromatography (hexanes/EtOAc = 6:1, 4:1) to give 4 (17.0 g, 87%) as a clear oil. 1H NMR (CDCl3) δ 7.71 (d, J = 0.9 Hz, 1 H), 7.43–7.31 (m, 10 H), 6.27 (t, J = 6.3 Hz, 1 H), 5.42 (ABq, J = 12.9 Hz, 2 H), 4.88 (s, 2 H), 4.21–4.13 (m, 2 H), 3.71 (dd, J = 7.8 Hz, 14.7 Hz, 1 H), 3.51 (dd, J = 3.9 Hz, 14.7 Hz, 1 H), 2.48 (ddd, J = 3.9 Hz, 6.0 Hz, 13.5 Hz, 1 H), 2.04–1.96 (m, 1 H), 1.91 (d, J = 1.2 Hz, 3 H), 1.50 (s, 9 H), 0.87 (s, 9 H), 0.035 (s, 3 H), 0.025 (s, 3 H); 13C NMR (CDCl3) δ 170.09, 156.72, 155.62, 139.79, 135.93, 135.17, 129.35, 128.59, 128.41, 128.01, 127.89, 104.46, 86.92, 83.76, 81.81, 77.11, 73.22, 68.54, 51.27, 41.30, 28.18, 25.64, 17.85, 11.99, −4.71, −4.98; IR (neat): 2,930, 1,674 cm−1; HRFABMS calcd. for C35H50N3O7Si (M+H)+: 652.3418, found: 652.3437.
3′-O-(tert-Butyldimethylsilyl)-5′-deoxy-5′-N-(O-benzyl)-hydroxylaminothymidine (5) Compound 4 (8.0 g, 0.012 mol) was dissolved in anhydrous dichloromethane (200 ml). Then trifluoroacetic acid (20 ml) was added. After stirring at rt for 2 h, a saturated aqueous NaHCO3 (200 ml) was added to quench the reaction at 0°C. The resulting solution was extracted with dichloromethane (2 × 100 ml). The organic layers were combined, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (hexanes/EtOAc = 1:1) to give 5 (5.6 g, 99%) as a clear oil. 1H NMR (CDCl3) δ 9.56 (s, 1 H), 7.36–7.31 (m, 5 H), 7.22 (d, J = 1.2 Hz, 1 H), 6.21 (t, J = 6.6 Hz, 1 H), 5.84 (br, 1 H), 4.74 (s, 2 H), 4.29 (m, 1 H), 4.02 (m, 1 H), 3.22 (dd, J = 4.2 Hz, 13.5 Hz, 1 H), 3.07 (dd, J = 6.9 Hz, 13.5 Hz, 1 H), 2.25 (ddd, J = 3.9 Hz, 6.6 Hz, 13.5 Hz, 1 H), 2.13–2.04 (m, 1 H), 1.88 (d, J = 0.9 Hz, 3 H), 0.89 (s, 9 H), 0.072 (s, 6 H); 13C NMR (CDCl3) δ 163.91, 150.28, 137.42, 135.60, 128.36, 128.33, 127.91, 111.00, 85.12, 83.94, 76.05, 73.13, 53.50, 40.33, 25.64, 17.85, 12.52, −4.71, −4.88; IR (neat): 3,184, 2,928, 1,699, 1,652 cm−1; HRFABMS calcd. for C23H36N3O5Si (M+H)+: 462.2424, found: 462.2399.
p-Nitrophenyl O4-benzyl-5′-deoxy-5′-N-(N-tert-butoxycarbonyl-O-benzyl)hydroxylaminothymidine 3′-carbonate (6) Compound 4 (7 g, 0.011 mol) was dissolved in THF (100 ml) and then 1.0 M TBAF in THF (12.9 ml) was added to the solution. The reaction mixture was stirred at rt for 20 min. Then THF was evaporated. The residue was redissolved in ethyl acetate (100 ml), washed with water (100 ml), 10% aqueous NH4Cl (2 × 100 ml) and water (100 ml). The ethyl acetate layer was separated, dried over Na2SO4, filtered, and evaporated to give the product as a white foam, which was used without further purification. H NMR (CDCl3) δ 7.80 (d, J = 0.9 Hz, 1 H), 7.43–7.31 (m, 10 H), 6.36 (t, J = 6.3 Hz, 1 H), 5.40 (s, 2 H), 4.90 (s, 2 H), 4.65 (br, 1 H), 4.36–4.28 (m, 2 H), 3.82 (dd, J = 8.1 Hz, 15 Hz, 1 H), 3.66 (dd, J = 4.2 Hz, 15 Hz, 1 H), 2.68 (ddd, J = 4.2 Hz, 5.7 Hz, 13.8 Hz, 1 H), 2.06–1.97 (m, 1 H), 1.89 (d, J = 0.9 Hz, 3 H), 1.50 (s, 9H); 13C NMR (CDCl3) δ 170.04, 156.67, 155.80, 139.89, 135.66, 134.87, 129.30, 128.44, 128.27, 128.24, 127.91, 127.77, 104.84, 86.73, 83.41, 81.73, 76.93, 72.07, 68.47, 50.98, 40.76, 28.05, 11.76; IR (neat): 3,372, 1,652 cm−1; HRFABMS calcd. for C29H36N3O7 (M+H)+: 538.2553, found: 538.2578. The crude compound was dissolved in anhydrous dichloromethane (100 ml). Then pyridine (1.62 ml, 0.02 mol) was added, followed by p-nitrophenyl chloroformate (2.6 g, 0.013 mol). The reaction solution was stirred at rt for 2 h. Then the solvent was evaporated. The residue was redissolved in ethyl acetate (100 ml), washed with water (100 ml), saturated aqueous NaHCO3 (3 × 100 ml) and water (100 ml). The ethyl acetate layer was separated, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (hexanes/EtOAc = 2:1, 1:1) to give 6 (7.12 g, 94% in two steps) as a clear oil. 1H NMR (CDCl3) δ 8.28 (d, J = 9.0 Hz, 2 H), 7.73 (d, J = 1.2 Hz, 1 H), 7.43–7.33 (m, 12 H), 6.42 (dd, J = 5.1 Hz, 8.7 Hz, 1 H), 5.44 (ABq, 2 H), 5.29 (d, J = 6.3 Hz, 1 H), 4.90 (s, 2 H), 4.47 (t, J = 5.1 Hz, 1 H), 3.88 (dd, J = 6.6 Hz, 15 Hz, 1 H), 3.78 (dd, J = 5.1 Hz, 15 Hz, 1 H), 2.81 (dd, J = 5.4 Hz, 13.8 Hz, 1 H), 2.18–2.04 (m, 1 H), 1.92 (d, J = 0.6 Hz, 3 H), 1.50 (s, 9 H); 13C NMR (CDCl3) δ 170.25, 156.30, 155.60, 155.04, 151.70, 145.39, 139.47, 135.65, 134.85, 129.34, 128.69, 128.43, 128.39, 128.07, 127.84, 125.21, 121.59, 105.24, 86.29, 82.12, 81.05, 80.18, 76.84, 68.67, 50.52, 37.57, 28.08, 11.88; IR (neat): 2,979, 1,767, 1,674 cm−1; HRFABMS calcd. for C36H38N4O11 (M+H)+: 703.2615, found: 703.2614.
Dimer 7 A mixture of the p-nitrophenylcarbonate 6 (5.5 g, 7.83 mmol), 5 (4.3 g, 9.33 mmol) and HOAt (5.33 g, 39 mmol) in dry DMF (50 ml) was treated with diisopropylethylamine (DIPEA) (6.82 ml, 39 mmol) and stirred at rt for 24 h. The solution was diluted with ethyl acetate (100 ml) and washed with water (2 × 100 ml), saturated aqueous NaHCO3 (3 × 100 ml) and brine (100 ml). The ethyl acetate layer was separated, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (hexanes/EtOAc = 2:1, 1:1, EtOAc) to give dimer 7 (7.06 g, 88%) as a clear oil. 1H NMR (CDCl3) δ 9.31 (s, 1 H), 7.73 (s, 1 H), 7.44–7.32 (m, 15 H), 7.18 (s, 1 H), 6.40 (dd, J = 5.1 Hz, 8.7 Hz, 1 H), 6.13 (t, 1 H), 5.44 (ABq, J = 12.9 Hz, 2 H), 5.16 (d, J = 6.6 Hz, 1 H), 4.91 (s, 2 H), 4.87 (s, 2 H), 4.32–4.26 (m, 2 H), 4.12 (m, 1 H), 3.90–3.73 (m, 3 H), 3.55 (dd, J = 4.2 Hz, 14.7 Hz, 1 H), 2.65 (dd, J = 5.4 Hz, 14.4 Hz, 1 H), 2.28–1.96 (m, 3 H), 1.92 (s, 3 H), 1.83 (s, 3 H), 1.48 (s, 9 H), 0.89 (s, 9 H), 0.052 (s, 6 H); 13C NMR (CDCl3) δ 170.19, 163.74, 156.55, 156.45, 155.66, 150.07, 139.70, 135.94, 135.79, 134.95, 134.62, 129.41, 129.37, 128.82, 128.59, 128.39, 128.03, 127.88, 110.87, 105.15, 86.26, 85.92, 83.15, 81.89, 81.53, 77.49, 76.71, 73.01, 68.64, 51.32, 50.39, 40.08, 37.60, 28.11, 25.58, 17.78, 12.23, 11.91, −4.78, −4.95; IR (neat): 2,931, 1,685 cm−1; HRFABMS calcd. for C53H69N6O13Si (M+H)+: 1,025.4692, found: 1,025.4739.
Dimer 8 Dimer 7 (3.0 g, 2.93 mmol) was dissolved in anhydrous dichloromethane (100 ml). Then trifluoroacetic acid (10 ml) was added. After stirring at rt for 2 h, a saturated aqueous NaHCO3 (100 ml) was added to quench the reaction at 0°C. The resulting solution was extracted with dichloromethane (2 × 100 ml). The organic layers were combined, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (EtOAc) to give dimer 8 (2.32 g, 95%) as a white foam. 1H NMR (CDCl3) δ 9.40 (s, 1 H), 9.37 (s, 1 H), 7.41–7.30 (m, 11 H), 7.18 (s, 1 H), 6.21 (dd, J = 6.0 Hz, 8.4 Hz, 1 H), 6.10 (t, 1 H), 5.22 (d, J = 6.3 Hz, 1 H), 4.89 (s, 2 H), 4.74 (s, 2 H), 4.29 (m, 1 H), 4.18 (m, 1 H), 4.10 (m, 1 H), 3.77 (dd, J = 8.1 Hz, 15 Hz, 1 H), 3.54 (d, J = 11.7 Hz, 1 H), 3.31–3.17 (m, 2 H), 2.37 (dd, J = 6.3 Hz, 14.1 Hz, 1 H), 2.20 (m, 3 H), 1.87 (s, 3 H), 1.86 (s, 3 H), 0.88 (s, 9 H), 0.043 (s, 6 H); 13C NMR (CDCl3) δ 164.01, 163.64, 156.86, 150.40, 150.19, 137.13, 136.49, 135.18, 134.83, 129.46, 128.93, 128.65, 128.47, 128.10, 111.52, 110.90, 86.22, 84.97, 83.11, 81.76, 77.43, 75.97, 73.12, 53.29, 51.27, 40.04, 36.65, 25.66, 17.87, 12.56, 12.27, −4.68, −4.87; IR (thin film): 3,188, 1,696 cm−1; HRFABMS calcd. for C41H55N6O11Si (M+H)+: 835.3698, found: 835.3690.
