

Abstract
In Duchenne muscular dystrophy (DMD) patients and the mouse model of DMD, mdx, dystrophin deficiency causes a decrease and mislocalization of muscle-specific neuronal nitric oxide synthase (nNOSμ), leading to functional impairments. Previous studies have shown that nitric oxide (NO) donation associated with anti-inflammatory action has beneficial effects in dystrophic mouse models. In this study, we have systematically investigated the effects of naproxcinod, an NO-donating naproxen derivative, on the skeletal and cardiac disease phenotype in mdx mice. Four-week-old mdx and C57BL/10 mice were treated with four different concentrations (0, 10, 21 and 41 mg/kg) of naproxcinod and 0.9 mg/kg of prednisolone in their food for 9 months. All mice were subjected to twice-weekly treadmill sessions, and functional and behavioral parameters were measured at 3, 6 and 9 months of treatment. In addition, we evaluated in vitro force contraction, optical imaging of inflammation, echocardiography and blood pressure (BP) at the 9-month endpoint prior to sacrifice. We found that naproxcinod treatment at 21 mg/kg resulted in significant improvement in hindlimb grip strength and a 30% decrease in inflammation in the fore- and hindlimbs of mdx mice. Furthermore, we found significant improvement in heart function, as evidenced by improved fraction shortening, ejection fraction and systolic BP. In addition, the long-term detrimental effects of prednisolone typically seen in mdx skeletal and heart function were not observed at the effective dose of naproxcinod. In conclusion, our results indicate that naproxcinod has significant potential as a safe therapeutic option for the treatment of muscular dystrophies.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is the most common X-linked inherited disease (1), affecting 1 in 3500 live male births in all populations with 30% of all cases resulting from de novo mutation (2). DMD is caused by a mutation in the dystrophin gene that leads to the absence of dystrophin protein in the skeletal muscle (3). Dystrophin is a subsarcolemmal protein involved in the link between the contractile machinery and the extracellular matrix; in the absence of dystrophin, the transmembrane dystrophin–glycoprotein complex (DGC) components are lost, and the stability of the sarcolemma is reduced (4,5). The loss of this force transmission system results in an increased fragility of the muscle fibers, which is believed to be responsible for the susceptibility of dystrophic muscle fibers to damage (6–9). Thus, it is thought that in dystrophic muscle, mechanical stress occurring during contraction can result in transient tears in the muscle sarcolemma (10), allowing an influx of extracellular calcium that lead to muscle necrosis and secondary inflammation (11–14). This mechanical damage results in elevated levels of creatine kinase (CK) that can be detected in the serum of both DMD patients and mdx mice (the mouse model of DMD) or by the presence of exogenous marker dye (Evans blue) within the muscles (15–17).
The loss of dystrophin also results in a significant reduction in another member of the DGC, neuronal nitric oxide synthase µ (nNOSµ) (18,19). Nitric oxide (NO) derived from nNOSμ plays a critical role in the physiology of the skeletal muscle, regulating force generation, muscle mass, fatigue, muscle repair from injury, oxidative stress and blood flow (20,21). Therefore, the loss of nNOSµ is believed to contribute significantly to dystrophic pathology (22–25). It has been recently reported that dystrophins carrying spectrin-like repeats 16 and 17 (R16/17) anchor nNOS to the sarcolemma (22,26). Restoring this portion of the dystrophin gene (R16/17) in mdx mice showed better efficacy in ameliorating histological muscle pathology and skeletal muscle function than all the mini-/microgene therapies that do not restore nNOS function (22).
Naproxcinod is a cyclooxygenase-inhibiting compound that has both analgesic and anti-inflammatory effects and was originally developed for the treatment of osteoarthritis (27). After absorption, the compound is metabolized to naproxen and nitroxybutyl ester, which allows it to also act as an NO donor. This mechanism of action makes naproxcinod the first in a new class of drugs, the cyclooxygenase-inhibiting nitric oxide donators (CINODs), which are postulated to have an analgesic efficacy similar to that of traditional non-steroidal anti-inflammatory drugs (NSAIDs), but with fewer gastrointestinal and cardiovascular side effects (28). In fact, lower cardiovascular side effects were demonstrated in a clinical trial in which 916 osteoarthritis patients were treated with naproxcinod for 13 weeks. These patients showed BP profiles similar to those of the patients receiving placebo, whereas the group treated with naproxen displayed elevations in BP (29). In addition, two Phase II randomized controlled trials showed a decrease in systolic BP of 2.1 mmHg when patients took naproxcinod (375 or 750 mg twice daily) for 6 weeks (28,30). Interestingly, it has also been reported that compounds belonging to the CINOD class lead to a significant, persistent recovery of muscle function in a number of models of muscular dystrophy. Specifically, long-term treatment of dystrophic mice (mdx or alpha-sarcoglycan-null mice) with two different CINODs, HCT 1026 (a NO-donating flurbiprofen) and NCX 320 (a NO-donating ibuprofen), substantially improved muscle morphology, and reduced muscle necrosis, inflammation and muscle ischemia (31–33). However, both HCT 1026 and NCX 320 are still in early stages of development and are not yet ready to be used in patients.
To date, the most effective form of medication for patients with DMD is represented by glucocorticoids (GCs) (34). Prednisone and its derivative prednisolone have been shown to provide some level of clinical benefit and are therefore the most widely used treatment for DMD (34,35). However, their mechanism of action remains unknown, and there are significant adverse side effects associated with their use (36,37). Therefore, alternative medications with reduced side effects are still needed.
Given the beneficial effects of the NO-donating ibuprofen and flurbiprofen derivatives on both heart and skeletal muscle, together with the reduction in the well-recognized adverse side-effect profile associated with the use of traditional NSAIDs, it is expected that naproxcinod will provide an effective therapy for DMD patients. The aim of the current study was to test the long-term use of naproxcinod versus prednisolone (currently the only standard of care for DMD patients) in young mdx mice (4 weeks of age). We found that long-term treatment of mdx mice with naproxcinod produced significant improvement in muscle strength, histology and heart function, with none of the side effects commonly associated with prednisolone.
RESULTS
Effect of naproxcinod treatment on the body weight (BW) of mdx mice
Since our drug was administered in food, we measured the average food intake/mouse/week. We found that all drug-treated mice consumed more food than did the mdx or wild-type (WT) vehicle-treated mice; however, both the 21 mg/kg/day naproxcinod- and prednisolone-treated groups had a significantly higher average food intake than did the mdx vehicle-treated mice at 42 weeks of age (9 months after the initiation of the treatments). No statistical differences were observed between the other groups at this time point (Fig. 1).
Figure 1.

Analysis of food consumption by WT and mdx mice at 42 weeks of age following 9 months of treatment. Data are expressed as means ± S.E.M. (n = 10 animals for the WT and n = 15 animals for the mdx vehicle-, naproxcinod- and prednisolone-treated mice). **P ≤ 0.01, comparing mdx vehicle-treated mice and mdx mice treated with 21 mg/kg/day naproxcinod or 0.9 mg/kg/day prednisolone.
