

Abstract
Osteogenesis imperfecta (OI), or brittle bone disease, is most often caused by dominant mutations in the collagen I genes COL1A1/COL1A2, whereas rarer recessive OI is often caused by mutations in genes encoding collagen I-interacting proteins. Recently, mutations in the gene for the proteinase bone morphogenetic 1 (BMP1) were reported in two recessive OI families. BMP1 and the closely related proteinase mammalian tolloid-like 1 (mTLL1) are co-expressed in various tissues, including bone, and have overlapping activities that include biosynthetic processing of procollagen precursors into mature collagen monomers. However, early lethality of Bmp1- and Tll1-null mice has precluded use of such models for careful study of in vivo roles of their protein products. Here we employ novel mouse strains with floxed Bmp1 and Tll1 alleles to induce postnatal, simultaneous ablation of the two genes, thus avoiding barriers of Bmp1−/− and Tll1−/− lethality and issues of functional redundancy. Bones of the conditionally null mice are dramatically weakened and brittle, with spontaneous fractures—defining features of OI. Additional skeletal features include osteomalacia, thinned/porous cortical bone, reduced processing of procollagen and dentin matrix protein 1, remarkably high bone turnover and defective osteocyte maturation that is accompanied by decreased expression of the osteocyte marker and Wnt-signaling inhibitor sclerostin, and by marked induction of canonical Wnt signaling. The novel animal model presented here provides new opportunities for in-depth analyses of in vivo roles of BMP1-like proteinases in bone and other tissues, and for their roles, and for possible therapeutic interventions, in OI.
INTRODUCTION
Osteogenesis imperfecta (OI) is a heritable connective tissue disorder characterized by deficits in bone material properties that result in bone fragility, and which can also result in low bone mass, bone deformities and impaired growth (1–3). More than 90% of OI cases are due to autosomal dominant mutations in COL1A1 and COL1A2, the genes encoding the two chains of collagen I. Collagen I is by far the most abundant protein of bone, in which it is of major structural importance and serves as the template upon which mineral is deposited. Recently, recessive OI mutations have been reported in genes encoding the proteins FKBP65 and HSP47, which participate in proper intracellular procollagen folding; in the gene for PEDF, an extracellular collagen I-binding protein and in genes encoding the three components of the collagen prolyl-3-hydroxylation complex (2,3).
Bone morphogenetic protein 1 (BMP1), the prototype of a group of structurally similar extracellular metalloproteinases that perform morphogenetic roles in a broad range of species (4,5), was first co-purified from bone extracts with the osteogenic TGFβ-like factors BMPs 2 and 4 (6). BMP1 and related proteinases were later shown to be capable of activating BMPs 2/4 by cleaving the extracellular antagonist chordin (Chd), in the context of embryonic dorsoventral patterning, in which BMPs 2/4 are major ventralizing proteins (7–9). Surprisingly, despite its initial inferred relationships with BMPs 2/4, the first demonstrated role for BMP1 was as a procollagen C-proteinase (pCP) (10), responsible for the biosynthetic cleavage of C-propeptides from procollagen precursors of the major fibrillar collagens I–III, a step necessary for the production of mature collagen monomers. Roles for BMP1-like proteinases in bone biology have thus been inferred by its association with osteoinductive factors BMPs 2/4, its demonstrated pCP activity; and such roles have also been inferred by findings of mutations at the procollagen I C-propeptide cleavage site that result in decreased cleavage efficiency, in cases of dominant OI (11). More recently, exome sequencing has identified the BMP1 gene as a candidate locus in two families with recessive OI (12,13).
There are four mammalian BMP1-like proteinases: BMP1 and mTLD (mammalian tolloid), which are encoded by alternatively spliced mRNAs from the same gene, and mTLL (mammalian tolloid-like) 1 and 2, which are each genetically distinct. In addition to providing pCP activity and activating BMP2/4, BMP1-like proteinases are reported to biosynthetically process various precursors into functional proteins involved in forming the extracellular matrix (ECM), and to activate additional growth factors (4,5). BMP1-like proteinase activities relevant to bone biology include activation of lysyl oxidase (an enzyme necessary to covalent cross-link formation in collagen fibrils) (14), biosynthetic processing of biglycan, osteoglycin and collagen V (all of which are thought to participate in regulating collagen I fibrillogenesis) (15), cleavage of dentin matrix protein 1 (DMP1) to produce peptides involved in ECM mineralization and osteocyte maturation (16,17) and activation of TGFβ1 (18), a critical protein in bone biology (19,20).
Mice null for the Bmp1 gene, which encodes both BMP1 and mTLD, are perinatal lethal from defects that include failure to close the ventral body wall (21). However, although Bmp1−/− embryos contain abnormal collagen fibrils, axial and appendicular skeletal elements are grossly normal, and only mild developmental delay in membranous bones of the skull is observed (21). Mice null for the mTLL1 gene (Tll1) are embryonic lethal at 13.5-dpc owing to cardiovascular defects, with no apparent defects in collagen fibrils (22), despite the finding that Tll1 is responsible for residual pCP activity in Bmp1−/− embryo fibroblasts (23). Early lethality has precluded further study of Bmp1−/− and Tll1−/− in vivo roles in bones and other tissues.
Tll1 and Bmp1 are co-expressed in various tissues, including bone (22,24–26), and have been shown to be additive/redundant in provision of cleavage activities against Chd, probiglycan and procollagen I in mouse embryo fibroblast (MEF) cultures (15,23). Overlapping/redundant activities may thus help explain the grossly normal skeletal morphology of Bmp1−/− embryos, and apparently normal collagen fibrils in Tll1−/− embryos. In contrast to Bmp1 and Tll1, the mTLL2 gene (Tll2) is expressed in developing skeletal muscle, but not bone (24), and Tll2-null mice present with only a mild muscle phenotype (27). Additionally, mTLL2 shows absent or reduced levels of activity against many ECM-related substrates [e.g. (16,23,24)]. Thus, it is unlikely that mTLL2 plays a major role in bone biology.
Here, we employ novel mouse strains with floxed Bmp1 and Tll1 alleles for induced postnatal, simultaneous ablation of the two genes, thus avoiding the barriers of Bmp1−/− and Tll1−/− lethality and issues of functional redundancy. In addition, a recent report that mTLD is a component of human plasma and may be involved in bone fracture repair (28) raised the potential issue that phenotypic effects of a tissue-specific knockdown of Bmp1 and/or Tll1 might to some extent be masked/compensated for by activities of circulating mTLD-related proteins. Thus, ubiquitous Bmp1 and Tll1 knockdown in all tissues was employed. The presented data demonstrate Bmp1/Tll1 products to be of profound importance to bone growth and modeling in mammals.
RESULTS
Creation of mice with ubiquitous knockdown of the Bmp1 and Tll1 genes
Toward conditional knockdown of the Tll1 gene, a mouse strain was produced with a floxed Tll1 allele, such that excision with Cre results in removal of exon 6 sequences that include the Zn2+-binding active site (Fig. 1A) and a frame shift, such that 20 amino acids downstream of the shift are translated in the new frame prior to a premature stop codon. Thus, the resulting deleted exon 6 allele (Tll1Δ6) is essentially null, producing either a severely truncated product comprising only 64 amino acid residues of mTLL1 that, in light of numerous structure-function studies performed on this family of proteinases, would be unlikely to interact with substrates, or an RNA with a premature stop codon that is rapidly degraded via nonsense-mediated decay. A mouse strain was produced with a floxed Bmp1 allele (Fig. 1E), such that excision with Cre results in removal of exons 3–6, thus removing sequences encoding the prodomain–protease domain junction and the majority of the protease domain, including the Zn2+-binding active site (all of which are shared by BMP1 and mTLD) and a frame shift, to create a null allele (Bmp1Δ3−6) in which either an extremely truncated protein containing only a few prodomain sequences is produced or in which RNA with premature stop codon is rapidly degraded via nonsense-mediated decay.
Figure 1.

Conditional disruption of Tll1 and Bmp1. (A) Structure of targeting vector and Tll1 locus before and after homologous recombination and subsequent Flp- and Cre-induced recombination to produce alleles Tll1loxP and Tll1Δ6, respectively. Arrows indicate directionality for transcription of neor (yellow box). Open box, TK selection gene. Green boxes represent Tll1 exons. Frt and loxP sequences are indicated by blue and red arrowheads, respectively. P, PstI. Orange and blue boxes represent 1.1-kb Eco72I-NheI and 496-bp HpaI-PstI 5′ and 3′ external Southern blot probes, respectively. (B) Genomic Southern blot analysis of PstI-restricted DNA from wild-type (WT) or correctly targeted (Tll1+/loxP−frt) ES clones. The 5′ probe detected 13.2-kb and 6.9-kb bands from wild-type and targeted alleles, respectively. The 3′ probe detected the 13.2-kb wild-type allele band, and a 5.3-kb band corresponding to the targeted allele. (C) Excision of the flrted neomycin resistance cassette. PCR genotyping of tail genomic DNA from mice heterozygous for the Tll1loxP−frt-targeted allele, and from mice heterozygous for the Tll1loxp allele, from which the flrted neor cassette has been excised via crossing with the ACTB:FLPe line of Flp ‘deleter’ mice. Bands of 663, 770 and 1130 bp correspond to wild type, Tll1loxP and Tll1loxP−frt alleles, respectively. (D) RT–PCR of total RNA from femora shows tamoxifen-induced excision of floxed Tll1 sequences. Bands of 179 and 544 bp correspond to TllΔ6 (excised) and Tllflox (unexcised control) alleles, respectively. (E) Green boxes and red arrowheads represent Bmp1 exons and LoxP sites, respectively, in floxed Bmp1 allele. (F) Screening of tamoxifen-induced excision of floxed alleles using PCR genotyping of ear genomic DNA shows extent of tamoxifen-induced excision of floxed Bmp1 sequences from a BTKO mouse. Bands of 233 and 298 bp correspond to Bmp1flox (unexcised) and Bmp1Δ3−6 (excised) alleles, respectively. BTKO mice with <75% excision were excluded from further study. (G) RT–PCR of total RNA from femora shows tamoxifen-induced excision of floxed Bmp1 sequences. Bands of 243 and 811 bp correspond to Bmp1Δ3−6 (excised) and Bmp1flox (unexcised control) alleles, respectively.
