

Abstract
Unsaturated fatty acids are metabolized to reactive products that can act as pro- or anti-inflammatory signaling mediators. Electrophilic fatty acid species, including nitro- and oxo-containing fatty acids, display salutary anti-inflammatory and metabolic actions. Electrophilicity can be conferred by both enzymatic and oxidative reactions, via the homolytic addition of nitrogen dioxide to a double bond or via the formation of α,β-unsaturated carbonyl and epoxide substituents. The endogenous formation of electrophilic fatty acids is significant and influenced by diet, metabolic, and inflammatory reactions. Transcriptional regulatory proteins and enzymes can sense the redox status of the surrounding environment upon electrophilic fatty acid adduction of functionally significant, nucleophilic cysteines. Through this covalent and often reversible posttranslational modification, gene expression and metabolic responses are induced. At low concentrations, the pleiotropic signaling actions that are regulated by these protein targets suggest that some classes of electrophilic lipids may be useful for treating metabolic and inflammatory diseases.
Keywords: Michael acceptors; α,β-unsaturated carbonyl; electrophiles; nitro-fatty acid; therapeutics
INTRODUCTION
The discovery of protein regulation by phosphorylation first revealed that posttranslational protein modifications (PTMs) are essential in the regulation of cell metabolism, differentiation, growth, and immune defense. At approximately the same time, it was discovered that the enzymatic oxygenation of unsaturated fatty acids forms autocrine and paracrine signaling mediators (prostaglandins and leukotrienes). Then, nitric oxide (•NO) was discovered to be a gaseous mediator of vessel relaxation that acts via heme iron nitrosation and the activation of guanylate cyclase. We now appreciate that •NO induces an even broader array of PTM reactions via the potent secondary oxidizing and nitrating species that this free radical generates. These advances in understanding led to Nobel Prizes in 1988, 1992, and 1998 and have catalyzed further discovery. In this regard, we discuss an emerging area of cell and organ regulation: the oxidation and nitration of unsaturated fatty acids to electrophilic products that induce PTM reactions. These redox-derived products modify protein function and alter patterns of gene expression.
Electrophilic fatty acids are products of both enzymatic and free radical reactions that target susceptible proteins containing evolutionarily conserved and functionally significant nucleophilic amino acids. The array of electrophile-sensitive proteins in cells confers organisms with the ability to sense and respond to endogenously generated reactive species. This property in turn links the metabolic and inflammatory status of an organism with differentiated cell and organ function, because many enzymes and transcriptional regulatory proteins that control metabolism and inflammatory responses contain functionally significant nucleophilic amino acids. Cysteine, one of the least abundant amino acids in proteins, is also the most reactive nucleophilic amino acid and is thus critical for defining the unique selectivity of target proteins for electrophilic PTMs.
This new perspective also suggests that electrophilic lipids and their homologs, if shown to be safe for therapeutic applications, may be useful as drugs for treating metabolic disorders and inflammation-related diseases that affect the cardiovascular and other systems. In the context of drug development, electrophilic drug candidates and electrophilic drug metabolites were once viewed as detrimental because of possible idiosyncratic drug responses and a potential to form irreversible covalent adducts with biomolecules, including DNA and protein. In this regard, covalent modifications have led to concerns about toxicity and mutagenesis. A classic example of this is the metabolism of acetaminophen by liver cytochrome P450s to the electrophile N-acetyl-p-benzoquinone. In certain cases, typically those involving an overdose, detoxification pathways are overwhelmed, and accumulation can lead to liver and renal failure. Safety concerns of this nature have led to a bias against covalent-acting drugs and metabolites in drug development, but at the same time, there are many examples of highly successful and safe drugs that act via covalent protein reactions (e.g., aspirin; penicillin; omeprazole; clopidogrel; and, most recently, dimethylfumarate) (1). With overwhelming evidence coming from both natural and synthetic electrophiles, only in recent years has the pharmaceutical industry begun to accept that some electrophiles at low concentrations will be beneficial as therapeutics because of their ability to regulate inflammatory and metabolic responses. Natural electrophiles, such as oxophytodecienoic acid, can be found in plants and are produced in response to environmental stress (2). Vegetables also contain sulforaphane, capsaicin, tripterpenoids, and isothiocyanates. These compounds represent a wide range of chemical classes and can act as antifungals and antibiotics, whereas others have antiproliferative and anti-inflammatory properties (3). This review focuses on the recent discovery of endogenously produced lipid electrophiles and discusses their actions in the context of being pleiotropic endogenous signaling mediators.
ELECTROPHILIC FATTY ACIDS AND PRECURSORS
Many different electrophiles or their precursors are found in plant- and animal-derived dietary sources or can be produced as metabolites of xenobiotics. An electrophile is a positively charged species that is attracted to an electron-rich center and accepts electron pairs to bond with nucleophiles. Electrophiles can be classified as Michael acceptors; a Michael addition is the reaction between a nucleophile and an electrophile containing an α,β-unsaturated carbonyl or a nitroalkene substituent (Figure 1). The electrophiles described herein are categorized as Michael acceptors, with the exception of polyunsaturated fatty acid (PUFA)-derived epoxides.
Figure 1.
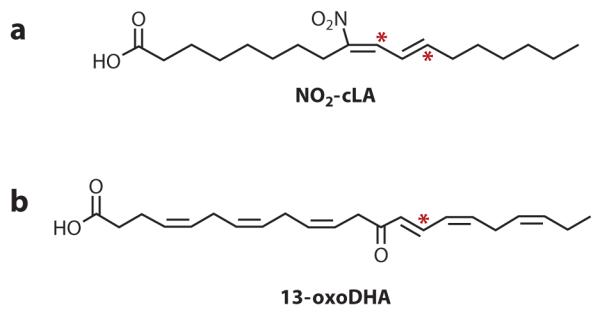
Endogenously detected electrophilic fatty acids. Two examples of electrophilic fatty acids that have been endogenously detected are (a) nitro-conjugated linoleic acid (NO2-cLA) and (b) 13-oxo-docosahexaenoic acid (13-oxoDHA). An asterisk indicates an electrophilic carbon.
The two essential fatty acids, α-linolenic acid (18:3n-3) and linoleic acid (LA) (18:2n-6), are not inherently synthesized in humans (4). Nonetheless, these fatty acids are the foundational building blocks for endogenous Ω-3 and Ω-6 fatty acids. LA is present in diverse plant oils and, through elongation and desaturation reactions, is converted to the abundant and metabolically essential Ω-6 arachidonic acid (AA). The precursor to Ω-3 fatty acids, α-linolenic acid, also undergoes elongation and desaturation to yield the anti-inflammatory docosahexaenoic (DHA) (22:6n-3) and eicosapentaenoic (EPA) (20:5n-3) Ω-3 fatty acids (4, 5). A third fatty acid species noted for beneficial health implications is conjugated linoleic acid (cLA) (6). Although cLA is found in meat and dairy products, nonruminants and humans produce cLA from the trans isomer of oleic acid (vaccenic acid). This cLA formation is mediated by the enterosalivary microbiome via bacterial Δ9-desaturase activity (6) (Figure 2). The Ω-3 and Ω-6 fatty acids can undergo nitration or oxygenation through both enzymatic and nonenzymatic reactions. PUFA oxidation and the addition of nitrogen dioxide (•NO2) to an alkene can result in the formation of electrophilic species. Many of these electrophilic fatty acids have been structurally characterized and described as downstream metabolites of Ω-3 and Ω-6 PUFAs, but not all have been defined with regard to their biological function, despite their abundance.
Figure 2.

Dietary sources of electrophilic fatty acid precursors. (a) Leafy vegetables and cured meats are high in nitrate (NO3−) and nitrite (NO2−). NO3− can be reduced by bacteria in the saliva to NO2−. NO2− in the stomach can form nitrous acid, HNO2, which decomposes to nitrogen dioxide (•NO2). •NO2 is able to form an adduct with free fatty acids. (b) This addition reaction happens preferentially with conjugated linoleic acid (cLA) to form the electrophilic fatty acid NO2-cLA. Meat and dairy products are rich sources of cLA. Additionally, bacteria can convert vaccenic acid, an isomer of oleic acid, to cLA. (c) The essential Ω-3 and Ω-6 fatty acids, α-linolenic acid (18:3n-3) and linoleic acid (18:2n-6), are also obtained through dietary sources, including various seeds and oils. These essential fatty acids are the precursors to the most-studied fatty acids, arachidonic acid (AA), docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA). Because humans are not very efficient at making DHA and EPA from α-linolenic acid, dietary sources of these Ω-3 fatty acids, such as fish, are also very important.
The determination of electrophilic fatty acid levels in biological samples is intrinsically imprecise because of rapid adduction to proteins and soluble thiols, incorporation into the lipid membrane and triglycerides, and further metabolic reactions such as desaturation and β-oxidation. Such imprecise measurements typically lead to an underestimation of actual concentrations (7–9). Additionally, analytical methods, such as enzyme-linked immunosorbent assays, lack sensitivity and specificity, as antibodies can cross-react with similar species. Liquid chromatography coupled with multiple reaction monitoring mass spectrometry is the method of choice for electrophilic lipid analysis, as this approach provides both sensitivity and specificity, especially when stable isotope dilution is employed (7, 10, 11).
NITRO-FATTY ACID FORMATION
Biomolecular targets of nitration include tyrosine, tryptophan, nucleic acids, and unsaturated fatty acids (12). •NO2 reacts readily with the olefinic carbons of unsaturated fatty acids to form nitro derivatives. These nitroalkenes are electrophilic and form Michael adducts with nucleophilic thiol species including glutathione (GSH) and protein cysteine residues, as well as with other nucleophilic species such as histidine. The electrophilic nitroalkenes that have been the subject of most studies include nitro-oleic acid (NO2-OA), nitro-linoleic acid (NO2-LA), nitro-conjugated linoleic acid (NO2-cLA), and nitro-arachidonic acid (NO2-AA) (11, 13).
•NO2 can form from •NO through auto-oxidation (Figure 3a). •NO easily diffuses into lipid membranes and lipoproteins because of its hydrophobic character, neutral charge, and small molecular radius. Although cell membranes make up only 3% of the total cell volume, 90% of •NO auto-oxidation occurs in this cellular compartment (13). Deemed the membrane lens effect, this favorable partitioning and increased concentration of •NO and oxygen (O2) inside the membrane facilitate the nitration and oxidation of PUFAs at the sn-2 position of phospholipids (11, 13). Alternatively, •NO2 can arise from the protonation of nitrite (NO2−) to nitrous acid (HNO2) or by NO2− oxidation by heme peroxidases (e.g., myeloperoxidase, cytochrome c, nitric oxide synthase, and NADPH oxidases), some of which are upregulated during inflammation. The conversion of dietary nitrate is one source of endogenous NO2−. NO2− can also be obtained directly from the diet and is abundant in biological fluids, especially saliva, ranging from low-nanomolar to high-micromolar concentrations, making such fluids a significant source of nitrating species (12, 13). These NO2− concentrations, combined with the low pH of the stomach and phagocytic lysosomes, also favor •NO2 formation via HNO2 generation. HNO2 is a nitrating agent itself, but it can also decompose to •NO, •NO2, and other oxides of nitrogen. Another significant mechanism of •NO2 formation involves peroxynitrite (ONOO−) and peroxynitrous acid (ONOOH). These species can readily diffuse through the membrane to mediate unsaturated fatty acid nitration and oxidation via homolysis of ONOOH to •NO2 and •OH (13). ONOO− also reacts with CO2 to form nitrosoperoxocarbonate (ONOOCO2), and like HNO2, this compound can undergo homolytic scission to form •NO2.
Figure 3.

(a) Reactive nitrogen and oxygen species formation of electrophilic fatty acids. Nitrate (NO3−) and nitrite (NO2−) are dietary sources of nitrogen dioxide (NO2•). NO3− is reduced to NO2− by enterosalivary bacteria. NO2−, combined with the low pH of the stomach, favors •NO2 formation via nitrous acid (HNO2) generation. Various oxides of nitrogen can form from the decomposition of HNO2 in the gut, including •NO2. •NO2 reacts with the π electrons of alkenes via an addition reaction, and a reaction with a second •NO2 results in double-bond reformation. In inflammation, •NO2 can arise from the protonation of NO2− to HNO2 or from NO2− oxidation by heme peroxidases. Another significant mechanism of •NO2 formation involves peroxynitrite (ONOO−) and peroxynitrous acid (ONOOH). These species mediate unsaturated fatty acid nitration and oxidation via homolysis of ONOOH to •NO2 and •OH. ONOO− also reacts with CO2 to form nitrosoperoxocarbonate (ONOOCO2), and like HNO2, this compound can undergo homolytic scission to form •NO2. Nonenzymatic formation of keto-fatty acids begins with initiation by free radical–mediated hydrogen atom abstraction. During the propagation reactions, molecular oxygen adds to a carbon-centered radical to form a peroxyl radical (COO•). This peroxyl radical is unstable and abstracts a hydrogen from another polyunsaturated fatty acid to form a peroxide. A peroxidase then converts the hydroperoxide to an hydroxyl group, which can be oxidized by a dehydrogenase to an α,β-unsaturated ketone. (b) Enzymatic electrophilic fatty acid formation. Ω-3 and Ω-6 fatty acids are oxidized by cyclooxygenase-2 (COX-2) or lipoxygenases (LOs) to hydroxylated lipid species. Therefore, LO or COX-2 hydroxylation at an olefinic carbon is the first step in the formation of an α,β-unsaturated carbonyl. Once the hydroxylated species is formed, one of a number of dehydrogenases can further oxidize the hydroxyl group to a ketone. COX-2 also metabolizes arachidonic acid (AA) to prostaglandin (PG)H2. A variety of PG synthases then form other PGs and thromboxane. PG synthases form PGD2 and PGE2, of which the two primary cyclopentenone prostaglandins, PGJ2 and PGA2, are metabolites, respectively. CYP450s also have epoxygenase abilities, and the CYP2C and CYP2J isoforms are primarily responsible for the formation of Ω-6-derived epoxides epoxyeicosatrienoic acids and Ω-3-derived epoxides. Although epoxides are electrophiles, they are not considered to be Michael acceptors. Asterisks indicate electrophilic carbons. Other abbreviations: cLA, conjugated linoleic acid; NO2-cLA, nitro-conjugated linoleic acid; NO2-OA, nitro-oleic acid; OA, oleic acid.