Consistent with their significantly higher food intake, the mdx group treated with 21 mg/kg/day naproxcinod showed a significantly higher mean BW (7.3%) than did the mdx vehicle-treated group. Prednisolone-treated mice, despite their increased food intake, showed a significantly lower BW (−10.1%) when compared with the mdx vehicle-treated group. There was no difference in BW between the mdx vehicle and WT groups at 42 weeks of age (Fig. 2). The same trend was also observed when we analyzed the percent change in BW from 5 to 42 weeks of age (Supplementary Material, Table S1).
Figure 2.

BW measurement of WT and mdx mice treated with either vehicle, or various concentrations of naproxcinod or prednisolone at 42 weeks of age following 9 months of treatment. Data are expressed as means ± S.E.M. (n = 10 animals for the WT mice and n = 15 animals for mdx vehicle-, naproxcinod- and prednisolone-treated mdx mice). *P ≤ 0.05 and **P ≤ 0.01, comparing mdx vehicle-treated mice and mdx mice treated with 21 mg/kg/day naproxcinod or 0.9 mg/kg/day prednisolone.
Effect of naproxcinod on muscle grip strength measurements (GSM) in mdx mice
Forelimb
As expected, the mdx vehicle-treated group showed a significant reduction in both absolute and normalized forelimb grip strength when compared with the WT vehicle-treated group at 42 weeks of age. Long-term treatment with prednisolone induced an additional significant reduction in forelimb grip strength. No statistically significant differences between the naproxcinod-treated groups and the mdx vehicle-treated group were observed at 42 weeks of age, or when the data were analyzed as the percentage change in absolute and normalized forelimb grip strength from 18 to 42 weeks of age (Supplementary Material, Table S2 and S3).
Hindlimb
Both the absolute and the normalized hindlimb grip strengths of the mdx vehicle-treated group were significantly lower than those of the WT group at 42 weeks of age. Like its effect on forelimb grip strength, prednisolone also significantly decreased the hindlimb grip strength in prednisolone-treated mice when compared with mdx vehicle-treated mice. No difference in either the absolute or normalized hindlimb grip strength was seen between any naproxcinod-treated groups and the mdx vehicle-treated group at 42 weeks of age (Supplementary Material, Table S2). However, the average percentage change in both the absolute and normalized hindlimb grip strength from 18 to 42 weeks of age was significantly higher in both the 10 and 21 mg/kg/day naproxcinod-treated groups than in the mdx vehicle-treated group, with the 21 mg/kg/day group exhibiting a higher percentage change than the 10 mg/kg/day group (Table 1).
Table 1.
Analysis of the percent change in absolute and normalized hindlimb GSM over time (each mdx drug-treated group compared only with mdx vehicle-treated mice). All models include only main effects, no interaction terms (models contain a quadratic term if appropriate).
| Measurement | mdx—pred. 0.9 mg | mdx—nap. 10 mg | mdx—nap. 21 mg | mdx—nap. 41 mg |
|---|---|---|---|---|
| % Change in GSM hindlimb | −0.837 (0.772) | 6.421 (0.025) | 9.226 (0.001) | 4.040 (0.165) |
| % Change in normalized GSM hindlimb | 3.169 (0.292) | 9.577 (0.001) | 12.336 (<0.001) | 5.489 (0.069) |
Effect of naproxcinod on cardiac function of mdx mice
At 42 weeks of age, the mdx mice had significantly lower BP (as measured by the non-invasive tail cuff) and compromised cardiac function (as assessed by fraction shortening and ejection fraction), than did the WT mice. Treatment with 10 and 21 mg/kg/day naproxcinod resulted in a significant 15.8 and 10.4% improvement in systolic BP, respectively, over that of the mdx vehicle-treated group (Fig. 3A). Furthermore, the fraction shortening (7.3 and 10.1% at 10 and 20 mg/kg/day respectively, versus the mdx vehicle-treated group) and ejection fraction (7.3% at 20 mg/kg/day versus the mdx vehicle-treated group) were also significantly improved following a 9-month treatment with naproxcinod (Fig. 3B and C). Conversely, the mdx prednisolone-treated group displayed significantly higher BP; a 54% increase over that of the mdx vehicle-treated group. This increase was also found to be significantly higher compared to WT mice (Fig. 3A). Also, both the fraction shortening (−9.0%) and ejection fraction (−7.4%) were significantly lower in the prednisolone-treated group than in the mdx vehicle-treated group and lower than either naproxcinod-treated group (Fig. 3B and C). These results indicate that long-term dosage with prednisolone has significant adverse side effects on heart function.
Figure 3.

BP (A) and echocardiography measurements (B and C) for WT and mdx mice treated with either vehicle, one of various concentrations of naproxcinod, or 0.9 mg/kg/day prednisolone at 42 weeks of age following 9 months of treatment. Data are expressed as means ± S.E.M. For systolic BP measurements: n = 8 animals for mdx vehicle-treated mice and 10 mg/kg/day naproxcinod-treated mice; n = 6 animals for 0.9 mg/kg/day prednisolone-, 21 mg/kg/day naproxcinod- and 41 mg/kg/day naproxcinod-treated mice; n = 10 animals for WT. For echocardiography: n = 8 animals for mdx vehicle-treated mice and 10 mg/kg/day naproxcinod-treated mice; n = 5 animals for 0.9 mg/kg/day prednisolone; n = 7 animals for 21 mg/kg/day naproxcinod- and 41 mg/kg/day naproxcinod-treated mice; n = 5 animals for WT. *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001, comparing mdx vehicle-treated mice and various treatment groups. The percentages above each of the plots indicate the percentage change from the mdx vehicle-treated group.
Effect of naproxcinod on inflammation in the limb muscles of mdx mice, as measured by optical imaging
To examine the effect of naproxcinod on inflammation in the forelimbs and hindlimbs of live treated and non-treated mice, we employed an optical imaging strategy. As expected, a significant increase in both fore- and hindlimb cathepsin activity was observed after 9 months of drug treatment in the mdx vehicle-treated mice when compared with the WT mice, indicating an increase in inflammation in the vehicle-treated mice. The naproxcinod-treated groups and the prednisolone-treated group showed a significant reduction in both fore- and hindlimb cathepsin activity when compared with the mdx vehicle-treated group, with the 21 mg/kg/day naproxcinod-treated group achieving the highest reduction in inflammation (Fig. 4A and B).
Figure 4.

Optical imaging of the forelimbs and hindlimbs of WT and mdx mice treated with either vehicle, various concentrations of naproxcinod or 0.9 mg/kg/day prednisolone at 42 weeks of age following 9 months of treatment. (A) Forelimb photon counts and (B) hindlimb photon counts were used as a direct measure of inflammation in live animals. Data are expressed as means ± S.E.M. (n = 7 animals for mdx vehicle-treated mice and 21 mg/kg/day naproxcinod-treated mice; n = 6 animals for 0.9 mg/kg/day prednisolone-, 10 mg/kg/day naproxcinod- and 41 mg/kg/day naproxcinod-treated mice; n = 5 animals for WT). ***P ≤ 0.001, comparing mdx vehicle-treated mice and various treatment groups. The percentages above each of the plots indicate the percentage change from the mdx vehicle-treated group.