The neor cassette was removed from floxed Tll1 alleles (Fig. 1A and C), and crosses were performed to produce mice doubly homozygous for the floxed Bmp1 and Tll1 alleles and hemizygous for a Cre-ERT2 fusion transgene driven by the human ubiquitin C promoter (29), for ubiquitous Cre activity upon treatment of mice with tamoxifen. Mice were administered tamoxifen for 6 days at 4 weeks of age, and for another 6 days at 6 weeks of age, to optimize excision rates and, no sooner than 3 weeks after completion of tamoxifen treatment, genomic DNA from ear punch samples were screened via PCR (Fig. 1F), and only mice showing >75% excision in the Bmp1floxed allele (97% of all tamoxifen-treated mice screened) were subjected to further analysis. Subsequent RT-PCR analysis of RNA from femora of these mice showed them to be doubly homozygous for excised Bmp1 and Tll1 alleles (BTKO mice), with un-excised, floxed Bmp1 and Tll1 sequences undetectable (Figs 1D and 2G), consistent with the conclusion that particularly high excision rates are achieved in BTKO bone.
Figure 2.

BTKO mice are runted compared with control littermates. (Top left) Representative image of control (Ctrl) and BTKO mice at 17 weeks of age. (Top right) Mice (male controls, n = 13; all others, n = 10) were weighed weekly, and both total body length (nose to anus) (bottom left) and femur length (bottom right) of the same animals were measured upon sacrificing. P-values: *< 0.05, **< 0.01, ***< 0.001.
BTKO mice are reduced in size and have bones with enlarged entheses and spontaneous fractures
Subsequent to tamoxifen-induced excision of Bmp1 and Tll1 sequences, BTKO mice show reduced weight gain compared with control littermates (Fig. 2), so that by 17 weeks of age (11 weeks after the final tamoxifen injection), they are significantly smaller, with an average weight 73% of control. Some of the difference in weight is due to a reduction in adipose tissue (Supplementary Material, Fig. S1), but BTKO mice also have a 13% reduction in length and a 5% reduction in femur length (Fig. 2), thus indicating impairment of skeletal growth. Other observed skeletal manifestations of the BTKO phenotype included bony protuberances found on numerous bones upon initial dissections of BTKO mice. To obtain a global view of the distribution of these abnormalities, complete skeletons of a small number of BTKO and control mice (n = 2, each) were cleared of soft tissues via Dermestid beetles. The bony protuberances were found on BTKO scapulae, long bones and pelvises (Fig. 3A–C) and corresponded to enlarged entheses, at which tendons and ligaments attach to bone. In addition, deformation of the pelvis was observed in the BTKO skeletons (Fig. 3C), as were calluses on ribs (Fig. 3D), some of which still contained cartilaginous cores (Fig. 3E), apparently marking sites of healing of spontaneous fractures.
Figure 3.

Enlarged entheses and spontaneous fractures in BTKO mice. Representative images of scapula (A), femur (B) and pelvis (C) of control (Ctrl) and BTKO mice at 17 weeks of age cleaned by Dermestid beetles. Enlarged entheses of BTKO mice are indicated by black arrows. In particular, note the enlargement of the third trochanter (arrow outline), the attachment site for the ascending tendon of the gluteus maximus muscle, on the BTKO femur (B), and the 90o distortion of the BTKO pelvis (C). Calluses are marked (asterisks) on the ribs of a BTKO mouse (D). Alizarin red/alcian blue staining of BTKO ribs found the presence of blue, cartilaginous cores in some calluses (E), indicating that they were the sites of spontaneous fracturing of the ribs. Scale bars: 2 mm.
Spontaneous fractures such as those seen in BTKO ribs can result from mild trauma and/or muscle contraction and are suggestive of profoundly weakened bone. Enlarged entheses can result from cycles of muscle contraction and relaxation that are accompanied by cycles of microfracturing and healing of bone at insertion sites, again consistent with the possibility of severely weakened bone. We note that mice null for the gene for myostatin (GDF8), an inhibitor of skeletal muscle growth that has been proposed to be activated by BMP1-like proteinases (30), have a doubling of skeletal muscle mass that is accompanied by enlarged entheses, thought to occur to accommodate the enlarged tendons at muscle attachment sites (31). However, we found BTKO muscle mass to be reduced compared with that of controls, proportionate to the reduced mass of the mice themselves (Supplementary Material, Fig. S2). Thus, if proteolytic activation of myostatin is decreased in BTKO mice, it is not affected to an extent sufficient to affect muscle mass, perhaps due in part to expression of the BMP1-like proteinase mTLL2 in skeletal muscle (24). We thus conclude spontaneous fractures and enlarged entheses to be due to substantially weakened bone, without contribution from enlargement or strengthening of skeletal muscle. As in the case of the enlarged entheses, it seems likely that the deformed pelvises may result from cycles of microfracturing and healing in response to cycles of muscle contraction and relaxation. Interestingly, enlarged entheses, a prominent feature of BTKO bones, have been shown in a hypomineralized phenotype resulting from loss of DMP1 (17), consistent with the possibility that enlarged BTKO entheses may result, at least in part, from the hypomineralization attendant upon decreased DMP1 cleavage.
BTKO femora have increased brittleness and decreased strength and stiffness
To determine the whole bone biomechanical characteristics of BTKO mice, both femora from 10 BTKO and 13 control males, and from 10 each BTKO and control females were subjected to three-point bending analysis. As can be seen from Figure 4, total displacement, the total amount of bone deformation prior to fracture, was markedly reduced in BTKO femora, as was post-yield displacement, a measure of ductility, thus showing BTKO femora to be significantly more brittle than controls. Maximum load sustained prior to fracture was also markedly reduced in BTKO femora, as was the energy necessary to fracture BTKO femora, measured as the area under the load-displacement curve. Thus, BTKO femora are shown to be more brittle and weaker and capable of absorbing less energy than those of controls, definitive characteristics of OI. Whole bone stiffness and yield load, the force necessary to cause permanent bone damage, were also significantly reduced in BTKO bone (Supplementary Material, Table S1). Representative load-displacement curves for control and BTKO demonstrate the almost total lack of post-yield displacement (ductility) prior to fracture in BTKO femora (Fig. 4).
Figure 4.

Whole bone mechanical performance comparison of BTKO and wild-type femora. Three-point bending analysis was performed on femora from 10 BTKO and 13 control males, and from 10 each BTKO and control females. Both femora from each animal were tested, and the averaged value taken as the femoral biomechanical performance for that mouse. P-values: *< 0.05, **< 0.01, ***< 0.001. Representative load-displacement curves for control and BTKO femora are shown. Yield points mark the beginnings of plastic deformation of bones.
Following fracture, specimen geometry in the plane of the fracture was assessed (see Materials and methods), allowing calculation of corresponding tissue-level mechanical properties, using standard beam equations (32,33). Calculated tissue-level properties (Supplementary Material, Table S1), which provide the material properties of the bone independent of size and shape, provided supporting evidence that BTKO femora are more brittle and weaker than controls. Values demonstrated a lower Young's modulus and post-yield strain for BTKO femora compared with controls, suggestive of inferior material properties for BTKO bone, with deficits in mineralization and bone ECM protein components, respectively; lowered toughness and yield stress (predicted strength) indicative of lower inherent resistance to fracture and lower bending strength, respectively; and lower maximum stress capable of causing fracture. The demonstrated impairment of strength, stiffness, post-yield displacement and energy to failure in BTKO femora represents a global deficit in biomechanical performance, affecting all its major elements.
Radiography and high-resolution microcomputed tomography
X-rayed BTKO femora showed signs of spontaneous fractures, thinned cortical bone, increased amounts of trabecular bone and, in some cases, flaring of the distal femur (Fig. 5A). In some BTKO mice, flaring of the proximal tibia was also seen (Supplementary Material, Fig. S3). This type of deformation of metaphyseal regions is suggestive of a softening of the bones owing to mineralization defects, although no bowing of diaphyseal regions was observed. Although only a small number of metaphyses were flared to the extent shown in Fig. 5A and Supplementary Material, Fig. S3, ∼2/3 of BTKO distal femora were observed to have some degree of flaring. Radiography also showed BTKO mice to have pronounced kyphosis at 4 months of age, which became even more severe at 6 months of age (Fig. 5B), characteristics that can be found in OI, osteoporosis or ligamentous disease.
Figure 5.

Radiography and high-resolution µ-CT comparisons of BTKO and control bone. (A) X-rayed hindlimbs show calluses at the sites of spontaneous fractures (asterisks), flaring of the distal femur (arrowhead), highly thinned cortical bone (white arrows) and increased trabecular bone (outlined arrow) in a representative BTKO specimen. (B) Radiography of four representative mice shows 4-month-old BTKO mice to have kyphosis and shows the kyphosis to become considerably more pronounced at 6 weeks of age. The individuals shown are female. (C) Representative µ-CT images of femoral diaphyses show BTKO cortical bone to have smaller total volume (TV) and bone volume (BV), increased porosity (BV/TV) and reduced tissue mineral density (TMD), compared with controls. Quantitation of the data is shown in histogram form (blue, controls; red, BTKO). (D) Representative µ-CT images of femoral metaphyses show lowered BV, BV/TV, reduced Tb.N and increased tracecular spacing (Tb.Sp), (but no significant difference in Tb.Th or TV) in BTKO trabecular bone, compared with controls. Quantitation of the data is shown in histogram form (blue, controls; red, BTKO). P-values: *< 0.05, **< 0.01, ***< 0.001. ns, not significant.
Analysis by microcomputed tomography (µ-CT) of BTKO and control femoral diaphyses (Fig. 5C) showed BTKO femora to have significantly smaller total volume (TV) and bone volume (BV), thus confirming the BTKO cortical bone in this region to be thinner than that of controls, whereas the reduced BV/TV ratio showed the BTKO diaphyseal cortical bone to have increased porosity as well. The same analysis also showed BTKO cortical bone to have reduced tissue mineral density compared with control bone. Analysis by µ-CT of metaphyses again showed a lowered BTKO BV/TV ratio and also showed reduced trabecular bone number (Tb.N) and increased trabecular spacing (Tb.Sp) (Fig. 5D). The thin, porous cortical bone and reduced Tb.N and increased Tb.Sp of BTKO bone are, together, somewhat reminiscent of the same features observed in osteoporotic bone.