Nitration of PUFAs depends on the surrounding O2 levels. At low O2 concentrations, nitration predominates, and at high O2 concentrations, lipid peroxidation is the major pathway. Although •NO2 can initiate lipid peroxidation in the presence of O2 through abstraction of a bisallylic hydrogen, •NO2 and •NO readily react with a carbon-centered radical to inhibit propagation (11). These reactions give rise to nitrosylated, nitrated, and nitrito-containing fatty acids. However, only the nitrated species are stable enough to be detected in biological samples (11). •NO2 reacts with the π electrons of alkenes via an addition reaction, and a reaction with a second •NO2 results in double-bond reformation. Like oxidation reactions, conjugated diene–containing PUFAs are especially susceptible to nitration, as opposed to methylene-interrupted species. This property makes cLA more susceptible than LA to nitration, a concept that has been affirmed in vitro and in vivo (14). Another product that has been detected during fatty acid nitration in vivo includes PUFAs containing both α,β-unsaturated keto and nitroalkenyl groups. These species are favored in the presence of O2 and involve double-bond rearrangement and reaction with O2 instead of a second •NO2 addition (11).
OXO-FATTY ACIDS: FORMATION THROUGH RADICAL OXIDATION
Oxidized fatty acids are formed via both enzymatic and nonenzymatic pathways. Enzymatic mechanisms are discussed for each class of oxo-fatty acids. Nonenzymatic formation begins with initiation by free radical–mediated hydrogen atom abstraction, followed by O2 addition to carbon radicals, peroxyl radical fragmentation and rearrangement, peroxyl radical addition to carbon-carbon double bonds, or cyclization and peroxyl-peroxyl termination (10) (Figure 3a). Free radicals can be generated by environmental agents such as air pollution, smoking, UV light, and ionizing radiation or endogenously by reactive species generated by NADPH oxidases, xanthine oxidase, myeloperoxidase, cytochrome P450s (CYP450s), uncoupled •NO synthase, lipoxygenases (LOs), and the mitochondrial electron transport chain (10, 15–18). During the propagation reactions, O2 adds to a carbon-centered radical to form a peroxyl radical (COO•). Addition of O2 to the C• occurs near diffusion-controlled rates at O2 pressures above 100 mm Hg (19), with the rate of O2 addition falling with O2 tension (10). The peroxyl radical is slower to react compared with other radicals, and it thus tends to be the predominant radical present in the acyl chain. The C-H bonds of bisallylic positions, such as C-11 in linoleate and C-7, C-10, and C-13 in arachidonate, are the weakest, and the H in these positions is preferentially abstracted by the peroxyl radical (20). The C-8 and C-11 of oleate have bond dissociation energies of ~10 kcal/mol higher than the C-H bonds of linoleate and arachidonate; therefore, they are less likely to undergo free radical oxidation (10). Intramolecular homolytic substitution on the peroxide bond frequently competes with the addition of O2. A β-peroxide can be converted to an alkoxyl radical and to an epoxide. Although epoxides are not classified as Michael acceptors, they are electrophilic and are prevalent products of free radical oxidation reactions (10). Conjugated alkenes, such as cLA, are more susceptible to epoxidation compared with the bisallylic configuration of LA. In addition to epoxidation, fatty acids containing three or more double bonds can also undergo peroxyl radical cyclization to a five-membered ring. Bicyclic endoperoxides, such as isoprostanes (isoPs), form in this way (10).
Important factors that affect fatty acid oxidation include the local reducing environment and O2 tension. When efficient hydrogen atom donors are replete, peroxyl radicals are readily trapped to form •OOH. Thus, formation of cyclization products, including isoPs, monocyclic peroxides, and isofurans, is suppressed (10). When hydrogen atom donors are not present, •OOH is not trapped, and O2 tension becomes the dominant factor. More cyclization occurs at low O2 tension, which is particularly relevant in diseased tissues. In the pathway of isoP formation, the second 5-exo cyclization involves a C• reaction with which the addition of O2 competes (10).
CYCLOPENTENONE PROSTAGLANDINS
Cyclopentenone prostaglandins (cyPGs) are formed by both enzymatic and nonenzymatic processes. Enzymatically, they are formed by AA conversion to the hydroperoxy endoperoxide prostaglandin G2 (PGG2) (Figure 3b). This reaction proceeds through hydrogen atom abstraction from AA by a tyrosine radical generated by the peroxidase active site. Two O2 molecules then react with the AA radical, yielding PGG2 (21). The peroxidase activity of COX then reduces PGG2 to PGH2. Thromboxane A2, PGI2, PGE2, PGD2, and PGF2α result from the further action of terminal synthases. PGA2 and PGJ2 are two cyPGs that are formed from the dehydration of PGE2 and PGD2, respectively. PGJ2 is further dehydrated through nonenzymatic pathways to form 15-deoxy-PGJ2 (15d-PGJ2) or Δ12-PGJ2 by albumin (22). COX can also form PGA1, the dehydrated product of PGE1, from dihomo-γ-linolenic acid (23). Nonenzymatic pathways of cyPG formation occur via the mechanisms described for isoP formation (24).
All cyPGs contain an electrophilic carbon in the cyclopentane ring; this carbon can form Michael adducts with soluble thiols. In addition to forming Michael adducts, cyPGs can also form a Schiff’s base with amino groups (8). The kinetics of Michael adduct formation and their reversibility depend on the overall structure of the cyPG, as some cyPGs contain electrophilic carbons in the side chains, which may provide more stability. Although the dienone reaction rate is six to eight times faster with soluble thiols, the Michael adducts are more labile; in contrast, immobilized thiols, as found in proteins, are viewed as irreversible under physiological conditions (9, 25). Carrier-mediated cyPG entry into cells promotes accumulation in the nucleus, as shown by a study using radioactive cyPGs (8, 9, 26, 27). Another study showed tracking to the mitochondria using the fluorescent mitochondrial dye BODIPY, which was attached to the carboxylic acid of 15d-PGJ2 (28). Detoxification pathways include (a) GSH adduct formation and export through multidrug resistance transporters and (b) reduction by alkenal reductases as observed for 15d-PGJ2 (29, 30).
KETO-FATTY ACIDS
PUFAs are also oxidized through specific enzymatic pathways to electrophilic keto-fatty acids. The initial enzymatic oxidation of fatty acids is mediated either by COX-2, -5, -12, or 15-LO or by CYP450 (31) (Figure 3b). The major oxygenation product formed is hydroxyl (ROH) species that can be further oxidized by a dehydrogenase to form the keto (C==O) (32–34). When the keto is adjacent to a double bond, an electrophilic α,β-unsaturated carbonyl is formed.
LA is oxygenated by 15-LO to 13-hydroxy- and hydroperoxy-9,11-(Z,E)-octadecadienoic acid (ODE). This molecule is further oxidized by NAD+-dependent 13-hydroxyoctadecadienoate dehydrogenase to the electrophilic 13-oxoODE (31, 32, 35). The initial addition of O2 to AA is catalyzed by COX-2, by a LO, or by CYP450. However, CYP450 oxidation of AA generally does not result in precursors of α,β-unsaturated ketone electrophiles. These enzymes dictate the position and stereochemistry of oxygenation, but many of the hydroxylated metabolites (HETEs) can be further oxidized by multiple dehydrogenases to form their electrophilic oxo-eicosatetraenoic acid (oxoETE) counterpart. For example, 5-HETE, a product of 5-LO, is further oxidized by 5-hydroxyeicosanoid dehydrogenase to 5-oxoETE (33). Also, 15(S)-HETE and 11(R)-HETE are oxidized by 15-hydroxyprostaglandin dehydrogenase (15-PGDH) to their corresponding oxoETEs (36, 37). 15-PGDH has an inverse relation with COX-2 expression (34) and oxidizes the proproliferative PGE2 to 15-ketoPGE2. Although often studied in the context of cancer, 15-PGDH is the dehydrogenase that forms the antiproliferative 15- and 11-oxoETE (36, 37). Notably, COX-2 in mouse and human macrophage cell lines oxygenates the Ω-3 fatty acids docosapentaenoic acid (DPA) and DHA, with preferential oxygenation at C-13 (38). In the presence of aspirin, COX-2 cyclooxygenase activity is inhibited, and its peroxidase activity increases to produce more hydroxylated metabolites with oxygenation primarily at C-17. A dehydrogenase, such as 15-PGDH, is also required for the formation of electrophilic species derived from the COX-2 oxidation products, hydroxy-DHA and hydroxy-DPA (38). The broad specificity of these dehydrogenases includes the di- and trihydroxylated AA and Ω-3 fatty acid derivatives (termed lipoxins, resolvins, and protectins) (39).
EPOXIDES
CYP450s are capable of different types of oxidation. The hydroxylase activity generates ω-hydroxylated HETEs from AA, whereas the epoxygenase activity of the CYP cysteine-heme ligand generates epoxides with a high degree of enantiofacial selectivity consisting of (R,S) and (S,R) enantiomers. This activity is typically associated with CYP2C and CYP2J family isoforms. Epoxidation results in the formation of the epoxyeicosatrienoic acids (EETs) 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET. CYP2C8 is the most prevalent human CYP isoform, and its activity results in equal amounts of 11,12-EET and 14,15-EET isomers. Relative to EET formation by CYP2C8, EET formation by CYP2J2 occurs less frequently but forms racemic 11,12-EET and 14,15-EET, with 76% being 14(R),15(S)-EET. Notably, CYP2J2 is the most abundant isoform in heart microsomes, and its product, 14(R),15(S)-EET, is the most reactive stereoisomer. In addition to CYP450 activity, epoxidation of AA is also catalyzed by hemoglobin in erythrocytes. The main pathways of EET metabolism include β-oxidation, ω-oxidation, chain elongation, and ring opening to form a less active vicinal diol by soluble epoxide hydrolase (40, 41).
Although not as extensively reported, epoxidation of EPA and DHA occurs through CYP-mediated reactions. Several studies reveal that EPA and DHA incubation with rodent renal and hepatic microsomes and recombinant CYP2C and CYP2J isoforms yields ω and ω-1 hydroxylated species and epoxide metabolites (42, 43). CYP2C and CYP2J isoforms are highly active for ω-3 fatty acids; however, preferential metabolite formation is altered compared with CYP-mediated AA metabolism. For instance, CYP1A1 and CYP2E1 typically hydroxylate AA at the ω-1 position, whereas they display highly specific epoxygenase activity toward EPA and DHA (43). The principal products from EPA metabolism are the ω and ω-1 EPAs, 19- and 20-HEPE (where HEPE denotes hydroxyeicosapentaenoic acid), and five regioisomeric epoxyeicosatetraenoic acids (EEQs). Similarly, CYP oxidation of DHA gives hydroxydocosaenoic acids, 21- and 22-hydroxy-DHA, and six regioisomeric epoxydocosapentaenoic acids (EDPs). The AA epoxygenases—CYP2C8, CYP2C9, CYP2C18, CYP2C19, and CYP2J2—also epoxygenate EPA and DHA. CYP2J2 and all CYP2C isoforms except for CYP2C8 show high stereoselectivity in producing (R,S) enantiomers of 17,18-EEQ and 19,20-EDP (42, 43).
In addition to the better-known LA-derived oxoODE and AA-derived oxoETE metabolites discussed above, other electrophilic metabolites of LA and AA can be formed, including the LA-derived epoxy-oxo-octadecenoic acid (EKODE) (44–46). This species and epoxy-hydroxyoctadecenoic acid are formed by initial conversion of LA to 13-LOOH and 9-LOOH via LO activity. A secondary oxidation requiring cysteine and iron yields the EKODE product 12,13-epoxy-9-oxo-10-trans-octadecenoic acid (44, 45). AA can also be metabolized to similar products, 11, 12, 15-hydroxyeicosatrienoic acid (THETA) and its unstable precursor 15-hydroxy-11,12-epoxyeicosatrienoic acid (HEETA), through a combination of CYP2J2 and 15-LO (47). In addition, a more stable HEETA stereoisomer is produced by oxygenation at C13–15, giving cis and trans products of 13-hydroxy-14,15-epoxyeicosatrienoic acid. Other hydroxyl-epoxide compounds, the hepoxilins, are formed by 12-LO metabolism of AA.