Effect of naproxcinod on histopathological features in the diaphragms of mdx mice
Histological assessment of hematoxylin and eosin (H&E)-stained sections for inflammation and other parameters (total fiber number, number of central nucleated muscle fibers, regeneration and degeneration) was performed on the diaphragm muscle after 9 months of drug treatment. Evaluation of the diaphragm showed that there was a significant reduction in inflammatory cell infiltration after treatment with 21 mg/kg/day naproxcinod (Fig. 5A and B). We did not detect any reduction in inflammation in the prednisolone-treated group. The 21 mg/kg/day naproxcinod-treated group also showed a significant reduction in the total fiber number and number of regenerating muscle fibers and a marked reduction in the number of central nucleated muscle fibers (−26.1%) in the diaphragm when compared with mdx vehicle-treated mice, but showed no change in the number of degenerating fibers (Fig. 6A–D).
Figure 5.
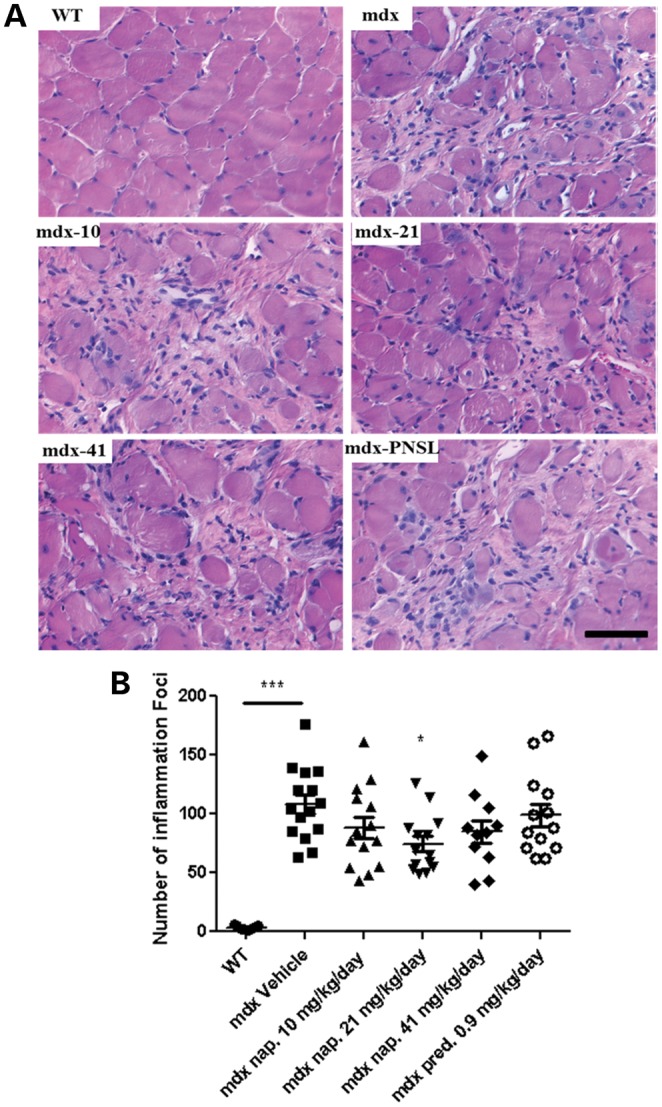
Quantification of inflammation in the diaphragm of WT and mdx mice treated with either vehicle, various concentrations of naproxcinod or 0.9 mg/kg/day prednisolone at 42 weeks of age following 9 months of treatment. (A) Representative histological images of the diaphragm and (B) quantification of the number of inflammatory foci within the diaphragm to determine the effects of the treatment on the inflammation. Data are expressed as means ± S.E.M. (n = 7 for mdx vehicle-treated mice and 21 mg/kg/day naproxcinod-treated mice; n = 6 for 0.9 mg/kg/day prednisolone-, 10 mg/kg/day naproxcinod- and 41 mg/kg/day naproxcinod-treated mice; n = 5 for WT). *P ≤ 0.05 and ***P ≤ 0.001, comparing mdx vehicle-treated mice and various treatment groups. Bar = 200 µm.
Figure 6.

Histological quantification of the total fiber number (A), number of centrally nucleated muscle fibers (B), number of degenerating muscle fibers (C) and number of regenerating fibers (D) in mdx vehicle-treated mice and mdx mice treated with 21 mg/kg/day naproxcinod. Data are expressed as means ± S.E.M. (n = 7 for mdx vehicle-treated mice and n = 10 for 21 mg/kg/day naproxcinod-treated mice). *P ≤ 0.05, comparing mdx vehicle-treated mice and mdx mice treated with 21 mg/kg/day.
Effect of naproxcinod on heart fibrosis in mdx mice
Since naproxcinod showed beneficial effects on the heart function of mdx mice, we performed Picro-Sirius red staining to assess the level of fibrosis in the hearts of these mice. We found a reduction in the level of heart fibrosis (−26%) in the 21 mg/kg/day-treated group when compared with the mdx vehicle-treated group, although this reduction did not reach statistical significance because of inter- and intra-group variation (Fig. 7A and B).
Figure 7.
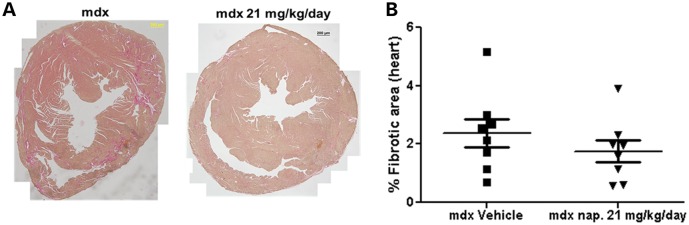
Quantification of the area of fibrosis in the heart of mdx mice and mdx mice treated with 21 mg/kg/day naproxcinod at 42 weeks of age following 9 months of treatment. (A) Representative images of the heart from mdx vehicle-treated mice and mdx mice treated with 21 mg/kg/day naproxcinod. (B) Quantification of the area of fibrosis in the heart, expressed as a percentage of the whole heart. There was a 26% mean reduction in the fibrosis in the hearts of mdx mice treated with 21 mg/kg/day naproxcinod as compared with mdx vehicle-treated mice but this difference did not reach statistical significance. Data are expressed as means ± S.E.M. (n = 8 for mdx vehicle-treated mice and 21 mg/kg/day naproxcinod-treated mice). Bar = 200 µm.