Dynamic histomorphometry
Sequential in vivo incorporation of fluorochromes calcein and alizarin red into newly calcifying bone showed the labeling surface (LS/BS, bone surfaces containing calcein, alizarin red or both) of BTKO mice to be much greater than those of controls, and the same is true for mineralizing surface [MS/BS, bone surfaces containing both calcein and alizarin + ½(surfaces containing either calcein or alizarin) the extent of bone surface that is actively mineralizing] (Table 1). A representative image illustrative of the difference in calcein and alizarin red incorporation into BTKO and control bone is shown in Figure 6A. There is no difference in mineral apposition rate (MAR, the distance between calcein and alizarin red labels divided by time between the labels) between BTKO and control bone; however, the bone formation rate (BFR, MAR × MS/BS) is seen to be twice that in BTKO bone as in control bone (Table 1). As the MAR is a measure of the rate of ECM deposition per osteoblast, whereas the BFR multiplies this rate by the fraction of bone surface with active osteoblasts (MS/BS, thus providing insights into the numbers of osteoblasts actively depositing ECM), it is apparent that there are much greater numbers of osteoblasts depositing mineralizing ECM within BTKO bone than in control bone.
Table 1.
Quantitative histomorphometry of mouse femur
| Ctrl | BTKO | P-value | |
|---|---|---|---|
| Dynamic histomorphometry | |||
| LS/BS | 26.90 ± 8.12 | 42.32 ± 1.80 | 0.004 |
| MS/BS | 14.83 ± 4.57 | 24.77 ± 1.63 | 0.001 |
| MAR | 0.76 ± 0.12 | 0.85 ± 0.24 | ns |
| BFR | 0.11 ± 0.04 | 0.22 ± 0.07 | 0.004 |
| Osteoblast activity | |||
| AP/BS | 49.76 ± 10.62 | 42.56 ± 2.46 | ns |
| AP_L/AP | 32.75 ± 8.52 | 47.8 ± 5.47 | 0.003 |
| AP_NL/AP | 62.22 ± 9.38 | 44.43 ± 6.11 | 0.002 |
| Osteoclast activity | |||
| TRAP/BS | 1.84 ± 1.73 | 5.73 ± 3.68 | 0.022 |
| TRAP_R/BS | 0.54 ± 0.67 | 2.13 ± 1.53 | 0.026 |
| TRAP_Only/BS | 1.18 ± 1.25 | 2.04 ± 1.43 | ns |
Figure 6.

Dynamic histomorphometric differences and cellular abnormalities in BTKO long bones. (A) In vivo double fluorochrome labeling with calcein (green) and alizarin red shows greatly increased labeled bone surfaces in the trabecular portion of BTKO femur, compared with a control. (B) TRAP staining for osteoclasts (red) is sharply increased in BTKO bone, compared with control, as shown in trabecular portions of the femur. Immunohistochemical staining (brown) performed on cortical femur showed higher levels of MEPE (C) and E-11 (D), and lower levels of sclerostin (E) for BTKO bone than those for controls. Scale bars: 50 µm.
Cellular abnormalities of BTKO bone
Interestingly, although staining for alkaline phosphatase (AP), a marker for active osteoblasts and quiescent bone lining cells on bone surfaces (AP/BS), was similar for BTKO and control bone, the proportion of active osteoblasts lining labeling surfaces (AP_L/AP), at which mineral is being deposited, was markedly increased in BTKO bone compared with controls, whereas the proportion of inactive osteoblasts lining non-labeling surfaces (AP_NL/AP) was markedly decreased in BTKO bone compared with controls (Table 1). Thus, increased numbers of active osteoblasts, rather than increased numbers of total osteoblasts, are shown to be responsible for the much greater deposition rate of mineralizing ECM detected by dynamic histomorphometry.
Staining for tartrate-resistant acid phosphatase (TRAP), a marker for osteoclasts, showed a sharp increase in numbers of osteoclasts lining bone surfaces (TRAP/BS) in both BTKO trabecular and cortical bone (Table 1), as show in Figure 6B. Whereas numbers of osteoclasts adjacent to non-mineralizing surfaces (TRAP_Only/BS) were not significantly higher in BTKO than in control bone, osteoclasts adjacent to mineralizing surfaces (TRAP_R/BS) were ∼4-fold greater in BTKO than in control bone (Table 1). Thus, the increased numbers of osteoblasts at mineralizing surfaces are amply matched by increased numbers of osteoclasts, explaining how, despite an elevated BFR indicative of increased numbers of active osteoblasts, BTKO cortical bone is thinner and there are decreased Tb.N and increased inter-trabecular spacing (Tb.Sp) in BTKO owing to resorption.
Immunohistochemical staining for MEPE (matrix extracellular phosphoglycoprotein, expressed in both osteoblasts and osteocytes) (Fig. 6C) and E-11/gp38 (a marker of early osteocytes) (Fig. 6D) was increased in the lacunae of BTKO cortical bone, compared with controls, whereas staining for sclerostin, a marker of mature osteocytes (Fig. 6E), seemed somewhat decreased in BTKO bone. These results suggest a deficit in osteocyte maturation and are somewhat reminiscent of similar results observed in DMP1-null mice (34), consistent with the possibility that part of the BTKO bone phenotype may result from decreased proteolytic activation of DMP1.
To further examine possible derangements of BTKO osteocytes, staining was performed with FITC, a small-molecular-weight dye that permeates the non-mineralized spaces in bone, thus providing a visual representation of the organization of non-mineralized cells, cellular processes and fibers under the confocal microscope. As can be seen from Figure 7A, BTKO osteocytes showed a relative loss of dendritic processes, by which mature osteocytes communicate with one another, receive mechanosensory signals and participate in the regulation of bone turnover (35); and a fraction of BTKO osteocytes seemed unable to maintain the integrity of the lacunae in which they were located, resulting in penetration of FITC through lacunae walls (Supplementary Material, Fig. S4).
Figure 7.
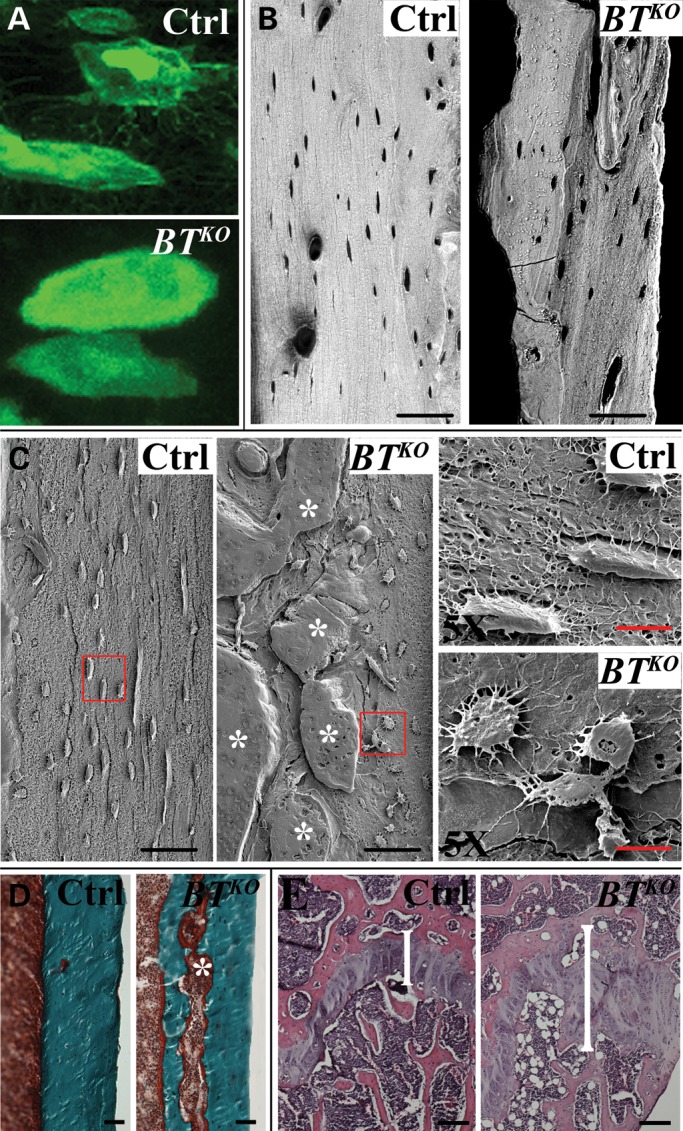
Morphological/structural defects in BTKO bone. (A) A representative high-magnification comparison is shown for FITC staining of cortical areas of femur that shows BTKO osteocytes (bottom panel) to have reduced numbers of dendritic processes than control osteocytes (top panel). (B) Representative backscattered SEM images contrast the homogeneous distribution of mineral in control cortical areas of femur with a heterogeneous distribution of areas of higher (white) and lower (grayish zones) levels of mineral in BTKO femur and also shows BTKO osteocytic lacunae to be fewer, generally more rounded and less spindle-shaped than in controls. (C) Illustrative acid-etched, resin-casted SEM images show in detail the malformed osteocytes and decreased numbers of dendrites in BTKO cortical femur. Asterisks denote areas of osteoid. (D) Representative Goldner's Masson trichrome staining demonstrates the type of osteoid seam (asterisk) present in BTKO cortical bone, but absent in controls. (E) H&E staining of femora shows BTKO marrow to contain markedly more adipocytes than controls and shows BTKO growth plates (brackets) to be relatively wider, compared with controls, and to have some disorganization in vertical columns. Black scale bars: 50 µm; red scale bar: 10 µm.
Backscattered scanning electron microscopy (SEM) contrasted the homogeneous distribution of mineral in control bone with a heterogeneous distribution of areas of higher and lower levels of mineral in BTKO bone (Fig. 7B). It also showed lacunae to be more rounded, less spindle-shaped and larger in BTKO than in control bone, consistent with a maturation defect in BTKO osteocytes. Acid-etched, resin-casted SEM images showed the malformed BTKO osteocytes and lesser numbers of dendrites in greater detail (Fig. 7C).
