

Abstract
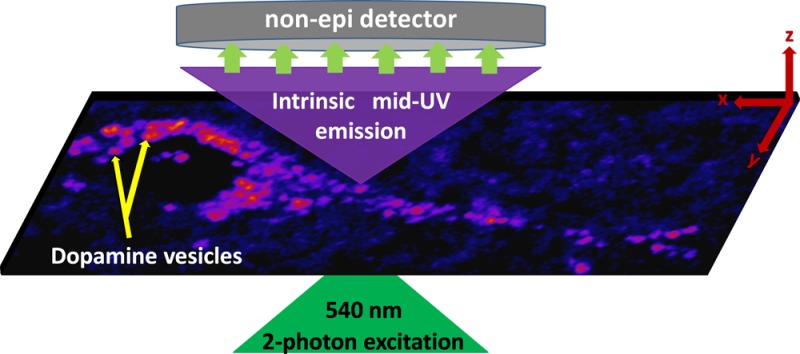
Dopaminergic neurotransmission has been investigated extensively, yet direct optical probing of dopamine has not been possible in live cells. Here we image intracellular dopamine with sub-micrometer three-dimensional resolution by harnessing its intrinsic mid-ultraviolet (UV) autofluorescence. Two-photon excitation with visible light (540 nm) in conjunction with a non-epifluorescent detection scheme is used to circumvent the UV toxicity and the UV transmission problems. The method is established by imaging dopamine in a dopaminergic cell line and in control cells (glia), and is validated by mass spectrometry. We further show that individual dopamine vesicles/vesicular clusters can be imaged in cultured rat brain slices, thereby providing a direct visualization of the intracellular events preceding dopamine release induced by depolarization or amphetamine exposure. Our technique opens up a previously inaccessible mid-ultraviolet spectral regime (excitation ∼ 270 nm, emission < 320 nm) for label-free imaging of native molecules in live tissue.
Keywords: Two-photon imaging, ultraviolet microscopy, neurotransmitter imaging, dopaminergic neurons, intrinsic fluorescence
Intracellular dopamine dynamics is the prelude to dopaminergic neurotransmission, which controls reward and motor circuits, and plays a major role in substance addiction and Parkinson’s disease.1 Yet there has been no optical method to probe it in live neurons. Radioactive dopamine uptake has provided low resolution maps,2 while amperometry and microdialysis have been used to monitor release in the extracellular space.3 Two recent developments promise more progress: first, a fluorescent dopamine-mimic which allows optical probing;4−6 and second, a dopamine ligand with enough magnetic resonance contrast which allows detection of released dopamine.7 However, how accurately any of these techniques report the underlying dopamine dynamics remains unclear.
The ideal alternative would be to directly visualize native dopamine with high resolution in living systems. Such attempts have so far been thwarted by the difficulty of harnessing the weak mid-ultraviolet (mid-UV, <320 nm) autofluorescence of dopamine. Despite the presence of a host of interesting biological molecules (e.g., dopamine, norepinephrine, tyrosine and tyrosine containing peptides, DOPA) that can fluoresce in mid-UV, there has been little success in live imaging of any chromophore since deep-UV excitation (∼270 nm) cannot penetrate living tissue and can cause extreme phototoxicity. In addition, the mid-UV emission is absorbed and hence cannot be collected efficiently using conventional microscope glass optics. A three-photon (or higher order) excitation strategy has enabled the visualization of serotonin, another UV chromophore and the only neurotransmitter that has been imaged directly so far.8−15 Similarly, two-photon excitation has been shown to work for imaging intracellular tryptophan.16 However, this approach fails for dopamine because of its deeper UV excitation and emission characteristics. Although deep-UV transmission imaging has been developed,17 it lacks specificity and penetration, and cannot image particular molecular species in live tissue at high resolution.
Taking cues from an earlier experiment with dopamine solution,18 we have now developed a technique which can provide dynamic high resolution images of intracellular dopamine in living cells in culture, and also in live rat brain slices. The optical requirements are met by a combination of two-photon excitation (at 540 nm) from an optical parametric oscillator (OPO) (which provides visible wavelength femtosecond pulses), a non-epifluorescent collection design (with an external detector placed directly on top of the specimen in an inverted microscope), and specific optical filters (Figure 1, see Methods). Our strategy allows the use of a conventional confocal microscope with some modifications, provided the instrument can accept an external visible laser source and can process signals from external detectors. A test with an aqueous solution of dopamine (2 mM) shows that a strong two-photon excited signal can be obtained by this method, even below saturating powers (25 mW at the back aperture of the objective, Supporting Information Figure S1). Experiments at lower concentrations show that our method can detect dopamine down to <100 μM (Supporting Information Figure S2). Since neuronal dopamine is packed at high concentrations in the vesicles (∼100s of mM19), this opens up the possibility of direct dopamine imaging, with sufficient signal-to-noise ratio over and above the generic UV-excited cellular background. We note that fluorescence from other mid-UV and near-UV fluorophores such as tyrosine, phenylalanine, or tryptophan residues in proteins will also get detected (see Supporting Information Figure S3 for absorption and emission spectra of these fluorophores). However, unlike neurotransmitters primarily stored in vesicles, proteins are distributed throughout the cell and are not localized in specific areas at high concentrations and hence these fluorophores only contribute to the diffuse background. We also note that second harmonic generation makes negligible contribution to our signal, because the detector sensitivity is nearly zero at wavelengths <280 nm.