Dimer 9 Dimer 7 (3.0 g, 2.93 mmol) was dissolved in THF (50 ml) and 1.0 M TBAF in THF (3.52 ml, 3.52 mmol) was added to the solution. The reaction mixture was stirred at rt for 1 h and THF was removed under reduced pressure. The residue was redissolved in ethyl acetate (100 ml), washed with 10% aqueous NH4Cl (3 × 100 ml) and water (100 ml). The organic layer was separated, dried over Na2SO4, filtered, and evaporated to dryness. Then it was dissolved in anhydrous dichloromethane (80 ml). Pyridine (0.44 ml, 5.84 mmol) was added, followed by p-nitrophenyl chloroformate (709 mg, 3.52 mmol). The reaction solution was stirred at rt for 2 h. Then it was quenched with saturated aqueous NaHCO3 (100 ml) and ethyl acetate (100 ml). The organic layer was washed with saturated aqueous NaHCO3 (2 × 100 ml) and water (100 ml). The ethyl acetate layer was separated, dried over Na2SO4, filtered, and evaporated. The residue was purified by flash column chromatography (hexanes/EtOAc = 1:1, EtOAc) to give dimer 9 (2.98 g, 95%) as a white foam. 1H NMR (CDCl3) δ 9.23 (s, 1 H), 8.27 (d, J = 9.3 Hz, 2 H), 7.72 (d, J = 0.9 Hz, 1 H), 7.44–7.31 (m, 17 H), 7.19 (s, 1 H), 6.38 (dd, J = 5.1 Hz, 9.0 Hz, 1 H), 6.19 (t, J = 6.7 Hz, 1 H), 5.44 (ABq, J = 12.6 Hz, 2 H), 5.37 (m, 1 H), 5.14 (d, J = 6.3 Hz, 1 H), 4.91 (s, 2 H), 4.86 (s, 2 H), 4.40 (m, 1 H), 4.34 (m, 1 H), 3.95–3.71 (m, 4 H), 2.66 (dd, J = 5.4 Hz, 14.4 Hz, 1 H), 2.54 (dd, J = 5.7 Hz, 12.9 Hz, 1 H), 2.46–2.39 (m, 1 H), 2.03–1.95 (m, 1 H), 1.92 (d, J = 1.2 Hz, 3 H), 1.84 (d, J = 1.2 Hz, 3 H), 1.49 (s, 9 H); 13C NMR (CDCl3) δ 170.29, 163.49, 156.51, 156.30, 155.71, 155.03, 151.80, 150.16, 145.49, 139.66, 135.82, 134.99, 134.42, 129.50, 129.41, 129.03, 128.70, 128.46, 128.11, 127.95, 125.30, 121.66, 111.52, 105.31, 86.29, 85.94, 82.01, 81.42, 80.74, 79.86, 77.74, 77.25, 76.81, 68.75, 50.66, 50.52, 37.62, 36.23, 28.18, 12.24, 11.98; IR (thin film): 1,767, 1,694 cm−1; HRFABMS calcd. for C54H58N7O17 (M+H)+: 1,076.3889, found: 1,076.3911.
Tetramer 10 A mixture of the p-nitrophenylcarbonate 9 (2.60 g, 2.42 mmol), dimer 8 (2.32 g, 2.78 mmol) and HOAt (1.64 g, 12 mmol) in dry DMF (30 ml) was treated with DIPEA (2.1 ml, 12 mmol) and stirred at rt for 24 h. The solution was diluted with ethyl acetate (100 ml) and washed with water (80 ml), saturated aqueous NaHCO3 (3 × 80 ml), water (80 ml) and brine (80 ml). The ethyl acetate layer was dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (EtOAc, CH2Cl2/MeOH = 10:1) to give tetramer 10 (4.07 g, 95%) as a white foam. 1H NMR (CDCl3) δ 9.58 (br, 2 H), 9.42 (s, 1 H), 7.73 (s, 1 H), 7.43–7.23 (m, 28 H), 6.40 (dd, J = 5.4 Hz, 9.0 Hz, 1 H), 6.18 (m, 2 H), 5.95 (br, 1 H), 5.42 (ABq, J = 12.9 Hz, 2 H), 5.18 (m, 1 H), 5.12 (m, 2 H), 4.88 (s, 8 H), 4.31 (m, 2 H), 4.20 (m, 2 H), 4.10 (m, 1 H), 3.95–3.75 (m, 7 H), 3.50 (d, J = 12 Hz, 1 H), 2.64 (dd, J = 4.2 Hz, 13.5 Hz, 1 H), 2.36–1.96 (m, 7 H), 1.91 (s, 3 H), 1.84 (s, 3 H), 1.82 (s, 3 H), 1.80 (s, 3 H), 1.48 (s, 9 H), 0.88 (s, 9 H), 0.03 (s, 6 H); 13C NMR (CDCl3) δ 170.24, 164.08, 163.78, 163.68, 156.81, 156.54, 156.39, 155.70, 150.46, 150.34, 150.07, 139.83, 136.82, 135.88, 135.66, 135.05, 134.86, 134.57, 134.49, 129.58, 129.53, 129.41, 128.93, 128.85, 128.66, 128.59, 128.46, 128.08, 127.95, 111.40, 111.35, 110.76, 105.22, 86.81, 86.34, 85.50, 82.88, 81.93, 81.57, 80.77, 77.68, 77.28, 77.20, 76.81, 73.03, 68.70, 51.43, 50.71, 50.55, 50.28, 39.91, 37.61, 36.33, 36.11, 28.18, 25.64, 17.82, 12.31, 12.23, 11.95, −4.69, −4.89; IR (thin film): 3,196, 1,700 cm−1; HRFABMS calcd. for C89H107N12O25Si (M+H)+: 1,772.98, found for (M+H)+: 1,773.18, (M+Na)+: 1,795.41.
Tetramer 11 Tetramer 10 (300 mg, 0.17 mmol) was dissolved in anhydrous dichloromethane (10 ml). Then trifluoroacetic acid (1 ml) was added. After stirring at rt for 1 h, a saturated aqueous NaHCO3 (80 ml) was added at 0°C to quench the reaction. The resulting solution was extracted with dichloromethane (3 × 50 ml). The organic layers were combined, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (EtOAc, CH2Cl2/MeOH = 10:1) to give tetramer 11 (250 mg, 93%) as a white foam. 1H NMR (CDCl3) δ 10.04 (s, 1 H), 9.98 (s, 1 H), 9.95 (s, 1 H), 9.90 (s, 1 H), 7.38–7.20 (m, 24 H), 6.23 (m, 2 H), 6.12 (br, 1 H), 5.98 (br, 1 H), 5.18 (br, 3 H), 4.88 (s, 6 H), 4.72 (s, 2 H), 4.16 (m, 5 H), 3.84 (m, 5 H), 3.51 (m, 1 H), 3.24 (m, 2 H), 2.35–2.19 (m, 8 H), 1.84 (s, 6 H), 1.82 (s, 3 H), 1.80 (s, 3 H), 0.87 (s, 9 H), 0.028 (s, 6 H); 13C NMR (CDCl3) δ 164.29, 164.06, 163.87, 156.82, 156.52, 150.49, 150.18, 137.23, 136.68, 135.98, 135.19, 134.84, 134.60, 134.56, 129.52, 129.48, 129.39, 128.84, 128.55, 128.36, 127.96, 111.34, 111.24, 110.76, 86.42, 85.32, 84.77, 82.78, 81.51, 80.70, 77.20, 75.88, 72.97, 53.32, 51.32, 50.46, 39.89, 36.52, 36.08, 25.61, 17.78, 12.51, 12.28, 12.19, 12.15, −4.73, −4.94; IR (thin film): 2,930, 1,694 cm−1; HRFABMS calcd. for C77H93N12O23Si (M+H)+: 1,581.62, found: 1,581.16.
Tetramer 12 Tetramer 10 (700 mg, 0.40 mmol) was dissolved in THF (20 ml) and 1.0 M TBAF in THF (0.47 ml, 0.47 mmol) was added to the solution. The reaction mixture was stirred at rt for 0.5 h. THF was then removed under reduced pressure. The residue was redissolved in ethyl acetate (100 ml), washed with water (100 ml), 10% aqueous NH4Cl (2 × 100 ml) and water (100 ml). The organic layer was dried over Na2SO4, filtered, and evaporated to dryness. Then the residue was dissolved in anhydrous dichloromethane (20 ml). Pyridine (0.06 ml, 0.80 mmol) was added, followed by p-nitrophenyl chloroformate (96 mg, 0.48 mmol). The reaction solution was stirred at rt overnight. Then solvent was removed under reduced pressure and the residue was redissolved in ethyl acetate (100 ml). The organic layer was washed with saturated aqueous NaHCO3 (3 × 100 ml) and water (100 ml). The ethyl acetate layer was separated, dried over Na2SO4, filtered, and evaporated. The residue was purified by flash column chromatography (hexanes/EtOAc = 1:1, EtOAc, CH2Cl2/MeOH = 10:1) to give tetramer 12 (585 mg, 81%) as a white foam. 1H NMR (CDCl3) δ 10.14 (s, 1 H), 10.02 (s, 1 H), 9.90 (s, 1 H), 8.24 (d, J = 8.7 Hz, 2 H), 7.74 (s, 1 H), 7.40–7.13 (m, 30 H), 6.41 (t, J = 6.9 Hz, 1 H), 6.20 (m, 3 H), 5.41 (s, 2 H), 5.37 (m, 1 H), 5.20–5.13 (m, 3 H), 4.88 (s, 8 H), 4.39–4.23 (m, 5 H), 3.86 (m, 7 H), 2.66–2.00 (m, 8 H), 1.91 (s, 3 H), 1.82 (s, 9 H), 1.48 (s, 9 H); 13C NMR (CDCl3) δ 170.13, 164.02, 163.90, 163.81, 156.42, 156.30, 155.59, 154.96, 151.72, 150.46, 150.42, 150.34, 145.34, 139.77, 136.22, 135.74, 135.42, 134.94, 134.49, 134.38, 129.43, 129.31, 128.83, 128.54, 128.34, 127.98, 127.80, 125.20, 121.58, 111.30, 111.22, 105.08, 86.24, 85.11, 81.84, 81.44, 80.58, 79.93, 77.59, 77.20, 76.69, 68.55, 50.43, 50.52, 37.53, 36.23, 35.90, 28.07, 12.14, 11.84; IR (thin film): 3,066, 1,694 cm−1; HRFABMS calcd. for C90H96N13O29 (M+H)+: 1,823.82, found: 1,823.51.