Goldner's Masson trichrome staining of cortical bone shows BTKO cortical bone to contain seams of unmineralized osteoid (Fig. 7D), which can also be seen via acid-etched SEM (Fig. 7C), another indication of a deficit in osteocyte function. Thus, a BTKO osteocyte maturation defect is supported by (1) increased osteoid seams, (2) enlarged osteocyte cell bodies, changes in cell shape, reduced numbers of dendrites and changes to the lacunar-canalicular system and (3) gene expression pattern changes.
Interestingly, another consistent cellular change noted in BTKO bone was a large increase in the numbers of adipocytes in the marrow (Fig. 7E). While it is not clear the extent to which this cellular change might contribute to the bone phenotype, it suggests the possibility of further derangements in cell differentiation pathways owing to loss of BMP1-like proteinase activity in BTKO mice.
Molecular analyses of the BTKO phenotype
The markedly reduced post-yield displacement of BTKO femur, showing much reduced ductility, is diagnostic of the type of impaired collagen I properties that lead to brittle bones (i.e. OI). We have previously shown that BMP1, mTLD and mTLL1 are all capable of removing the procollagen I C-propeptide in vitro and that products of endogenous Bmp1 and Tll1 genes are together responsible for processing endogenous procollagen I C-propeptides in cultures of MEFs (10,23,24). To determine the extent to which Bmp1 and Tll1 ablation might affect procollagen I processing in BTKO bone, western blots of BTKO and control femur extracts were probed with anti-procollagen I C-propeptide antibody. As can be seen from Figure 8A, procollagen C-propeptide cleavage is much reduced in BTKO bone, although low levels of residual processing were observed upon overexposure of the blot (not shown). Surprisingly, although transmission electron microscopy (TEM) previously showed the collagen fibrils in Bmp1−/− (21) and Bmp1−/−/Tll1−/− doubly null (23) embryos to have a ‘barbed wire’ appearance, with barb-like projections on fibril surfaces apparently associated with retained C-propeptides (36), TEM showed no discernable barbs associated with the collagen fibrils of BTKO tendons (Fig. 8D) or bone (Supplementary Material, Fig. S5). In fact, measurement of tendon fibrils (measurements were made in tendon rather than bone, as the parallel fibrils of tendon greatly facilitates such measurements) found the only discernable difference to be a shift in BTKO fibril diameter distributions to larger diameters, with a reduction in the subpopulation of less mature fibrils with diameters centered in the 50- to 55-nm range, a shift that may represent some loss of fine control in the lateral growth of collagen fibrils via lateral fusion of smaller diameter fibrils (Fig. 8D). In addition, there was a higher density of fibrils in control tendon than in BTKO tendon, in which diameters were larger. No change in D-spacing was detected between BTKO and control collagen fibrils.
Figure 8.

BTKO bones show decreased processing of procollagen I and DMP1 and increased canonical Wnt signaling. Shown are western blots of extracts from femora of 17-week-old (A and B) and 10-week-old (C) BTKO and control (Ctrl) mice. (A) Anti-procollagen I C-propeptide antibodies show decreased procollagen I processing in BTKO femur. (B) Anti-DMP1 antibodies indicate a reduction in processing of DMP1 to its active 57-kDa form in BTKO femur. (C) Anti-β-catenin antibodies provide evidence of higher canonical Wnt-signaling levels in BTKO femur. All western blots were repeated using at least three different BTKO and control samples, each from a different mouse. Representative images are shown. (D) TEM shows BTKO tendon to have collagen fibrils that are grossly normal in appearance, but which have a shift in fibril diameter distributions to larger diameters. First quartile (Q1) values demonstrate a reduction in the small diameter subpopulation (vertical arrows) in BTKO tendon, compared with control (62 nm versus 70 nm). In addition, the median, first quartile (Q1) and third quartile (Q3) values are all increased in the mutant tendon, indicating a general shift to larger diameter fibrils in BTKO tendon. This increase in fibril diameter is consistent with the significant decrease in fibril density observed in BTKO tendon relative to control (58/mm2 versus 65/mm2). X, mean; sd, standard deviation.
A number of the defects observed in the BTKO bone phenotype are reminiscent of defects seen in mice null for the DMP1 gene (Dmp1). Such defects include osteomalacia, shortened long bones with flared metaphyses, an abnormal osteocyte lacunocanalicular system and defective osteocyte maturation (17,37). As DMP1 has been proposed as a substrate for BMP1-like proteinases (16), and as processing of DMP1 to produce active C-terminal 57-kDa fragment is necessary to DMP1 function (34,38), we determined whether such processing was actually impaired in BTKO bone. As can be seen from Figure 8B, conversion of full-length ∼120-kDa DMP1 to a 57-kDa C-terminal fragment is reduced in femur extracts of BTKO femur, compared with control femur. Thus, it can be concluded that BMP1-like proteinases do participate in the in vivo processing/activation of DMP1 in bone. However, it is not clear from the western blots the extent to which DMP1 cleavage is reduced in BTKO bone. It has previously been reported that Dmp1-null mice have malformed, expanded growth plates, in which the normally vertical columns of chondrocytes have become disorganized, and a dramatically expanded hypertrophic zone is observed (37). There does appear to be a relatively minor defect in BTKO growth plates as documented by expanded cartilage zones, with mild disorganization in vertical columns (Fig. 7E), and the reduction of trabecular BV in the metaphysis (Fig. 6) is also suggestive of a deficit in endochondral bone formation. However, the growth plate defects are much milder than those in Dmp1-null mice. In addition, Dmp1-null mice are severely hypophosphatemic and mildly hypocalcemic (39), whereas there were no significant differences in serum phosphate (Pi: BTKO 0.8749 ± 0.0494 mmol/ml, n = 8; control 0.8734 ± 0.0604 mmol/ml, n = 8) or serum ionized calcium (Ca2+: BTKO 7.741 ± 0.196 mg/dl, n = 8; control 7.793 ± 0.203 mg/dl, n = 8) between BTKO and control mice. Thus, there appears to be sufficient residual DMP1 cleavage in BTKO mice to avoid the full-blown growth plate and serum electrolyte phenotypes observed in Dmp1-null mice.
Although there have been no previous reports of the involvement of BMP1-like proteinases in the regulation of Wnt signaling, the observed decrease in BTKO bone of staining for sclerostin (above), which is an inhibitor of Wnt signaling (40), and the recent importance that canonical Wnt signaling has been shown to play in bone modeling and mineral density (41), prompted us to determine whether levels of canonical Wnt signaling in BTKO bone might differ from those of controls. Interestingly, consistent with the decreased sclerostin staining in BTKO femora (Fig. 7F), the femora of 10-week-old BTKO mice showed marked increases in the levels of β-catenin (Fig. 8C), indicative of marked increases in the levels of canonical Wnt signaling.
DISCUSSION
Here, we present novel, floxed Bmp1 and Tll1 mouse strains that have enabled us to overcome the barriers of Bmp1−/− and Tll1−/− lethality and issues of functional redundancy, to begin in-depth characterization of in vivo functions of a family of proteinases hypothesized to play key roles in vertebrate development and homeostasis and, in particular, in ECM formation and maintenance (4,5). The association of mutations that disrupt procollagen I C-propeptide processing with cases of dominant OI (11) and recent association of BMP1 amino acid changes with cases of recessive OI (12,13) prompted us to focus initial analysis of BTKO mice on the skeletal system.
Three-point bending analysis showed BTKO bone to be markedly weak and brittle, the definitive characteristics of OI. In particular, impaired post-yield deformation, or ductility, implied an ECM protein defect, consistent with the possibility that the BTKO bone phenotype is, like cases of OI (1–3), impacted by deficits in the formation and/or properties of collagen I fibrils, as such fibrils are thought to provide most of the tensile strength of bone. As is also the case in OI, at least some of the mineralization defects in BTKO bone are presumed to arise secondarily to deficits in collagen I fibrils, which are thought to be intimately involved in the nucleation and growth of the nanocrystals that constitute bone mineralization (42). Dynamic histomorphometry results and the markedly increased levels of active osteoblasts and osteoclasts at mineralizing surfaces in BTKO bone are all consistent with a model in which BTKO osteoblasts deposit an inferior quality ECM, resulting in dramatically increased bone turnover in an ultimately futile attempt to repair/replace the inferior bone.
Despite the ECM defect implied by mechanical testing, and the pronounced deficit in procollagen C-propeptide cleavage, BTKO collagen fibrils were surprisingly normal in appearance via TEM, with the only obvious morphological abnormality being a shift to larger fibril diameter distributions, perhaps representing some loss of fine control in collagen fibril lateral growth. In contrast, TEM has shown Bmp1-null (21) and Bmp1−/−Tll1−/− doubly null (23) embryos to have distinctly abnormal collagen fibrils with barb-like projections, demonstrated by immunohistochemistry to be associated with retained C-propeptides (36). It is possible that morphological defects in BTKO collagen fibrils would be more pronounced if tamoxifen-induced excision of Bmp1/Tll1 sequences was pre- rather than post-natal. However, TEM examination of dermal collagen fibrils of OI patients with C-propeptide cleavage site mutations also showed a lack of barbs (11) (collagen fibrils of OI patients with BMP1 missense mutations (12,13) have yet to be characterized via TEM). We thus suggest that C-propeptide-associated barbs may become detectable via TEM only when C-propeptide processing activity falls below some threshold. This suggests that C-propeptide processing activity is lower in Bmp1-null embryos than that in BTKO tissues, which is plausible, as Bmp1 function is totally absent in the former, but low residual Bmp1 expression remains in the latter, and, although Tll1 expression is much reduced in BTKO tissues, Bmp1 may provide higher levels of in vivo pCP activity than does Tll1 (22). It is also possible that the appearance of barbs on Bmp1−/− embryo fibrils and their absence on fibrils of BTKO mice or OI patients with C-propeptide processing defects may be influenced by differences in embryonic and postnatal processes of collagen fibrillogenesis, which have been documented in some tissues (43). Future experiments, involving use of Bmp1floxed/− mice, compound heterozygous for floxed and null Bmp1 alleles (thus providing lower levels of residual Bmp1 activity upon tamoxifen treatment than those found in BTKO mice) and both pre- and post-natal ablation of Bmp1 activity, will provide additional insights into how levels of pCP activity and pre- and post-natal mechanisms of fibrillogenesis may contribute to formation of C-propeptide-associated barbs on collagen fibrils.