ELECTROPHILIC FATTY ACID PRECURSORS
Inflammation-resolving fatty acids derived from AA, EPA, and DHA are dihydroxy or trihydroxy in nature. The AA-derived lipoxins and the EPA- and DHA-derived resolvins, protectins, and maresins are produced by dual enzyme reactions during acute inflammation and are proposed to mediate resolution (39). These mediators block neutrophil recruitment, promote infiltration and activation of monocytes, and induce phagocytosis and lymphatic clearance of apoptotic neutrophils by activated macrophages (39). These polyhydroxylated species require transcellular biosynthesis or sequential actions of (a) LOs (i.e., 5-LO/12-LO or 15-LO/5-LO) or by a series of reactions involving (b) COX-2 and CYP450 or (c) COX-2 and LO (39, 48). Although these species are proposed to bind to and activate specific G protein–coupled receptors (GPCRs), they are also substrates for electrophile generation. 15-PGDH, which was proposed to inactivate binding to GPCRs and account for a putative short half-life, converts these dihydroxy and trihydroxy compounds to electrophiles containing α,β-unsaturated carbonyls. For example, the 15-PGDH-derived metabolite of lipoxin A4 (LXA4), 15-oxoLXA4, induces the expression of a broad array of electrophile-regulated genes. Thus, whereas the oxidation of the hydroxy groups on diverse bioactive lipids is typically viewed as a metabolic inactivation reaction, this reaction can convert nonelectrophilic species that activate GPCRs to electrophilic products that act via more pleiotropic mechanisms.
PLEIOTROPIC SIGNALING ACTIONS OF ELECTROPHILIC FATTY ACIDS
Electrophilic fatty acids function by PTM of proteins and transcription factors. Multiple classes of electrophilic signaling molecules are expected to have unique patterns of downstream signaling, including those formed from oxidation reactions (e.g., α,β-unsaturated ketones) and nitration reactions (e.g., nitroalkenes). Through Michael addition, electrophilic fatty acids will adduct susceptible protein cysteine and histidine residues, inducing alterations in protein structure, function, and subcellular distribution (49). The targets for electrophilic fatty acid modification are thus diverse, yielding pleiotropic and sometimes reversible effects on an array of key signaling pathways (Figure 4). Although dependent on concentration, target specificity, cell type, metabolism, and reversibility (50, 51), many of these effects provide beneficial outcomes in the contexts of inflammation and other disease processes (52). Importantly, the endogenous production of these molecules is often a result of pro-oxidative and stress conditions, thus providing a rheostat mechanism for resolving the inflammatory environment.
Figure 4.

Transcriptional regulators as targets of electrophilic fatty acid modification. The endogenous production and exogenous administration of electrophilic fatty acids target multiple redox-sensing transcriptional regulators. In the cytoplasm, nitroalkenes (a) putatively bind HSF-1, releasing HSP70 and driving HSF-1-dependent gene transcription, and (b) covalently adduct Keap1, causing dissociation from and translocation of Nrf2 to induce ARE gene transcription. (c) The cyPG 15d-PGJ2 modifies the p65 subunit of NF-κB, sustaining inhibition by IκB and blocking p50/p65-dependent gene transcription. In the nucleus, (d) α,β-unsaturated electrophilic fatty acids covalently bind and act as partial PPARγ agonists, stimulating gene transcription. Abbreviations: 15d-PGJ2, 15-deoxy-prostaglandin J2; ARE, antioxidant-response element; cyPG, cyclopentenone prostaglandin; HSF-1, heat shock factor 1; HSP70, heat shock protein 70; IκB, inhibitor of κB; Keap1, kelch-like ECH-associated protein 1; NF-κB, nuclear factor-κB; Nrf2, nuclear factor (erythroid-derived 2)-like 2; PPARγ, peroxisome proliferator–activated receptor γ.
Nrf2-Keap1
Nrf2 [nuclear factor (erythroid-derived 2)-like 2], a responder to oxidative stress and inducer of phase II enzymes through the antioxidant-response element (ARE), regulates the expression of ~100 genes, including those encoding heme oxygenase-1 (HO-1), NAD(P)H dehydrogenase quinone-1 (NQO1), γ-glutamylcysteine synthase, GSH peroxidase, and copper/zinc superoxide dismutase. Nrf2 is bound in the cytoplasm to the inhibitor Keap1 (kelch-like ECH-associated protein 1). Under basal conditions, Nrf2 is expressed at low levels to maintain cell homeostasis; however, most Nrf2 is ubiquitinated by cullin-based E3 ubiquitin ligase (Cul3), which is in complex with Keap1, resulting in proteasomal targeting and degradation. Inducers such as xenobiotics, pathogens, and other stressors protect Nrf2 from Keap1-dependent ubiquitination (53). Instead, Keap1 disassembly from Nrf2 allows for nuclear translocation, promoting transcriptional activation and the subsequent expression of phase II genes.
Electrophilic fatty acids adduct Keap1, causing the dissociation of this inhibitor from Nrf2. Specifically, electrophilic adduction of Cys273 and Cys288 in Keap1 facilitates Nrf2 activation and transcriptional upregulation of ARE-regulated genes (54, 55). Some studies also suggest that modification of Cys151 is necessary for dissociation of Keap1 from Cul3 (56) and for Nrf2 activation (57); however, these findings are controversial (58). Variability in signaling modifications exist within this molecular complex because of the gamut of cysteines available (27 cysteines in Keap1) and the plasticity of responses defined by the redox environment and the electrophile—a phenomenon deemed the cysteine code (57). In addition, cysteines within Nrf2 may be targets of electrophilic modification for nuclear translocation and gene induction (59). The interplay between Nrf2 and Keap1 thiol reactivities may thus contribute to the downstream effects mediated by electrophilic fatty acids.
Nitroalkenes and α,β-unsaturated ketones similarly activate Nrf2 (60). Keap1 adduction by both electrophilic fatty acid classes occurs through Cys273. NO2-LA-mediated Nrf2 activation was reported in human aortic endothelial cells and vascular smooth muscle cells, upregulating HO-1 protein and decreasing proliferation via cell cycle arrest through p27kip1 expression, respectively (61, 62). Similarly, NO2-OA administration both in vitro and in vivo leads to upregulation of HO-1 expression and activity (63, 64). Besides NO2-OA and NO2-LA, cholesteryl-nitrolinoleate (CLNO2) also induces HO-1 expression in macrophages (65). Furthermore, HO-1 induction can occur in an Nrf2-independent manner following NO2-LA treatment, highlighting the ability of other transcriptional mediators, such as cyclic AMP (cAMP) response element (CRE), CRE binding protein-1 (CREB-1), activator protein-1 (AP-1), and E-box sequences (66), to help elicit the anti-inflammatory effects of nitroalkenes.
The α,β-unsaturated ketones—such as the electrophilic, COX-derived 15d-PGJ2—also activate Nrf2-dependent gene expression (67). Although the relevance of this species to in vivo signaling has been questioned, when very low rates of production are combined with facile reactions of 15d-PGJ2, even undetectable levels of 15d-PGJ2 are proposed to accumulate to an extent to which adduction to Keap1 eventually induces HO-1 and GSH production (68). This finding provides a rationale for why free 15d-PGJ2 and presumably other electrophilic species are measured in vivo at far lower concentrations than those required to modulate signaling pathways in vitro (69). In mouse peritoneal macrophages, 15d-PGJ2 and possibly the generation of other electrophilic lipids by carrageenan-induced pleurisy increase HO-1 and peroxiredoxin expression in an Nrf2-dependent manner (70). Enzymatically formed oxo derivatives of ω-3 fatty acids also induce nuclear accumulation of Nrf2 and activation of Nrf2-dependent gene expression (71). Specifically, treatment of macrophages with 17-oxoDPA and 17-oxoDHA leads to induction of HO-1. Finally, by using an ARE-luciferase reporter assay in IMR-32 neuroblastoma cells, electrophilic LA metabolites, such as the EKODEs, induced transcription and protein levels of ARE-regulated genes such as NQO1 (72).
In addition to increasing GSH levels following electrophilic fatty acid–mediated Nrf2 activation, 15d-PGJ2 can decrease intracellular GSH via covalent thiol adduction. Although electrophilic fatty acids can mitigate inflammation, the reaction of 15d-PGJ2 with GSH can also reverse the suppression of IL-6-dependent intracellular adhesion molecule (ICAM)-1 expression in endothelial cells (73). Likewise, NO2-OA upregulates Nrf2-dependent genes such as BACH1, which represses transcription of ARE-regulated genes (74). These mechanisms may thus induce a counteracting regulation to the anti-inflammatory effects of electrophilic fatty acids that would contribute to the overall cellular homeostasis. Furthermore, other electrophilic fatty acids, such as oxidized phospholipids, activate Nrf2 but, in doing so, enhance vascular endothelial growth factor expression and proangiogenic actions (75).
NF-κB
Nuclear factor-κB (NF-κB) controls cell survival; proliferation; and a host of inflammatory mediators, including cytokines, chemokines, and adhesion molecules (76). Thus, NF-κB is essential for the immune response and clearance of infection, yet is easily exploited under chronic inflammatory conditions. Dimerization of two of the subunits (p50, p65, p52, RelB, or c-Rel) must occur for NF-κB to bind DNA and activate gene transcription. However, NF-κB is sequestered in the cytoplasm by inhibitors of κB (IκB) proteins, which mask the nuclear localization sequence of the subunits. Upon stimulating signals, IκB kinases (IKKs) phosphorylate IκB, targeting it for ubiquitination and proteasomal degradation and allowing NF-κB to be translocated into the nucleus to activate transcription. The blockade of this pathway diminishes proinflammatory responses.
Through inhibition of DNA binding, nitroalkenes reduce NF-κB activation and subsequent gene transcription. NO2-LA and NO2-OA blunt cytokine responses via alkylation of the p65 subunit of NF-κB (77). Upon nitroalkene administration, decreases in cytokine production following lipopolysaccharide (LPS) stimulation of macrophages correspond to IC50 values ranging from 0.4 to 1.9 μM for IL-6, TNFα, and MCP-1. Additionally, NO2-LA treatment prevents monocyte-endothelial cell interaction under static and physiological flow conditions by reducing expression of vascular cell adhesion molecule (VCAM)-1, another target of NF-κB regulation. In vivo administration of NO2-OA, yielding nanomolar concentrations in murine plasma, inhibits leukocyte recruitment to the vascular endothelium and reduces aortic expression of adhesion molecules. Further in vitro studies show that NO2-OA inhibits the recruitment of TLR4 and TNF receptor–associated factor 6 (TRAF6) to membrane lipid raft compartments (78). Inhibition of NF-κB occurs only after pretreatment of macrophages with NO2-LA and CLNO2 20 h prior to LPS stimulation, suggesting a potential need for the induction of other anti-inflammatory mediators, such as HO-1, to aid in inflammation resolution (65).
15d-PGJ2 also inhibits NF-κB activation and subsequent upregulation of proinflammatory molecules via glutathionylation of p65, restricting DNA binding. Specifically, GSH upregulation via 15d-PGJ2-induced Nrf2 activation, as discussed above, promotes the modification of p65 at Cys38, Cys160, or Cys216, each of which is a target of electrophilic glutathionylation (79). Beyond inhibiting p65 DNA binding, 15d-PGJ2 also inhibits degradation of IκB-α, leading to the sequestration of NF-κB in the cytoplasm (80, 81). Likewise, the DHA-derived cyclopentenone neuroprostane A4-NP also suppresses NF-κB activation by protecting IκB-α from Ser32/36 phosphorylation, in part through thiol modification of the Cys179 residue present in IKK (82). Analogous cysteine adduction occurs following electrophilic cyPG administration (83). In aggregate, these data affirm that electrophilic oxo- and nitro-fatty acids are potent NF-κB inhibitors that act at multiple sites within the signaling cascade; such inhibitors show potential for alleviating this component of proinflammatory signaling.
PPARγ
Peroxisome proliferator–activated receptor γ (PPARγ) is a nuclear receptor that regulates lipid homeostasis, inflammatory signaling, and adipocyte differentiation. Importantly, PPARγ regulates the partitioning of lipid stores away from peripheral sites (e.g., liver, muscle) and reduces adipokine expression, thus facilitating increased insulin sensitivity. Moreover, PPARγ activation in myeloid cells instigates lipid clearance while suppressing expression of proinflammatory mediators like IFNγ and inducible nitric oxide synthase (iNOS or NOS2) (84).
Both nitroalkenes and α,β-unsaturated ketones are PPARγ ligands. The nitro group of NO2-LA was shown through structural characterization to specifically interact with Arg288 and Glu343 of PPARγ, facilitating ligation (85). Other studies confirm this interaction and describe an additional alkylation of Cys285 in the PPARγ ligand–binding domain by both NO2-LA and NO2-OA (38, 86). NO2-LA-dependent PPARγ activation stimulates macrophage CD36 expression, the receptor for oxidized low-density lipoprotein (oxLDL), and promotes adipocyte differentiation (87). NO2-OA is a more potent agonist for PPARγ activation than is NO2-LA; NO2-OA and NO2-LA have EC50 values of 1,900 and 870 nM, respectively (38). Reporter gene construct analyses show that NO2-OA stimulates all PPARs, with PPARγ activation predominating; such stimulation limits adipogenesis (PPARγ2 and activating protein-2 expression) and promotes glucose uptake in preadipocytes (88). NO2-OA also upregulates genes such as those encoding Glut4, CBP-1, and PGC-1α (38), all of which increase insulin sensitivity and reduce fat accumulation and proinflammatory cytokines. Unlike full PPARγ agonists such as the thiazolidinedione insulin sensitizer rosiglitazone, NO2-LA and NO2-OA act as partial agonists (88–90) and may reduce the risk of peripheral edema, weight gain, and adverse cardiovascular events. However, these in vitro observations of NO2-OA-mediated PPARγ activation using reporter gene–based constructs have not been convincingly verified in vivo. For example, the addition of a PPARγ antagonist had no effect on the ability of NO2-OA to lower blood pressure in a murine hypertension model (91). Also, PPARγ-regulated gene expression was not significantly induced upon acute intravenous administration of NO2-OA in mice (78).
