

Abstract
The copper (Cu) exporter ATP7B mediates cellular resistance to cisplatin (cDDP) by increasing drug efflux. ATP7B binds and sequesters cDDP in into secretory vesicles. Upon cDDP exposure ATP7B traffics from the trans-Golgi network (TGN) to the periphery of the cell in a manner that requires the cysteine residues in its metal binding domains (MBD). To elucidate the role of the various domains of ATP7B in its cDDP-induced trafficking we expressed a series of mCherry-tagged variants of ATP7B in HEK293T cells and analyzed their subcellular localization in basal media and after a 1 h exposure to 30 μM cDDP. The wild type ATP7B and a variant in which the cysteines in the CXXC motifs of MBD 1-5 were converted to serines trafficked out of the trans-Golgi (TGN) when exposed to cDDP. Conversion of the cysteines in all 6 of the CXXC motifs to serines, or in only the sixth MBD, rendered ATP7B incapable of trafficking on exposure to cDDP. Truncation of MBD1-5 or MBD1-6 resulted in the loss of TGN localization. Addition of the first 63 amino acids of ATP7B to these variants restored TGN localization to a great extent and enabled the MBD1-5 variant to undergo cDDP-induced trafficking. A variant of ATP7B in which the aspartate 1027 residue in the phosphorylation domain was converted to glutamine localized to the TGN but was incapable of cDDP-induced trafficking. These results demonstrate that the CXXC motif in the sixth MBD and the catalytic activity of ATP7B are required for cDDP-induced trafficking as they are for Cu-induced redistribution of ATP7B; this provides further evidence that cDDP mimics Cu with respect to the molecular mechanisms by they control the subcellular distribution of ATP7B.
Introduction
Copper (Cu) transporters CTR1, ATP7A, ATP7B and the metallochaperone ATOX1 are known to regulate the cellular pharmacology of Pt containing drugs by modulating their uptake, efflux and subcellular distribution.1-7 The P1B-type ATPases, ATP7A (Menkes disease protein) and ATP7B (Wilsons disease protein) alter the cellular pharmacology of the Pt drugs by mediating their vesicular sequestration and eventual efflux through the secretory compartment.5-7 The mechanism by which ATP7A and ATP7B regulate the sequestration and efflux of the Pt-containing drugs was proposed to be similar to that for Cu but recent data have demonstrated that substantial differences exists.8 ATP7A and ATP7B reside in the trans-Golgi network (TGN). ATP7B was shown to co-localize with TGN markers TGN38,9 p23010 and syntaxin 6.11 ATP7B has also been reported to localize in late endosomes.12 The six N-terminal metal binding domains (MBDs) of ATP7B play an essential role in the transport of Cu. They receive Cu1+ from ATOX1 via the core CXXC motif found in each MBD and the sixth MDB may propel it to the metal binding residues within the channel for eventual ATP-dependent passage across the vesicle membrane.13 Each of the 6 MBDs in both ATP7A and ATP7B has a ferredoxin βαββαβ fold similar to that found in ATOX1.14,15 The sixth MBD of ATP7A and ATP7B, which lies adjacent to the trans-membrane pore, was shown to be essential for the transport of Cu.16-19 The CXXC motif in the MBDs of ATOX1 and ATP7B have been shown to interact with cDDP which can cause structural denaturation and multimerization of the MBDs at high concentrations.8,20,21
Previous work from this laboratory showed that the CXXC motifs within the MBDs are important to the capacity of ATP7B to traffic from the TGN to peripheral vesicles in response to cDDP as well as to its ability to mediate cDDP resistance.8 Conversion of the CXXC motif of each MDB to SXXS did not alter subcellular localization of ATP7B but did prevent it from mediating cDDP resistance. Deletion of either the first 5 or all 6 of the MBDs both prevented TGN localization and disabled its ability to mediate resistance. This data is in agreement with the previous finding that the first 63 amino acids of ATP7B contain residues that mediate the retention of ATP7B in the TGN compartment.22
Very little is known about the role of other domains of ATP7B in with respect to its ability to regulate the cellular pharmacology of the Pt-containing drugs. We have reported that the CPC motif in the sixth transmembrane segment is required for ATP-dependent transport of cDDP.6 In addition, a role for the phosphorylation domain of ATP7B in the transport of cDDP was shown by demonstrating that cDDP induced the phosphorylation of ATP7B which is known to occur on aspartate 1027.23,24 In the current study we examined the effect of making mutations in the MBDs, the TGN localization domain and the phosphorylation domain of ATP7B on its ability to undergo cDDP-induced trafficking in HEK293T cells. We show that cDDP induced trafficking of ATP7B from the TGN and that both the CXXC motif of the sixth MBD and the catalytic activity of ATP7B are essential to this effect. We confirm that a TGN localization signal resides in the first 63 amino acids of this transporter, and that the interaction of cDDP with ATP7B is transient and non-denaturing based on the ability of the trafficked protein to return to the TGN after cDDP is removed.