Effect of naproxcinod treatment on behavioral activities as determined by open-field activity
No significant difference was observed between the mdx vehicle-treated group and the WT group in terms of behavioral activity evaluated at 42 weeks of age, except for vertical activity, in which the mdx vehicle-treated group performed significantly worse than the WT group. No difference was detected in behavioral activity at 42 weeks of age between any of the naproxcinod-treated groups and the mdx vehicle-treated group. Conversely, the 0.9 mg/kg/day prednisolone-treated group showed significantly higher activity levels for all the parameters than did the mdx vehicle-treated group (Supplementary Material, Fig. S1A–E). Although the changes were not statistically significant because of the large inter- and intra-group variation at 42 weeks of age, the 21 mg/kg/day naproxcinod-treated mdx mice exhibited the highest positive percentage change when compared with the mdx vehicle-treated mice in term of horizontal activity (21.5%), total distance traveled (32.9%), vertical activity (48%), movement time (33%) and the lowest rest time (−2.6%; Supplementary Material, Fig. S1A–E). When the percentage change in the activity levels from 18 to 42 weeks of age was considered, we saw no significant difference between any of the various treatment groups and the mdx vehicle-treated group, or between the mdx vehicle-treated group and the WT group (Supplementary Material, Table S4).
Effect of naproxcinod on the in vitro force of the extensor digitorum longus (EDL) muscle and serum CK of mdx mice
At 42 weeks of age, naproxcinod-treated mice showed a significant increase in EDL muscle mass when compared with mdx vehicle-treated mice (Supplementary Material, Fig. S2A). Despite this increase in muscle mass, there was no significant difference in maximal (Supplementary Material, Fig. S2B) or specific force (Supplementary Material, Fig. S2C) between naproxcinod-treated mdx mice and mdx vehicle-treated mice. However, both the 10 and 41 mg/kg/day naproxcinod-treated mice showed a trend toward increased strength, with a 6.4 and 9.1% improvement in the maximal EDL force over the mdx vehicle-treated mice, respectively. Conversely, the prednisolone-treated mice showed a significant reduction in the maximal force (−22.9%) when compared with the mdx vehicle-treated group (Supplementary Material, Fig. S2B). Comparison of in vitro force measurements between WT and mdx vehicle-treated mice showed a significant decrease in the specific force of the mdx vehicle-treated mice when compared with WT mice (Supplementary Material, Fig. S2C).
The EDL muscle was also subjected to a fatigue protocol to assess its level of fatigue resistance and recovery in response to naproxcinod treatment. There was no difference in the induced decline in measured force between the drug-treated mdx groups and the mdx vehicle-treated group. However, as expected, the EDL muscles from the mdx vehicle-treated mice fatigued at a significantly faster rate than did the EDL muscles from the WT mice. In contrast, there was no improvement in the rate of recovery between the mdx vehicle-treated mice and the WT mice, or between the mdx drug-treated groups and the mdx vehicle-treated group (Supplementary Material, Fig. S2D).
Serum CK was significantly elevated in the mdx vehicle-treated group when compared with the WT group. However, there was no improvement in the CK levels when the mdx mice were treated with any of the concentrations of naproxcinod or with prednisolone (Supplementary Material, Fig. S3).
DISCUSSION
In this study, we investigated the effect of long-term treatment with naproxcinod on both skeletal and cardiac function in the mdx mouse model of DMD. The results presented here indicate that long-term treatment of mdx mice with naproxcinod significantly improves the disease phenotype. In addition, these beneficial effects persisted throughout the disease state (9 months of treatment), without the adverse side effects observed with prednisolone.
Treatment of the mdx mice with naproxcinod for 9 months resulted in an increase in BW (a simple measure of the overall drug effect on the phenotype of a mouse) and food consumption in these mice. Increased food consumption suggests an increase in appetite as the result of the treatment, perhaps as a result of an improvement in skeletal muscle function and thereby in their behavioral activity. The maximal food consumption was observed in the mdx group treated with 21 mg/kg/day naproxcinod, along with the maximal increase in BW observed for this group. Apart from the 21 mg/kg/day naproxcinod-treated group, the prednisolone-treated mice consumed a greater amount of food than any other group; however, they weighed significantly less at the end of the study (42 weeks of age). These findings suggest that prednisolone may increase the appetite of the mice, but it adversely affect them, leading to a significant reduction in BW. A decrease in BW and increase in appetite as the result of prednisone/prednisolone treatment has been previously documented (37–41). One of the known mechanisms of GCs is the induction of catabolic effects in skeletal muscle through the activation of genes encoding ubiquitin ligases such as Muscle RING Finger 1 (MuRF1) and Muscle Atrophy F-box (MAFbx), which in turn induce muscle atrophy (42,43). This finding, together with stunted growth, another well-documented side effect of GCs (41), may have outweighed the effect of increased food intake and resulted in weight loss rather than weight gain in the prednisolone-treated mice.
The beneficial effect of naproxcinod also extended to an improvement in muscle function. Hindlimb grip strength was significantly improved in the mdx naproxcinod-treated mice (10 and 21 mg/kg/day groups) when compared with the mdx mice treated with vehicle. The improvement in hindlimb function may be explained by the ability of NO donors to alleviate the muscle ischemia, a defect often associated with the loss of sarcolemmal nNOS in the dystrophic muscle fibers (32). Sarcolemma-targeted nNOS attenuates α-adrenergic vasoconstriction in contracting muscles and improves muscle perfusion during exercise (22). This process is defective in both mdx mice and patients with DMD (23,24), thus promoting fatigue and injury of dystrophic muscles. In addition, the optical imaging results (a direct measure of inflammation in a live animal) indicated that there was a significant reduction in inflammation in both the fore- and hindlimb muscles of the naproxcinod-treated mice. This finding may also explain the improvement in the hindlimb grip strength measurement. Despite the significant reduction in inflammation in the forelimb muscles, we did not detect any significant improvement in their grip strength. It is possible that the compound affects different skeletal muscle groups differently. Prednisolone-treated mice, on the other hand, performed significantly worse in both fore- and hindlimb grip strength at all-time points (18–42 weeks) despite having a significant reduction in inflammation, as measured by optical imaging. The effect of prednisolone/prednisone on the grip strength of mdx mice has been extensively studied. Previous studies have indicated that low-dose and short-term treatment with prednisolone/prednisone appears to improve grip strength in mdx mice (40,44–46). We believe that in our study, the deterioration in grip strength in the prednisolone-treated mice is a result of the negative side effects associated with long-term treatment with prednisolone. One of the side effects of prednisolone is the induction of muscle atrophy (47), which may lead to a reduction in force production.