15d-PGJ2 is also a PPARγ ligand; however, it does not promote as robust a response as does NO2-LA (87). Nonetheless, through direct binding to PPARγ, 15d-PGJ2 induces differentiation of C3H10T1/2 fibroblasts to adipocytes (92). Like the nitroalkenes, other electrophilic oxo derivatives of ω-3 fatty acids, including 4-oxoDHA, 5-oxoEPA, 17-oxoDPA, and 17-oxoDHA, also covalently adduct PPARγ at Cys285 (93) and are partial agonists for PPARγ (71). Potent AA derivatives, such as 12-oxoETE and 15-oxoETE, are likewise considered to be partial PPARγ agonists on the basis of in-depth elucidation of structural binding and conformational changes within the ligand-binding domain and the loop region (86).
As a means of controlling signaling, both NO2-LA and α,β-unsaturated ketones form intracellular conjugates with GSH, similar to what has been shown with Nrf2 (73). In these instances, removal of the conjugates from the cell via efflux transporters, such as multidrug resistance–associated protein-1, inhibits PPARγ-dependent transcription (29, 94), suggesting a cell-autonomous limitation mechanism.
Heat Shock Proteins
To initiate the stress response, heat shock proteins (HSPs) are induced to alleviate unfolded protein accumulation and endoplasmic reticulum stress. Most HSPs function as chaperones or proteases. Microarray transcriptome analyses showed that NO2-OA treatment of human umbilical vein endothelial cells promotes the expression of both Nrf2-regulated and heat shock factor (HSF)-1-and HSF-2-regulated target genes. For example, there is a profound upregulation of HSPA6, HPA1A (inducible HSP70), DNAJA4, and HSPB8. More specifically, HSP70 mRNA and protein levels increase in a dose-dependent manner following nitroalkene administration (63). CyPGs also increase the levels of HSP70 expression along with the expression levels of other molecular chaperones (95). HSF-1 binds to HSP70, inhibiting functional activity. Thus, electrophilic fatty acid adduction of critical cysteines within HSF-1 may facilitate HSP70 release, activating the heat shock response and resolving cellular stress.
Additional Inflammation-Related Proteins, Enzymes, and Ion Channels
Downstream of cytokine and growth factor ligation, signal transducers and activators of transcription (STATs) are involved in regulating cell survival, differentiation, and proliferation. Upon receptor-ligand interaction, cytoplasmic Janus kinases ( JAKs) are activated, leading to the phosphorylation of STAT substrates, nuclear translocation, and gene transcription (96). Prior to nuclear accumulation, combinations of STAT subunits dimerize to control different genes. STAT1, for example, can form homodimers or heterodimers with STAT3 to induce the expression of genes stimulated by type II interferon (IFNγ) or type I interferon (IFNα or IFNβ), respectively. STATs are therefore crucial in mounting antiviral and cytotoxic immune responses. LPS-stimulated macrophages treated with NO2-LA or NO2-OA demonstrate reduced phosphorylation of STAT1 in comparison with macrophages stimulated only by LPS without NO2-LA or NO2-OA treatment (97). This result is likely due to augmentation of mitogen-activated protein kinase (MAPK) phosphatase-1 activity, which limits proinflammatory STAT1 activity (98). Similarly, 15d-PGJ2 abrogates IFNγ-induced STAT1 and STAT3 phosphorylation via PPARγ-independent mechanisms, including the upregulation of suppressor of cytokine signaling proteins that deactivate JAKs (99). Specifically, 15d-PGJ2 reduces phosphorylation of STAT3 at Tyr705, inhibiting IL-6-dependent stimulation of endothelial cells (73).
In addition to transcription factor modification, electrophilic fatty acids also affect the activity of inflammation-related enzymes. For example, NOS2, which is controlled by NF-κB activation, is suppressed following NO2-LA, CLNO2, and NO2-AA administration to macrophages, resulting in decreased inflammatory •NO generation (65, 100). NOS2 expression is also reduced following treatment with 17-oxoDPA, 17-oxoDHA (71), and A4-NP (82). Although NOS2-derived •NO is important for pathogen clearance, chronic activation contributes to deleterious consequences, such as autoimmunity, ischemic injury, hypotension, and neurodegeneration (101). Thus, attenuation of this enzyme by electrophilic fatty acids lessens chronic inflammation. In contrast, endothelial NOS (NOS1), in association with HO-1, is enhanced in mouse aortas following in vivo treatment of NO2-OA. NOS1 expression and activity also increase, in part due to NO2-OA-stimulated phosphorylation of NOS1 Ser1179 (102). Unlike NOS2, NOS1 is responsible for limiting vasoconstriction and inflammatory cell margination, which may alleviate vascular injury. The ability of nitroalkenes to promote opposing effects on these two NOS isoforms highlights the pleiotropic actions of these molecules.
Similarly, cyclooxygenases provoke inflammation-related responses through the generation of prostaglandins from AA. Two isoforms, COX-1 and COX-2, exist. These isoforms mediate prostaglandin production during homeostasis (COX-1) or upon induction of COX-2. NO2-AA administration inhibits the prostaglandin endoperoxide hydrogen synthase of activated platelets (103). Likewise, A4-NP hinders COX-2 expression in stimulated macrophages (82). As a feedback mechanism for alleviating proinflammatory conditions, COX-2 generates anti-inflammatory oxo derivatives of DHA within macrophages, causing reduced cytokine- and NOS2-derived •NO production and enhancing Nrf2 and PPARγ activation (71). The search for specific COX-2 inhibitors over pan-COX inhibitors, such as nonsteroidal anti-inflammatory drugs (NSAIDs), is extensive (104), with the goal of alleviating the nonspecific gastrointestinal side effects of NSAIDs. More in-depth studies of electrophilic fatty acid effects on COX-2 may help to identify potential candidates.
Sheddases, such as matrix metalloproteinases (MMPs), also contribute to inflammation by cleaving proinflammatory molecules, leading to the release of cytokines, atherosclerotic plaque instability, and cancer metastasis (105, 106). MMPs are present within the cell as latent zymogens, consisting of a cysteine thiol-Zn2+ interaction linking the propeptide domain with the catalytic subunit. For MMPs to be activated, the thiol interaction must be dissociated. Through thiol alkylation of pro-MMP, high concentrations of NO2-OA reduce the proteolytic activity of MMP-7 and MMP-9, whereas lower concentrations concomitantly increase activity while decreasing MMP transcription, limiting inflammatory progression in a biphasic manner. Moreover, in vivo administration of NO2-OA lowers MMP-9 expression in atherosclerotic lesions of ApoE−/− mice (107). The net effect of electrophilic fatty acid action is therefore to limit MMP expression and function.
Although the protein reactions of electrophilic fatty acids are often reversible, the inhibition of xanthine oxidoreductase (XOR), another contributor to inflammation, is irreversible and highly specific for NO2-OA. Xanthine oxidase–derived uric acid production and partially reduced reactive oxygen species generation can be pathological upon XOR activation. Because high concentrations of added thiol reductants do not restore activity, the pterin dithiolene of the XOR molybdenum cofactor may be the adduction site of NO2-OA (108). In contrast, 15d-PGJ2 administration to human lymphocytes stimulates the generation of reactive oxygen species by XOR (109), illustrating the opposing responses of this locus of oxidant generation to electrophilic fatty acids.
The proinflammatory actions of 15d-PGJ2 are further exemplified in lymphocytes, endothelial cells, and smooth muscle cells, where micromolar concentrations of this electrophile result in the upregulation of MAPK and COX-2 and promote cytokine/chemokine production (110, 111). Similar concentrations of 15d-PGJ2 enhance reactive oxygen species and induce human neuronal cell death through the modification of Cys35 and Cys69 within thioredoxin, an enzyme important for protection against oxidative stress (112). In contrast, lower concentrations of cyPG administration afford astrocyte-dependent neuroprotection in a GSH depletion model of cell death (113). Thus, excessive α,β-unsaturated ketone concentrations impart proinflammatory effects, impacting disease pathophysiology (114, 115) and emphasizing the need for strict control of the levels of these mediators in vivo.
Lastly, electrophilic fatty acids can also mediate the activation of neurological ion channels. In particular, transient receptor potential (TRP) channels are involved in neurogenic pain, inflammation, and overreactivity. DHA and EPA activate mammalian TRPA1 channels, stimulating the secretion of hormones from enteroendocrine cells and characterizing TRPA1 channels as endogenous PUFA sensors (116). NO2-OA also activates sensory neurons (117) and bladder afferent nerve TRP channels (118), which are rich in cysteines and are thus targets for electrophile modulation (119). A biphasic activation of TRP channels is suggested whereby nitroalkene modulation of the nociceptor TRPA1 channel occurs at Cys619, Cys639, Cys663, and Lys708 (120). This initial stimulation by electrophilic fatty acids leads to calcium influx, depolarization, and firing, whereas the secondary response at higher NO2-OA concentrations can promote the desensitization of TRP channels, thus alleviating proinflammatory and pathophysiological events.
PHYSIOLOGICAL AND THERAPEUTIC IMPLICATIONS
On the basis of the pleiotropic signaling actions of electrophilic fatty acids and the apparent safety of many electrophilic FDA-approved drugs, the potential benefits of the in vivo administration of these reactive lipid mediators have been evaluated in a plethora of murine models of disease. Diseases or pathologies that are discussed below include diabetes and metabolic syndrome, nephropathy, ischemia-reperfusion injury, cardiovascular disease, pulmonary inflammation, and chronic inflammatory disorders.
Diabetes and Metabolic Syndrome
With the rising epidemic of obesity, more efficacious therapeutic strategies are needed to address the complex pathophysiological events that lead to metabolic syndrome. Because of the cardiovascular and cancer-related risks associated with the thiazolidinedione insulin sensitizers rosiglitazone and pioglitazone, the use of these drugs to treat type 2 diabetes is significantly diminished (121), and alternative treatment options are needed. Electrophilic fatty acids, including ω-3 PUFA oxo derivatives and nitroalkenes, show promise in alleviating glucose intolerance and insulin resistance. By using leptin receptor–deficient ob/ob−/− mice and obese Zucker rats, a 4-keto ester derivative of DHA diminished blood glucose levels and prevented any unwanted side effects, such as those seen with the thiazolidinediones (122). In addition, administration of purified EPA/DHA to type 2 diabetes patients improves vascular function compared with function following placebo treatment. Specifically, the ω-3 fatty acids are hypothesized to reduce oxidant generation, thus augmenting the sensitivity of vascular smooth muscle cells to •NO-mediated vasodilation (123). The ω-3-derived resolvin D1 also improves insulin sensitivity by resolving the chronic underlying inflammation associated with obesity (lowered IL-6 expression) while increasing adiponectin levels (124). In addition, ω-6 AA metabolites, such as hepoxilins, act as insulin sensitizers and promote the release of insulin from pancreatic islet cells (125). Nitroalkenes also show promise in alleviating metabolic dysfunction in obese animals. Treatment of ob/ob−/− mice (38) and high-fat diet–fed mice with NO2-OA reduces glucose intolerance and insulin resistance. Unlike rosiglitazone, which also acts through PPARγ, NO2-OA does not increase weight gain. Additionally, nitroalkene administration to obese Zucker rats significantly lowers body weight, with associated decreases in plasma triglycerides and lipid peroxidation products and increases in the anti-inflammatory high-density lipoprotein (126), overall attenuating insulin resistance.
Nephropathy
Renal inflammation and kidney failure are often the outcomes of chronic inflammatory conditions such as diabetes and hypertension but can also be triggered following acute injury or kidney transplantation. In a murine adriamycin-induced model of focal glomerular sclerosis, pretreatment with NO2-OA reduces creatinine levels, glomerulosclerosis, tubulointerstitial fibrosis, urinary lipid peroxidation products, and renal inflammation (127). Renal TNFα, MCP-1, ICAM-1, VCAM-1, and PGE2 are also attenuated following NO2-OA treatment in a multiorgan endotoxemia model (128). Furthermore, multiple indices of renal protection are also evident in mice treated with NO2-OA after 30 min of bilateral ischemia-reperfusion (129). In LPS-treated kidney mesangial cells, abrogation of renal MCP-1 is detected after 15d-PGJ2 treatment (80). In addition, known Nrf2 inducers protect renal cells from oxidative stress (130). Therefore, multiple lines of evidence show that electrophilic fatty acid activation of Nrf2 and attenuation of NF-κB-dependent gene expression promote renal protection.