Experimental
Reagents
The lentiviral vector, pLVX-mCherry-C1, In-Fusion cloning kit, packaging kit and anti-mCherry antibodies were from Clontech, Palo Alto, CA. Restriction enzymes were from New England Biolabs, Ipswich, MA. Secondary antibodies, fluorescent reagents, Superscript III, PCR kits, Genetailor mutagenesis kit, PCR2.1-TOPO vector, media and sera were from Invitrogen, Carlsbad, CA. P230 polyclonal antibody to mCherry was from BioVision Inc. Milpitas, California, monoclonal antibody to p230 was from BD Biosciences, San Diego, CA, polyclonal antibody to syntaxin 6 was from Cell Signaling Technology, Inc. Danvers MA, monoclonal antibody to actin was from Santa Cruz Biotechnology, Santa Cruz CA. Cisplatin was from Teva Parental Inc., Irvine, CA. Complete protease inhibitor was from Roche, Nutley NJ. Electrophoresis gels and Bradford reagent were from Bio Rad, Richmond, CA. Other chemicals were from Sigma, St. Louis, MO.
ATP7B variants
The wild type ATP7B (mC-WT), mC-Δ1-5 (in which MBDs 1-5 spanning amino acids 1-539 were truncated), mC-Δ1-6 (in which MBDs 1-6 spanning amino acids 1-599 were truncated) constructs were generated from a 2008 cervical carcinoma cell cDNA library and the construction of mC-MBD1-6C/S in which cysteines of all 6 CXXC motifs were converted to serines were described previously.8 mC-LΔ1-5 and mC-LΔ1-6 were constructed by PCR amplification of the first 63 amino acids of ATP7B (1-63) and inserting them into the Xho1 and BamH1 site of pLVX-mCherry upstream of ATP7B-Δ1-5 and ATP7B-Δ1-6 using infusion procedure. Forward and reverse primers used for PCR amplification of the first 63 amino acids were respectively, GGACTCAGATCTCGAGTTATGCCTGAGCAGGAGAGACAG and TAGATCCGGTGGATCCACA TGA CTG GCA AGT CAT GCC. Variants mC-D1027E, mC-MBD1-5C/S and mC-MBD6C/S (plasmids pCMB414, pCMB399, pCMB514) were generously provided by Dr S. La Fontaine and Dr J. F. Mercer (University of Melbourne, Melbourne Australia).18 These constructs were amplified with PCR using forward primer GGA CTC AGA TCT CGA GTT ATGCCTGAGCAGGAGAGACAG and reverse primer TAGATCCGGTGGATCCTCAGATGTACTGCTCCTCATC and subcloned in the pLVX-mCherry-C1 vector using the In-Fusion cloning kit.
Cell culture and expression of lentiviral constructs of ATP7B
HEK293T cells were cultured in DMEM with 4 mM l-glutamine, 4.5 g L −1 glucose, 1 mM sodium pyruvate, 1.5 g L −1 sodium bicarbonate and 10% fetal bovine serum. 2008 cells were obtained from Dr Philip DeSaia25 and cultured in RPMI and 10% fetal bovine serum. Cells were incubated at 37 °C, 5% CO2. Lentiviral stocks of ATP7B variants were produced in HEK293T cells and used to transduce fresh cultures of the HEK293T cells.26 Selection was made with 10 μg mL −1 puromycin. A pool of cells expressing high levels of the fluorescent mCherry tag was obtained by three rounds of FACS sorting.
Assay of cytotoxicity
Sensitivity to cDDP was determined using the Cell Count Kit 8 (CCK-8) colorimetric assay (Dojindo, Gaithersburg, MD) as previously reported.8
Fluorescence microscopy
Cells were cultured on coverslips coated with 5 μg mL −1 gelatin. They were allowed to attach overnight and then they were exposed for 15 min to either 30 μM cDDP at 37 °C or medium alone. Cells were quickly rinsed with cold PBS and fixed with 3.7% formaldehyde in PBS for 5 min, permeabilized with 0.3% Triton × 100 in PBS for another 5 min, blocked with 5% BSA in PBS for 1 h, incubated successively with primary and secondary antibodies for 1 h each, for 30 min with Hoechst 33342 and FITC-tagged phalloidin, washed for 15 min three more times and then mounted on slides with Gelvatol. Imaging was performed using a DeltaVision deconvolution microscope system (Applied Precision, Inc., Issaquah, WA.) at the UCSD Cancer Center’s Digital Imaging Shared Resource Facility.