The next, and likely most important, aspect of the search for a better DMD treatment is to find a way to improve cardiac function, since patients with DMD eventually die of cardiac failure. The progression of the DMD disease in cardiac muscle is much slower than in skeletal muscle and is characterized by reduced systolic function and cardiac arrhythmias (48). Unlike skeletal muscle, cardiac muscle is incapable of undergoing regeneration, since it lacks stem cells akin to the satellite cells of skeletal muscle (49). Consistent with previous findings, the mdx mice in our study had significantly reduced cardiac function when compared with WT mice (50–53). With naproxcinod treatment (10 or 21 mg/kg/day), the systolic BP, fraction shortening and ejection fraction were significantly improved over those of the mdx vehicle-treated mice, reaching the values obtained for WT mice. In addition, there was a 26% reduction in cardiac fibrosis in the naproxcinod-treated mice when compared with mdx vehicle-treated mice as measured by Picro-Sirius red staining. This reduction is consistent with the improvement in cardiac function that we observed with naproxcinod treatment. Conversely, prednisolone treatment resulted in a 54% increase in systolic BP over the mdx vehicle-treated group, yielding pressures that were also significantly higher than those in WT mice. Steroid treatment can result in retention of sodium that leads to fluid accumulation, which can result in high BP. In addition, prednisolone treatment caused a significant decrease in both the fraction shortening and ejection fraction when compared with vehicle treatment of mdx mice, leading to an exacerbation of the cardiomyopathy associated with mdx mice. This increased cardiomyopathy can be the result of an increase in cardiac fibrosis, which usually results in a dilation of the heart that decreases its ability to shorten and contract. Consistent with this explanation the observation that prednisolone treatment results in decreased cardiac function and increased cardiac fibrosis in mdx mice (53).
Apart from cardiomyopathy, patients with DMD also experience respiratory failure, which can also cause death in these patients (2,54,55). Therefore, improvement in the diaphragm is also of benefit. Histological examination of the diaphragm indicated that there was a significant reduction in inflammation as a result of naproxcinod treatment (21 mg/kg/day). In addition, there was a reduction in the total fiber number, central nucleated fibers and regenerating fibers, all of which are consistent with reduced damage and regeneration. Inflammation plays a hefty role in the pathogenesis of DMD and, together with fibrotic replacement of myofibers contributes to muscle weakness. Inflammatory cells are not only responsible for clearing up debris but can also produce enzymes and toxic molecules that can damage even healthy bystander muscle (56). A reduction in inflammatory cells can, therefore, prevent the extensive damage caused by such cells.
We did not detect any significant difference in behavioral activity with an open-field Digiscan apparatus, in in vitro force measurements, or in serum CK levels as the result of naproxcinod treatments. There are several explanations for these observations: (1) there was significant intra- and inter-group variation in many of these parameters, which may have affected the outcomes; and/or (2) the compound may have affected different muscles or tissues differently. Although the changes were not statistically significant, naproxcinod had positive behavioral effects, especially at 21 mg/kg/day. At this concentration all the behavioral parameters tested showed an improvement over that in mdx vehicle-treated mice, to a level similar to WT mice. The improvement in behavioral effects is likely influenced by the improvement in the pathological features of the diaphragm, cardiac and skeletal muscles.
Although several studies have examined the effects of NO-donor compounds on skeletal muscle function, none have examined the effects of these compounds in a single muscle in an in vitro setting (31–33). With our method, there was an improvement in the maximal force (10 and 41 mg/kg/day of naproxcinod), but a reduction in the specific contractile force of the EDL muscles of the naproxcinod-treated mice when compared with the mdx vehicle-treated group. The decrease in the EDL specific force at all naproxcinod concentrations tested may be a result of the fact that naproxcinod-treated EDL muscles exhibited a significant increase in EDL muscle mass when compared with the mdx vehicle-treated muscles. The increase in muscle mass results in an increase in cross-sectional area of the muscle. Since the specific force is a measure of maximal force corrected for cross-sectional area, the significant increase in cross-sectional area resulted in a reduction in specific force. Why naproxcinod treatment leads to an increase in EDL muscle mass remains to be elucidated. Consistent with previous findings (57), we observed that the specific force of the EDL muscles in the mdx mice was significantly lower than that of the WT mice. In the prednisolone-treated group, there was a significant reduction in the maximal force when compared with the mdx vehicle-treated group, perhaps because the prednisolone caused a significant reduction in the EDL muscle mass and thereby induced muscle atrophy and a subsequent reduction in force generation.
When we measured serum CK levels, we found that the levels were significantly higher in mdx mice than in WT mice. Although the comparison between mdx and WT mice showed the expected trend, the question still remains as to whether there is any correlation between serum CK levels and structural damage. In other words, is the serum CK level a true predictor of the skeletal muscle damage? Several studies have indicated that while CK levels are used as a marker of muscle damage, especially in disease states like DMD, the levels do not always correlate with the degree of structural damage (58–61).
Overall, our findings indicate that naproxcinod has significant potential as a safe and effective therapeutic option for muscular dystrophy especially for long-term treatment. It was effective in improving muscle strength and cardiac function as well as histopathological features (especially in limiting inflammation). The compound did not show the same adverse side effects and was more potent and beneficial in correcting the secondary defect in mdx mice than was the traditionally used GC, prednisolone. A naproxcinod dose of 21 mg/kg/day (the dose at which the compound exerted the maximal beneficial effects in most of our tests) appears to be an effective dose. This is one of the few compounds that we have tested that has shown promising results, with improvement in most of the parameters tested. Therefore, we believe our results point to the direction of a possible alternative to the currently used prednisolone, with reduced adverse side effects and a more beneficial battery of outcomes in patients with DMD.
MATERIALS AND METHODS
Animals
Mice were handled according to local Institutional Animal Care and Use Committee guidelines. Male mdx and C57BL/10ScSnJ WT mice, 4 weeks of age, were purchased from the Jackson laboratory (Bar Harbor, Maine, USA).
Dose of naproxcinod
The actual per-day intake of naproxcinod by a mouse deviated slightly from the predicted amounts of 10, 20, 40 and 0.75 mg/kg/day. The actual intake was calculated using the following formula: [(average of food intake/day/mouse) × (amount of drug/kg)]/BW in kg/1000. Using this formula, the actual intake amounts of naproxcinod were determined to be 10, 21, 41 and 0.9 mg/kg/day (rounded off to a whole number, except for prednisolone).
Study design
The study involved five groups of mdx mice (15 mice/group) and one group of WT control mice (n = 10; C57BL/10SnJ), as follows: (1) WT mice (treated with vehicle), (2) mdx mice (vehicle), (3) mdx mice (naproxcinod, 10 mg/kg/day), (4) mdx mice (naproxcinod, 21 mg/kg/day), (5) mdx mice (naproxcinod, 41 mg/kg/day) and (6) mdx mice (prednisolone, 0.9 mg/kg/day). Vehicle, naproxcinod and prednisolone were administered orally in food at the concentrations aforementioned every day for 9 months. The mice were acclimatized to the animal facility for 1 week upon arrival and then randomly assigned to their respective treatment groups based on their BW. Starting at 5 weeks of age, all groups of mice were subjected to treadmill exercise twice weekly for 7 months. Treadmill exercise was performed to unmask the mdx mouse phenotype. Due to the loss of trial animals to both old age and exercise, we did not perform the treadmill exercise for the full 9 months. Since the deaths were not specific to any particular treatment group, therefore, they were not the result adverse side effects of the drug(s) after long-term treatment. For the functional measurements (Digiscan and grip strength measurements), all animals were acclimatized to all the instruments before the actual readings were recorded at 3, 6 and 9 months of treatment. All the functional measurements were performed in the morning hours and in a blinded fashion.