Ischemia-Reperfusion Injury
Ischemia-reperfusion injury occurs after blood flow returns to an area deprived of O2. Restoration of blood flow results in polymorphonuclear cell (PMN) infiltration, inflammatory cytokine expression, and the generation of reactive oxygen and nitrogen species. Markedly, murine myocardial ischemia-reperfusion leads to endogenous NO2-LA and NO2-OA formation, and administration of nitroalkene during ischemia decreases myocardial infarct size and protects left ventricular function (90). NO2-OA treatment of rats upon bilateral renal ischemia-reperfusion injury protects against PMN infiltration and expression of inflammatory mediators, including ICAM-1, IL-1β, TNFα, and the NADPH oxidase subunits p47phox and gp91phox (129). Ischemic preconditioning, a mechanism to protect a tissue against ischemia-reperfusion injury, also generates nitrated fatty acids within the mitochondria. NO2-LA causes mild mitochondrial uncoupling through electrophilic modification of adenine nucleotide translocase and uncoupling protein 2, both of which protect against ischemia-reperfusion injury (131, 132). Additionally, ω-3 PUFA dietary supplementation prior to hepatic ischemia-reperfusion injury alleviates increased liver necrosis; plasma alanine transaminase; and production of NOS2, COX-2, and high-mobility-group box-1 (133). Likewise, 15d-PGJ2 and other α,β-unsaturated ketones mitigate ischemia-reperfusion injury in a number of organs (134). Electrophilic fatty acids thus lessen the severity of damage following O2 return.
Cardiovascular Disease
Adverse effects of activated platelets, monocytes, and neutrophils can also mediate vascular occlusion in association with atherosclerosis. Progressive plaque deposition, myocardial infarction, peripheral vascular disease, and stroke ensue. Vascular inflammation, characterized by leukocyte adhesion, endothelial activation of NF-κB, and activation of TLR4 signaling, is reduced following intravenous injection of NO2-OA (135). Upon treatment with NO2-LA, aggregation of thrombin-stimulated healthy human platelets is reduced via •NO-independent mechanisms associated with an elevation of cAMP. Also in a •NO-independent manner, NO2-LA reduces platelet P-selectin expression and enhances vasodilator-stimulated phosphoprotein phosphorylation, thus resulting in decreased adherence and elevated antiaggregatory properties, respectively (136). NO2-AA also inhibits platelet aggregation by decreasing prostaglandin endoperoxide hydrogen synthase–dependent thromboxane B2 formation (103).
Moreover, NO2-LA inhibits activated neutrophil superoxide generation in a •NO- and NADPH oxidase–independent, cAMP-dependent manner. Furthermore, expression of CD11b and elastase release is significantly downregulated after nitroalkene treatment, mitigating adhesion and degranulation of neutrophils and associated sequelae (137). Inhibition of neutrophil function may limit tissue damage, maintaining cell integrity. These beneficial effects on platelets and neutrophil function may be of direct benefit in preventing the progression of atherosclerosis, and further investigation is warranted.
In addition to those features described above, leukocyte accumulation and plaque instability within atherosclerotic lesions occur as a consequence of neutrophil and platelet activation (138). Importantly, macrophages are recruited to plaques, and in the presence of reactive oxygen species, LDL is oxidized, providing the impetus for macrophage conversion into atherogenic foam cells. NO2-OA treatment attenuates lesion formation in ApoE−/− mice, a model of atherosclerosis, by reducing inflammatory cell accumulation and chemokine production. Specifically, NO2-OA blocks oxLDL-mediated STAT1 phosphorylation, which lowers lipid uptake in macrophages and subsequent foam cell formation (139).
A complication from the use of angioplasty to clear plaque obstructions in the vasculature is restenosis due to vessel wall injury and neointimal cell proliferation. Systemic administration of NO2-OA inhibits this neointimal hyperplasia after wire injury of the femoral artery in a murine model of restenosis after angioplasty (64). Activation of Nrf2-regulated HO-1 expression accounts for much of the vascular protection because inhibition of HO-1 by Sn(IV)-protoporphyrin or genetic ablation reverses the protective effects of NO2-OA.
Myocardial infarction and hypertension are also limited by electrophilic fatty acids. Oxo derivatives of DHA and EPA may induce cardioprotection by acting as PPARγ agonists in a mechanism similar to that of nitro fatty acids (71, 140). 15d-PGJ2 and EETs protect against myocardial infarction via ligation of PPARγ and activation of ATP channels, respectively (141, 142). EETs contribute to vascular and cardiac physiology through regulating vascular dilation, promoting angiogenesis, and decreasing inflammation and platelet aggregation. In the heart, EETs regulate myocyte contraction and increase coronary blood flow through several mechanisms that involve the activation of TRP ion channels leading to an influx of calcium and through an unidentified receptor that stimulates cAMP production (40). In contrast, 15d-PGJ2 reduces NF-κB activation, enhances HSP70 expression, and increases phosphorylation of the prosurvival kinase Akt; all these actions can reduce myocardial injury (143). ω-6 metabolites of AA, like the HEETAs, induce vasorelaxation in small mesenteric arteries of rabbits (47). Furthermore, in patients who survive myocardial infarction, daily dietary supplementation of ω-3 PUFA is associated with event-free survival (144). Finally, angiotensin II (Ang II)-induced hypertension is alleviated by systemic NO2-OA administration through a PPARγ-independent mechanism in which NO2-OA binds the Ang II receptor and inhibits inositol triphosphate and calcium mobilization without inhibiting Ang II binding to the receptor. NO2-OA binding results in the attenuation of vasoconstriction in murine mesenteric vessels (91). These data indicate that the potential benefits of electrophilic fatty acids for cardiovascular health are vast and promising for clinical utility.
Pulmonary Inflammation
The positive effects of electrophilic fatty acids on inflammation and stress may also provide beneficial outcomes in pulmonary diseases. Lung disorders, such as asthma, chronic obstructive pulmonary disease (COPD), pulmonary hypertension, and acute injury, commonly have a high component of inflammation-mediated damage. Although evidence of proinflammatory activation has been shown following nitroalkene treatment of pulmonary type II epithelial cells in vitro (145), in vivo treatment following intratracheal administration of LPS reduces acute lung injury (146). Treated mice show diminished bronchial alveolar lavage (BAL) fluid protein, neutrophil infiltration, and oxidative stress. In addition to oxidative inflammatory conditions, decreased •NO production sets the stage for endothelial dysfunction and vascular cell proliferation and remodeling, all of which increase susceptibility to hypertension. In vitro and in vivo studies demonstrated that NO2-OA increased HO-1 and NOS3 in the vasculature, resulting in protective effects associated with enhanced •NO production (102). Studies have shown that increasing •NO production can partially reverse PAH and vessel remodeling once established (146a). The ω-3-derived resolvins, protectins, and maresins also display potent proresolving actions in a variety of cell types and in in vivo models of inflammatory diseases, including asthma, acute lung injury, and cystic fibrosis (147–151). Moreover, 15d-PGJ2 resolves chronic eosinophilia following allergen challenge, decreasing airway inflammation (152). Similarly, 15d-PGJ2 treatment of carrageenan-induced acute lung injury reduces BAL leukocyte and albumin levels in an Nrf2-dependent manner (153). The disruption of Nrf2 activation and the subsequent antioxidant response are associated with both COPD and asthma (154, 155), pointing to the potential for electrophilic fatty acid alleviation of pulmonary inflammatory disorders.
Chronic Inflammatory Disorders
Electrophilic fatty acids, as modulators of NF-κB and STATs, likely alter immune responses underlying chronic inflammation. Evidence of protection against models of cerebrospinal injury, gastrointestinal injury, pancreatitis, multiple sclerosis, and rheumatoid arthritis exists for 15d-PGJ2 (156). Physiological and biochemical manifestations of inflammatory bowel disease are also attenuated by NO2-OA in a murine model (157). The suppression of proinflammatory cytokines and prostaglandins is the primary mechanism behind the alleviation of these disorders. CyPGs such as PGJ2 also affect cell cycle regulation and could be investigated as suppressors of tumorigenesis (158). Specifically, the antiproliferative activity of cyPGs has been linked to the blockade of multiple features related to aberrant tumor growth, including cell cycle progression, activation of mitochondrial apoptotic signaling, induction of oxidative stress, disruption of the cytoskeleton, activation of MAPKs, and downregulation of human telomerase reverse transcriptase.
Studies have also demonstrated the antiviral properties of cyPGs through induction of HSPs, inhibition of viral replication, and viral protein glycosylation. In addition to such antiviral properties, cyPG targets include cytoskeletal proteins (vimentin, actin, and tubulin) as well as estrogen receptor α, Keap1, UCH-L1, Ras, GSTP-1, PPARγ, thioredoxin, various components of the NF-κB signaling pathway, and soluble epoxide hydrolase (8). Many of these play critical roles in regulating redox homeostasis.
Although the above data focus on cyPGs as anti-inflammatory molecules that control disease pathogenesis, in some instances, these molecules have been linked to the onset of disease. 15d-PGJ2 has been linked to human arthritis, and other cyPGs have been linked to neurodegenerative diseases. Like many endogenous compounds, including •NO, cyPGs may have biphasic modes of action based on concentration, cellular context, and the presence of targets (8).
In summary, the actions of electrophilic fatty acids depend on their concentration, reaction environment, β-oxidation, adduction to nucleophilic proteins, metabolic reactions that inactivate the electrophilic center, GSH reaction, and the export of GSH-electrophile adducts from the cell (52, 94). The endogenous concentrations of these species in tissues, blood, and urine, along with the in vivo responses to 5- to 25-fold-higher concentrations achieved after administration of these mediators, indicate that electrophilic fatty acids are pleiotropic modulators of metabolic and inflammatory responses. The array of beneficial actions observed in animal model studies and the apparent safety of so-called soft electrophiles suggest the potential for broad therapeutic utility.
SUMMARY POINTS.
Electrophilic fatty acids are formed by the addition of nitrogen dioxide to an olefinic carbon or by the oxidation of an unsaturated fatty acid to an α,β-unsaturated carbonyl or epoxide.
Oxidation of unsaturated fatty acids is mediated by enzymatic pathways or through free radical reactions of reactive oxygen species.
Electrophilic fatty acids covalently bind nucleophiles, including soluble thiols such as GSH, and nucleophilic amino acids, including cysteine and histidine.
Many transcriptional regulatory proteins have a critical cysteine to which electrophilic fatty acids are able to bind, thus altering gene transcription.
The therapeutic potential of electrophilic fatty acids is evident for a broad range of inflammatory conditions and diseases, as electrophilic fatty acid signaling targets multiple redox-sensing transcriptional regulators of inflammation.
Glossary
- Electrophile
a positively or partial positively charged species attracted to an electron-rich center. Electrophiles are Michael acceptors
- Nucleophile
an electron-rich molecule that donates an electron pair to form a covalent bond with an electrophile
- Michael addition
the addition of a nucleophile to an α,β-unsaturated carbonyl
- α,β-Unsaturated carbonyl
has the general structure −(O = C)−Cα = Cβ−. In this compound, the carbonyl group is conjugated with an alkene
- Ω-3 fatty acids
polyunsaturated fatty acids with a double bond at the third carbon atom from the end of the carbon chain
- Ω-6 fatty acids
polyunsaturated fatty acids with a double bond at the sixth carbon atom from the end of the carbon chain
Footnotes
DISCLOSURE STATEMENT The authors acknowledge a financial interest in Complexa, Inc.
LITERATURE CITED
- 1.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011;10:307–17. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 2.Bottcher C, Pollmann S. Plant oxylipins: plant responses to 12-oxo-phytodienoic acid are governed by its specific structural and functional properties. FEBS J. 2009;276:4693–704. doi: 10.1111/j.1742-4658.2009.07195.x. [DOI] [PubMed] [Google Scholar]
- 3.Gersch M, Kreuzer J, Sieber SA. Electrophilic natural products and their biological targets. Nat. Prod. Rep. 2012;29:659–82. doi: 10.1039/c2np20012k. [DOI] [PubMed] [Google Scholar]
- 4.Lands B. Consequences of essential fatty acids. Nutrients. 2012;4:1338–57. doi: 10.3390/nu4091338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singer P, Shapiro H, Theilla M, Anbar R, Singer J, Cohen J. Anti-inflammatory properties of omega-3 fatty acids in critical illness: novel mechanisms and an integrative perspective. Intensiv. Care Med. 2008;34:1580–92. doi: 10.1007/s00134-008-1142-4. [DOI] [PubMed] [Google Scholar]
- 6.Churruca I, Fernandez-Quintela A, Portillo MP. Conjugated linoleic acid isomers: differences in metabolism and biological effects. Biofactors. 2009;35:105–11. doi: 10.1002/biof.13. [DOI] [PubMed] [Google Scholar]
- 7.Woodcock SR, Bonacci G, Gelhaus SL, Schopfer FJ. Nitrated fatty acids: synthesis and measurement. Free Radic. Biol. Med. 2013;59:14–26. doi: 10.1016/j.freeradbiomed.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garzon B, Oeste CL, Diez-Dacal B, Perez-Sala D. Proteomic studies on protein modification by cyclopentenone prostaglandins: expanding our view on electrophile actions. J. Proteomics. 2011;74:2243–63. doi: 10.1016/j.jprot.2011.03.028. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki M, Mori M, Niwa T, Hirata R, Furuta R, et al. Chemical implications for antitumor and antiviral prostaglandins: reaction of Δ7-prostaglandin A1 and prostaglandin A1 methyl esters with thiols. J. Am. Chem. Soc. 1997;119:2376–85. [Google Scholar]
- 10.Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem. Rev. 2011;111:5944–72. doi: 10.1021/cr200084z. An encompassing review of free radical oxidation of lipids and condition-dependent product formation.