Western blotting
Fifty μg protein was analyzed on 4–15% polyacrylamide gels, blotted onto PVDP paper and reacted with primary and secondary antibodies as described previously.8
Statistical analysis
All two-group comparisons utilized Student’s t-test with the assumption of unequal variance. Data are presented as mean ± SEM.
Results
Construction and expression of mCherry-tagged ATP7B variants in HEK293T cells
Fig. 1 presents a schematic drawing of the various variant forms of ATP7B that were used in this study. These variant forms of mCherry-tagged ATP7B, and the empty pLVX-mCherry-C1 vector, were stably expressed in HEK293T cells; successfully infected cell expressing the ATP7B variants were recovered by FACS. These included: mC-WT (mCherry tagged wild type ATP7B); mC-MBD1-6C/S (cysteines in all 6 metal binding domains converted to serines); mC-MBD1-5C/S (cysteines in MBDs 1-5 converted to serines); mC-MBD6C/S (cysteines in just the sixth MBD converted to serines); and mC-D1027E (aspartate 1027 in the phosphorylation domain converted to glutamate). Fig. 2 shows a Western blot of lysates from HEK-293T cells expressing these variants. Endogenous ATP7B was detected at 150 kDa. The mCherry produced by the empty vector was detected at 30 kDa (not shown), and the full length wild type mC-ATP7B was detected at ~180 kDa as were the variants without truncation. As shown in Fig. 2, the mC-ATP7B variants mC-Δ1-6, mC-Δ1-5, mC-LΔ1-6 and mC-LΔ1-5 were each detected at their expected sizes.
Fig. 1.

Schematic presentation of ATP7B variants used in this study. mC, mCherry; L, sequence spanning amino acids 1-63; CXXC, metal binding motifs in MBDs; SXXS, metal binding cysteines converted to serines; mC-WT, mCherry tagged wild type ATP7B; mC-MBD1-6C/S, CXXC in MBD1-6 converted to SXXS; mC-MBD1-5C/S, n CXXC MBD1-5 converted to SXXS; mC-MBD6C/S, CXXC in only MBD6 converted to SXXS; mC-Δ1-5, MBD1-5 truncated; mC-LΔ1-5, the first 63 amino acids of ATP7B added back to mC-Δ1-5; mC-Δ1-6, MBDs 1-6 truncated; mC-LΔ1-6, the first 63 amino acids of ATP7B added back to mC-Δ1-6; mC-D1027E, aspartate 1027 in the phosphorylation domain converted to glutamate.
Fig. 2.
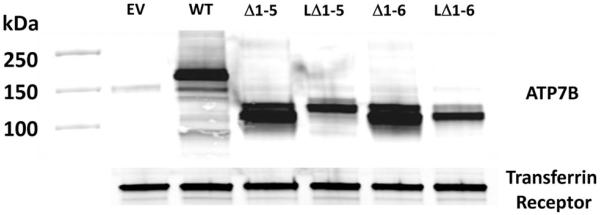
Western blot analysis of HEK293T lysates expressing the mCherry-tagged ATP7B variants. The blot was probed with monoclonal antibodies against ATP7B and the transferrin receptor.
Subcellular localization of mCherry-tagged ATP7B variants in HEK293T cells
To ensure that the mCherry tag did not affect the function of ATP7B by altering its localization, the HEK293-mC cells that expressed only the mCherry tag were analyzed by fluorescence microscopy. As seen in Fig. 3, the mCherry peptide was dispersed uniformly throughout the cell body and nucleus and did not relocate after exposure of cells to 30 μM cDDP for 1 h. In contrast, all the full length ATP7B variants showed clear TGN localization as evidenced by high degrees of co-localization with TGN markers syntaxin 6 and p230 (Fig. 4A-C). Co-localization of ATP7B variants with syntaxin 6 was found only in the juxta-nuclear regions; the syntaxin 6 positive vesicles in the periphery were devoid of ATP7B. Thus, under basal conditions all of the full length ATP7B variants localized to TGN regardless of their metal binding or catalytic activities.
Fig. 3.

Subcellular distribution of mCherry in untreated and cisplatin treated HEK293T cells. Nuclei were stained with Hoechst 33342 (blue) and a FITC-tagged phalloidin (green) was used to stain actin; mCherry is red.
Fig. 4.