GSM
Forelimb and hindlimb grip strength were measured using a grip strength meter (Columbus Instruments, Columbus, OH, USA) as previously described (62). The maximum values for each measurement were used for subsequent analysis. The grip strength measurements were collected over a 5-day period.
Open-field behavioral activity measurements (Digiscan)
Locomotor activity was measured using an open-field Digiscan apparatus (Omnitech Electronics, Columbus, OH, USA) as previously described (62). The locomotor activity data were recorded every 10 min for 1 h over a 4-day period.
Treadmill exercise
Mice were not acclimatized to the treadmill prior to the start of the exercise. They were placed on the treadmill set at a 0° incline and run for 30 min at a maximum speed of 12 m/min, twice a week as previously described (62).
BP measurements
The mice were sedated using 1.5% isofluorane with constant monitoring of the plane of anesthesia and maintenance of the body temperature at 36.5–37.5°C. The heart rate was maintained at 450–550 beats/min. A BP cuff was placed around the tail, and the tail was then placed in a sensor assembly for non-invasive BP monitoring (SC1000, Hatteras Instruments, Cary, NC, USA) during anesthesia. Ten consecutive BP measurements were taken. Qualitative and quantitative measurements of tail BP, including systolic pressure, diastolic pressure and mean pressure, were made offline using analytic software.
Echocardiography
Echocardiography was performed on anesthetized mice using a high-frequency ultrasound probe over a 20 min period as previously described (62,63). The quantitative measurements were made offline using analytic software (VisualSonics, Toronto, Ontario, Canada).
Optical imaging
Cathepsin activity was measured from the optical imaging scanning of the limb muscles using caged near infrared substrate as previously described (64).
In vitro force measurements
In vitro force measurement of the EDL muscle from the right hindlimb was performed as previously described (40). The protocol to fatigue the EDL muscle was as follows: 60 isometric contractions for 300 ms each, once every 5 s. The EDL muscles were stimulated at 100 Hz. The force was recorded every minute during the repetitive contractions and again at 5 and 10 min afterward to measure recovery.
Histological evaluations
At the end of the trial, directly following euthanasia, the muscles (diaphragm and heart) were removed and fixed in 10% formalin for later processing. Both diaphragm and heart were sectioned and stained for H&E by Histoserv (Germantown, MD, USA). Five random digital images were taken using an Eclipse E800 (Nikon, Japan) microscope, and blinded analysis was done using Image J (NIH) as previously reported (40,62).
Picro-Sirius red staining was performed to measure the degree of fibrosis in the hearts of the trial mice. In brief, paraffin sections were deparaffinized in xylene followed by nuclear staining with Weigert's hematoxylin for 8 min. They were then washed and then stained with Picro-Sirius red (0.5 g of Sirius red F3B, saturated aqueous solution of picric acid) for an additional 30 min. The sections were cleared in three changes of xylene and mounted in Permount.
Serum CK
Blood samples were taken via cardiac puncture when the animals were euthanized, and the serum collected was used for the measurement of muscle CK levels as previously described (53).
Statistical analysis
Normality assessment of each measurement using both the Shapiro–Wilk normality test and visual examination of histograms determined that BW, grip strength measurements and average food eaten were normally distributed, while Digiscan measures were not. Therefore, parametric tests were used for all measures other than Digiscan, for which non-parametric tests were used. For each measurement at each time point, two separate comparisons were done. A comparison between mdx vehicle and WT vehicle groups used either t-tests or Wilcoxon rank sum tests, depending on the distribution. A further comparison between the mdx vehicle-treated group and each of the mdx treatment groups was also performed. For parametric measurements, pair-wise t-tests were used to compare the vehicle group to each of the treated groups, with the resulting P-values adjusted for multiple comparisons using the Sidak method. For non-parametric measurements, pair-wise Wilcoxon rank sum tests were used to compare the vehicle-treated group to each of the drug-treated groups, again with P-values adjusted for multiple comparisons using the Sidak method. Measurements were further tested longitudinally using mixed effects linear models to account for the repeated nature of the data. Only the main effects were tested with no interaction, and each model was assessed for fit. The mdx vehicle-treated group was compared with the WT vehicle group using a log-rank test to the equality of the survival functions. Log-rank tests with post hoc adjustment for multiple comparisons were used to compare the mdx vehicle group to all other mdx-treated groups. No overall adjustments of the type 1 error rate (multiple testing) were made, and statistical tests were considered significant at the P < 0.05 level. All analyses were performed using Stata V11 (College Station, TX, USA).
SUPPLEMENTARY MATERIAL
FUNDING
This work is supported by the following: National Institutes of Health grants (1K26RR032082, 1P50AR060836-01, 1U54HD071601 and 2R24HD050846-06), Department of Defense grants (W81XWH-11-1-0330, W81XWH-11-1-0782, W81XWH-10-1-0659, W81XWH-11-1-0809 and W81XWH-09-1-0599) and a translational research grant from the Muscular Dystrophy Association.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr Deborah McClellan for editing this manuscript.
Conflict of Interest statement. The authors have declared that no conflict of interest exists pertaining to this study. The study design, data collection, interpretation and data analysis were performed by the team of scientists at the preclinical testing facility at Children's National Medical Center as part of a fee-per-service agreement. The study design and the manuscript were made available for comments and suggestions by the Nicox Research Institute scientists. The Nicox Research Institute scientists had no role in data collection, interpretation or analysis. However, they made the final decision to publish the manuscript. D.M. and E.O. are affiliated with the Nicox Research Institute.