Hexamer 13 Procedure 1: A mixture of the p-nitrophenylcarbonate 9 (100 mg, 0.093 mmol), tetramer 11 (150 mg, 0.095 mmol) and HOAt (127 mg, 0.93 mmol) in dry DMF (5 ml) was treated with DIPEA (0.16 ml, 0.92 mmol) and stirred at rt for 24 h. The solution was diluted with ethyl acetate (80 ml) and washed with saturated aqueous NaHCO3 (3 × 50 ml), water (50 ml) and brine (50 ml). The ethyl acetate layer was separated, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (CH2Cl2/MeOH = 20:1, 10:1) to give hexamer 13 (216 mg, 92%).
Procedure 2: A mixture of the p-nitrophenylcarbonate 12 (200 mg, 0.11 mmol), dimer 8 (200 mg, 0.24 mmol) and HOAt (75 mg, 0.55 mmol) in dry DMF (5 ml) was treated with DIPEA (0.1 ml, 0.57 mmol) and stirred at rt for 18 h. The solution was diluted with ethyl acetate (100 ml) and washed with saturated aqueous NaHCO3 (3 × 100 ml), water (100 ml) and brine (100 ml). The ethyl acetate layer was dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (EtOAc, CH2Cl2/MeOH = 10:1) to give hexamer 13 (230 mg, 83%) as a foam. 1H NMR (CDCl3) δ 9.97 (br, 5 H), 7.73 (s, 1 H), 7.40–7.22 (m, 40 H), 6.40 (t, J = 6.6 Hz, 1 H), 6.22–6.14 (m, 5 H), 5.42 (s, 2 H), 5.12 (m, 5 H), 4.87 (s, 12 H), 4.30–3.85 (m, 18 H), 3.48 (d, J = 12.6 Hz, 1 H), 2.63 (d, J = 9.0 Hz, 1 H), 2.35–2.00 (m, 11 H), 1.91 (s, 3 H), 1.82–1.80 (m, 15 H), 1.47 (s, 9 H), 0.87 (s, 9 H), 0.022 (s, 6 H); 13C NMR (CDCl3) δ 170.18, 164.35, 164.09, 163.95, 156.84, 156.50, 156.42, 155.62, 150.45, 150.09, 139.82, 135.83, 135.45, 134.99, 134.87, 134.57, 134.41, 129.50, 129.44, 129.36, 128.80, 128.60, 128.52, 128.39, 128.02, 127.87, 111.31, 111.23, 111.03, 110.67, 105.08, 86.27, 85.15, 82.65, 81.86, 81.50, 80.66, 77.20, 76.75, 72.91, 68.61, 51.40, 50.49, 39.82, 37.54, 36.01, 28.12, 25.60, 17.76, 12.30, 12.18, 11.90, −4.73, −4.96; IR (thin film): 3,196, 1,694 cm−1; matrix assisted laser desorption mass spectrometry (MALDI-MS) calcd. for C125H144N18O37SiNa (M+Na)+: 2,541.70, found: 2,542.27.
Hexamer 18 Hexamer 13 (200 mg, 0.079 mmol) was dissolved in THF (10 ml) and 1.0 M TBAF in THF (0.095 ml, 0.095 mmol) was added to the solution. The reaction mixture was stirred at rt for 0.5 h. THF was then removed in vacuo. The residue was redissolved in ethyl acetate (50 ml), washed with 10% aqueous NH4Cl (3 × 50 ml) and water (50 ml). The organic layer was separated, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (CH2Cl2/MeOH = 10:1, 5:1) to give the product (180 mg, 94%) as a white foam, which was used directly in next step without further purification. 1H NMR (CDCl3) δ 10.09 (br, 5 H), 7.73 (s, 1 H), 7.39–7.20 (m, 40 H), 6.39 (t, 1 H), 6.18 (m, 5 H), 5.41 (s, 2 H), 5.11 (m, 5 H), 4.87 (s, 12 H), 4.30–3.82 (m, 19 H), 2.62 (d, 1 H), 2.36–2.00 (m, 11 H), 1.90 (s, 3 H), 1.80–1.73 (m, 15 H), 1.47 (s, 9 H); 13C NMR (CDCl3) δ 170.23, 164.05, 156.53, 156.45, 155.70, 150.50, 139.85, 135.83, 135.03, 134.60, 134.45, 129.48, 129.40, 128.88, 128.66, 128.56, 128.44, 128.08, 127.92, 111.28, 110.76, 105.19, 86.32, 85.13, 81.93, 81.53, 80.66, 77.68, 77.20, 76.80, 72.21, 68.67, 50.52, 39.29, 37.63, 36.19, 28.16, 12.22, 11.94; IR (neat): 3,196, 1,694, 1,682 cm−1; MALDI-MS calcd. for C119H130N18 O37Na (M+Na)+: 2,427.4420, found: 2,428.19.
To the resulting hexamer (90 mg, 0.037 mmol) in 5:1 methanol/methylene chloride (10 ml) was added 10% Pd-C (10 mg). The reaction solution was stirred under a H2 atmosphere for 3 days. The catalyst was filtered off through Celite® and the filtrate was evaporated and purified by C18 reverse-phase flash column chromatography (H2O/MeOH = 1:1) to give 18 (62 mg, 93%) as a white solid. Mp 208°C (decomp.); 1H NMR (CDCl3+CD3OD, 600 MHz) δ 7.50 (d, J = 0.6 Hz, 1 H), 7.44 (br, 5 H), 6.28 (dd, J = 5.4 Hz, 9.0 Hz, 1 H), 6.24 (m, 5 H), 5.28 (d, J = 5.4 Hz, 5 H), 4.34 (m, 6 H), 4.17 (m, 1 H), 3.90–3.78 (m, 12 H), 2.44–2.24 (m, 12 H), 1.94 (d, J = 1.2 Hz, 3 H), 1.92 (d, J = 1.2 Hz, 12 H), 1.91 (s, 3 H), 1.48 (s, 9 H); 13C NMR (CDCl3+CD3OD, 150 MHz) δ 164.56, 164.42, 156.91, 156.46, 156.42, 150.53, 150.49, 150.46, 136.36, 135.96, 135.61, 111.03, 110.98, 110.63, 85.43, 85.11, 84.42, 82.62, 81.44, 81.19, 80.95, 71.50, 51.96, 51.60, 38.80, 36.03, 35.86, 27.62, 11.70, 11.64; IR (KBr): 3,436, 1,694 cm−1; FABMS calcd. for C70H88N18O37Na (M+Na)+: 1,796.5646, found: 1,796.35.
Octamer 14 A mixture of the p-nitrophenylcarbonate 12 (350 mg, 0.19 mmol), tetramer 11 (450 mg, 0.28 mmol) and HOAt (131 mg, 0.96 mmol) in dry DMF (10 ml) was treated with DIPEA (0.17 ml, 0.97 mmol) and stirred at rt for 24 h. The solution was diluted with ethyl acetate (100 ml) and washed with water (100 ml), saturated aqueous NaHCO3 (3 × 100 ml), water (100 ml) and brine (100 ml). The ethyl acetate layer was separated, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (EtOAc, CH2Cl2/MeOH = 10:1, 5:1), followed by a size exclusion Sephadex LH-20 column (CH2Cl2/MeOH = 1:1) to give octamer 14 (510 mg, 81%) as a white solid. Mp 195°C (decomp.); 1H NMR (CDCl3) δ 10.09 (br, 7 H), 7.73 (s, 1 H), 7.40–7.27 (m, 52 H), 6.40 (t, 1 H), 6.20 (m, 7 H), 5.42 (s, 2 H), 5.12 (m, 7 H), 4.88 (s, 16 H), 4.31–3.84 (m, 25 H), 2.60–1.99 (m, 16 H), 1.91 (s, 3 H), 1.82–1.79 (m, 21 H), 1.48 (s, 9 H), 0.87 (s, 9 H), 0.022 (s, 6 H); 13C NMR (CDCl3) δ 170.15, 164.44, 164.12, 156.83, 156.46, 156.38, 155.58, 150.40, 150.07, 139.79, 135.81, 134.97, 134.85, 134.55, 134.39, 129.43, 129.33, 128.78, 128.50, 128.36, 127.99, 127.84, 111.26, 111.08, 110.60, 105.04, 86.25, 85.15, 82.57, 81.82, 81.48, 80.68, 77.20, 76.73, 72.87, 68.57, 50.46, 39.76, 37.56, 36.09, 28.09, 25.57, 17.74, 12.15, 11.87, −4.76, −4.97; IR (thin film): 3,195, 1,715, 1,694, 1,682, 1,652 cm−1; MALDI-MS calcd. for C161H182N24O49SiNa (M+Na)+: 3,288.44, found: 3,287.19.
Octamer 15 Octamer 14 (200 mg, 0.061 mmol) was dissolved in anhydrous dichloromethane (10 ml). Then trifluoroacetic acid (1 ml) was added. After stirring at rt for 2 h, a saturated aqueous NaHCO3 (80 ml) was added to quench the reaction at 0°C. The resulting solution was extracted with dichloromethane (3 × 50 ml). The organic layers were combined, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (CH2Cl2, CH2Cl2/MeOH = 10:1, 5:1) to give octamer 15 (160 mg, 85%) as a foam. 1H NMR (10% CD3OD in CDCl3) δ 10.15 (br, 8 H), 7.32 (m, 48 H), 6.20 (m, 8 H), 5.14 (m, 7 H), 4.86 (s, 14 H), 4.70 (s, 2 H), 4.17 (m, 11 H), 3 83 (m, 11 H), 3.49 (d, 1 H), 3.21 (m, 2 H), 2.26 (m, 16 H), 1.82-1.79 (m, 24 H), 0.86 (s, 9 H), 0.018 (s, 6 H); 13C NMR (10% CD3OD in CDCl3) δ 164.13, 156.81, 156.37, 150.37, 150.10, 137.15, 136.03, 135.24, 134.76, 134.52, 129.42, 128.83, 128.52, 128.35, 127.95, 111.27, 111.15, 110.69, 85.63, 84.79, 82.70, 81.57, 80.71, 77.20, 75.84, 72.91, 53.19, 51.30, 50.46, 39.86, 36.05, 25.56, 17.74, 12.40, 12.08, −4.78, −5.00; IR (thin film): 3,207, 1,684, 1,682, 1,655 cm−1; MALDI-MS calcd. for C149H169N24O47Si (M+H)+: 3,076.21, found: 3,076.63 (M+H)+, 3,096.62 (M+Na)+.