Subcellular localization of mCherry tagged variants of ATP7B in HEK293T cells in the absence of exposure to cDDP. (A) Subcellular localization of wild type mC-WT and variants mC-MBD1-6C/S, mC-MBD1-5C/S and mC-MBD6C/S. (B) Subcellular localization of variants mC-Δ1-5, mC-LΔ1-5, mC-Δ1-6 and mC-LΔ1-6. (C) Subcellular localization of the phosphorylation mutant mC-D1027E. mCherry appears as red. A rabbit monoclonal antibody to syntaxin 6 and a mouse monoclonal antibody to P230 were used as TGN markers; secondary antibodies were conjugated with AlexaFluor 488 and AlexaFluor 647 for respective detection of syntaxin 6 and p230 (both are shown in green); nuclei were stained with Hoechst 33342 (blue).
ATP7B variants mC-Δ1-6 and mC-Δ1-5 that lacked all or part of the N-terminal domain, localized mainly in the periphery of the cells and in these regions they showed some degree of co-localization with the two TGN markers. This suggested that these two variants were defective in trafficking from the cell periphery back to the TGN. Addition of the first 63 amino acids of ATP7B to these variants restored TGN localization to some extent as previously shown.22 The mC-LΔ1-5 and mC-LΔ1-6 variants localized mainly in internal vesicles and had a higher degree of co-localization with at syntaxin 6 and p230 TGN (Fig. 4B). The mC-D1027E variant localized to the TGN as described above.
Role of metal binding domains of ATP7B in cDDP-induced trafficking
To determine the role of metal binding and phosphorylation domains, we used variants in which the cysteine residues in MBDs 1-6, MBDs 1-5 and MBD6 only were converted to serines, or in which aspartate 1027 in the phosphorylation domain was converted to glutamate (D1027E). HEK293T cells expressing these and the wild type (mC-WT) variants of ATP7B were exposed for 1 h to 30 μM cDDP and then fixed and processed for microscopy. Fig. 5A shows that, whereas mC-WT had TGN localization under basal conditions, it became dispersed throughout the cell following a 1 h exposure to cDDP. The data is quantified in Fig. 5B which shows that 89.7 ± 4.5 (SD)% of the HEK293T cells had ATP7B localized in the TGN under basal conditions, but only 18.5 ± 5.7 (SD)% of cells showed TGN localization following cDDP exposure (p = 0.00001). A similar analysis was carried out in the 2008 cells and this showed that 89.0 ± 5.6 (SD)% of cells had TGN localization in basal medium but only 9.0 ± 4.1 (SD)% after a 1 h cDDP treatment (p = 0.000007). The histograms presented in Fig. 5B also document the reversibility of the cDDP effect. There was a rapid increase in the fraction of cells with TGN localization over the first 15 min and then a slower increase that reached 60–70% by 6 h. Conversion of the CXXC to SXXS in all 6 MBDs in mC-MBD1-6C/S rendered the ATP7B incapable of trafficking following cDDP exposure as we previously reported.8 This indicated that binding of cDDP to one or more of the MBD CXXC motifs is essential for triggering the trafficking of ATP7B (Fig. 6A).
Fig. 5.

Return of mC-WT to the TGN following termination of cDDP exposure in HEK293T cells. (A) Top, mC-WT distribution in the absence of cDDP exposure; middle, mC-WT distribution after 1 h treatment with 30 μM cDDP; bottom, mC-WT distribution 3 h after removal of cDDP. mC-WT is red, nuclei are blue and actin is green. (B) Top panel, percent of HEK293T cells in which mC-WT was localized to TGN as a function of time following cDDP removal; bottom panel, percent of 2008 cells in which mC-WT was localized to TGN as a function of time following cDDP removal. * p = 0.00001; ** p = 0.000007.
Fig. 6.

Effect of exposure to 30 μM cDDP for 1 h on the distribution of mCherry tagged ATP7B variants in HEK293T cells. (A) mC-MBD1-6C/S; (B) mC-MBD1-5C/S; (C) mC-mBD6C/S; (D) mC-D1027E. Nuclei were stained with Hoechst 33342 (blue), and a FITC-tagged phalloidin (green) was used to stain actin; mCherry is red.
The role of the sixth MBD was examined by studying HEK293T cells expressing the mCMBD1-5C/S and mC-MBD6C/S variants. Fig. 6B shows that conversion of the cysteines in MBDs1-5 to serines while keeping MBD6 intact resulted in normal trafficking of ATP7B following exposure to 30 μM cDDP for 1 h. This indicates that the CXXC motif in MBD6 is sufficient to trigger trafficking in response to cDDP and that the CXXC motifs in the other MBDs are not essential. This was confirmed by the fact that the mC-MBD6C/S variant, in which the CXXC motif in only the sixth MBD was modified, did not traffic upon exposure to cDDP (Fig. 6C).