REFERENCES
- 1.Roses A.D., Herbstreith M.H., Appel S.H. Membrane protein kinase alteration in Duchenne muscular dystrophy. Nature. 1975;254:350–351. doi: 10.1038/254350a0. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman E.P., Schwartz L. Dystrophin and disease. Mol Aspects Med. 1991;12:175–194. doi: 10.1016/0098-2997(91)90001-3. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman E.P., Brown R.H., Jr, Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 4.Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 5.Ohlendieck K., Campbell K.P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1991;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutter O.F., Burton F.L., Bovell D.L. Mechanical properties of normal and mdx mouse sarcolemma: bearing on function of dystrophin. J Muscle Res Cell Motil. 1991;12:585–589. doi: 10.1007/BF01738447. [DOI] [PubMed] [Google Scholar]
- 7.Menke A., Jockusch H. Decreased osmotic stability of dystrophin-less muscle cells from the mdx mouse. Nature. 1991;349:69–71. doi: 10.1038/349069a0. [DOI] [PubMed] [Google Scholar]
- 8.Menke A., Jockusch H. Extent of shock-induced membrane leakage in human and mouse myotubes depends on dystrophin. J Cell Sci. 1995;108(Pt 2):727–733. doi: 10.1242/jcs.108.2.727. [DOI] [PubMed] [Google Scholar]
- 9.Pasternak C., Wong S., Elson E.L. Mechanical function of dystrophin in muscle cells. J Cell Biol. 1995;128:355–361. doi: 10.1083/jcb.128.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deconinck N., Dan B. Pathophysiology of Duchenne muscular dystrophy: current hypotheses. Pediatr Neurol. 2007;36:1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 12.Emery A.E., Burt D. Intracellular calcium and pathogenesis and antenatal diagnosis of Duchenne muscular dystrophy. Br Med J. 1980;280:355–357. doi: 10.1136/bmj.280.6211.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fong P.Y., Turner P.R., Denetclaw W.F., Steinhardt R.A. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science. 1990;250:673–676. doi: 10.1126/science.2173137. [DOI] [PubMed] [Google Scholar]
- 14.Turner P.R., Westwood T., Regen C.M., Steinhardt R.A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature. 1988;335:735–738. doi: 10.1038/335735a0. [DOI] [PubMed] [Google Scholar]
- 15.McArdle A., Edwards R.H., Jackson M.J. Time course of changes in plasma membrane permeability in the dystrophin-deficient mdx mouse. Muscle Nerve. 1994;17:1378–1384. doi: 10.1002/mus.880171206. [DOI] [PubMed] [Google Scholar]
- 16.Emery A.E. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 17.Matsuda R., Nishikawa A., Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J Biochem. 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- 18.Brenman J.E., Chao D.S., Xia H., Aldape K., Bredt D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- 19.Chang W.J., Iannaccone S.T., Lau K.S., Masters B.S., McCabe T.J., McMillan K., Padre R.C., Spencer M.J., Tidball J.G., Stull J.T. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci USA. 1996;93:9142–9147. doi: 10.1073/pnas.93.17.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stamler J.S., Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- 21.De Palma C., Clementi E. Nitric oxide in myogenesis and therapeutic muscle repair. Mol Neurobiol. 2012;46:682–692. doi: 10.1007/s12035-012-8311-8. [DOI] [PubMed] [Google Scholar]
- 22.Lai Y., Thomas G.D., Yue Y., Yang H.T., Li D., Long C., Judge L., Bostick B., Chamberlain J.S., Terjung R.L., et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119:624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sander M., Chavoshan B., Harris S.A., Iannaccone S.T., Stull J.T., Thomas G.D., Victor R.G. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2000;97:13818–13823. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas G.D., Sander M., Lau K.S., Huang P.L., Stull J.T., Victor R.G. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci USA. 1998;95:15090–15095. doi: 10.1073/pnas.95.25.15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li D., Yue Y., Lai Y., Hakim C.H., Duan D. Nitrosative stress elicited by nNOSmicro delocalization inhibits muscle force in dystrophin-null mice. J Pathol. 2011;223:88–98. doi: 10.1002/path.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai Y., Zhao J., Yue Y., Duan D. alpha2 and alpha3 helices of dystrophin R16 and R17 frame a microdomain in the alpha1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci USA. 2013;110:525–530. doi: 10.1073/pnas.1211431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schnitzer T.J., Kivitz A.J., Lipetz R.S., Sanders N., Hee A. Comparison of the COX-inhibiting nitric oxide donator AZD3582 and rofecoxib in treating the signs and symptoms of osteoarthritis of the knee. Arthritis Rheum. 2005;53:827–837. doi: 10.1002/art.21586. [DOI] [PubMed] [Google Scholar]
- 28.Wallace J.L., Viappiani S., Bolla M. Cyclooxygenase-inhibiting nitric oxide donators for osteoarthritis. Trends Pharmacol Sci. 2009;30:112–117. doi: 10.1016/j.tips.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 29.White W.B., Schnitzer T.J., Fleming R., Duquesroix B., Beekman M. Effects of the cyclooxygenase inhibiting nitric oxide donator naproxcinod versus naproxen on systemic blood pressure in patients with osteoarthritis. Am J Cardiol. 2009;104:840–845. doi: 10.1016/j.amjcard.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 30.Karlsson J., Pivodic A., Aguirre D., Schnitzer T.J. Efficacy, safety, and tolerability of the cyclooxygenase-inhibiting nitric oxide donator naproxcinod in treating osteoarthritis of the hip or knee. J Rheumatol. 2009;36:1290–1297. doi: 10.3899/jrheum.081011. [DOI] [PubMed] [Google Scholar]
- 31.Brunelli S., Sciorati C., D’Antona G., Innocenzi A., Covarello D., Galvez B.G., Perrotta C., Monopoli A., Sanvito F., Bottinelli R., et al. Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci USA. 2007;104:264–269. doi: 10.1073/pnas.0608277104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas G.D., Ye J., De Nardi C., Monopoli A., E., Victor R.G. Treatment with a nitric oxide-donating NSAID alleviates functional muscle ischemia in the mouse model of Duchenne muscular dystrophy. PLoS One. 2012;7:e49350. doi: 10.1371/journal.pone.0049350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sciorati C., Miglietta D., Buono R., Pisa V., Cattaneo D., Azzoni E., Brunelli S., Clementi E. A dual acting compound releasing nitric oxide (NO) and ibuprofen, NCX 320, shows significant therapeutic effects in a mouse model of muscular dystrophy. Pharmacol Res. 2011;64:210–217. doi: 10.1016/j.phrs.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manzur A.Y., Kinali M., Muntoni F. Update on the management of Duchenne muscular dystrophy. Arch Dis Child. 2008;93:986–990. doi: 10.1136/adc.2007.118141. [DOI] [PubMed] [Google Scholar]
- 35.Collins C.A., Morgan J.E. Duchenne’s muscular dystrophy: animal models used to investigate pathogenesis and develop therapeutic strategies. Int J Exp Pathol. 2003;84:165–172. doi: 10.1046/j.1365-2613.2003.00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher I., Abraham D., Bouri K., Hoffman E.P., Muntoni F., Morgan J. Prednisolone-induced changes in dystrophic skeletal muscle. FASEB J. 2005;19:834–836. doi: 10.1096/fj.04-2511fje. [DOI] [PubMed] [Google Scholar]