Octamer 16 Octamer 14 (200 mg, 0.061 mmol) was dissolved in THF (10 ml) and 1.0 M TBAF in THF (0.074 ml, 0.074 mmol) was added to the solution. The reaction mixture was stirred at rt for 1.5 h. THF was then removed under reduced pressure. The residue was redissolved in ethyl acetate (80 ml), washed with water (50 ml), 10% aqueous NH4Cl (3 × 50 ml) and water (50 ml). The organic layer was separated, dried over Na2SO4, filtered, and evaporated to dryness. Then the residue was dissolved in anhydrous dichloromethane (10 ml). Pyridine (0.01 ml, 0.13 mmol) was added, followed by p-nitrophenyl chloroformate (19 mg, 0.094 mmol). The reaction solution was stirred at rt for 1 h. Then the reaction mixture was quenched with saturated aqueous NaHCO3 (80 ml). The aqueous layer was extracted with methylene chloride (2 × 50 ml). The organic layers were combined, dried over Na2SO4, filtered, and evaporated. The residue was purified by flash column chromatography (EtOAc, CH2Cl2/MeOH = 10:1, 5:1) to give octamer 16 (192 mg, 94%) as a foam. 1H NMR (CDCl3) δ 10.03 (s, 7 H), 8.23 (d, J = 9.0 Hz, 2 H), 7.73 (s, 1 H), 7.39–7.20 (m, 54 H), 6.40 (t, 1 H), 6.19 (m, 7 H), 5.41 (s, 2 H), 5.37 (m, 1 H), 5.12 (m, 7 H), 4.87 (s, 16 H), 4.31–4.20 (m, 10 H), 3.84 (m, 14 H), 2.62 (d, 1 H), 2.32 (m, 15 H), 1.90 (s, 3 H), 1.80–1.78 (m, 21 H), 1.47 (s, 9 H); 13C NMR (CDCl3) δ 170.20, 164.14, 156.52, 156.43, 155.65, 155.03, 151.80, 150.45, 145.42, 139.81, 135.84, 135.01, 134.59, 134.44, 129.47, 129.39, 128.88, 128.63, 128.56, 128.43, 128.06, 127.90, 125.28, 121.66, 111.22, 105.12, 86.31, 85.13, 81.92, 81.51, 80.62, 77.20, 76.79, 68.64, 50.52, 37.59, 36.12, 28.15, 12.19, 11.92; IR (thin film): 3,196, 1,694 cm−1; MALDI-MS calcd. for C162H171N25O53Na (M+Na)+: 3,339.28, found: 3,337.84.
16mer 19 A mixture of the p-nitrophenylcarbonate 16 (150 mg, 0.045 mmol), octamer 15 (140 mg, 0.046 mmol) and HOAt (31 mg, 0.23 mmol) in dry DMF (5 ml) was treated with DIPEA (0.04 ml, 0.23 mmol) and stirred at rt for 24 h. The solution was diluted with ethyl acetate (80 ml) and washed with water (50 ml), saturated aqueous NaHCO3 (3 × 50 ml), water (50 ml) and brine (50 ml). The ethyl acetate layer was separated, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (EtOAc, CH2Cl2/MeOH = 10:1, 5:1), followed by a size exclusion Sephadex LH-20 column chromatography (CH2Cl2/MeOH = 1:1) to give a mixture of 16mer 17 and octamers. The crude product was dissolved in THF (10 ml) and 1.0 M TBAF in THF (0.07 ml, 0.07 mmol) was added to the solution. The reaction mixture was stirred at rt for 3 h. THF was then removed in vacuo. The residue was redissolved in ethyl acetate (80 ml), washed with 10% aqueous NH4Cl (3 × 50 ml) and water (50 ml). The organic layer was dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (CH2Cl2/MeOH = 10:1, 5:1), followed by C18 reverse-phase chromatography (THF/H2O = 6:4, 7:3) to give the desilylated 16mer (145 mg, 52%) as a white solid. 1H NMR (CDCl3+CD3OD, 600 MHz) δ 7.79 (s, 1 H), 7.44–7.32 (m, 100 H), 6.32 (t, 1 H), 6.19 (m, 15 H), 5.41 (ABq, J = 12.6 Hz, 2 H), 5.18 (m, 15 H), 4.91–4.88 (m, 32 H), 4.30–4.23 (m, 18 H), 3.90–3.73 (m, 31 H), 2.58 (d, 1 H), 2.36–2.03 (m, 31 H), 1.92 (s, 3 H), 1.81 (s, 45 H), 1.48 (s, 9 H); IR (KBr): 3,480, 1,704, 1,694, 1,682 cm−1; MALDI-MS calcd. for C299H320N48O97Na (M+Na)+: 6,161.11, found: 6,154.7.
The resulting 16mer (8 mg, 1.3 μmol) was dissolved in 1:1 THF/H2O solution (10 ml) and 10% Pd-C (1 mg) was added. The reaction solution was stirred under H2 atmosphere for 3 days. The catalyst was filtered over Celite® and washed with 1:1 THF/H2O. The filtrate was evaporated and purified by C18 reverse-phase flash column chromatography (H2O/THF = 5:5, 4:6) and a Sephadex LH-20 column to give 19 (4 mg, 67%) as a white solid. MALDI-MS calcd. for C180H218N48O97Na (M+Na)+: 4,628.98, found: 4,626.72.
p-Nitrophenyl 5′-O-(p,p′-dimethoxytrityl)thymidine 3′-carbonate (22) 5′-O-(p,p′-Dimethoxytrityl)thymidine 21 (2.0 g, 3.68 mmol) was dissolved in anhydrous dichloromethane (30 ml). Then pyridine (0.554 ml, 7.35 mmol) was added, followed by p-nitrophenyl chloroformate (890 mg, 4.42 mmol). The reaction solution was stirred at rt overnight. Then the solution was quenched with saturated aqueous NaHCO3 (100 ml) and extracted with methylene chloride (2 × 100 ml). The organic layers were combined, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (hexanes/EtOAc = 2:1, 1:1) to give 22 (2.53 g, 97%) as a white solid. Mp 118–120°C; 1H NMR (CDCl3) δ 9.97 (s, 1 H), 8.26 (d, J = 9.0 Hz, 2 H), 7.65 (s, 1 H), 7.42–7.26 (m, 11 H), 6.85 (m, 4 H), 6.55 (dd, J = 5.4 Hz, 9.3 Hz, 1 H), 5.48 (d, J = 5.1 Hz, 1 H), 4.35 (s, 1 H), 3.78 (s, 6 H), 3.62–3.49 (m, 2 H), 2.73–2.50 (m, 2 H), 1.44 (s, 3 H); 13C NMR (CDCl3) δ 163.90, 158.62, 154.95, 151.69, 150.60, 145.34, 143.93, 135.01, 134.89, 134.85, 129.92, 128.98, 127.91, 127.60, 127.11, 125.19, 121.57, 113.18, 112.90, 111.72, 87.15, 84.15, 83.31, 80.27, 63.49, 55.07, 37.66, 11.55; IR (neat): 3,186, 3,058, 2,933, 1,767, 1,693, 1,253 cm−1; HRFABMS calcd. for C38H35N3O11 (M)+: 709.2272, found: 709.2290.
Dimer 23 A mixture of p-nitrophenylcarbonate 22 (750 mg, 1.06 mmol), 5 (443 mg, 0.96 mmol) and HOAt (654 mg, 4.80 mmol) in dry DMF (10 ml) was treated with DIPEA (0.84 ml, 4.82 mmol) and stirred at rt for 24 h. The solution was diluted with ethyl acetate (80 ml) and washed with water (50 ml), saturated aqueous NaHCO3 (3 × 50 ml) and brine (50 ml). The ethyl acetate layer was separated, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (hexanes/EtOAc = 1:1, EtOAc) to give dimer 23 (820 mg, 83%) as a clear oil. 1H NMR (CDCl3) δ 10.30 (s, 1 H), 10.28 (s, 1 H), 7.60 (s, 1 H), 7.37–7.25 (m, 15 H), 6.83 (m, 4 H), 6.46 (t, 1 H), 6.18 (t, 1 H), 5.42 (m, 1 H), 4.87 (s, 2 H), 4.29 (m, 1 H), 4.11 (s, 2 H), 3.76 (s, 3 H), 3.75 (s, 3 H), 3.61–3.45 (m, 3 H), 2.68–2.21 (m, 5 H), 1.78 (s, 3 H), 1.33 (s, 3 H), 0.87 (s, 9 H), 0.86 (s, 9 H), 0.029 (s, 6 H); 13C NMR (CDCl3) δ 164.13, 163.94, 158.43, 156.39, 150.59, 150.24, 143.86, 135.85, 135.01, 134.88, 134.79, 134.49, 129.81, 129.21, 128.62, 128.34, 127.84, 127.71, 126.91, 113.00, 111.40, 111.71, 86.83, 85.61, 84.04, 83.65, 82.88, 77.16, 77.00, 72.79, 63.42, 54.90, 50.86, 39.78, 37.57, 25.41, 17.57, 11.97, 11.31, −4.95, −5.15; IR (neat): 3,185, 3,061, 2,931, 1,694 cm−1; FABMS calcd. for C55H65N5O13Si (M)+: 1,032.2425, found: 1,032.2.
Dimer 20 Dimer 23 (700 mg, 0.68 mmol) was dissolved in THF (20 ml) and then 1.0 M TBAF in THF (0.815 ml) was added to the solution. The reaction mixture was stirred at rt for 30 min. Then THF was removed under reduced pressure. The residue was redissolved in ethyl acetate (100 ml) and washed with water (100 ml), 10% aqueous NH4Cl (2 × 100 ml) and water (100 ml). The ethyl acetate layer was separated, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (EtOAc, CH2Cl2/MeOH = 10:1) to give the product (590 mg, 95%). 1H NMR (CDCl3) δ 10.04 (br, 2 H), 7.53 (s, 1 H), 7.40–7.23 (m, 15 H), 6.84 (d, J = 8.7 Hz, 4 H), 6.42 (t, J = 6.6 Hz, 1 H), 6.19 (t, J = 6.3 Hz, 1 H), 5.35 (s, 1 H), 4.85 (s, 2 H), 4.16–4.09 (m, 3 H), 3.77 (s, 6 H), 3.42 (m, 3 H), 2.54–2.14 (m, 5 H), 1.79 (s, 3 H), 1.34 (s, 3 H); 13C NMR (CDCl3) δ 164.09, 158.61, 156.52, 150.94, 144.13, 136.06, 135.16, 135.08, 134.62, 130.02, 129.54, 128.83, 128.49, 128.02, 127.93, 127.08, 113.20, 111.12, 86.94, 85.40, 84.06, 83.46, 82.76, 77.20, 72.25, 63.70, 55.15, 51.14, 39.42, 37.76, 12.18, 11.59; HRFABMS calcd. for C49H52N5O13 (M+H)+: 918.3562, found: 918.3512.