Role of phosphorylation domain of ATP7B in cDDP-induced trafficking
To determine whether catalytic activity of ATP7B is required for cDDP-induced trafficking we expressed the D1027E variant in HEK293T cells. In this variant the aspartate 1027, which becomes acylphosphoryated in the presence of Cu and cDDP, was replaced by glutamic acid which disables the Cu transport function of ATP7B. This variant localized to the TGN under basal conditions but failed to traffic after 1 h exposure to 30 μM cDDP (Fig. 6D). Thus, in addition to the CXXC motif in MBD6, the catalytic activity of the protein is also important for its ability traffic in response to cDDP.
The role of first 63 amino acids of ATP7B in cDDP-induced trafficking
As described above and shown previously,8 the mC-Δ1-5 variant that lacks MBD1-5, and mC-Δ1-6 that lacks all 6 MBDs, were dispersed throughout the cell even in the absence or presence of cDDP (Fig. 7A and B). This reconfirmed our previous observation that cDDP had no detectable effect on the distribution of these variants in HEK293 cells.8 In addition to missing either the first 5 or all 6 of the MDB, these variants are also missing the 63 amino acids N-terminal to the first MBD. Interestingly, the mC-Δ1-6 variant seemed to localize mainly at the surface of the cell where it co-localized with plasma membrane actin (Fig. 7B). The first 63 amino acids of ATP7B were added back to these proteins producing the mC-LΔ1-5 and mC-LΔ1-6 variants. In both cases this markedly altered their localization causing them to reside in the central part of the cell and in the TGN. Exposure of HEK293T cells expressing the mC-LΔ1-5 variant to cDDP caused the dispersal of this variant throughout the cell (Fig. 7A); cDDP-failed to induce equivalent trafficking of the mC-LΔ1-6 variant. These studies not only establish a role for the first 63 amino acids of ATP7B in TGN localization but also confirm the importance of the sixth MBD as a trigger for trafficking in response to cDDP.
Fig. 7.

Effect of restoring the first 63 amino acids of ATP7B to variants in which either the first 5 or all 6 MBD were removed by truncation. (A) Distribution of mC-Δ1-5 (top two panels) and mC-LΔ1-5 (bottom two panels) variants in HEK293T cells in basal medium and following incubation for 1 h with 30 μM cDDP. (B) Distribution of mC-Δ1-6 (top two panels) and mC-LΔ1-6 (bottom two panels) variants in HEK293T cells in basal medium and following incubation for 1 h with 30 μM cDDP. Nuclei were stained with Hoechst 33342 (blue), and a FITC-tagged phalloidin (green) was used to stain actin; mCherry is red.
Reversibility of the effects of cisplatin on localization of mCherry-ATP7B
Previous in vitro data showing that ATP7B becomes multimerized in the presence of cDDP suggested the possibility that cDDP binding to the CXXC motifs might irreversibly denature the protein. To determine whether the effect of cDDP on ATP7B is reversible, we asked whether the cDDP-induced redistribution of wild type ATP7B was reversible. HEK293T and cervical carcinoma 2008 cells expressing mC-WT ATP7B were exposed to 30 μM cDDP for 1 h and then allowed to recover for up to 6 h in growth medium that contained 10 μg mL −1 cycloheximide to block new protein synthesis. In both cells recovery of the dispersed protein toward the TGN was detected after 15 min at which time the mC-WT ATP7B was in the TGN region in 40.4 ± 13.3% of HEK293/mC-WT cells and 37.4 ± 9.7% of 2008/mC-WT cells. At the end of 6 h, mC-WT ATP7B was localized to the TGN in 71.2 ± 11.3% of HEK293 and 65.9 ± 13% of 2008 cells. Thus, in both these cell types ATP7B that had trafficked out of the TGN in response to cDDP returned to the TGN once the drug was removed.
Discussion
Expression of mCherry-tagged ATP7B and its variants in HEK293T cells provided an excellent model system for documentation of the ability of cDDP to trigger relocalization of the protein and identification of domains essential to this trafficking. The fact that mCherry itself distributed randomly throughout the cell but that the mCherry tagged wild type ATP7B was localized to the region of the TGN indicates that mCherry is a suitable tag for these types of studies. We have previously studied the effect of making changes to the domain structure of ATP7B in human cervical carcinoma 2008 cells.8 One of the advantages of the 2008 cells is that they do not express organic cation transporters which are known to transport cDDP (reviewed in ref. 27), and therefore they are useful for studies of the effect of ATP7B on cDDP transport and cytotoxicity. However, the 2008 cells are so small that they are suboptimal for studies of trafficking, and a major advantage of the HEK293T cells is that they are large and flat making changes in distribution easier to quantify. A disadvantage of the HEK293T cells is that, being kidney cells, they do expression the organic cation transports which renders them questionable for studies of ATP7B-dependent changes in transport or cDDP sensitivity.