- 11.Schopfer FJ, Cipollina C, Freeman BA. Formation and signaling actions of electrophilic lipids. Chem. Rev. 2011;111:5997–6021. doi: 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lundberg JO, Weitzberg E. Biology of nitrogen oxides in the gastrointestinal tract. Gut. 2013;62:616–29. doi: 10.1136/gutjnl-2011-301649. [DOI] [PubMed] [Google Scholar]
- 13.Trostchansky A, Bonilla L, Gonzalez-Perilli L, Rubbo H. Nitro-fatty acids: formation, redox signaling, and therapeutic potential. Antioxid. Redox Signal. 2013;19(11):1257–65. doi: 10.1089/ars.2012.5023. [DOI] [PubMed] [Google Scholar]
- 14.Bonacci G, Baker PR, Salvatore SR, Shores D, Khoo NK, et al. Conjugated linoleic acid is a preferential substrate for fatty acid nitration. J. Biol. Chem. 2012;287:44071–82. doi: 10.1074/jbc.M112.401356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stockl J. Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 2010;12:1009–59. doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Upston JM, Neuzil J, Witting PK, Alleva R, Stocker R. Oxidation of free fatty acids in low density lipoprotein by 15-lipoxygenase stimulates nonenzymic, α-tocopherol-mediated peroxidation of cholesteryl esters. J. Biol. Chem. 1997;272:30067–74. doi: 10.1074/jbc.272.48.30067. [DOI] [PubMed] [Google Scholar]
- 17.Upston JM, Witting PK, Brown AJ, Stocker R, Keaney JF., Jr Effect of vitamin E on aortic lipid oxidation and intimal proliferation after arterial injury in cholesterol-fed rabbits. Free Radic. Biol. Med. 2001;31:1245–53. doi: 10.1016/s0891-5849(01)00721-3. [DOI] [PubMed] [Google Scholar]
- 18.Witting PK, Mohr D, Stocker R. Assessment of prooxidant activity of vitamin E in human low-density lipoprotein and plasma. Methods Enzymol. 1999;299:362–75. doi: 10.1016/s0076-6879(99)99036-5. [DOI] [PubMed] [Google Scholar]
- 19.Pratt DA, Mills JH, Porter NA. Theoretical calculations of carbon-oxygen bond dissociation enthalpies of peroxyl radicals formed in the autoxidation of lipids. J. Am. Chem. Soc. 2003;125:5801–10. doi: 10.1021/ja034182j. [DOI] [PubMed] [Google Scholar]
- 20.Porter NA, Wolf RA, Yarbro EM, Weenen H. The autoxidation of arachidonic acid: formation of the proposed SRS-A intermediate. Biochem. Biophys. Res. Commun. 1979;89:1058–64. doi: 10.1016/0006-291x(79)92115-6. [DOI] [PubMed] [Google Scholar]
- 21.Hamberg M, Svensson J, Samuelsson B. Prostaglandin endoperoxides. A new concept concerning the mode of action and release of prostaglandins. Proc. Natl. Acad. Sci. USA. 1974;71:3824–28. doi: 10.1073/pnas.71.10.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K. 15-Deoxy-Δ12,14-prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes. J. Biol. Chem. 2002;277:10459–66. doi: 10.1074/jbc.M110314200. [DOI] [PubMed] [Google Scholar]
- 23.Ziboh VA, Miller CC, Cho Y. Metabolism of polyunsaturated fatty acids by skin epidermal enzymes: generation of antiinflammatory and antiproliferative metabolites. Am. J. Clin. Nutr. 2000;71:S361–66. doi: 10.1093/ajcn/71.1.361s. [DOI] [PubMed] [Google Scholar]
- 24.Hardy KD, Cox BE, Milne GL, Yin H, Roberts LJ., 2nd Nonenzymatic free radical–catalyzed generation of 15-deoxy-Δ12,14-prostaglandin J2–like compounds (deoxy-J2-isoprostanes) in vivo. J. Lipid Res. 2011;52:113–24. doi: 10.1194/jlr.M010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bickley JF, Ciucci A, Evans P, Roberts SM, Ross N, Santoro MG. Reactions of some cyclopentenones with selected cysteine derivatives and biological activities of the product thioethers. Bioorg. Med. Chem. 2004;12:3221–27. doi: 10.1016/j.bmc.2004.03.061. [DOI] [PubMed] [Google Scholar]
- 26.Narumiya S, Fukushima M. Site and mechanism of growth inhibition by prostaglandins. I. Active transport and intracellular accumulation of cyclopentenone prostaglandins, a reaction leading to growth inhibition. J. Pharmacol. Exp. Ther. 1986;239:500–5. [PubMed] [Google Scholar]
- 27.Narumiya S, Ohno K, Fujiwara M, Fukushima M. Site and mechanism of growth inhibition by prostaglandins. II. Temperature-dependent transfer of a cyclopentenone prostaglandin to nuclei. J. Pharmacol. Exp. Ther. 1986;239:506–11. [PubMed] [Google Scholar]
- 28.Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ. Res. 2006;99:924–32. doi: 10.1161/01.RES.0000248212.86638.e9. [DOI] [PubMed] [Google Scholar]
- 29.Paumi CM, Wright M, Townsend AJ, Morrow CS. Multidrug resistance protein (MRP) 1 and MRP3 attenuate cytotoxic and transactivating effects of the cyclopentenone prostaglandin, 15-deoxy-Δ12,14prostaglandin J2 in MCF7 breast cancer cells. Biochemistry. 2003;42:5429–37. doi: 10.1021/bi027347u. [DOI] [PubMed] [Google Scholar]
- 30.Yu X, Egner PA, Wakabayashi J, Wakabayashi N, Yamamoto M, Kensler TW. Nrf2-mediated induction of cytoprotective enzymes by 15-deoxy-Δ12,14-prostaglandin J2 is attenuated by alkenal/one oxidoreductase. J. Biol. Chem. 2006;281:26245–52. doi: 10.1074/jbc.M604620200. [DOI] [PubMed] [Google Scholar]
- 31.Mesaros C, Lee SH, Blair IA. Targeted quantitative analysis of eicosanoid lipids in biological samples using liquid chromatography–tandem mass spectrometry. J. Chromatogr. B. 2009;877:2736–45. doi: 10.1016/j.jchromb.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bronstein JC, Bull AW. Substrate specificity and characterization of partially purified rat liver 13-hydroxyoctadecadienoic acid (13-HODE) dehydrogenase. Arch. Biochem. Biophys. 1997;348:219–25. doi: 10.1006/abbi.1997.0364. [DOI] [PubMed] [Google Scholar]
- 33.Erlemann KR, Cossette C, Grant GE, Lee GJ, Patel P, et al. Regulation of 5-hydroxyeicosanoid dehydrogenase activity in monocytic cells. Biochem. J. 2007;403:157–65. doi: 10.1042/BJ20061617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Na HK, Park JM, Lee HG, Lee HN, Myung SJ, Surh YJ. 15-Hydroxyprostaglandin dehydrogenase as a novel molecular target for cancer chemoprevention and therapy. Biochem. Pharmacol. 2011;82:1352–60. doi: 10.1016/j.bcp.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Murphy RC, Zarini S. Glutathione adducts of oxyeicosanoids. Prostaglandins Other Lipid Mediat. 2002;68–69:471–82. doi: 10.1016/s0090-6980(02)00049-7. [DOI] [PubMed] [Google Scholar]
- 36.Wei C, Zhu P, Shah SJ, Blair IA. 15-oxo-Eicosatetraenoic acid, a metabolite of macrophage 15-hydroxyprostaglandin dehydrogenase that inhibits endothelial cell proliferation. Mol. Pharmacol. 2009;76:516–25. doi: 10.1124/mol.109.057489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, Zhang S, Arora JS, Snyder NW, Shah SJ, Blair IA. 11-Oxoeicosatetraenoic acid is a cyclooxygenase-2/15-hydroxyprostaglandin dehydrogenase–derived antiproliferative eicosanoid. Chem. Res. Toxicol. 2011;24:2227–36. doi: 10.1021/tx200336f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, et al. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat. Chem. Biol. 2010;6:433–41. doi: 10.1038/nchembio.367. The first to show the formation of endogenous α,β-unsaturated carbonyls derived from COX-2 metabolism of Ω-3 fatty acids.
- 39.Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem. Rev. 2011;111:5922–43. doi: 10.1021/cr100396c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol. Rev. 2012;92:101–30. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arnold C, Markovic M, Blossey K, Wallukat G, Fischer R, et al. Arachidonic acid–metabolizing cytochrome P450 enzymes are targets of ω-3 fatty acids. J. Biol. Chem. 2010;285:32720–33. doi: 10.1074/jbc.M110.118406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konkel A, Schunck WH. Role of cytochrome P450 enzymes in the bioactivation of polyunsatu-rated fatty acids. Biochim. Biophys. Acta. 2011;1814:210–22. doi: 10.1016/j.bbapap.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 44.Gardner HW, Jursinic PA. Degradation of linoleic acid hydroperoxides by a cysteine. FeCl3 catalyst as a model for similar biochemical reactions. I. Study of oxygen requirement, catalyst and effect of pH. Biochim. Biophys. Acta. 1981;665:100–12. doi: 10.1016/0005-2760(81)90238-1. [DOI] [PubMed] [Google Scholar]
- 45.Gardner HW, Kleiman R. Degradation of linoleic acid hydroperoxides by a cysteine. FeCl3 catalyst as a model for similar biochemical reactions. II. Specificity in formation of fatty acid epoxides. Biochim. Biophys. Acta. 1981;665:113–24. doi: 10.1016/0005-2760(81)90239-3. [DOI] [PubMed] [Google Scholar]
- 46.Bruder ED, Ball DL, Goodfriend TL, Raff H. An oxidized metabolite of linoleic acid stimulates corticosterone production by rat adrenal cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003;284:R1631–35. doi: 10.1152/ajpregu.00753.2002. [DOI] [PubMed] [Google Scholar]
- 47.Chawengsub Y, Gauthier KM, Nithipatikom K, Hammock BD, Falck JR, et al. Identification of 13-hydroxy-14,15-epoxyeicosatrienoic acid as an acid-stable endothelium-derived hyperpolarizing factor in rabbit arteries. J. Biol. Chem. 2009;284:31280–90. doi: 10.1074/jbc.M109.025627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Serhan CN, Fredman G, Yang R, Karamnov S, Belayev LS, et al. Novel proresolving aspirin-triggered DHA pathway. Chem. Biol. 2011;18:976–87. doi: 10.1016/j.chembiol.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Batthyany C, Schopfer FJ, Baker PR, Duran R, Baker LM, et al. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J. Biol. Chem. 2006;281:20450–63. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin D, Saleh S, Liebler DC. Reversibility of covalent electrophile-protein adducts and chemical toxicity. Chem. Res. Toxicol. 2008;21:2361–69. doi: 10.1021/tx800248x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rudolph TK, Freeman BA. Transduction of redox signaling by electrophile-protein reactions. Sci. Signal. 2009;2:re7. doi: 10.1126/scisignal.290re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rudolph V, Schopfer FJ, Khoo NK, Rudolph TK, Cole MP, et al. Nitro-fatty acid metabolome: saturation, desaturation, β-oxidation, and protein adduction. J. Biol. Chem. 2009;284:1461–73. doi: 10.1074/jbc.M802298200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 54.Kansanen E, Kivela AM, Levonen AL. Regulation of Nrf2-dependent gene expression by 15-deoxy-Δ12,14-prostaglandin J2. Free Radic. Biol. Med. 2009;47:1310–17. doi: 10.1016/j.freeradbiomed.2009.06.030. [DOI] [PubMed] [Google Scholar]
- 55.Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, et al. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006;26:221–29. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem. Res. Toxicol. 2008;21:705–10. doi: 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- 57.Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, et al. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 2009;29:493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kansanen E, Bonacci G, Schopfer FJ, Kuosmanen SM, Tong KI, et al. Electrophilic nitro-fatty acids activate NRF2 by a KEAP1 cysteine 151–independent mechanism. J. Biol. Chem. 2011;286:14019–27. doi: 10.1074/jbc.M110.190710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He X, Ma Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1–dependent ubiquitination-proteasomal degradation, and transcription activation. Mol. Pharmacol. 2009;76:1265–78. doi: 10.1124/mol.109.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsujita T, Li L, Nakajima H, Iwamoto N, Nakajima-Takagi Y, et al. Nitro-fatty acids and cyclopentenone prostaglandins share strategies to activate the Keap1-Nrf2 system: a study using green fluorescent protein transgenic zebrafish. Genes Cells. 2011;16:46–57. doi: 10.1111/j.1365-2443.2010.01466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wright MM, Schopfer FJ, Baker PR, Vidyasagar V, Powell P, et al. Fatty acid transduction of nitric oxide signaling: Nitrolinoleic acid potently activates endothelial heme oxygenase 1 expression. Proc. Natl. Acad. Sci. USA. 2006;103:4299–304. doi: 10.1073/pnas.0506541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villacorta L, Zhang J, Garcia-Barrio MT, Chen XL, Freeman BA, et al. Nitro-linoleic acid inhibits vascular smooth muscle cell proliferation via the Keap1/Nrf2 signaling pathway. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H770–76. doi: 10.1152/ajpheart.00261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kansanen E, Jyrkkanen HK, Volger OL, Leinonen H, Kivela AM, et al. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: identification of heat shock response as the major pathway activated by nitro-oleic acid. J. Biol. Chem. 2009;284:33233–41. doi: 10.1074/jbc.M109.064873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cole MP, Rudolph TK, Khoo NK, Motanya UN, Golin-Bisello F, et al. Nitro-fatty acid inhibition of neointima formation after endoluminal vessel injury. Circ. Res. 2009;105:965–72. doi: 10.1161/CIRCRESAHA.109.199075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferreira AM, Ferrari MI, Trostchansky A, Batthyany C, Souza JM, et al. Macrophage activation induces formation of the anti-inflammatory lipid cholesteryl-nitrolinoleate. Biochem. J. 2009;417:223–34. doi: 10.1042/BJ20080701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wright MM, Kim J, Hock TD, Leitinger N, Freeman BA, Agarwal A. Human haem oxygenase-1 induction by nitro-linoleic acid is mediated by cAMP, AP-1 and E-box response element interactions. Biochem. J. 2009;422:353–61. doi: 10.1042/BJ20090339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Levonen AL, Landar A, Ramachandran A, Ceaser EK, Dickinson DA, et al. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 2004;378:373–82. doi: 10.1042/BJ20031049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oh JY, Giles N, Landar A, Darley-Usmar V. Accumulation of 15-deoxy-Δ12,14-prostaglandin J2 adduct formation with Keap1 over time: effects on potency for intracellular antioxidant defence induction. Biochem. J. 2008;411:297–306. doi: 10.1042/bj20071189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, et al. Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J. Biol. Chem. 2005;280:27244–50. doi: 10.1074/jbc.M502551200. [DOI] [PubMed] [Google Scholar]
- 70.Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, et al. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Δ12,14-prostaglandin J2. Mol. Cell. Biol. 2004;24:36–45. doi: 10.1128/MCB.24.1.36-45.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chaves SS, Groeger J, Helgenberger L, Auerbach SB, Bialek SR, et al. Improved anamnestic response among adolescents boosted with a higher dose of the hepatitis B vaccine. Vaccine. 2010;28:2860–64. doi: 10.1016/j.vaccine.2010.01.059. [DOI] [PubMed] [Google Scholar]
- 72.Wang R, Kern JT, Goodfriend TL, Ball DL, Luesch H. Activation of the antioxidant response element by specific oxidized metabolites of linoleic acid. Prostaglandins Leukot. Essent. Fatty Acids. 2009;81:53–59. doi: 10.1016/j.plefa.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wung BS, Wu CC, Hsu MC, Hsieh CW. 15-Deoxy-Δ12,14-prostaglandin J2 suppresses IL-6-induced STAT3 phosphorylation via electrophilic reactivity in endothelial cells. Life Sci. 2006;78:3035–42. doi: 10.1016/j.lfs.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 74.Jyrkkanen HK, Kuosmanen S, Heinaniemi M, Laitinen H, Kansanen E, et al. Novel insights into the regulation of antioxidant-response-element-mediated gene expression by electrophiles: induction of the transcriptional repressor BACH1 by Nrf2. Biochem. J. 2011;440:167–74. doi: 10.1042/BJ20110526. [DOI] [PubMed] [Google Scholar]
- 75.Afonyushkin T, Oskolkova OV, Philippova M, Resink TJ, Erne P, et al. Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependent mechanism: novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler. Thromb. Vasc. Biol. 2010;30:1007–13. doi: 10.1161/ATVBAHA.110.204354. [DOI] [PubMed] [Google Scholar]
- 76.Tak PP, Firestein GS. NF-κB: a key role in inflammatory diseases. J. Clin. Investig. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, et al. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J. Biol. Chem. 2006;281:35686–98. doi: 10.1074/jbc.M603357200. Nitroalkylation of the p65 subunit of NF-κB by nitroalkenes inhibits DNA binding and subsequent proinflammatory gene expression.