The resulting dimer (600 mg, 0.65 mmol from multiple preparations described earlier) was dissolved in anhydrous methylene chloride (15 ml). At 0°C, DIPEA (0.23 ml, 1.32 mmol) and chlorophosphoramidite (0.22 ml, 0.99 mmol) were added to the solution. The reaction mixture was stirred at 0°C for 5 min and then at rt for 3 h. Then the solution was quenched with 5% aqueous NaHCO3 (100 ml) and methylene chloride (100 ml). The organic layer was washed with saturated aqueous NaHCO3 (100 ml) and brine (100 ml). The organic layer was separated, dried over Na2SO4, filtered, evaporated and purified by flash column chromatography (EtOAc/CH2Cl2/Et3N = 45:45:10) to give the desired phosphoramidite 20 (653 mg, 89%). 1H NMR (CDCl3) δ 7.59 (s, 1 H), 7.40–7.24 (m, 15 H), 6.84 (d, J = 8.7 Hz, 4 H), 6.45 (t, J = 6.3 Hz, 1 H), 6.21 (t, 1 H), 5.41 (s, 1 H), 4.88 (m, 2 H), 4.48 (m, 1 H), 4.27 (m, 1 H), 4.11 (m, 1 H), 3.79 (s, 6 H), 3.83–3.40 (m, 5 H), 2.63–2.38 (m, 8 H), 2.21 (m, 1 H), 1.80 (s, 3 H), 1.34 (s, 3 H), 1.19–1.15 (m, 12 H); 13C NMR (CDCl3) δ 164.09, 163.93, 158.58, 156.51, 156.41, 150.61, 150.36, 150.28, 143.96, 135.96, 135.16, 135.02, 134.92, 134.62, 134.55, 129.94, 129.34, 128.73, 128.44, 127.97, 127.86, 127.06, 117.67, 117.58, 113.14, 111.54, 111.06, 86.98, 85.63, 84.14, 83.76, 82.14, 81.74, 77.30, 77.14, 74.46, 74.24, 74.07, 73.84, 63.54, 58.09, 58.03, 57.85, 57.78, 55.08, 50.93, 50.67, 38.54, 37.67, 24.42, 24.36, 24.32, 24.26, 20.24, 20.15, 20.06, 19.21, 12.05, 11.42; 31P NMR (CDCl3) δ 149.90, 149.76; IR (neat): 2,984, 2,730, 1,694, 1,470, 1,216 cm−1; HRFABMS calcd. for C58H67 N7O14P: 1,116.4484, found: 1,116.4456.
Syntheses of the oligodeoxynucleotides Oligode–oxynucleotides 24, T@-1, 2, 3, dT16, dA16, and the 32mer were synthesized on a Beckman Oligo 1000M DNA-synthesizer using commercial phosphoramidites and dimer 20. Oligomers were prepared on a 1.0 μmol scale employing the standard synthesis cycle. The efficiency of each coupling step was monitored by the release of the dimethoxyltrityl cation. The average coupling efficiency of dimer 20 was of 99.5%. Removal of dT16, dA16, and the 32mer from the solid support and deprotection was performed using the standard procedure (5 min in ammonia/methylamine for cleavage, 10 min at 65°C for deprotection). Removal of T@-1, 2, 3 and oligomer 24 from the solid support and deprotection was carried out at rt in concentrated aqueous ammonia solution for 1 h. After concentration, the oligothymidylate analogs T@-1, 2, 3 and oligomer 24 were redissolved in triethylammonium acetate buffer solution (1 ml, pH 7.15, 0.1 M) and 10% Pd-C (1 mg) was added. The mixture was stirred at rt for 6 h under a H2 atmosphere. The catalyst was filtered off through Celite®. The filtrate was concentrated under reduced pressure to give oligonucleotides T*-1, 2, 3 and oligomer 25. The dry oligomers were resuspended in distilled water (1 ml) prior to use. Their concentrations were determined by UV absorbance at 254 nm, using extinction coefficients E254 of d(A16) (221.76), d(T16) and T*-1, 2, 3 (126.72), 32mer (377.28) (Eckstein 1991).
MALDI-TOF mass spectroscopy confirmed the structure of the oligomers. MALDIMS calcd. for dT16: C160H209N32O110P15Na (M+Na)+: 4,828.2, found: 4,828.9; for T@-1: C168H215N33O109P14Na (M+Na)+: 4,897.4, found: 4,898.2; for T@-2: C176 H221N34O108P13Na (M+Na)+: 4,966.5, found: 4,966.8; for T@-3: C184H227N35O107P132Na (M+Na)+: 5,035.7, found: 5,037.6; for 24: C94H110N17O44P3Na (M+Na)+: 2,297.9, found: 2,297; for T*-1: C161H210N33O109P14 (M+H)+: 4,785.2, found: 4,785.9; for T*-2: C162H209 N34O108P13Na (M+Na)+: 4,786.3, found: 4,791.1; for 26: C73H89N17O41P3FeNa (M+Na)+: 2,032.4, found: 2,034.
Purity of the samples was confirmed by HPLC on a C18 3.9 × 150 mm column using a linear gradient of acetonitrile from 1 to 50% in 0.1 M triethylammonium acetate buffer (pH 7.15) over 60 min at a flow rate of 1.0 ml min−1.
Circular dichroism spectra
Circular dichroism (CD) spectra were obtained on a Varian CD spectrophotometer. Scans were run from 320 to 200 nm taking a measurement every 1 nm. The integration time for each data point was 1.3 s. Three scans were made with each sample and then averaged. Samples were held in a 0.2 cm path-length cuvette and the temperature was maintained at 25 or 5°C as indicated in the Figures.
Melting experiments
Thermal denaturation plots were recorded on a Varian CD spectrophotometer. Experiments were performed with equimolar mixtures of complementary oligonucleotides each at a concentration of 10 μm in 100 mM NaCl, 0.1 mM EDTA, 10 mM phosphate buffer, pH 7.0. The cell compartment was flushed with dry nitrogen during measurements. Melting curves were obtained by observing the CD at 250 nm of a solution of the oligomers in a 0.2 cm path-length cuvette as the temperature was raised 0.5°C min−1 from 5 to 90°C. Data was recorded every 0.5°C. All samples had been previously annealed by cooling from 90°C to rt overnight. For Tm determinations hyperchromicity was used. %Hyperchromicity (wavelength) = 100[D(T)–D0]/D0 with D(T) = absorption at temperature T and D0 = lowest absorption in the temperature interval. Tm values were determined from the maxima of the first derivative plots of absorbance versus temperature.
Enzymatic hydrolysis
The oligonucleotides (20 μM) were incubated at 37°C in 1 ml of 50 mM sodium acetate buffer (pH 4.5), 300 mM sodium chloride, 100 mM zinc acetate, and 50 μl of water containing 20 units of nuclease S1. Every 10 min, 100 μl of the solution was removed, frozen and thawed right before HPLC analysis. Aliquots were analyzed by analytical HPLC on a C18 column using a linear gradient of acetonitrile from 1 to 25% in 0.1 M triethylammonium acetate buffer (pH 7.15) over 30 min at a flow rate of 1.0 ml min−1. The relative concentrations of the full-length oligomers were determined from quantification of the corresponding HPLC signals.
59FeCl3 chelation study
Each oligonucleotide (30 μM) was incubated overnight at 25°C with 1 eq. (~55,000 cpm) of radioactive 59FeCl3 in 5 μl of buffer containing 50 mM Tris-HCl pH 7.5 and 70 mM KCl. A volume of 1 μl of 6× sucrose dye was added and then loaded onto a 20%, pH 7.3 denaturing polyacrylamide gel. The gel was run at 125 V for 1 h and exposed to X-ray film.
Preparation of 5′-end-labeled 32mer
To a mixture of 32mer (1 μg), T4 polynucleotide kinase 109 buffer (1 ll) and DEPC water (5 ll), was added γ-[32P-ATP] (1 ll, 166 lCi), followed by T4 polynucleotide kinase (1 ll, 5 U). The mixture was incubated at 37°C for 30 min, then heated to 90°C to inactivate the kinase for 2 min. The crude oligonucleotide was purified using denaturing polyacrylamide gel electrophoresis. It was diluted with formamide dye solution (10 ll) and loaded onto a 20% denaturing polyacrylamide gel using a Tris-borate/EDTA running buffer (50 mM Tris free base, 50 mM boric acid, and 1 mM EDTA, pH 8.3), running for 4 h at 350 V. The desired 5′-end-labeled 32mer was visualized by autoradiography, excised from the gel, crushed and eluted at 37°C overnight with a solution of 500 mM ammonium acetate, 1 mM EDTA and 0.1% SDS. The solute DNA was precipitated with cold ethanol and washed with cold 70% ethanol. The DNA pellet was dried and brought up in water to a concentration of about 250 nM. The oligomer was then further desalted and purified by loading onto a Sephadex G-25 spin column and eluting.
Binding studies of 32mer with oligothymidylate analogs
Binding studies for CD analysis were performed in a Tris-HCl buffer solution (50 mM Tris, 50 mM NaCl, pH 7.45) with a final volume of 5 ll. Oligonucleotides d(T16) or T*-1, 2, 3 (0.1 pmol, 1 μl of 100 μM) were incubated with or without FeCl3 (0.2 pmol, 1 μl of 200 μM) at rt overnight. 5′-32P-32mer (1 ll, 4.2 × 104 cpm) was added to the solution and heated at 70°C for 2 min and then slowly cooled to 4°C for 2 h. All samples were treated with sucrose dye (1 ll) and loaded onto a 15% native polyacrylamide gel using a Tris-borate/EDTA running buffer (50 mM Tris free base, 50 mM boric acid, and 1 mM EDTA, pH 8.3), running at 4°C for 2 h at 125 V. Binding was visualized by autoradiography.