The wild type ATP7B and all the variants that retained the first 63 amino acids N-terminal to the first MBD were localized to the TGN compartment of the HEK293T cells in the absence of cDDP exposure. Thus, neither conversion of the CXXC motifs to SXXS in either the first 5 or all 6 MBDs, nor disabling the enzymatic activity by changing aspartate 1027 to glutamic acid, affected TGN localization. This indicates that the information for TGN localization is stored in sequences that are independent of MBD binding or catalytic activities. Truncating the protein by removing the first 63 amino acids N-terminal to the MBD, and either the first 5 or all 6 MBD, resulted in failure of TGN localization indicating that the TGN localization signal sequence resides somewhere N-terminal to the C-terminal end of MBD6. This was confirmed by showing that adding back just the first 63 amino acids to the mC-Δ1-5 and mC-Δ1-6 variants without adding back the missing MBD restored their TGN localization establishing that the localization signal lies somewhere in the first 63 amino acids and that it functions even in the absence of all 6 MBD consistent previous data.22
The importance of the CXXC motifs in the MBDs to the ability of cDDP to induce trafficking away from the TGN was explored by converting these to SXXS in either just the first 5 MBD or all 6 MBD. It turned out that only the MBD6 had to be intact for cDDP to trigger relocalization. MBD6 was also sufficient to mediate cDDP-induced relocalization of the variant in which the first 5 MBD had been completely removed but the first 63 amino acids added back to restore residence in the TGN. This result obtained with cDDP mirrors results previously reported for Cu-induced trafficking from the TGN.28
The invariant aspartate (Asp1027) in the phosphorylation domain (DKTGTIT) is essential for Cu-transporting catalytic activity of ATP7B.18 Mutation of this residue to glutamate did not affect the TGN localization of this variant under basal conditions as the mC-D1027E variant was detected in the perinuclear region along with the TGN markers syntaxin 6 and p230. This variant has previously been shown to be incapable of Cu-induced trafficking29,30 indicating that that the ability to transport Cu is essential to its ability to trigger trafficking.18,31 The fact that the D1027E variant failed to traffic upon exposure to cDDP indicates that the same is true for cDDP. It is likely that a specific conformation associated with the acyl-phosphate intermediate exposes a trafficking signal or obscures a TGN retention sequence. Vanderwerf et al.32 reported that there is a correlation between kinase-dependent phosphorylation of ATP7B and its trafficking to cytosolic vesicles, and that hyper-phosphorylation of ATP7B is associated with increased vesicle trafficking.
The high-affinity Cu influx transporter CTR1 mediates the accumulation of cDDP in cells that do not express organic cation transporters, and we have previously proposed that cDDP may be distributed throughout the cell through binding to Cu chaperones and transporters.33 Essential to such a concept is that the interaction of cDDP with the Cu binding protein must be transient, and that it not involve the loss of the ammine ligands of cDDP since these are clearly still present in the adducts that cDDP forms with DNA. Using recombinant MBS6 produced in bacteria we showed that cDDP can cause denaturing aggregation that is dependent on the CXXC motif being intact,8 and others have shown that cDDP forms at least one type of complex with the Cu chaperone ATOX1 in which the ammine ligands are lost,34 both of which might argue against the ability of cDDP to transiently interact with Cu binding proteins and remain intact in the process. However, the finding that the cDDP-induced trafficking of ATP7B is reversible, and that in the absence of new ATP7B synthesis the protein that was induced to leave the TGN returns to it within a relatively short period of time, suggests that short-lived and non-denaturing interactions between cDDP and Cu binding proteins do occur in the whole cell. Indeed, some of the complexes that cDDP forms with ATOX1 in vitro retain the ammine ligands suggesting the possibility that ATOX1 may be able to donate cDDP to another Cu binding protein.21,34
Conclusions
The results of this study indicate that cDDP and Cu behave in a similar manner with respect to induction of the trafficking of ATP7B which is consistent with the concept that both are effluxed by this transporter through the same molecular mechanism. While cDDP may bind to the CXXC motifs in the first 5 MBDs of ATP7B, these interactions are not essential to the ability of cDDP to trigger trafficking of ATP7B from the TGN. However, the CXXC motif in the sixth MBD is essential suggesting that, as for Cu, the binding of cDDP at this site produces a change in the structure of the protein that facilitates its loading into vesicles destined for export from the TGN. Like Cu, cDDP triggers acyl phosphorylation of aspartate 1027 and this was found to be essential to the ability of cDDP to stimulate relocalization of ATP7B. Since acyl phosphorylation is also required for ATP7B to pump Cu into vesicles in the secretory pathway, this indicates that only transport-proficient forms of ATP7B can exit the TGN in response to cDDP. Finally, we confirm that a TGN localization signal resides in the first 63 amino acids as has been noted before.22
Acknowledgements
The authors thank Drs Sharon La Fontaine and Julian F. Mercer for providing vectors and advice. This work was supported by the National Institutes of Health CA095298. Additional support for core laboratories was from grants P30 NS047101 for the UCSD Neuroscience Microscopy Shared Facility and the UCSD Cancer Center Specialized Support Grant P30 CA23100.