Although dermal collagen fibrils of OI patients with C-propeptide cleavage site mutations were reported to have irregular profiles with ‘blebs’ on fibril surfaces, upon cross section (11), such irregular cross sections with blebs were not detected in BTKO fibrils, and thus do not appear to be consistent features of fibrils in tissues with lowered C-propeptide processing levels.
Analysis by µ-CT showed BTKO bones to have a generalized decrease in mineral density, although backscattered SEM showed mineral to be heterogeneously distributed, with areas of low and high density. In contrast, a generalized increase in bone mineral density was reported for the two mild OI patients with C-propeptide cleavage site mutations and for a sib pair with mild OI and a BMP1 signal peptide missense mutation that impairs secretion (11,12). A generalized increase in bone mineral density was also reported for zebrafish with mutations in the BMP1 gene bmp1a (12). However, despite the apparent contrast between generalized decreases and increases in mineral density in BTKO bones and these other skeletal phenotypes, respectively, a feature at least some of these reported phenotypes share with the BTKO phenotype is heterogeneity in the distribution of bone mineral. In the zebrafish model, for example, although adult vertebrae had increased mineral density, mineral density was reduced in fin bones and in larval vertebrae (12). In the OI patients with C-propeptide cleavage mutations, although DXA analysis indicated elevated bone mineral density and Fourier transform infrared imaging showed an increased mineral/ECM ratio, both patients had radiographic osteopenia, and pronounced heterogeneity of mineralization was documented in one of the patients (11). Importantly, a sib pair with a BMP1 protease domain missense mutation and a severe OI-like phenotype was, like BTKO mice, reported to have generalized decreases in bone density (distribution of mineral was not analyzed) (13). We interpret these various observations to mean that diminished BMP1-like proteinase activity, or even just a deficit in the levels of C-propeptide cleavage, leads to deranged ECM–mineral interactions that can have varying effects on mineral deposition, leading to heterogeneity in mineral density not only between different skeletal phenotypes but also within a given bone. Variables that affect whether mineral levels will be increased or decreased in a given context by these types of mutations remain to be determined.
Proteolytic cleavage of DMP1 is reduced in BTKO bone, consistent with the osteomalacia, shortened long bones with flared metaphyses, enlarged entheses, abnormal osteocyte lacunocanalicular system and defective osteocyte morphology/maturation seen in these bones, all of which defects have been described in Dmp1−/− mice and in human patients with autosomal recessive hypophosphatemic rickets caused by mutations in the DMP1 gene (17). However, unlike Dmp1−/− mice, which have rickets, grossly broadened and deranged growth plates and are severely hypophosphatemic, BTKO mice are not rachitic and have relatively minor growth plate defects and normal serum phosphate levels. Interestingly, both rickets and growth plate defects in Dmp1−/− mice are secondary to hypophosphatemia, whereas osteomalacia is, for the most part, not (17). We thus speculate that sufficient DMP1 is activated by residual Bmp1 and/or Tll1 expression in BTKO mice to avoid hypophosphatemia and attendant major growth plate defects and rickets, whereas the diminution in proteolytic activation of DMP1 is sufficient to contribute to the mineralization defects and derangements in osteocytic biology seen in these mice—derangements that appear to contribute in a substantial way to the BTKO skeletal phenotype.
Although BTKO and Dmp1−/− bone are similar in having increased numbers of AP-positive osteoblasts, they differ in that BTKO bone has increased, whereas Dmp1−/− bone has decreased numbers of TRAP-positive osteoclasts (44). The reduced numbers of osteoclasts in Dmp1−/− bone may be, to some extent, a secondary effect of hypophosphatemia (44), which does not occur in BTKO mice, whereas the increased numbers of osteoclasts in BTKO bone may be influenced, to some degree, by the abnormal collagen of BTKO bone, as defective bone collagen has been suggested to contribute to elevation of osteoclast levels in OI (45). The extent to which reduced cleavage of proposed substrates of BMP1-like proteinases other than DMP1 and procollagen I might contribute to the BTKO skeletal phenotype remains to be determined.
Interestingly, and consistent with decreased staining in BTKO femora for sclerostin, a marker for mature osteocytes and an inhibitor of canonical Wnt signaling, femora of 10-week-old BTKO mice showed markedly increased β-catenin levels, indicative of markedly increased canonical Wnt signaling. This result may seem counterintuitive, as increased canonical Wnt signaling has previously been associated, by numerous studies, with increased bone mass and strength (41), and as both of the latter are reduced in BTKO mice. Nevertheless, we surmise that the lower sclerostin levels, resulting from deficits in osteocyte maturation, result in increased canonical Wnt signaling that helps drive the increased numbers of active osteoblastic cells involved in the accelerated bone remodeling observed in BTKO bone. In addition, it is thought that bone bending in response to strain results in decreased sclerostin expression by osteocytes, thus locally increasing Wnt signaling to increase bone mass as part of the adaptive response of bone to strain (46,47). Because the BTKO bones are less stiff than the control bones, the BTKO bones would experience more strain in response to a given load than would control bones, and this might be expected to contribute to decreased sclerostin production, except in cases where the maturation deficits of BTKO osteocytes compromise their usual role as mechanosensors (48). The observation of high Wnt signaling in BTKO bone is germane to current efforts to develop new bone-building therapies for increasing bone mass via positively modulating canonical Wnt signaling (41), as they demonstrate an example of low bone mass and high bone fragility in which canonical Wnt signaling is already at high levels, such that up-modulation of such signaling might not be expected to effectively ameliorate the bone phenotype. Such therapies can be tested in the BTKO mouse, to determine whether this is the case.
Wnt signaling is thought to favor the differentiation of mesenchymal stem cells (MSCs) down the osteoblastic, rather than the adipocytic, pathway (49), consistent with the increased numbers of active osteoblasts observed in BTKO bone. In this context, however, it was surprising that BTKO bone also contained markedly more adipocytes than did control bones (e.g. Fig. 7E). We note that BMP1-like proteinases are thought to be involved in regulating a number of signaling pathways (e.g. BMP signaling) (4) that can interact with the Wnt-signaling pathway in complex ways to affect MSC differentiation (49). Moreover, as interactions with ECM components doubtless affect MSC differentiation, deficits in processing of various ECM components in BTKO bone may contribute to the abnormal distribution/numbers of cell types seen in this tissue. Future experiments employing cultures containing BTKO and control bone MSCs will attempt to identify the factors involved in producing the abnormal distribution/numbers of MSC-derived cell types found in BTKO bone.
It has been reported that human and rat plasma contain mTLD (50), one of the two alternatively spliced proteinases encoded by the gene that also encodes BMP1, and that i.v. injection of recombinant mTLD or anti-mTLD antibody enhances or delays, respectively, bone fracture repair in rats (28). Clearly, the BTKO mouse model system presented here provides the opportunity for determining the extent to which circulating mTLD may normally affect bone growth and modeling, via injecting recombinant mTLD into BTKO mice and determining the extent to which this may ameliorate the bone phenotype. Such studies could provide a direct readout of the portion of the BTKO bone phenotype owing to loss of circulating mTLD and would also provide insights into the potential for therapeutic interventions via administrating exogenous BMP1-like proteinases to OI patients with deficiencies in endogenous BMP1-like proteinases (12,13), or C-propeptide cleavage site mutations that delay procollagen processing (11,51). Comparable studies could employ recombinant BMP1 and mTLL1, or cocktails comprising combinations of the three proteinases, to determine the most efficacious therapeutic approach.
MATERIALS AND METHODS
Tll1- and Bmp1-floxed mice
129/Sv genomic DNA from the 5′ homology arm of a previously described Tll1 targeting vector (22) was cleaved with BglII, partially filled with dATP and dGTP, and recut with Xba I to produce a 759-bp fragment containing exon VI. This fragment was ligated into vector ploxPNT (52,53), which had been cut with SalI, partially filled with dCTP and dTTP, and then recut with XbaI. In resulting plasmid, ploxPNTVI, Tll1 exon VI replaced the neor cassette of the original ploxPNT vector, as an insert flanked by loxP sites. A 3.2-kb EcoRI-XbaI genomic fragment previously used as a Tll1 5′ homology arm (22) was placed into pBluescript II KS+ (Stratagene), and the resulting plasmid was cut with KpnI and ClaI, removing the pBluescript polylinker ClaI site, but retaining the polylinker KpnI site, now fused to the 5′ end of the genomic insert. Oligomers 5′ CTAGACTGCAGAATCGATGC 3′ and 5′ GGCCGCATCGATTCTGCAGT 3′ were annealed to form a linker that was ligated to the 3′ end of the 3.2-kb insert to introduce PstI and ClaI sites. The modified insert was then excised with KpnI and ClaI and ligated into ploxPNTVI, which had been cut with KpnI-ClaI. The resulting plasmid was cut with XhoI and NotI, and the Tll1 3.6-kb 3′ homology arm described in Clark et al. (22) was inserted to produce ploxTll1. The latter was cut with NheI and treated with calf intestine AP. Plasmid J35 (54) was cut with EagI, self-ligated, partially digested with BamHI and then recut with BagI and BglII, to remove sequences flanked by the frt sites. The remaining vector backbone was then ligated to the Pgk1 promoter-neor cassette that had been cut from vector pPNT (52) with BamHI and NotI. The resulting plasmid, containing an frt-flanked neor cassette, was cut with NotI and blunt-ended with Klenow fragment (to remove a NotI site between the neor cassette and one of the frt sites that would have interfered with linearization of the final targeting vector) and then self-ligated to form plasmid pJFRTneo. Plasmid pJFRTneo was cut with BssHII and NcoI and ligated to a fragment formed via annealing of oligonucleotides 5′ CGCGCATGCCCGACGGCGAAGATCTCGTCGTGACC 3′ and 5′ CATGGGTCACGACGAGATCTTCGCCGTCGGGCATG 3′, which corrected an Asp to Glu mutation in the original pPNT neor cassette. The resulting plasmid was then cut with NheI, to release the frt-flanked neor cassette, which was then ligated into an NheI site of ploxTll1 to form the finished targeting vector (Fig. 1A).