- 78.Villacorta L, Chang L, Salvatore SR, Ichikawa T, Zhang J, et al. Electrophilic nitro-fatty acids inhibit vascular inflammation by disrupting LPS-dependent TLR4 signalling in lipid rafts. Cardiovasc. Res. 2012;98:116–24. doi: 10.1093/cvr/cvt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lin YC, Huang GD, Hsieh CW, Wung BS. The glutathionylation of p65 modulates NF-κB activity in 15-deoxy-Δ12,14-prostaglandin J2–treated endothelial cells. Free Radic. Biol. Med. 2012;52:1844–53. doi: 10.1016/j.freeradbiomed.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 80.Rovin BH, Lu L, Cosio A. Cyclopentenone prostaglandins inhibit cytokine-induced NF-κB activation and chemokine production by human mesangial cells. J. Am. Soc. Nephrol. 2001;12:1659–67. doi: 10.1681/ASN.V1281659. [DOI] [PubMed] [Google Scholar]
- 81.Castrillo A, Diaz-Guerra MJ, Hortelano S, Martin-Sanz P, Bosca L. Inhibition of IκB kinase and IκB phosphorylation by 15-deoxy-Δ12,14-prostaglandin J2 in activated murine macrophages. Mol. Cell. Biol. 2000;20:1692–98. doi: 10.1128/mcb.20.5.1692-1698.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Musiek ES, Brooks JD, Joo M, Brunoldi E, Porta A, et al. Electrophilic cyclopentenone neuroprostanes are anti-inflammatory mediators formed from the peroxidation of the omega-3 polyunsatu-rated fatty acid docosahexaenoic acid. J. Biol. Chem. 2008;283:19927–35. doi: 10.1074/jbc.M803625200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IκB kinase. Nature. 2000;403:103–8. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 84.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARγ. Annu. Rev. Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 85.Li Y, Zhang J, Schopfer FJ, Martynowski D, Garcia-Barrio MT, et al. Molecular recognition of nitrated fatty acids by PPARγ. Nat. Struct. Mol. Biol. 2008;15:865–67. doi: 10.1038/nsmb.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Waku T, Shiraki T, Oyama T, Fujimoto Y, Maebara K, et al. Structural insight into PPARγ activation through covalent modification with endogenous fatty acids. J. Mol. Biol. 2009;385:188–99. doi: 10.1016/j.jmb.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 87.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, et al. Nitrolinoleic acid: an endogenous peroxisome proliferator–activated receptor γ ligand. Proc. Natl. Acad. Sci. USA. 2005;102:2340–45. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, et al. Fatty acid transduction of nitric oxide signaling: Multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator–activated receptor ligands. J. Biol. Chem. 2005;280:42464–75. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baker PR, Schopfer FJ, Sweeney S, Freeman BA. Red cell membrane and plasma linoleic acid nitration products: synthesis, clinical identification, and quantitation. Proc. Natl. Acad. Sci. USA. 2004;101:11577–82. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rudolph V, Rudolph TK, Schopfer FJ, Bonacci G, Woodcock SR, et al. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc. Res. 2010;85:155–66. doi: 10.1093/cvr/cvp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang J, Villacorta L, Chang L, Fan Z, Hamblin M, et al. Nitro-oleic acid inhibits angiotensin II–induced hypertension. Circ. Res. 2010;107:540–48. doi: 10.1161/CIRCRESAHA.110.218404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator–activated receptor γ and promotes adipocyte differentiation. Cell. 1995;83:813–19. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 93.Itoh T, Fairall L, Amin K, Inaba Y, Szanto A, et al. Structural basis for the activation of PPARγ by oxidized fatty acids. Nat. Struct. Mol. Biol. 2008;15:924–31. doi: 10.1038/nsmb.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alexander RL, Bates DJ, Wright MW, King SB, Morrow CS. Modulation of nitrated lipid signaling by multidrug resistance protein 1 (MRP1): Glutathione conjugation and MRP1-mediated efflux inhibit nitrolinoleic acid–induced, PPARγ-dependent transcription activation. Biochemistry. 2006;45:7889–96. doi: 10.1021/bi0605639. [DOI] [PubMed] [Google Scholar]
- 95.Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med. Res. Rev. 2001;21:185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- 96.Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J. Cell Sci. 2004;117:1281–83. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 97.Ichikawa T, Zhang J, Chen K, Liu Y, Schopfer FJ, et al. Nitroalkenes suppress lipopolysaccharide-induced signal transducer and activator of transcription signaling in macrophages: a critical role of mitogen-activated protein kinase phosphatase 1. Endocrinology. 2008;149:4086–94. doi: 10.1210/en.2007-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Venema RC, Venema VJ, Eaton DC, Marrero MB. Angiotensin II–induced tyrosine phosphor-ylation of signal transducers and activators of transcription 1 is regulated by Janus-activated kinase 2 and Fyn kinases and mitogen-activated protein kinase phosphatase 1. J. Biol. Chem. 1998;273:30795–800. doi: 10.1074/jbc.273.46.30795. [DOI] [PubMed] [Google Scholar]
- 99.Park EJ, Park SY, Joe EH, Jou I. 15d-PGJ2 and rosiglitazone suppress Janus kinase–STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J. Biol. Chem. 2003;278:14747–52. doi: 10.1074/jbc.M210819200. [DOI] [PubMed] [Google Scholar]
- 100.Trostchansky A, Souza JM, Ferreira A, Ferrari M, Blanco F, et al. Synthesis, isomer characterization, and anti-inflammatory properties of nitroarachidonate. Biochemistry. 2007;46:4645–53. doi: 10.1021/bi602652j. [DOI] [PubMed] [Google Scholar]
- 101.Salvemini D, Kim SF, Mollace V. Reciprocal regulation of the nitric oxide and cyclooxygenase pathway in pathophysiology: relevance and clinical implications. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013;304:R473–87. doi: 10.1152/ajpregu.00355.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Khoo NK, Rudolph V, Cole MP, Golin-Bisello F, Schopfer FJ, et al. Activation of vascular endothelial nitric oxide synthase and heme oxygenase-1 expression by electrophilic nitro-fatty acids. Free Radic. Biol. Med. 2010;48:230–39. doi: 10.1016/j.freeradbiomed.2009.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Trostchansky A, Bonilla L, Thomas CP, O’Donnell VB, Marnett LJ, et al. Nitroarachidonic acid, a novel peroxidase inhibitor of prostaglandin endoperoxide H synthases 1 and 2. J. Biol. Chem. 2011;286:12891–900. doi: 10.1074/jbc.M110.154518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chakraborti AK, Garg SK, Kumar R, Motiwala HF, Jadhavar PS. Progress in COX-2 inhibitors: a journey so far. Curr. Med. Chem. 2010;17:1563–93. doi: 10.2174/092986710790979980. [DOI] [PubMed] [Google Scholar]
- 105.Newby AC. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc. Med. 2007;17:253–58. doi: 10.1016/j.tcm.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004;4:617–29. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 107.Bonacci G, Schopfer FJ, Batthyany CI, Rudolph TK, Rudolph V, et al. Electrophilic fatty acids regulate matrix metalloproteinase activity and expression. J. Biol. Chem. 2011;286:16074–81. doi: 10.1074/jbc.M111.225029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kelley EE, Batthyany CI, Hundley NJ, Woodcock SR, Bonacci G, et al. Nitro-oleic acid, a novel and irreversible inhibitor of xanthine oxidoreductase. J. Biol. Chem. 2008;283:36176–84. doi: 10.1074/jbc.M802402200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Alvarez-Maqueda M, El Bekay R, Alba G, Monteseirin J, Chacon P, et al. 15-Deoxy-Δ12,14-prostaglandin J2 induces heme oxygenase-1 gene expression in a reactive oxygen species–dependent manner in human lymphocytes. J. Biol. Chem. 2004;279:21929–37. doi: 10.1074/jbc.M400492200. [DOI] [PubMed] [Google Scholar]
- 110.Harris SG, Smith RS, Phipps RP. 15-Deoxy-Δ12,14-PGJ2 induces IL-8 production in human T cells by a mitogen-activated protein kinase pathway. J. Immunol. 2002;168:1372–79. doi: 10.4049/jimmunol.168.3.1372. [DOI] [PubMed] [Google Scholar]
- 111.Meade EA, McIntyre TM, Zimmerman GA, Prescott SM. Peroxisome proliferators enhance cyclooxygenase-2 expression in epithelial cells. J. Biol. Chem. 1999;274:8328–34. doi: 10.1074/jbc.274.12.8328. [DOI] [PubMed] [Google Scholar]
- 112.Shibata T, Yamada T, Ishii T, Kumazawa S, Nakamura H, et al. Thioredoxin as a molecular target of cyclopentenone prostaglandins. J. Biol. Chem. 2003;278:26046–54. doi: 10.1074/jbc.M303690200. [DOI] [PubMed] [Google Scholar]
- 113.Haskew-Layton RE, Payappilly JB, Xu H, Bennett SA, Ratan RR. 15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) protects neurons from oxidative death via an Nrf2 astrocyte-specific mechanism independent of PPARγ. J. Neurochem. 2013;124:536–47. doi: 10.1111/jnc.12107. [DOI] [PubMed] [Google Scholar]
- 114.LoPachin RM, Barber DS, Gavin T. Molecular mechanisms of the conjugated α,β-unsaturated carbonyl derivatives: relevance to neurotoxicity and neurodegenerative diseases. Toxicol. Sci. 2008;104:235–49. doi: 10.1093/toxsci/kfm301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kumagai T, Matsukawa N, Kaneko Y, Kusumi Y, Mitsumata M, Uchida K. A lipid peroxidation–derived inflammatory mediator: identification of 4-hydroxy-2-nonenal as a potential inducer of cyclooxygenase-2 in macrophages. J. Biol. Chem. 2004;279:48389–96. doi: 10.1074/jbc.M409935200. [DOI] [PubMed] [Google Scholar]
- 116.Motter AL, Ahern GP. TRPA1 is a polyunsaturated fatty acid sensor in mammals. PLoS ONE. 2012;7:e38439. doi: 10.1371/journal.pone.0038439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sculptoreanu A, Kullmann FA, Artim DE, Bazley FA, Schopfer F, et al. Nitro-oleic acid inhibits firing and activates TRPV1- and TRPA1-mediated inward currents in dorsal root ganglion neurons from adult male rats. J. Pharmacol. Exp. Ther. 2010;333:883–95. doi: 10.1124/jpet.109.163154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Artim DE, Bazely F, Daugherty SL, Sculptoreanu A, Koronowski KB, et al. Nitro-oleic acid targets transient receptor potential (TRP) channels in capsaicin sensitive afferent nerves of rat urinary bladder. Exp. Neurol. 2011;232:90–99. doi: 10.1016/j.expneurol.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, et al. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541–45. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- 120.Taylor-Clark TE, Ghatta S, Bettner W, Undem BJ. Nitrooleic acid, an endogenous product of nitrative stress, activates nociceptive sensory nerves via the direct activation of TRPA1. Mol. Pharmacol. 2009;75:820–29. doi: 10.1124/mol.108.054445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Woodcock J, Sharfstein JM, Hamburg M. Regulatory action on rosiglitazone by the US Food and Drug Administration. N. Engl. J. Med. 2010;363:1489–91. doi: 10.1056/NEJMp1010788. [DOI] [PubMed] [Google Scholar]