Abbreviations
- BME
β-Mercaptoethanol
- DTT
Dithiothreitol
- MDB
Metal binding domain
Nomenclature
- ATP7B
ATPase, Cu++ transporting, beta polypeptide; Gene-Bank ID: ENSG00000123191; PDB ID: P35670; EC 3.6
Footnotes
Disclosure
The authors report no conflicts of interest.
References
- 1.Larson CA, Blair BG, Safaei R, Howell SB. Mol. Pharmacol. 2009;75:324–330. doi: 10.1124/mol.108.052381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holzer AK, Samimi G, Katano K, Naerdemann W, Lin X, Safaei R, Howell SB. Mol. Pharmacol. 2004;66:817–823. doi: 10.1124/mol.104.001198. [DOI] [PubMed] [Google Scholar]
- 3.Holzer AK, Manorek GH, Howell SB. Mol. Pharmacol. 2006;70:1390–1394. doi: 10.1124/mol.106.022624. [DOI] [PubMed] [Google Scholar]
- 4.Song I, Savaraj N, Siddik Z, Liu P, Wei Y, Wu C, Kuo M. Mol. Cancer Ther. 2004;3:1543–1549. [PubMed] [Google Scholar]
- 5.Samimi G, Katano K, Holzer AK, Safaei R, Howell SB. Mol. Pharmacol. 2004;66:25–32. doi: 10.1124/mol.66.1.25. [DOI] [PubMed] [Google Scholar]
- 6.Safaei R, Larson BJ, Otani S, Rasmussen ML, Howell SB. Mol. Pharmacol. 2008;73:461–468. doi: 10.1124/mol.107.040980. [DOI] [PubMed] [Google Scholar]
- 7.Katano K, Safaei R, Samimi G, Holzer A, Rochdi M, Howell SB. Mol. Pharmacol. 2003;64:466–473. doi: 10.1124/mol.64.2.466. [DOI] [PubMed] [Google Scholar]
- 8.Safaei R, Adams PL, Maktabi MH, Mathews RA, Howell SB. J. Inorg. Biochem. 2012;110:8–17. doi: 10.1016/j.jinorgbio.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta A, Bhattacharjee A, Dmitriev OY, Nokhrin S, Braiterman L, Hubbard AL, Lutsenko S. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5390–5395. doi: 10.1073/pnas.1014959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van den Berghe PV, Stapelbroek JM, Krieger E, de Bie P, van de Graaf SF, de Groot RE, van Beurden E, Spijker E, Houwen RH, Berger R, Klomp LW. Hepatology. 2009;50:1783–1795. doi: 10.1002/hep.23209. [DOI] [PubMed] [Google Scholar]
- 11.Guo Y, Nyasae L, Braiterman LT, Hubbard AL. Am. J. Physiol.: Gastrointest. Liver Physiol. 2005;289:G904–G916. doi: 10.1152/ajpgi.00262.2005. [DOI] [PubMed] [Google Scholar]
- 12.Yanagimoto C, Harada M, Kumemura H, Abe M, Koga H, Sakata M, Kawaguchi T, Terada K, Hanada S, Taniguchi E, Ninomiya H, Ueno T, Sugiyama T, Sata M. Hepatol. Res. 2011;41:484–491. doi: 10.1111/j.1872-034X.2011.00788.x. [DOI] [PubMed] [Google Scholar]
- 13.Solioz M, Vulpe C. Trends Biochem. Sci. 1996;21:237–241. [PubMed] [Google Scholar]
- 14.Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, Hao L, He S, Hurwitz DI, Jackson JD, Ke Z, Krylov D, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Thanki N, Yamashita RA, Yin JJ, Zhang D, Bryant SH. Nucleic Acids Res. 2007;35:D237–D240. doi: 10.1093/nar/gkl951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnesano F, Banci L, Bertini I, Ciofi-Baffoni S, Molteni E, Huffman DL, O’Halloran TV. Genome Res. 2002;12:255–271. doi: 10.1101/gr.196802. [DOI] [PubMed] [Google Scholar]