129/S6 embryonic stem (ES) cells were electroporated with NotI-linearized targeting vector and subjected to selection in G418 and gancyclovir, producing 287 doubly resistant colonies. Four correctly targeted lines were identified by Southern blotting (Fig. 1B). ES cells from the four independently targeted cell lines were injected into C57BL/6 blastocysts at the University of Wisconsin Biotechnology Center Transgenic mouse facility. Chimeras were crossed with C57BL/6 mice (Jackson Laboratories), and analysis of agouti offspring by Southern blotting for the targeted allele found germ line transmission from two of the independently targeted ES cell lines, leading to establishment of two separate mouse lines with allele Tll1loxP−frt. For excision of the Pgk1 promoter-neor cassette, to produce allele Tll1loxP, Tll1loxP−frt mice were crossed with the ACTB:FLPe line (55).
Generation of the Bmp1-floxed mice will be described elsewhere.
All mice were housed and treated in accordance with NIH guidelines, using protocols approved by the Research Animal Resources Center of the University of Wisconsin-Madison.
Induced excision of Bmp1 and Tll1 in juvenile mice
To excise floxed Bmp1 and Tll1 sequences, a mouse carrying the Cre-ERT2 transgene under control of the human ubiquitin C promoter (29) was crossed onto a Bmp1flox/flox; Tll1flox/flox background, to produce Bmp1flox/flox; Tll1flox/flox; Cre-ERT2 mice. Bmp1flox/flox; Tll1flox/flox littermate mice lacking the Cre transgene were used as controls in this study. Tamoxifen (Sigma Life Science) was administered to both Bmp1flox/flox; Tll1flox/flox; Cre-ERT2 mice and Bmp1flox/flox; Tll1flox/flox controls through IP injection at a concentration of 100 mg/kg body weight (solubilized in 98% corn oil, 2% ethanol) once per day for 6 days at 4 weeks and then again for 6 days at 6 weeks of age. Bmp1flox/flox; Tll1flox/flox; Cre-ERT2 mice from which floxed sequences have been excised consequent to treatment with tamoxifen are hereafter referred to as BTKO mice. Mice, unless otherwise indicated in the text, were sacrificed at 17 weeks of age for analysis.
Preparation and staining of whole skeletons
To obtain a global view of skeletal abnormalities, soft tissues were removed from intact skeletons using Dermestid beetles, at the University of Wisconsin-Madison Zoological Museum. Rib fractures were verified through alizarin red/alcian blue staining of whole mouse skeletons, as described previously (56). Briefly, mouse skeletons were eviscerated and fixed in 95% ethanol overnight. The preparations were then stained 3 days in alizarin red/alcian blue and then cleared through sequential incubations in 2% KOH, 25% glycerol/2% KOH, 50% glycerol/2% KOH, 75% glycerol/2% KOH and 100% glycerol.
Biomechanical testing
Femora were harvested from 10 BTKO and 13 control males, and from 10 each BTKO and control females. After sacrifice, femora were disarticulated and soft tissues were removed. Biomechanical performance of femoral diaphyses was tested using quasi-static three-point bending under displacement control with a support span of 7.5 mm, as previously described (57). Bones were loaded at a constant rate of 0.3 mm/sec until failure occurred. Both femora from each animal were tested, and the averaged value taken as the femoral biomechanical performance for that mouse. Geometric properties of the femora tested were obtained from analysis (SigmaScan) of digital images of the fracture cross section, and this information, along with the whole bone mechanical testing data obtained from the three-point bending experiment, was used to calculate material properties of the bone tissue according to standard beam theory equations (32,33).
X-ray radiography and high-resolution microcomputed tomography
Bones were dissected free of skeletal muscles for radiography with a Faxitron MX-20 specimen radiography system (Faxitron X-ray Corp.) as described previously (37). For high-resolution microcomputed tomography (µ-CT) analyses, femora were scanned at 3.5-μm resolution using a µ-CT35 imaging system (Scanco Medical). The µ-CT analyses included: (1) high-resolution scanning of the femur mid-shaft region (100 slices along the mid-shaft between the two epiphyses along the cranial–caudal axis and (2) high-resolution scanning of the femur metaphyseal region proximal to the distal growth plate for trabecular bone evaluations. In trabecular bone evaluations, 400 slices were chosen in the bone center, with the cortical shell excluded. Data obtained from high-resolution µ-CT scanning were analyzed via Scanco software, which provided: the bone volume-to-total volume ratio (BV/TV), Tb.N, trabecular thickness (Tb.Th), trabecular spacing (Tb.Sp) and tissue mineral density.
Dynamic and cellular histomorphometry of bones
To prepare bone samples for histomorphometric analysis, mice received intraperitoneal injections of 10 mg/kg calcein (Sigma C-0875) and 30 mg/kg alizarin complexone (Sigma A-3882) at 7 and 2 days, respectively, prior to sacrifice, at 17 weeks of age. Whole hindlimbs were dissected from the hip, and skin and non-adherent muscle and connective tissues were removed from the bone, without scraping periosteal surfaces. Proximal femur and distal tibial bone was then removed to expose the marrow space, and the bone samples, with knee joints intact, were placed in 10% formalin at 4°C for 2–3 days with slow agitation. Samples were then subjected to computer-automated dynamic and cellular bone histomorphometry, essentially as described by Hong et al. (58).
Histology
Bones were fixed in 10% formalin at 4°C, decalcified and embedded in paraffin, and 4-µm-thick sections were used for H&E and TRAP staining, and immunohistochemistry, as described previously (17). For immunohistochemical analyses, the following antibodies were used: anti-MEPE (59) (gift from Larry Fisher, National Institutes of Health, Bethesda, MD, USA), diluted 1:400; anti-sclerostin (R&D Biosystems), diluted 1:400; and anti-E-11 [(60), gift from Lynda Bonewald], diluted 1:50, polyclonal antibodies. Un-demineralized specimens were embedded in methylmethacrylate, and 6 µm sections were subjected to Goldner's Masson trichrome staining, as previously described (17).
Backscattered scanning electron microscopy, acid etching SEM and FITC imaging
Long bones were dissected and fixed in 70% ethanol at room temperature for 24 h. Tissue specimens were then dehydrated in ascending concentrations of ethanol (from 70 to 100%), embedded in MMA without decalcification and sectioned using a water-cooled diamond-impregnated circular saw (Isomet, Buehler). The cut surface was polished using 1-, 0.3- and 0.05-μm alumina alpha micropolish II solutions (Buehler) on a soft cloth rotating wheel (61). Each sample was placed in an ultrasonic bath between steps and immediately following the polishing steps. The dehydrated specimens were then sputter-coated with carbon and scanned with a backscattered electron detector in a JEOL JSM-6010LA scanning electron microscope. Parameters were kept constant while the backscattered SEM images were taken under the scanning electron microscope, as described previously (17,62).
Staining with FITC (fluorescein isothiocyanate) was performed essentially as described (63,64). Briefly, long bones, immediately after harvesting and dissection, were placed in 10% formalin at 4°C for 2 days, followed by embedding in MMA bone blocks. A cross section (1–2 mm thick) was cut with a diamond-bladed saw (Buehler), and the cortical sections were then sanded and ground to a final thickness of ∼100 μm for confocal imaging.
Western blot analysis
Disarticulated femora, with soft tissue removed, were frozen and homogenized to a fine powder in liquid nitrogen, using a porcelain mortar and pestle. The powder was then transferred into T-PER Tissue Protein Extraction Reagent with Halt protease inhibitor (Thermo Scientific). Samples were then sonicated 30 s and centrifuged, and supernatants were stored at −80°C until use; at which time, 50 µg aliquots/lane were subjected to SDS–PAGE and then transferred to nitrocellulose filters (one DMP1 western blot in Figure 8B was transferred to PVDF membrane), which were then blocked in 5% nonfat dry milk. Blots were probed with polyclonal antibody LF-41, directed against the pro-α1(I) C-propeptide (65), a gift from Larry Fisher; rabbit polyclonal antibody 857, specific for the C-terminal region of DMP1 (66), the kind gift of Chunlin Qin (Baylor College of Dentistry, Dallas, TX, USA); mouse monoclonal antibody against mouse β-catenin (BD Biosciences), rabbit polyclonal GAPDH (Sigma) and mouse monoclonal antibody to α-tubulin (Millipore). Secondary anti-rabbit and anti-mouse IgG antibodies both were from BioRad. Primary and secondary antibodies were diluted 1:1000 and 1:5000, respectively. Procollagen and α-tubulin blots, and the DMP1 blot on PVDF membrane, were visualized using the ECL kit, whereas all other blots were visualized using the ECL Plus kit (both kits were from GE Healthcare). All western blots were repeated using at least 3 different BTKO and control samples, each from a different mouse. Representative images are shown.
Transmission electron microscopy
Achilles tendons were fixed in 4% paraformaldehyde, 2.5% glutaraldehyde, 0.1 m sodium cacodylate and 8 mm CaCl2, adjusted to pH 7.4 with NaOH. After 15 min at room temperature, fixation continued at 4°C for 2 h, followed by transfer to buffer containing 0.4% paraformaldehyde, 0.25% glutaraldehyde, 10% sucrose and 0.1 m sodium cacodylate at 4°C. Tendons were then rinsed with cacodylate buffer and post-fixed 1 h with 1% osmium tetroxide, dehydrated in an ethanol series followed by 100% propylene oxide, infiltrated and embedded in a mixture of Embed 812, nadic methyl anhydride, dodecenylsuccinic anhydride and DMP-30 (EM Sciences) and polymerized overnight at 60°C. Ultra-thin, ∼90 nm sections were prepared using a Leica ultramicrotome and post-stained with 2% aqueous uranyl acetate and 1% phosphotungstic acid, pH 3.2. Sections were examined and imaged at 80 kV using a JEOL 1400 transmission electron microscope equipped with a Gatan Orius widefield side mount CC Digital camera. Bone was processed similarly, except that after fixation specimens were rinsed with cacodylate buffer and transferred to decalcification solution (10% EDTA in PBS, pH 7.4), containing 0.4% paraformaldehyde and 0.25 glutaraldehyde on a rocker at 4°C. Decalcification solution was changed every 2–3 days for 12 days.
Tendon diameter analysis was obtained from pooled data from one tendon from each of three mice of same genotype. Five or ten digital images from each tendon were taken from non-overlapping areas at ×50 000. Images were randomized and masked before fibril diameters were measured using a RM Biometrics-Bioquant Image Analysis System. A region of interest (ROI) of appropriate size was determined within the image so that a minimum of 60 fibrils were measured from each image. All fibrils in the ROI were measured per image. Fibril diameters were measured along the minor axis of the fibril cross section. Tendon diameter measurements were pooled into groups by genotype.