- 122.Yamamoto K, Itoh T, Abe D, Shimizu M, Kanda T, et al. Identification of putative metabolites of docosahexaenoic acid as potent PPARγ agonists and antidiabetic agents. Bioorg. Med. Chem. Lett. 2005;15:517–22. doi: 10.1016/j.bmcl.2004.11.053. [DOI] [PubMed] [Google Scholar]
- 123.Stirban A, Nandrean S, Gotting C, Tamler R, Pop A, et al. Effects of n-3 fatty acids on macro-and microvascular function in subjects with type 2 diabetes mellitus. Am. J. Clin. Nutr. 2010;91:808–13. doi: 10.3945/ajcn.2009.28374. [DOI] [PubMed] [Google Scholar]
- 124.Gonzalez-Periz A, Horrillo R, Ferre N, Gronert K, Dong B, et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB J. 2009;23:1946–57. doi: 10.1096/fj.08-125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pace-Asciak CR, Mizuno K, Yamamoto S, Granstrom E, Samuelsson B. Oxygenation of arachidonic acid into 8,11,12- and 10,11,12-trihydroxyeicosatrienoic acid by rat lung. Adv. Prostaglandin Thromboxane Leukot. Res. 1983;11:133–39. [PubMed] [Google Scholar]
- 126.Wang H, Liu H, Jia Z, Guan G, Yang T. Effects of endogenous PPAR agonist nitro-oleic acid on metabolic syndrome in obese Zucker rats. PPAR Res. 2010;2010:601562. doi: 10.1155/2010/601562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu S, Jia Z, Zhou L, Liu Y, Ling H, et al. Nitro-oleic acid protects against adriamycin-induced nephropathy in mice. Am. J. Physiol. Ren. Physiol. 2013;305(11):F11533–41. doi: 10.1152/ajprenal.00656.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang H, Liu H, Jia Z, Olsen C, Litwin S, et al. Nitro-oleic acid protects against endotoxin-induced endotoxemia and multiorgan injury in mice. Am. J. Physiol. Ren. Physiol. 2010;298:F754–62. doi: 10.1152/ajprenal.00439.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu H, Jia Z, Soodvilai S, Guan G, Wang MH, et al. Nitro-oleic acid protects the mouse kidney from ischemia and reperfusion injury. Am. J. Physiol. Ren. Physiol. 2008;295:F942–49. doi: 10.1152/ajprenal.90236.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhu H, Zhang L, Amin AR, Li Y. Coordinated upregulation of a series of endogenous antioxidants and phase 2 enzymes as a novel strategy for protecting renal tubular cells from oxidative and electrophilic stress. Exp. Biol. Med. 2008;233:753–65. doi: 10.3181/0801-RM-5. [DOI] [PubMed] [Google Scholar]
- 131.Nadtochiy SM, Baker PR, Freeman BA, Brookes PS. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: implications for cardioprotection. Cardiovasc. Res. 2009;82:333–40. doi: 10.1093/cvr/cvn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nadtochiy SM, Zhu Q, Urciuoli W, Rafikov R, Black SM, Brookes PS. Nitroalkenes confer acute cardioprotection via adenine nucleotide translocase 1. J. Biol. Chem. 2012;287:3573–80. doi: 10.1074/jbc.M111.298406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kim K, Jung N, Lee K, Choi J, Kim S, et al. Dietary omega-3 polyunsaturated fatty acids attenuate hepatic ischemia/reperfusion injury in rats by modulating Toll-like receptor recruitment into lipid rafts. Clin. Nutr. 2013;32(5):855–62. doi: 10.1016/j.clnu.2012.11.026. Fish oil inhibited downstream TLR4 signaling mechanisms important for inflammation and damage following ischemia/reperfusion injury.
- 134.Giaginis C, Tsantili-Kakoulidou A, Theocharis S. Peroxisome proliferator–activated receptor-γ ligands: potential pharmacological agents for targeting the angiogenesis signaling cascade in cancer. PPAR Res. 2008;2008:431763. doi: 10.1155/2008/431763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Villacorta L, Chang L, Salvatore SR, Ichikawa T, Zhang J, et al. Electrophilic nitro-fatty acids inhibit vascular inflammation by disrupting LPS-dependent TLR4 signalling in lipid rafts. Cardiovasc. Res. 2013;98:116–24. doi: 10.1093/cvr/cvt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Coles B, Bloodsworth A, Eiserich JP, Coffey MJ, McLoughlin RM, et al. Nitrolinoleate inhibits platelet activation by attenuating calcium mobilization and inducing phosphorylation of vasodilator-stimulated phosphoprotein through elevation of cAMP. J. Biol. Chem. 2002;277:5832–40. doi: 10.1074/jbc.M105209200. [DOI] [PubMed] [Google Scholar]
- 137.Coles B, Bloodsworth A, Clark SR, Lewis MJ, Cross AR, et al. Nitrolinoleate inhibits superoxide generation, degranulation, and integrin expression by human neutrophils: novel antiinflammatory properties of nitric oxide–derived reactive species in vascular cells. Circ. Res. 2002;91:375–81. doi: 10.1161/01.res.0000032114.68919.ef. [DOI] [PubMed] [Google Scholar]
- 138.Della Bona R, Cardillo MT, Leo M, Biasillo G, Gustapane M, et al. Polymorphonuclear neutrophils and instability of the atherosclerotic plaque: a causative role? Inflamm. Res. 2013;62:537–50. doi: 10.1007/s00011-013-0617-0. [DOI] [PubMed] [Google Scholar]
- 139.Rudolph TK, Rudolph V, Edreira MM, Cole MP, Bonacci G, et al. Nitro-fatty acids reduce atherosclerosis in apolipoprotein E–deficient mice. Arterioscler. Thromb. Vasc. Biol. 2010;30:938–45. doi: 10.1161/ATVBAHA.109.201582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nadtochiy SM, Redman EK. Mediterranean diet and cardioprotection: the role of nitrite, polyun-saturated fatty acids, and polyphenols. Nutrition. 2011;27:733–44. doi: 10.1016/j.nut.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gross GJ, Hsu A, Falck JR, Nithipatikom K. Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J. Mol. Cell. Cardiol. 2007;42:687–91. doi: 10.1016/j.yjmcc.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wayman NS, Hattori Y, McDonald MC, Mota-Filipe H, Cuzzocrea S, et al. Ligands of the peroxisome proliferator–activated receptors (PPAR-γ and PPAR-α) reduce myocardial infarct size. FASEB J. 2002;16:1027–40. doi: 10.1096/fj.01-0793com. [DOI] [PubMed] [Google Scholar]
- 143.Zingarelli B, Hake PW, Mangeshkar P, O’Connor M, Burroughs TJ, et al. Diverse cardioprotective signaling mechanisms of peroxisome proliferator–activated receptor-γ ligands, 15-deoxy-Δ12,14-prostaglandin J2 and ciglitazone, in reperfusion injury: role of nuclear factor-κB, heat shock factor 1, and Akt. Shock. 2007;28:554–63. doi: 10.1097/shk.0b013e31804f56b9. [DOI] [PubMed] [Google Scholar]
- 144.Marchioli R, Barzi F, Bomba E, Chieffo C, Di Gregorio D, et al. Early protection against sudden death by n-3 polyunsaturated fatty acids after myocardial infarction: time-course analysis of the results of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI)-Prevenzione. Circulation. 2002;105:1897–903. doi: 10.1161/01.cir.0000014682.14181.f2. [DOI] [PubMed] [Google Scholar]
- 145.Guo CJ, Schopfer FJ, Gonzales L, Wang P, Freeman BA, Gow AJ. Atypical PKCζ transduces electrophilic fatty acid signaling in pulmonary epithelial cells. Nitric Oxide. 2011;25:366–72. doi: 10.1016/j.niox.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Reddy AT, Lakshmi SP, Reddy RC. The nitrated fatty acid 10-nitro-oleate diminishes severity of LPS-induced acute lung injury in mice. PPAR Res. 2012;2012:617063. doi: 10.1155/2012/617063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146a.Schermuly RT, Stasch JP, Pullamsetti SS, Middendorff R, Muller D, et al. Expression and function of soluble guanylate cyclase in pulmonary arterial hypertension. Eur. Respir. J. 2008;32:881–91. doi: 10.1183/09031936.00114407. [DOI] [PubMed] [Google Scholar]
- 147.El Kebir D, Jozsef L, Pan W, Wang L, Petasis NA, et al. 15-Epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am. J. Respir. Crit. Care Med. 2009;180:311–19. doi: 10.1164/rccm.200810-1601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Karp CL, Flick LM, Park KW, Softic S, Greer TM, et al. Defective lipoxin-mediated anti-inflammatory activity in the cystic fibrosis airway. Nat. Immunol. 2004;5:388–92. doi: 10.1038/ni1056. [DOI] [PubMed] [Google Scholar]
- 149.Karp CL, Flick LM, Yang R, Uddin J, Petasis NA. Cystic fibrosis and lipoxins. Prostaglandins Leukot. Essen. Fatty Acids. 2005;73:263–70. doi: 10.1016/j.plefa.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 150.Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, et al. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A4. Nat. Med. 2002;8:1018–23. doi: 10.1038/nm748. [DOI] [PubMed] [Google Scholar]
- 151.Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, et al. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J. Immunol. 2007;178:496–502. doi: 10.4049/jimmunol.178.1.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Farnesi-de-Assuncao TS, Alves CF, Carregaro V, de Oliveira JR, da Silva CA, et al. PPAR-γ agonists, mainly 15d-PGJ2, reduce eosinophil recruitment following allergen challenge. Cell. Immunol. 2012;273:23–29. doi: 10.1016/j.cellimm.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 153.Mochizuki M, Ishii Y, Itoh K, Iizuka T, Morishima Y, et al. Role of 15-deoxy 12,14 prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am. J. Respir. Crit. Care Med. 2005;171:1260–66. doi: 10.1164/rccm.200406-755OC. [DOI] [PubMed] [Google Scholar]
- 154.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Suzuki M, Betsuyaku T, Ito Y, Nagai K, Nasuhara Y, et al. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2008;39:673–82. doi: 10.1165/rcmb.2007-0424OC. [DOI] [PubMed] [Google Scholar]
- 156.Surh YJ, Na HK, Park JM, Lee HN, Kim W, et al. 15-DeoxyΔ12,14-prostaglandin J2, an electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem. Pharmacol. 2011;82:1335–51. doi: 10.1016/j.bcp.2011.07.100. [DOI] [PubMed] [Google Scholar]
- 157.Borniquel S, Jansson EA, Cole MP, Freeman BA, Lundberg JO. Nitrated oleic acid up-regulates PPARγ and attenuates experimental inflammatory bowel disease. Free Radic. Biol. Med. 2010;48:499–505. doi: 10.1016/j.freeradbiomed.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Knopfova L, Smarda J. The use of Cox-2 and PPARγ signaling in anti-cancer therapies. Exp. Ther. Med. 2010;1:257–64. doi: 10.3892/etm_00000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
RELATED RESOURCES
- 1.Fritz KS, Petersen DR. An overview of the chemistry and biology of reactive aldehydes. Free Radic. Biol. Med. 2013;59:85–91. doi: 10.1016/j.freeradbiomed.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishida M, Sawa T, Kitajima N, Ono K, Inoue H, et al. Hydrogen sulfide anion regulates redox signaling via electrophile sulfhydration. Nat. Chem. Biol. 2012;8:714–24. doi: 10.1038/nchembio.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]