- 16.Achila D, Banci L, Bertini I, Bunce J, Ciofi-Baffoni S, Huffman DL. Proc. Natl. Acad. Sci. U. S. A. 2006;103:5729–5734. doi: 10.1073/pnas.0504472103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forbes JR, Hsi G, Cox DW. J. Biol. Chem. 1999;274:12408–12413. doi: 10.1074/jbc.274.18.12408. [DOI] [PubMed] [Google Scholar]
- 18.Cater MA, Forbes J, La Fontaine S, Cox D, Mercer JF. Biochem. J. 2004;380:805–813. doi: 10.1042/BJ20031804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iida M, Terada K, Sambongi Y, Wakabayashi T, Miura N, Koyama K, Futai M, Sugiyama T. FEBS Lett. 1998;428:281–285. doi: 10.1016/s0014-5793(98)00546-8. [DOI] [PubMed] [Google Scholar]
- 20.Arnesano F, Banci L, Bertini I, Felli IC, Losacco M, Natile G. J. Am. Chem. Soc. 2011;133:18361–18369. doi: 10.1021/ja207346p. [DOI] [PubMed] [Google Scholar]
- 21.Palm ME, Weise CF, Lundin C, Wingsle G, Nygren Y, Bjorn E, Naredi P, Wolf-Watz M, Wittung-Stafshede P. Proc. Natl. Acad. Sci. U. S. A. 2011;108:6951–6956. doi: 10.1073/pnas.1012899108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braiterman L, Nyasae L, Guo Y, Bustos R, Lutsenko S, Hubbard A. Am. J. Physiol.: Gastrointest. Liver Physiol. 2009;296:G433–G444. doi: 10.1152/ajpgi.90489.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huster D, Lutsenko S. J. Biol. Chem. 2003;278:32212–32218. doi: 10.1074/jbc.M305408200. [DOI] [PubMed] [Google Scholar]
- 24.Leonhardt K, Gebhardt R, Mossner J, Lutsenko S, Huster D. J. Biol. Chem. 2009;12:7793–7802. doi: 10.1074/jbc.M805145200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Disaia PJ, Sinkovics JG, Rutledge FN, Smith JP. Am. J. Obstet. Gynecol. 1972;114:979–989. doi: 10.1016/0002-9378(72)90109-3. [DOI] [PubMed] [Google Scholar]
- 26.Larson CA, Adams PL, Blair BG, Safaei R, Howell S. Mol. Pharmacol. 2010;78:333–339. doi: 10.1124/mol.110.064766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burger H, Loos WJ, Eechoute K, Verweij J, Mathijssen RH, Wiemer EA. Drug Resist. Updates. 2011;14:22–34. doi: 10.1016/j.drup.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Payne AS, Gitlin JD. J. Biol. Chem. 1998;273:3765–3770. doi: 10.1074/jbc.273.6.3765. [DOI] [PubMed] [Google Scholar]
- 29.Terada K, Aiba N, Yang XL, Iida M, Nakai M, Miura N, Sugiyama T. FEBS Lett. 1999;448:53–56. doi: 10.1016/s0014-5793(99)00319-1. [DOI] [PubMed] [Google Scholar]
- 30.La Fontaine S, Theophilos MB, Firth SD, Gould R, Parton RG, Mercer JF. Hum. Mol. Genet. 2001;10:361–370. doi: 10.1093/hmg/10.4.361. [DOI] [PubMed] [Google Scholar]
- 31.Mercer JFB, Camakaris J. Met. Ions Gene Regul. 1997:250–276. [Google Scholar]
- 32.Vanderwerf SM, Cooper MJ, Stetsenko IV, Lutsenko S. J. Biol. Chem. 2001;276:36289–36294. doi: 10.1074/jbc.M102055200. [DOI] [PubMed] [Google Scholar]
- 33.Howell SB, Safaei R, Larson CA, Sailor MJ. Mol. Pharmacol. 2010;77:887–894. doi: 10.1124/mol.109.063172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boal AK, Rosenzweig AC. J. Am. Chem. Soc. 2009;131:14196–14197. doi: 10.1021/ja906363t. [DOI] [PMC free article] [PubMed] [Google Scholar]