Reverse transcription PCR for detected excision of Bmp1 and Tll1 sequences
Disarticulated femora, soft tissue- and marrow-free, were frozen and homogenized to a fine powder in liquid nitrogen, using a porcelain mortar and pestle. RNA was extracted from the powder using TRIzol reagent (Life Sciences Technology). Reverse transcription (RT)–PCR was performed using primers for Tll1 (5′ CAGAGAGCCATGTTCAAGCA 3′, forward; and 5′ CTTGCAGGGTTTCTCCACAT 3′, reverse) andBmp1 (5′ TGGATGAGGAGGACTTGAGG 3′, forward; and 5′ GTGCTGTCTTGGAGGGTCTC 3′, reverse).
Serum biochemistry
Serum calcium was measured in 17-week-old male mice using a colorimetric calcium kit (Pointe Scientific, Inc.) following the manufacturer's instructions. Serum inorganic phosphorus was measured in 17-week-old male mice by the ammonium molybdate–ascorbic acid method of Chen et al. (67).
Statistical analysis
Where appropriate, data were analyzed using Fischer's t-test and P < 0.05 was considered significant. All data are recorded as average ± SEM. For mechanical testing, as well as length measurements, left and right femora of each mouse were analyzed separately and the values averaged.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants AR60636 and AR59685 (to S.-J.L.), AR54753 (to R.D.B.), AR44745 (to D.E.B.), DE018486 (to J.F.), and AR53815 and AR47746 (to D.S.G.) from the National Institutes of Health.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Laura Halverson Monahan of the University of Wisconsin-Madison Zoological Museum for her assistance in the preparation of skeletons using Dermestid beetles, and the laboratory of Ray Vanderby, of the Department of Orthopedics and Rehabilitation, for access to and assistance with their three-point bending apparatus.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Sillence D.O., Senn A., Danks D.M. Genetic heterogeneity in osteogenesis imperfecta. J. Med. Genet. 1979;16:101–116. doi: 10.1136/jmg.16.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byers P.H., Pyott S.M. Recessively inherited forms of osteogenesis imperfecta. Annu. Rev. Genet. 2012;46:475–497. doi: 10.1146/annurev-genet-110711-155608. [DOI] [PubMed] [Google Scholar]
- 3.Forlino A., Cabral W.A., Barnes A.M., Marini J.C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 2011;7:540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muir A., Greenspan D.S. Metalloproteinases in Drosophila to humans that are central players in developmental processes. J. Biol. Chem. 2011;286:41905–41911. doi: 10.1074/jbc.R111.299768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hopkins D.R., Keles S., Greenspan D.S. The bone morphogenetic protein 1/Tolloid-like metalloproteinases. Matrix Biol. 2007;26:508–523. doi: 10.1016/j.matbio.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wozney J.M., Rosen V., Celeste A.J., Mitsock L.M., Whitters M.J., Kriz R.W., Hewick R.M., Wang E.A. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242:1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 7.Piccolo S., Agius E., Lu B., Goodman S., Dale L., De Robertis E.M. Cleavage of Chordin by Xolloid metalloprotease suggests a role for proteolytic processing in the regulation of Spemann organizer activity. Cell. 1997;91:407–416. doi: 10.1016/S0092-8674(00)80424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blader P., Rastegar S., Fischer N., Strahle U. Cleavage of the BMP-4 antagonist chordin by zebrafish tolloid. Science. 1997;278:1937–1940. doi: 10.1126/science.278.5345.1937. [DOI] [PubMed] [Google Scholar]
- 9.Jasuja R., Voss N., Ge G., Hoffman G.G., Lyman-Gingerich J., Pelegri F., Greenspan D.S. bmp1 and mini fin are functionally redundant in regulating formation of the zebrafish dorsoventral axis. Mech. Dev. 2006;123:548–558. doi: 10.1016/j.mod.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Kessler E., Takahara K., Biniaminov L., Brusel M., Greenspan D.S. Bone morphogenetic protein-1: the type I procollagen C-proteinase. Science. 1996;271:360–362. doi: 10.1126/science.271.5247.360. [DOI] [PubMed] [Google Scholar]
- 11.Lindahl K., Barnes A.M., Fratzl-Zelman N., Whyte M.P., Hefferan T.E., Makareeva E., Brusel M., Yaszemski M.J., Rubin C.J., Kindmark A., et al. COL1 C-propeptide cleavage site mutations cause high bone mass osteogenesis imperfecta. Hum. Mutat. 2011;32:598–609. doi: 10.1002/humu.21475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asharani P.V., Keupp K., Semler O., Wang W., Li Y., Thiele H., Yigit G., Pohl E., Becker J., Frommolt P., et al. Attenuated BMP1 function compromises osteogenesis, leading to bone fragility in humans and zebrafish. Am. J. Hum. Genet. 2012;90:661–674. doi: 10.1016/j.ajhg.2012.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Glez V., Valencia M., Caparros-Martin J.A., Aglan M., Temtamy S., Tenorio J., Pulido V., Lindert U., Rohrbach M., Eyre D., et al. Identification of a mutation causing deficient BMP1/mTLD proteolytic activity in autosomal recessive osteogenesis imperfecta. Hum. Mutat. 2012;33:343–350. doi: 10.1002/humu.21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uzel M.I., Scott I.C., Babakhanlou-Chase H., Palamakumbura A.H., Pappano W.N., Hong H.H., Greenspan D.S., Trackman P.C. Multiple bone morphogenetic protein 1-related mammalian metalloproteinases process pro-lysyl oxidase at the correct physiological site and control lysyl oxidase activation in mouse embryo fibroblast cultures. J. Biol. Chem. 2001;276:22537–22543. doi: 10.1074/jbc.M102352200. [DOI] [PubMed] [Google Scholar]
- 15.Scott I.C., Imamura Y., Pappano W.N., Troedel J.M., Recklies A.D., Roughley P.J., Greenspan D.S. Bone morphogenetic protein-1 processes probiglycan. J. Biol. Chem. 2000;275:30504–30511. doi: 10.1074/jbc.M004846200. [DOI] [PubMed] [Google Scholar]
- 16.Steiglitz B.M., Ayala M., Narayanan K., George A., Greenspan D.S. Bone morphogenetic protein-1/Tolloid-like proteinases process dentin matrix protein-1. J. Biol. Chem. 2004;279:980–986. doi: 10.1074/jbc.M310179200. [DOI] [PubMed] [Google Scholar]
- 17.Feng J.Q., Ward L.M., Liu S., Lu Y., Xie Y., Yuan B., Yu X., Rauch F., Davis S.I., Zhang S., et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006;38:1310–1315. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge G., Greenspan D.S. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J. Cell Biol. 2006;175:111–120. doi: 10.1083/jcb.200606058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonewald L.F., Mundy G.R. Role of transforming growth factor-beta in bone remodeling. Clin. Orthop. Relat. Res. 1990;250:261–276. [PubMed] [Google Scholar]
- 20.Erlebacher A., Filvaroff E.H., Ye J.Q., Derynck R. Osteoblastic responses to TGF-beta during bone remodeling. Mol. Biol. Cell. 1998;9:1903–1918. doi: 10.1091/mbc.9.7.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki N., Labosky P.A., Furuta Y., Hargett L., Dunn R., Fogo A.B., Takahara K., Peters D.M., Greenspan D.S., Hogan B.L. Failure of ventral body wall closure in mouse embryos lacking a procollagen C-proteinase encoded by Bmp1, a mammalian gene related to Drosophila tolloid. Development. 1996;122:3587–3595. doi: 10.1242/dev.122.11.3587. [DOI] [PubMed] [Google Scholar]
- 22.Clark T.G., Conway S.J., Scott I.C., Labosky P.A., Winnier G., Bundy J., Hogan B.L., Greenspan D.S. The mammalian Tolloid-like 1 gene, Tll1, is necessary for normal septation and positioning of the heart. Development. 1999;126:2631–2642. doi: 10.1242/dev.126.12.2631. [DOI] [PubMed] [Google Scholar]
- 23.Pappano W.N., Steiglitz B.M., Scott I.C., Keene D.R., Greenspan D.S. Use of Bmp1/Tll1 doubly homozygous null mice and proteomics to identify and validate in vivo substrates of bone morphogenetic protein 1/tolloid-like metalloproteinases. Mol. Cell Biol. 2003;23:4428–4438. doi: 10.1128/MCB.23.13.4428-4438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott I.C., Blitz I.L., Pappano W.N., Imamura Y., Clark T.G., Steiglitz B.M., Thomas C.L., Maas S.A., Takahara K., Cho K.W., et al. Mammalian BMP-1/tolloid-related metalloproteinases, including novel family member mammalian Tolloid-like 2, have differential enzymatic activities and distributions of expression relevant to patterning and skeletogenesis. Dev. Biol. 1999;213:283–300. doi: 10.1006/dbio.1999.9383. [DOI] [PubMed] [Google Scholar]
- 25.Takahara K., Lyons G.E., Greenspan D.S. Bone morphogenetic protein-1 and a mammalian tolloid homologue (mTld) are encoded by alternatively spliced transcripts which are differentially expressed in some tissues. J. Biol. Chem. 1994;269:32572–32578. [PubMed] [Google Scholar]
- 26.Takahara K., Brevard R., Hoffman G.G., Suzuki N., Greenspan D.S. Characterization of a novel gene product (mammalian tolloid-like) with high sequence similarity to mammalian tolloid/bone morphogenetic protein-1. Genomics. 1996;34:157–165. doi: 10.1006/geno.1996.0260. [DOI] [PubMed] [Google Scholar]
- 27.Lee S.J. Genetic analysis of the role of proteolysis in the activation of latent myostatin. PLoS One. 2008;3:e1628. doi: 10.1371/journal.pone.0001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grgurevic L., Macek B., Mercep M., Jelic M., Smoljanovic T., Erjavec I., Dumic-Cule I., Prgomet S., Durdevic D., Vnuk D., et al. Bone morphogenetic protein (BMP)1–3 enhances bone repair. Biochem. Biophys Res. Commun. 2011;408:25–31. doi: 10.1016/j.bbrc.2011.03.109. [DOI] [PubMed] [Google Scholar]