- 37.Griggs R.C., Moxley R.T., 3rd, Mendell J.R., Fenichel G.M., Brooke M.H., Pestronk A., Miller J.P., Cwik V.A., Pandya S., Robison J., et al. Duchenne dystrophy: randomized, controlled trial of prednisone (18 months) and azathioprine (12 months) Neurology. 1993;43:520–527. doi: 10.1212/wnl.43.3_part_1.520. [DOI] [PubMed] [Google Scholar]
- 38.Moxley R.T., 3rd, Ashwal S., Pandya S., Connolly A., Florence J., Mathews K., Baumbach L., McDonald C., Sussman M., Wade C., et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2005;64:13–20. doi: 10.1212/01.WNL.0000148485.00049.B7. [DOI] [PubMed] [Google Scholar]
- 39.Drachman D.B., Toyka K.V., Myer E. Prednisone in Duchenne muscular dystrophy. Lancet. 1974;2:1409–1412. doi: 10.1016/s0140-6736(74)90071-3. [DOI] [PubMed] [Google Scholar]
- 40.Sali A., Guerron A.D., Gordish-Dressman H., Spurney C.F., Iantorno M., Hoffman E.P., Nagaraju K. Glucocorticoid-treated mice are an inappropriate positive control for long-term preclinical studies in the mdx mouse. PLoS One. 2012;7:e34204. doi: 10.1371/journal.pone.0034204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huynh T., Uaesoontrachoon K., Quinn J.L., Tatem K.S., Heier C.R., Van Der Meulen J.H., Yu Q., Harris M., Nolan C.J., Haegeman G., et al. Selective modulation through the glucocorticoid receptor ameliorates muscle pathology in mdx mice. J Pathol. 2013;231:223–235. doi: 10.1002/path.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bodine S.C., Latres E., Baumhueter S., Lai V.K., Nunez L., Clarke B.A., Poueymirou W.T., Panaro F.J., Na E., Dharmarajan K., et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 43.Shin Y.S., Fink H., Khiroya R., Ibebunjo C., Martyn J. Prednisolone-induced muscle dysfunction is caused more by atrophy than by altered acetylcholine receptor expression. Anesth Analg. 2000;91:322–328. doi: 10.1097/00000539-200008000-00017. [DOI] [PubMed] [Google Scholar]
- 44.Keeling R.M., Golumbek P.T., Streif E.M., Connolly A.M. Weekly oral prednisolone improves survival and strength in male mdx mice. Muscle Nerve. 2007;35:43–48. doi: 10.1002/mus.20646. [DOI] [PubMed] [Google Scholar]
- 45.Granchelli J.A., Pollina C., Hudecki M.S. Pre-clinical screening of drugs using the mdx mouse. Neuromuscul Disord. 2000;10:235–239. doi: 10.1016/s0960-8966(99)00126-1. [DOI] [PubMed] [Google Scholar]
- 46.Huynh T., Uaesoontrachoon K., Quinn J.L., Tatem K.S., Heier C.R., Van Der Meulen J.H., Yu Q., Harris M., Nolan C.J., Haegeman G., et al. Selective modulation through the glucocorticoid receptor ameliorates muscle pathology in mdx mice. J Pathol. 2013;231:223–235. doi: 10.1002/path.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schakman O., Kalista S., Barbe C., Loumaye A., Thissen J.P. Glucocorticoid-induced skeletal muscle atrophy. Int J Biochem Cell Biol. 2013;45:2163–2172. doi: 10.1016/j.biocel.2013.05.036. [DOI] [PubMed] [Google Scholar]
- 48.McNally E.M. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu Rev Med. 2007;58:75–88. doi: 10.1146/annurev.med.58.011706.144703. [DOI] [PubMed] [Google Scholar]
- 49.Rumyantsev P.P. Interrelations of the proliferation and differentiation processes during cardiac myogenesis and regeneration. Int Rev Cytol. 1977;51:186–273. [PubMed] [Google Scholar]
- 50.Van Erp C., Irwin N.G., Hoey A.J. Long-term administration of pirfenidone improves cardiac function in mdx mice. Muscle Nerve. 2006;34:327–334. doi: 10.1002/mus.20590. [DOI] [PubMed] [Google Scholar]
- 51.Bostick B., Yue Y., Long C., Marschalk N., Fine D.M., Chen J., Duan D. Cardiac expression of a mini-dystrophin that normalizes skeletal muscle force only partially restores heart function in aged Mdx mice. Mol Ther. 2009;17:253–261. doi: 10.1038/mt.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu B., Moulton H.M., Iversen P.L., Jiang J., Li J., Spurney C.F., Sali A., Guerron A.D., Nagaraju K., Doran T., et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci USA. 2008;105:14814–14819. doi: 10.1073/pnas.0805676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guerron A.D., Rawat R., Sali A., Spurney C.F., Pistilli E., Cha H.J., Pandey G.S., Gernapudi R., Francia D., Farajian V., et al. Functional and molecular effects of arginine butyrate and prednisone on muscle and heart in the mdx mouse model of Duchenne Muscular Dystrophy. PLoS One. 2010;5:e11220. doi: 10.1371/journal.pone.0011220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilroy J., Cahalan J.L., Berman R., Newman M. Cardiac and pulmonary complications in Duchenne’s progressive muscular dystrophy. Circulation. 1963;27:484–493. doi: 10.1161/01.cir.27.4.484. [DOI] [PubMed] [Google Scholar]
- 55.Melacini P., Vianello A., Villanova C., Fanin M., Miorin M., Angelini C., Dalla Volta S. Cardiac and respiratory involvement in advanced stage Duchenne muscular dystrophy. Neuromuscul Disord. 1996;6:367–376. doi: 10.1016/0960-8966(96)00357-4. [DOI] [PubMed] [Google Scholar]
- 56.Tidball J.G. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288:R345–R353. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- 57.Lynch G.S., Hinkle R.T., Chamberlain J.S., Brooks S.V., Faulkner J.A. Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J Physiol. 2001;535:591–600. doi: 10.1111/j.1469-7793.2001.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van der Meulen J.H., Kuipers H., Drukker J. Relationship between exercise-induced muscle damage and enzyme release in rats. J Appl Physiol. 1991;71:999–1004. doi: 10.1152/jappl.1991.71.3.999. [DOI] [PubMed] [Google Scholar]
- 59.Komulainen J., Vihko V. Exercise-induced necrotic muscle damage and enzyme release in the four days following prolonged submaximal running in rats. Pflugers Arch. 1994;428:346–351. doi: 10.1007/BF00724517. [DOI] [PubMed] [Google Scholar]
- 60.Friden J., Lieber R.L. Serum creatine kinase level is a poor predictor of muscle function after injury. Scand J Med Sci Sports. 2001;11:126–127. doi: 10.1034/j.1600-0838.2001.011002126.x. [DOI] [PubMed] [Google Scholar]
- 61.Dahlqvist J.R., Voss L.G., Lauridsen T., Krag T.O., Vissing J. A pilot study of muscle plasma protein changes following exercise. Muscle Nerve. 2013;49:261–266. doi: 10.1002/mus.23909. [DOI] [PubMed] [Google Scholar]
- 62.Spurney C.F., Gordish-Dressman H., Guerron A.D., Sali A., Pandey G.S., Rawat R., Van Der Meulen J.H., Cha H.J., Pistilli E.E., Partridge T.A., et al. Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve. 2009;39:591–602. doi: 10.1002/mus.21211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spurney C.F., Knoblach S., Pistilli E.E., Nagaraju K., Martin G.R., Hoffman E.P. Dystrophin-deficient cardiomyopathy in mouse: expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart. Neuromuscul Disord. 2008;18:371–381. doi: 10.1016/j.nmd.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baudy A.R., Sali A., Jordan S., Kesari A., Johnston H.K., Hoffman E.P., Nagaraju K. Non-invasive optical imaging of muscle pathology in mdx mice using cathepsin caged near-infrared imaging. Mol Imaging Biol. 2011;13:462–470. doi: 10.1007/s11307-010-0376-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.