Binding for gel shift and sequence-selective cleavage assays was performed in 50 mM Tris pH 7.5 and 70 mM KCl with a final volume of 10 ll. A volume of 150 μm T*-3 was incubated with 1 eq. of either FeCl3 or Fe(acac)3 in the buffer earlier mentioned for 8-24 h at rt. A volume of 1 μl of 250 nM 5′-32P end-labeled 32mer (~200,000 cpm) was added to the bottom of a 1.7 ml microcentrifuge tube and then 1 μl of 12.5-fold molar excess competitor (in competition experiments) added to that droplet. A volume of 1 μl of 250 nM T16 or T*-3 was added to the side of the tube and then washed into the 32mer with an appropriate amount of binding buffer. Reactions were heated to 70°C and allowed to slow cool to 25°C and then transferred to ice for 10 min. Reactions were then either treated with sucrose dye and electrophoresed on a 20% native gel as earlier or subjected to hydroxyl radical cleavage.
Fenton chemistry induced cleavage experiment
Binding reactions (10 ll) were allowed to equilibrate on ice for at least 10 min prior to the Fenton reaction. Cleavage was initiated by adding 2 μl of 325 mM Na ascorbate to the inside lid of the microcentrifuge tube, 1 μl of 0.42% H2O2 to the wall and a brief spin to mix. The reactions took place on ice for 1 h and were quenched with 2.5 μl of a solution containing 100 mM thiourea, 1 mM EDTA and 2 μg of carrier tRNA. Reactions were then diluted with 9 μl of 2× formamide dye (98% deionized formamide, 0.015% bromophenol blue, 0.015% xylene cyanol), boiled at 70°C and loaded onto an 18% sequencing gel. Cleavage fragments were separated by electrophoresis at 1,500 V for about 4 h and then exposed to X-ray film at −80°C.
Results and discussion
Syntheses of oligothymidylate analogs with hydroxamate linkages
The retrosynthetic analysis of the hydroxamate-linked oligonucleotides indicated that 5′-deoxy-5′-N-hydroxylaminonucleosides could serve as the common building blocks for these oligomers. A general and efficient route for the conversion of the 5′-hydroxyl group of a nucleoside into the corresponding 5′-Nhydroxylamino substituted derivatives was developed using Mitsunobu reactions of N-(tert-butoxycarbonyl)-O-(benzyloxycarbonyl)hydroxylamine (BocNHOCbz) with nucleosides (Li and Miller 1999). Careful control of reactant concentrations and solvent composition allowed minimal use of protecting groups for adenosine, uridine, cytidine and guanosine. This methodology was applied to thymidine for the syntheses of thymidine oligomers with hydroxamate linkages.
Starting from 3′-O-tert-butyldimethylsilyl-5′-O-dimethoxytritylthymidine 1, a benzyl group was introduced to the O4-position of the thymidine in two steps (Hayakawa et al. 1993) by reaction with p-toluenesulfonyl chloride in dichloromethane, followed by treatment of the resulting sulfonate with benzyl alcohol, to give 2 in 88% overall yield. Removal of the dimethoxytrityl group in a dichloroacetic acid dichloromethane solution afforded the desired free 5′-hydroxy compound 3 in 87% yield. Mitsunobu reaction of 3 with N-(tert-butoxycarbonyl)-O-(benzyl)-hydroxylamine (BocNHOBn) in the presence of di-tert-butyl azodicarboxylate (DBAD) and triphenylphosphine in THF gave an 87% yield of the desired protected hydroxylamino derivative 4 (Scheme 1; Li and Miller 2000).
Scheme 1.

Synthesis of hydroxylamino derivatized thymidine monomer
When thymidine monomer 4 was exposed to 10% trifluoroacetic acid in dichloromethane, both the Boc and the O4-benzyl protecting groups were removed to give 5 in 99% yield (Scheme 1). After treating 4 with n-tetrabutylammonium fluoride (TBAF) in THF to remove the silyl group, the p-nitrophenyl (PNP) carbonate 6 was obtained in 94% yield by reaction with nitrophenyl chloroformate (Scheme 2). Since direct reaction of 5 with 6 was sluggish, 1-hydroxy-7-azabenzotriazole (HOAt) was used to form a more active carbonate reaction intermediate (Dubowchik et al. 1997) for the coupling reaction and generated hydroxamate linked dimer 7 in 89% yield.
Scheme 2.

Synthesis of thymidine dimer with hydroxamate linkage
Applying the same coupling methodology, thymidine tetramer 10 was obtained from the coupling of dimers 8 and 9. Hexamer 13 was obtained from the coupling of the dimer 8 and tetramer 12, or coupling of dimer 9 and tetramer 11. Octamer 14 was obtained from the coupling of tetramers 11 and 12, 16mer 17 was obtained from the coupling of octamers 15 and 16. After treating 13 and 17, respectively, with TBAF in THF to cleave the silyl group and hydrogenolysis conditions to remove the benzyl protecting groups, fully unprotected thymidine hexamer 18 and 16mer 19 were obtained (Fig. 2).
Fig. 2.

Thymidine oligomers with hydroxamate linkages
Three non-adjacent hydroxamate linkages are required for effective Fe(III) chelation and target DNA binding
Since oligonucleotides with hydroxamate linkages are designed to sequence-selectively cleave DNA or mRNA sense strands using Fenton chemistry, these oligomers are required to bind with both iron and sense strands with reasonable affinity. Oligomers 18 and 19 were used as models for the binding studies with Fe(III) and complementary poly(dA) strand, respectively.
Since Fe(III) binds with three hydroxamate functionalities to form a stable hexadentate octahedral complex, designed oligonucleotides with three or more hydroxamate linkages were envisioned to be effective Fe(III) chelators. However, modeling studies indicated that two adjacent internucleosidic hydroxamate linkages are too closely positioned to bind to the same Fe(III). Iron binding studies of oligomers of various lengths demonstrated that hexamer 18 is the shortest oligomer that could form a stable iron complex. After 18 was treated with ferric acetylacetonate (Dionis et al. 1989), Fe(acac)3, the resulting iron complex was analyzed by mass spectroscopy. The mass spectrum showed a strong iron complex signal and no signal from unchelated hexamer 18, indicating the formation of a stable iron complex [Fe(18)]. Since there are six hydroxamate functionalities in the hexamer, three nonadjacent linkages can be used for binding to the same iron and form a stable complex.
Binding studies between hydroxamate linked thymidine 16mer d(T*16) 19 (* represents hydroxamate linkages), and normal phosphate containing deoxyadenosine 16mer d(A16) were carried out using CD spectroscopy. Figure 3 shows the CD spectra of hydroxamate linked thymidine 16mer d(T*16) 19, deoxyadenosyl 16mer d(A16) and duplex d(T*16) · d(A16). After the oligothymidylate analog d(T*16) was incubated with complementary d(A16), no significant change was observed in the CD spectra. This result indicated there were no conformational changes (i.e., single strands to duplex) of oligonucleotides d(T*16) and d(A16) in solution. Apparently, these two oligomers still existed in single-stranded forms and were not associated with each other. Moreover, the CD spectrum of fully hydroxamate linked oligonucleotide d(T*16) exhibited very weak signals, indicating little interaction between neighboring bases. This suggested that the rigidity of the hydroxamate linkages prevented formation of a helical structure and base stacking.
Fig. 3.

CD spectra of d(T*16), d(A16) and duplex d(T*16) ·d(A16). Solution conditions: 20 μm oligomers, 100 mM NaCl, 0.1 mM EDTA, 10 mM phosphate buffer, pH 7.0 at 25°C
Syntheses of oligothymidylate analogs with mixed linkages
As mentioned earlier, binding studies showed that the hydroxamate linked oligothymidine can effectively chelate with Fe(III). However, CD spectral analyses (Rodger and Norden 1997) of the fully modified hydroxamate-containing oligonucleotides indicated that the hydroxamate linkage was too rigid and the oligothymidylate analog (16mer) could not bind to the complementary DNA d(A16). In order to overcome this problem, thymidine oligomers with alternating hydroxamate and phosphodiester linkages were designed to take advantage of the more flexible phosphodiester linkages. Such hybrid compounds were anticipated to allow the incorporation of both desired properties, iron binding and duplex formation. Syntheses of mix-linked thymidine oligomers involved the incorporation of a phosphoramidite dimer 20 into automatic DNA syntheses. The synthesis of dimer 20 is shown in Scheme 3.
The phosphoramidite dimer 20 was incorporated near the center of thymidine 16mers in one, two and three positions using phosphoramidite chemistry on solid support according to a standard automated 1 μmol synthesis cycle (Eckstein 1991), No difference in activity between the dimer 20 and a regular phosphoramidite was observed, as monitored by the release of the dimethoxytrityl cation after each coupling step. The average coupling efficiency of dimer 20 was an impressive 99.5%. After solid phase synthesis, oligonucleotides T@-1, 2, 3 with O-benzyl-N-hydroxamate internucleosidic linkages were obtained with one, two and three modifications in the middle of the sequence. Due to the observed instability of the O-benzyl-N-hydroxamate linkage in concentrated aqueous ammonia solution at high temperature, these oligonucleotides were deblocked and removed from the solid support separately by treatment with concentrated aqueous ammonia solution at rt for 1 h. Then, the benzyl protecting groups were removed by hydrogenolysis in triethylammonium acetate buffer (0.1 M, pH 7.15) to give T*-1, T*-2 and T*-3, respectively (Scheme 4). The composition of these oligothymidylate analogs was verified by MALDI-MS, which has become a powerful method for the mass analysis of oligonucleotides (Pieles et al. 1993) The purity of these modified thymidine oligomers was indicated by reverse-phase HPLC on a C18 column. Oligomers with O-benzyl-N-hydroxamate linkages, obtained directly from solid phase synthesis, had sufficient purity and were used for the subsequent hydrogenolysis reactions immediately after they were cleaved from the solid support.
Scheme 4.
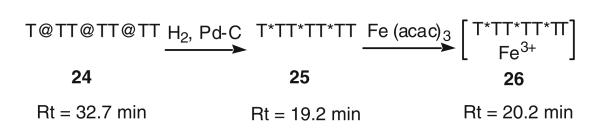
Iron complex synthesis of hydroxamate-linked oligothymidine
Nuclease stability assays
Resistance of hydroxamate linked oligonucleotides towards nuclease S1 (endonuclease) was investigated by comparison to nuclease susceptibility of unmodified d(T)16 (Peyrottes et al. 1994) Analytical HPLC was used to determine the relative concentrations of the full-length oligomers after they were incubated with nuclease for a certain time period (Morvan et al.1991). The results indicated that hydroxamate-containing oligonucleotides were less susceptible to nuclease hydrolysis relative to the all natural d(T)16. Thus, due to the modified linkage, a stabilizing effect was observed. The half-lives of the oligonucleotides increased with increased incorporation of hydroxamate linkages in the sequence (Table 1).