- 29.Ruzankina Y., Pinzon-Guzman C., Asare A., Ong T., Pontano L., Cotsarelis G., Zediak V.P., Velez M., Bhandoola A., Brown E.J. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfman N.M., McPherron A.C., Pappano W.N., Davies M.V., Song K., Tomkinson K.N., Wright J.F., Zhao L., Sebald S.M., Greenspan D.S., et al. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc. Natl. Acad. Sci. USA. 2003;100:15842–15846. doi: 10.1073/pnas.2534946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamrick M.W., McPherron A.C., Lovejoy C.O., Hudson J. Femoral morphology and cross-sectional geometry of adult myostatin-deficient mice. Bone. 2000;27:343–349. doi: 10.1016/S8756-3282(00)00339-2. [DOI] [PubMed] [Google Scholar]
- 32.Solverson P., Murali S.G., Litscher S.J., Blank R.D., Ney D.M. Low bone strength is a manifestation of phenylketonuria in mice and is attenuated by a glycomacropeptide diet. PLoS One. 2012;7:e45165. doi: 10.1371/journal.pone.0045165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner C.H., Burr D.B. Basic biomechanical measurements of bone: a tutorial. Bone. 1993;14:595–608. doi: 10.1016/8756-3282(93)90081-K. [DOI] [PubMed] [Google Scholar]
- 34.Lu Y., Yuan B., Qin C., Cao Z., Xie Y., Dallas S.L., McKee M.D., Drezner M.K., Bonewald L.F., Feng J.Q. The biological function of DMP-1 in osteocyte maturation is mediated by its 57-kDa C-terminal fragment. J. Bone Miner. Res. 2011;26:331–340. doi: 10.1002/jbmr.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klein-Nulend J., Bakker A.D., Bacabac R.G., Vatsa A., Weinbaum S. Mechanosensation and transduction in osteocytes. Bone. 2013;54:182–190. doi: 10.1016/j.bone.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 36.Steiglitz B.M., Kreider J.M., Frankenburg E.P., Pappano W.N., Hoffman G.G., Meganck J.A., Liang X., Hook M., Birk D.E., Goldstein S.A., et al. Procollagen C proteinase enhancer 1 genes are important determinants of the mechanical properties and geometry of bone and the ultrastructure of connective tissues. Mol. Cell Biol. 2006;26:238–249. doi: 10.1128/MCB.26.1.238-249.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye L., Mishina Y., Chen D., Huang H., Dallas S.L., Dallas M.R., Sivakumar P., Kunieda T., Tsutsui T.W., Boskey A., et al. Dmp1-deficient mice display severe defects in cartilage formation responsible for a chondrodysplasia-like phenotype. J. Biol. Chem. 2005;280:6197–6203. doi: 10.1074/jbc.M412911200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y., Prasad M., Gao T., Wang X., Zhu Q., D'Souza R., Feng J.Q., Qin C. Failure to process dentin matrix protein 1 (DMP1) into fragments leads to its loss of function in osteogenesis. J. Biol. Chem. 2010;285:31713–31722. doi: 10.1074/jbc.M110.137059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ling Y., Rios H.F., Myers E.R., Lu Y., Feng J.Q., Boskey A.L. DMP1 depletion decreases bone mineralization in vivo: an FTIR imaging analysis. J. Bone Miner. Res. 2005;20:2169–2177. doi: 10.1359/JBMR.050815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X., Zhang Y., Kang H., Liu W., Liu P., Zhang J., Harris S.E., Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005;280:19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 41.Baron R., Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 2013;19:179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 42.Eyre D.R., Weis M.A. Bone collagen: new clues to its mineralization mechanism from recessive osteogenesis imperfecta. Calcif Tissue Int. 2013;93:338–347. doi: 10.1007/s00223-013-9723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Canty E.G., Lu Y., Meadows R.S., Shaw M.K., Holmes D.F., Kadler K.E. Coalignment of plasma membrane channels and protrusions (fibripositors) specifies the parallelism of tendon. J. Cell Biol. 2004;165:553–563. doi: 10.1083/jcb.200312071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang R., Lu Y., Ye L., Yuan B., Yu S., Qin C., Xie Y., Gao T., Drezner M.K., Bonewald L.F., et al. Unique roles of phosphorus in endochondral bone formation and osteocyte maturation. J. Bone Miner. Res. 2011;26:1047–1056. doi: 10.1002/jbmr.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uveges T.E., Collin-Osdoby P., Cabral W.A., Ledgard F., Goldberg L., Bergwitz C., Forlino A., Osdoby P., Gronowicz G.A., Marini J.C. Cellular mechanism of decreased bone in Brtl mouse model of OI: imbalance of decreased osteoblast function and increased osteoclasts and their precursors. J. Bone Miner. Res. 2008;23:1983–1994. doi: 10.1359/jbmr.080804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frost H.M. Bone ‘mass’ and the ‘mechanostat’: a proposal. Anat. Rec. 1987;219:1–9. doi: 10.1002/ar.1092190104. [DOI] [PubMed] [Google Scholar]
- 47.Tu X., Rhee Y., Condon K.W., Bivi N., Allen M.R., Dwyer D., Stolina M., Turner C.H., Robling A.G., Plotkin L.I., et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50:209–217. doi: 10.1016/j.bone.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonewald L.F., Johnson M.L. Osteocytes, mechanosensing and Wnt signaling. Bone. 2008;42:606–615. doi: 10.1016/j.bone.2007.12.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin G.L., Hankenson K.D. Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J. Cell Biochem. 2011;112:3491–3501. doi: 10.1002/jcb.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grgurevic L., Macek B., Healy D.R., Brault A.L., Erjavec I., Cipcic A., Grgurevic I., Rogic D., Galesic K., Brkljacic J., et al. Circulating bone morphogenetic protein 1–3 isoform increases renal fibrosis. J. Am. Soc. Nephrol. 2011;22:681–692. doi: 10.1681/ASN.2010070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pollitt R., McMahon R., Nunn J., Bamford R., Afifi A., Bishop N., Dalton A. Mutation analysis of COL1A1 and COL1A2 in patients diagnosed with osteogenesis imperfecta type I-IV. Hum. Mutat. 2006;27:716. doi: 10.1002/humu.9430. [DOI] [PubMed] [Google Scholar]
- 52.Tybulewicz V.L., Crawford C.E., Jackson P.K., Bronson R.T., Mulligan R.C. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-M. [DOI] [PubMed] [Google Scholar]
- 53.Shalaby F., Rossant J., Yamaguchi T.P., Gertsenstein M., Wu X.F., Breitman M.L., Schuh A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 54.Struhl G., Basler K. Organizing activity of wingless protein in Drosophila. Cell. 1993;72:527–540. doi: 10.1016/0092-8674(93)90072-X. [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez C.I., Buchholz F., Galloway J., Sequerra R., Kasper J., Ayala R., Stewart A.F., Dymecki S.M. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- 56.McLeod M.J. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology. 1980;22:299–301. doi: 10.1002/tera.1420220306. [DOI] [PubMed] [Google Scholar]
- 57.Saless N., Litscher S.J., Lopez Franco G.E., Houlihan M.J., Sudhakaran S., Raheem K.A., O'Neil T.K., Vanderby R., Demant P., Blank R.D. Quantitative trait loci for biomechanical performance and femoral geometry in an intercross of recombinant congenic mice: restriction of the Bmd7 candidate interval. FASEB J. 2009;23:2142–2154. doi: 10.1096/fj.08-118679. [DOI] [PubMed] [Google Scholar]
- 58.Hong S.H., Jiang X., Chen L., Josh P., Shin D.G., Rowe D. Computer-automated static, dynamic and cellular bone histomorphometry. J. Tissue Sci. Eng. 2012;S1:004. doi: 10.4172/2157-7552.S1-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogbureke K.U., Fisher L.W. Expression of SIBLINGs and their partner MMPs in salivary glands. J. Dent. Res. 2004;83:664–670. doi: 10.1177/154405910408300902. [DOI] [PubMed] [Google Scholar]
- 60.Zhang K., Barragan-Adjemian C., Ye L., Kotha S., Dallas M., Lu Y., Zhao S., Harris M., Harris S.E., Feng J.Q., et al. E11/gp38 selective expression in osteocytes: regulation by mechanical strain and role in dendrite elongation. Mol. Cell Biol. 2006;26:4539–4552. doi: 10.1128/MCB.02120-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller S.C., Omura T.H., Smith L.J. Changes in dentin appositional rates during pregnancy and lactation in rats. J. Dent. Res. 1985;64:1062–1064. doi: 10.1177/00220345850640080701. [DOI] [PubMed] [Google Scholar]
- 62.Martin D.M., Hallsworth A.S., Buckley T. A method for the study of internal spaces in hard tissue matrices by SEM, with special reference to dentine. J. Microsc. 1978;112:345–352. doi: 10.1111/j.1365-2818.1978.tb00085.x. [DOI] [PubMed] [Google Scholar]
- 63.Ascenzi M.G., Roe A.K. The osteon: the micromechanical unit of compact bone. Front. Biosci. (Landmark Ed) 2012;17:1551–1581. doi: 10.2741/4003. [DOI] [PubMed] [Google Scholar]
- 64.Cao Z., Zhang H., Zhou X., Han X., Ren Y., Gao T., Xiao Y., de Crombrugghe B., Somerman M.J., Feng J.Q. Genetic evidence for the vital function of Osterix in cementogenesis. J. Bone Miner. Res. 2012;27:1080–1092. doi: 10.1002/jbmr.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fisher L.W., Stubbs J.T., 3rd, Young M.F. Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta. Orthop. Scand. Suppl. 1995;266:61–65. [PubMed] [Google Scholar]
- 66.Maciejewska I., Cowan C., Svoboda K., Butler W.T., D'Souza R., Qin C. The NH2-terminal and COOH-terminal fragments of dentin matrix protein 1 (DMP1) localize differently in the compartments of dentin and growth plate of bone. J. Histochem. Cytochem. 2009;57:155–166. doi: 10.1369/jhc.2008.952630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen P.S., Toribara T.Y., Warner H. Microdetermination of phosphorus. Anal. Chem. 1956;28:1756–1758. doi: 10.1021/ac60119a033. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.