Table 1.
Half-lives of oligothymidylate analogs in the presence of nuclease S1
| Compounds | Ta1/2 (min) |
|---|---|
| d(T)16 | 25.3 |
| TTTTTTTT*TTTTTTTT T*-1 | 29.1 |
| TTTTTTT*TT*TTTTTTT T*-2 | 32.7 |
| TTTTTT*TT*TT*TTTTTT T*-3 | 44.4 |
50 mM sodium acetate buffer (pH 4.5) containing 300 mM sodium chloride and 100 mM zinc acetate, 37°C
Iron binding studies
Binding of these mix-linked thymidine oligomers with Fe(III) was studied in two ways. The first study used a model heptathymidylate analog 25, which was synthesized through the same solid phase DNA synthesis and hydrogenolysis procedure mentioned before (Scheme 4). Second, metallation of T*-1, T*-2 and T*-3 analogs with 59FeCl3 was used. Incorporation of phosphoramidite dimer 20 into solid phase DNA synthesis produced heptamer 24, which was deprotected and removed from the solid support by treatment with concentrated aqueous ammonia at rt for 1 h. Reverse-phase HPLC analysis gave a retention time of 32.7 min. The benzyl groups were then removed by hydrogenolyses in triethylammonium acetate buffer solution (pH 7.15) to give heptamer T*TT*TT*TT, 25, with a retention time of 19.2 min under the same analytical conditions. When heptamer 25 was treated with Fe(III)-acetylacetonate, Fe(acac)3, HPLC analysis showed a complete conversion of 25 to the corresponding iron complex 26 with a retention time of 20.2 min. MALDI-MS analysis of oligomer 26 gave a molecular mass of 2,034 Da (calcd. 2,032.4 Da for M+Na), confirming the effective stoichiometric chelation between 25 and Fe(III).
Effective Fe(III) binding by T*-3 was also confirmed by individually incubating d(T16), T*-1, T*-2 and T*-3 with radioactive 59FeCl3 and loading the metallated thymidylate oligomers onto a denaturing polyacrylamide gel with subsequent autoradiography. Only the T*-3 lane on the film had a band corresponding to a 16 nt oligomer (data not shown), indicating T*-3 is the only oligomer capable of chelating Fe(III).
Mixed-linked oligothymidylate analogs bind complementary DNA sequence-specifically although with reduced affinity
Binding studies between the mix-linked oligothymidylate analogs with d(A16) were carried out using two approaches: first using CD spectroscopy and second, native PAGE mobility-shift assays. Figure 4 shows the CD spectra of single strand oligomers. CD profiles of oligothymidylate analogs T*-1, 2, 3 were similar to those of normal dT16 (Fig. 5). These CD profiles are consistent with typical polymeric thymidyl DNA CD spectra, which constitute a positive CD band at 285 nm and a negative CD band at 250 nm, indicating the helical structure of polymeric thymidyl DNA in the solution.
Fig. 4.

CD spectra of d(T16), d(A16) and oligothymidylate analogs T*-1, 2, 3. Solution conditions: 10 μM oligomers, 100 mM NaCl, 0.1 mM EDTA, 10 mM phosphate buffer, pH 7.0 at 5°C
Fig. 5.

CD spectra of duplex d(T16)·d(A16) and duplexes of d(A16) with T*-1, 2, 3. Solution conditions: 10 μM oligomers, 100 mM NaCl, 0.1 mM EDTA, 10 mM phosphate buffer, pH 7.0 at 5°C
After single strand oligothymidylate analogs T*-1, 2, 3 were annealed with complementary DNA d(A16), their CD shifted from characteristic single strand helical spectra into distinct characteristic duplex spectra, similar to the CD of natural phosphate linked oligomer d(T16) with d(A16) (Rodger and Norden 1997; Fig. 5). These duplex CD spectra did not match the sums of the CD spectra of the individual single strands, indicating conformational changes (i.e., single strand to duplex) of the oligomers due to their association.
DNA 32mer, TCTGCAAGA16GTCGACTA, was synthesized by standard solid phase DNA chemistry using phosphoramidite methodology and was used to study the proposed sequence-selective Fenton cleavage reaction. The central A16 region of the 32mer is where oligothymidylate analogs T*-1, 2, 3 were anticipated to selectively bind. The binding of this 32mer with both d(T16) and T*-3 was studied by a polyacrylamide gel shift experiment to check both affinity and sequence-selectivity of binding (Fig. 5; Slater et al. 1996; Kneale 1994). The DNA 32mer was 32P-radiolabeled at the 5′-end by phosphorylation with γ-[32P-ATP]. After annealing the 32mer with thymidine oligomers from 70°C to rt and equilibrating at 4°C for 10 min, thymidine oligomers bound with the 32mer and the resulting duplexes were subjected to polyacrylamide gel electrophoresis. The gel was then analyzed by autoradiography of radiolabeled 32mer. Binding of the thymidine oligomers to the 32mer resulted in duplexes that have retarded electrophoretic mobility relative to the single strand 32mer on the native gel (Fig. 6, lane 2–6). It can be seen from Fig. 5 that the T*-3 complex binds sequence-selectively although with lower affinity than unmodified d(T16). T*-3 loaded with Fe(III) also binds selectively and with slightly lower affinity than unmetallated T*-3. This may be due to some bending in the oligomer to accommodate chelation of the Fe(III). Sequence-selective binding was demonstrated by competing the T*-3+ Fe(III) off the labeled 32mer with an excess of unlabeled 32mer (Fig. 6, lane 5) while challenging the T*-3+ Fe(III)-labeled 32mer complex with an excess of random sequence primer (Fig. 5, lane 6) had no significant effect on binding.
Fig. 6.

Autoradiograph of a nondenaturing 20% polyacrylamide gel at 4°C. Lane 1 5′-32P end-labeled 32mer d(TCTGCAAGA16GTCGACTA); lane 2 5′-32P end-labeled 32mer annealed with d(T16); lane 3 5′-32P end-labeled 32mer with T*-3; lane 4 5′-32P end-labeled 32mer with T*-3+ Fe(III); lane 5 5′-32P end-labeled 32mer with 12.5-fold molar excess of unlabeled 32mer+ T*-3+ Fe(III); lane 6 5′-32P end-labeled 32mer with 12.5-fold molar excess of random sequence primer+ T*-3+ Fe(III). Solution conditions: 25 nM oligomers, 25 nM FeCl3, 70 mM KCl, 50 mM Tris–HCl buffer, pH 7.5
T*-3 site-specifically cleaves complementary DNA
From previously discussed studies, T*-3 was shown to be an effective iron chelator that also could bind with a complementary DNA strand with reasonable affinity in the presence of iron. Therefore, it was anticipated that this iron binding “antisense” T*-3 would hybridize to the complementary strand and bring iron into the close proximity of the A16 region of the 32mer. Indeed, under Fenton chemistry conditions, sequence-selective cleavage within the A16 region occurred (Fig. 7). The experiment was performed on T*-3 and 5′-32P end-labeled 32mer in the presence of Fe(III), H2O2 and ascorbate. At equal concentration of T*-3 (relative to the concentration of the 32mer), sequence-selective cleavage occurred within the 32mer A16 region as indicated in lane 6 (Fig. 7).
Fig. 7.

Autoradiograph showing the cleavage pattern produced by T*-3 iron complex with 5′-32P end-labeled 32mer d(TCTGCAAGA16GTCGACTA). Lane 1 5′-32P end-labeled 32mer subjected to Maxam–Gilbert G+ A sequencing (Maxam and Gilbert 1980); lane 2 25 nM 5′-32P end-labeled 32mer, 100 μm (NH4)2Fe(SO4)2·6H2O, 200 μM EDTA, 50 mM ascorbate, 0.03% H2O2; lane 3 25 nM 5′-32P end-labeled 32mer; lane 4 25 nM 5′-32P end-labeled 32mer, 50 mM ascorbate, 0.03% H2O2; lane 5 25 nM 5′-32P end-labeled 32mer, 25 nM T*-3; lane 6 25 nM 5′-32P end-labeled 32mer, 25 nM T*-3, 25 nM Fe3+, 50 mM ascorbate, 0.03% H2O2. Solution conditions: 70 mM KCl, 50 mM Tris–HCl buffer, pH 7.5
Extensive additional studies might be necessary to determine the exact mode of the observed cutting. However, as planned, one possibility involves Fenton chemistry of iron bound T*-3 binding to the 32mer. The binding of the iron complex to the 32mer brought iron in close to the DNA16 strand. Under Fenton chemistry conditions, hydroxyl radicals were generated near the polyA strands and sequence-selective cleavage occurred (Fig. 7, lane 6). The cutting site generated by the T*-3 iron complex covers six nucleotides, with maximum cleavage in the center (Fig. 8). This cutting range is consistent with previous studies involving specific Fenton-type cleavage of nucleic acids (Dreyer and Dervan 1985; Floreancig et al. 1999). Once the hydroxamate functionalities of T*-3 chelated with Fe(III) and formed a hexadentate octahedral iron complex, the conformation of this complex was no longer the same as the T*-3. Presumably, there were then fewer than 16 contiguous thymidine residues available to pair with A16, enabling an unanticipated binding site spanning both the A16 region and a small part of the 3′ flanking sequence (Figs. 8, 9). This resulted in a specific although 3′ shifted cleavage pattern. The slight amount of cleavage resulting from the general EDTA-Fe reaction (lane 2, Fig. 7) is both nonselective and inefficient as compared with the cleavage resulting from the T*-3 reaction (lane 6, Fig. 6). This demonstrates the ability of T*-3 to efficiently chelate and target the iron to the template DNA.
Fig. 8.

Graph of the superimposition of densitometric scans of lanes 2 and 6 from Fig. 7 showing the density distribution of cleavage patterns from Fe-EDTA ladder and T*-3 cleavage. The bracket indicates the region of sequence-specific cleavage
Fig. 9.

Conceptual model of T*-3 bound to the 32mer target. In this model, two T–G wobble pairs are proposed to form in addition to the seven Watson–Crick A–T base pairs. The duplex is strained and this conformation would explain both the 3′-shifted cleavage pattern and the observed low thermal stability
Certainly several other explanations are possible for the cleavage pattern observed in Fig. 7. Extensions of the studies reported here would help elucidate these important details. It is hypothesized that incorporation of the mixed-linked hydroxamate analogs at one end of an antisense oligonucleotide would promote normal octahedral coordination of the Fe(III) while allowing the rest of the sequence to assume standard B-form duplex conformation. This would form a shepard’s crook-like structure where the crook is twisted around the iron (or other metal) and the shaft is free to completely base pair with its target.
Conclusions and outlook
Hydroxamate linked oligonucleotides represent a new class of backbone modified antisense oligonucleotides. Thymidine oligomers with fully modified linkages were synthesized through efficient coupling steps. Thymidine 16mer analogs with alternating phosphodiester and hydroxamate linkages were also synthesized through solid phase DNA synthesis with one to three modifications in the middle of the sequence. Applying the same methodology, other hydroxamate-linked oligonucleotides with different base sequences are envisioned after appropriate protecting group modifications.
Replacement of negatively charged phosphodiester linkages by hydroxamate linkages only somewhat weakened duplex stability of the oligonucleotide analogs when hybridized with a complementary poly(dA) strand. Additionally, nuclease stability assays demonstrated that hydroxamate linkages are resistant to nuclease hydrolysis. The resistance of hydroxamate linked oligonucleotides towards endonuclease is improved when the modifications were in the middle of the sequence. Incorporation of the hydroxamate linkages at the end of the nucleotide sequence should prevent the degradation of oligonucleotides from exonucleases.
The iron binding of the thymidine oligomer with three hydroxamate linkages was confirmed by MALDI-MS and CD analysis. A thymidine 16mer with three hydroxamate linkages incorporated in the center of the sequence can bind with both iron and the corresponding target polyA strand. Under Fenton chemistry conditions, this novel iron binding oligothymidylate analog cleaved the complementary DNA strand sequence-selectively. End-modified oligonucleotides containing hydroxamate linkages could give different cleavage patterns.
In conclusion, use of hydroxamate internucleosidic linkages provides a promising antisense oligonucleotide modification. It offers an alternative sequence-selective inhibition mechanism for antisene therapeutics. Also, it is useful as a nuclease resistant probe. Moreover, it might have the potential to assist microbial cell membrane transport through iron uptake systems.
Scheme 3.

Synthesis of phosphoramidite dimer 20
Acknowledgments
We gratefully acknowledge the NIH (AI 30988) and Kimeragen, Inc. for the support of this research. H.L. is grateful to the Department of Chemistry and Biochemistry, University of Notre Dame for financial support in the form of a Reilly fellowship. C.A.F. is grateful to the Department of Chemistry and Biochemistry, University of Notre Dame for financial support in the form of a Podrasnik Fellowship. We appreciate the use of the Bioscience Core facility for the solid phase DNA synthesis, Mass Spectrometry and NMR facilities at the Department of Chemistry and Biochemistry, University of Notre Dame. We are particularly indebted to Prof. P. Huber for assistance with electrophoresis studies and helpful discussions. We also thank Ms. Maureen Metcalf for assistance with the manuscript.
References
- Aboul-Fadl T. Antisense oligonucleotides: the state of the art. Curr Med Chem. 2005;12:2193–2214. doi: 10.2174/0929867054864859. doi:10.2174/092 9867054864859. [DOI] [PubMed] [Google Scholar]
- Agrawal S, editor. Antisense therapeutics. Humana Press; Totowa: 1996. [Google Scholar]
- Agrawal S. Antisense oligonucleotides: a new therapeutic principle. Biochim Biophys Acta. 1999;1489:53–68. doi: 10.1016/s0167-4781(99)00141-4. [DOI] [PubMed] [Google Scholar]
- Arano Y, Uezono T, Akizawa H, Ono M, Wakisaka K, Nakayama M, Sakahara H, Konishi J, Yokoyama A. Reassessment of diethylenetriaminepentaacetic acid (DTPA) as a chelating agent for Indium-111 labeling of polypeptides using a newly synthesized monoreactive DTPA derivative. J Med Chem. 1996;39:3451–3460. doi: 10.1021/jm950949+. doi: 10.1021/jm950949+ [DOI] [PubMed] [Google Scholar]
- Chu CK, Baker DC, editors. Nucleosides and nucleotides as antitumor and antiviral agents. Plenum Press; New York: 1993. [Google Scholar]
- Crooke ST, Lebleu B, editors. Antisense research and applications. CRC Press; Boca Raton: 1993. [Google Scholar]
- Dionis JB, Jenny HB, Peter HH. Synthesis and analytical characterization of a major desferrioxamine B metabolite. J Org Chem. 1989;54:5623–5627. doi:10.1021/jo00284a044. [Google Scholar]
- Dreyer GB, Dervan PB. Sequence-specific cleavage of single-stranded DNA: oligodeoxynucleotide-EDTA × Fe(II) Proc Natl Acad Sci USA. 1985;82:968–972. doi: 10.1073/pnas.82.4.968. doi:10.1073/pnas.82.4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubowchik GM, King HD, Pham-Kaplita K. Efficient mitomycin C coupling with stable p-nitrophenyl-benzyl carbonates using n-hydroxybenzotriazole as a catalytic additive. Tetrahedron Lett. 1997;38:5261–5264. doi:10.1016/S0040-4039(97)01159-3. [Google Scholar]
- Eckstein F, editor. Oligonucleotides and analogues: a practical approach. IRL Press; Oxford: 1991. [Google Scholar]
- Fischman AJ, Babich JW, Strauss HW. A ticket to ride: peptide radiopharmaceuticals. J Nucl Med. 1993;34:2253–2263. [PubMed] [Google Scholar]
- Floreancig PE, Swalley SE, Trauger JW, Dervan PB. Recognition of the minor groove of DNA by hairpin polyamides containing α-substituted-β-amino acids. J Am Chem Soc. 1999;122:6342–6350. doi:10.1021/ja000509u. [Google Scholar]
- Ghosh M, Lambert LJ, Huber PW, Miller MJ. Synthesis, Bioactivity, and DNA-Cleaving Ability of Desferrioxamine β-Nalidixic Acid and Anthraquinone Carboxylic Acid Conjugates. Bioorg Med Chem Lett. 1995;5:2337–2340. doi:10.1016/0960-894X(95)00412-M. [Google Scholar]
- Hashimoto S, Nakamur Y. Nuclease activity of a hydroxamic acid derivative in the presence of various metal ions. J Chem Soc Chem Comm. 1995:1413–1414. [Google Scholar]
- Hayakawa Y, Hirose M, Noyori R. J Org Chem. 1993;58:5551–5555. doi:10.1021/jo00072a 050. [Google Scholar]
- Kneale GG, editor. DNA-protein interactions principles and protocols. Humana Press; Totowa: 1994. [Google Scholar]
- Lebedeva I, Stein CA. Antisense oligonucleotides: promise and reality. Annu Rev Pharmacol Toxicol. 2001;41:403–419. doi: 10.1146/annurev.pharmtox.41.1.403. doi:10.1146/annurev.pharmtox.41.1.403. [DOI] [PubMed] [Google Scholar]
- Li H, Miller MJ. Syntheses of 5′-deoxy-5′-N-hydroxyla-minopyrimidine and purine nucleosides: building-blocks for novel antisense oligonucleosides with hydroxamate linkages. J Org Chem. 1999;64:9289–9293. doi:10.1021/jo991153b. [Google Scholar]
- Li H, Miller MJ. Syntheses and binding studies of oligonucleotides containing N-hydroxycarbamate linkages: potential DNA cleaving antisense oligomers. Tetrahedron Lett. 2000;41:4323–4327. doi:10.1016/S0040-4039(00)00662-6. [Google Scholar]
- Manoharan M. Oligonucleotide conjugates as potential antisense drugs with improved uptake, biodistribution, targeted delivery and mechanism of action. Antisense Nucleic Acid Drug Dev. 2002;12:103–128. doi: 10.1089/108729002760070849. doi:10.1089/1087 29002760070849. [DOI] [PubMed] [Google Scholar]
- Mansoor M, Melendez AJ. Advances in antisense oligonucleotide development for target identification, validation, and as novel therapeutics. Gene Regul Syst Biol. 2008;2:275–295. doi: 10.4137/grsb.s418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–553. doi: 10.1016/s0076-6879(80)65059-9. doi:10.1016/S0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- Minshull J, Hunt Y. The use of single-stranded DNA and RNase H to promote quantitative ‘hybrid arrest of translation’ of mRNA/DNA hybrids in reticulocyte lysate cell-free translations. Nucleic Acids Res. 1986;14:6433–6444. doi: 10.1093/nar/14.16.6433. doi: 10.1093/nar/14.16.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morvan F, Porumb H, Degols G, Lefebvre I, Pompon A, Sproat BS, Rayner B, Malvy C, Lebleu B, Imbach JL. Comparative evaluation of seven oligonucleotide analogs as potential antisense agents. J Med Chem. 1991;36:280–287. doi: 10.1021/jm00054a013. doi:10.1021/jm00054a013. [DOI] [PubMed] [Google Scholar]
- Peyrottes S, Vasseur JJ, Imbach JL, Rayner B. Synthesis, base pairing properties and nuclease resistance of oligothymidylate analogs containing methoxyphosphoramidate internucleoside linkages. Nucleosides Nucleotides. 1994;13:2135–2149. doi:10.1080/15257779408013 213. [Google Scholar]
- Pieles U, Zurcher W, Schar M, Moser HE. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry: a powerful tool for the mass and sequence analysis of natural and modified oligonucleotides. Nucleic Acids Res. 1993;21:3191–3196. doi: 10.1093/nar/21.14.3191. doi:10.1093/nar/21.14.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodger A, Norden B, editors. Circular dichroism and linear dichroism. Oxford University Press; New York: 1997. [Google Scholar]
- Roosenberg JMII, Lin YM, Lu Y, Miller MJ. Studies and syntheses of siderophore, microbial iron chelators, and analogs as potential drug delivery agents. Curr Med Chem. 2000;7:159–197. doi: 10.2174/0929867003375353. [DOI] [PubMed] [Google Scholar]
- Sanghvi YS, Cook PC, editors. Carbohydrate modifications in antisense research. 1994. ACS symposium series 580. [Google Scholar]
- Slater GW, Mayer P, Drouin G. Migration of DNA through gels. Methods Enzymol. 1996;270:272–295. doi: 10.1016/s0076-6879(96)70014-9. doi:10.1016/S0076-6879(96)70014-9. [DOI] [PubMed] [Google Scholar]
- Stein CA. Two problems in antisense biotechnology: in vitro delivery and the design of antisense experiments. Biochim Biophys Acta. 1999;1489:45–52. doi: 10.1016/s0167-4781(99)00143-8. [DOI] [PubMed] [Google Scholar]
- Uhlmann E, Peyman A. Antisense oligonucleotides: a new therapeutic principle. Chem Rev. 1990;90:544–584. doi: 10.1021/cr00102a001. [Google Scholar]
- Wickstrom E, editor. Prospects for antisense nucleic acid therapy of cancer and AIDS. Wiley-Liss; New York: 1991. [Google Scholar]