Figure 1.
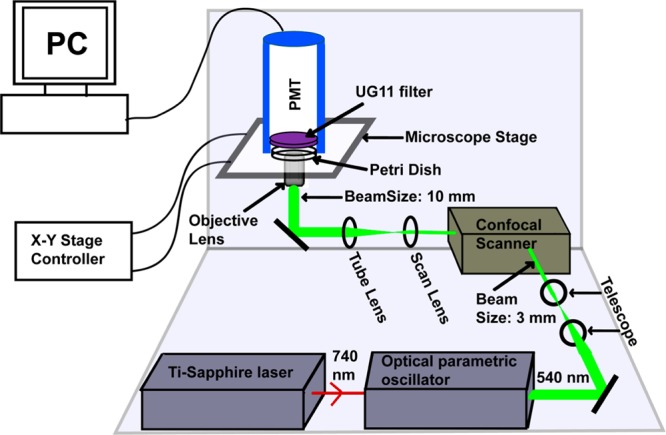
Schematic representation of the optical setup for dopamine imaging (not to scale). Optical parametric oscillator synchronously pumped by mode-locked tunable Ti-Sapphire laser at 740 nm produces 540 nm ∼100 fs pulses. The collimated beam (3 mm in diameter, achieved using a telescope setup) is routed through a confocal scan-box to an inverted microscope equipped with a 60× water immersion objective lens (numerical aperture, NA = 1.2). The beam diameter is 10 mm at the back of the objective lens and has an average power of 30 mW. The emission is collected in the forward direction, with an external photomultiplier tube (PMT) placed directly above the sample. A pair of special glass based absorptive filters (UG11) is placed in front of the PMT which transmits the mid-UV fluorescence but efficiently blocks the 540 nm excitation light.
We establish the technique by imaging the dopaminergic cell line MN9D.20,21 Figure 2a shows a two-photon image of MN9D cells (a single optical slice). The cells appear bright, with darker region representing the nucleus. Some regions do appear brighter than the rest, although no clear punctate structures are seen. In comparison, cultured glial cells (obtained from rat brain, not expected to contain dopamine) appear much less fluorescent under identical imaging conditions (Figure 2b, 3× brightened). The integrated intensity per cell (arbitrary units) for MN9D cells (92 ± 4, no. of cells, n = 35) is significantly higher (p < 0.001) than that for glia (22 ± 1, n = 24). Further, we test if the observed intensity is sensitive to conditions which reduce or increase the level of dopamine in these cells. We treat the MN9D cells with para-chloroamphetamine (PCA), a nonaddictive amphetamine and a powerful monoamine-releasing drug, in order to deplete intracellular dopamine content.22 Figure 2d shows the same set of MN9D cells shown in Figure 2a, but after 15 min of 100 μM PCA treatment. The brightness reduces by approximately 34 ± 5% (n = 18), while the vehicle treated control set of cells (Figure 2c, 0 min; Figure 2f, 15 min) shows negligible change (1 ± 3%, n = 23). This reduction is statistically significant (p < 0.001). In the complementary approach, the dopamine levels are increased in the glial cells by dopamine infusion, presumably via glial plasma membrane transporters (which may or may not be specific for dopamine).23 As expected, glial cells incubated with 10 mM dopamine in the media for 4 h show a clear increase in brightness (Figure 2e, 3× brightened). The cells were washed before imaging to get rid of extracellular dopamine. We note that in this experiment it was not possible to observe the same cells before and after treatment as the treatment duration is 4 h, during which the glia move, grow, and divide. The mean intensity (arbitrary units) of the dopamine treated glial cells (32 ± 1, n = 58) (Figure 2e) is significantly higher (p < 0.001) than that of control cells (22 ± 1, n = 24) (Figure 2b). These results suggest that the fluorescence signal indeed originates from intracellular dopamine. We note that the background fluorescence contributed by other UV-fluorophores may lead to an underestimation of the amount of changes in dopamine levels.
Figure 2.

Two-photon autofluorescence of dopamine in MN9D cells and glia. (a, d) Collection of dopaminergic MN9D cells before (a) and 15 min after (d) treatment with 100 μM of PCA. The signal intensity decreases by 34 ± 5%. Vehicle-treated control cells are shown under identical imaging conditions in (c) (before) and (f) (after). The control cells show 1 ± 3% increase in the same period. (b, e) Glia treated with vehicle (b) and 10 mM dopamine for 4 h (e). Fluorescence intensities are false color coded. For (b) and (e), the brightness is 3× enhanced. Scale bar = 10 μm.
To validate the identity of the imaged fluorophore, we then quantitatively analyze the dopamine content of the MN9D and glial cells, and also glial cells treated with dopamine (10 mM for 4 h) using a HPLC coupled mass spectrometer. The cellular dopamine amount is calibrated against a known concentration of isotopically labeled dopamine added to the cell extracts and scaled with the dry mass of the cellular specimens. Dopamine (protonated mass of 154) and isotopically labeled dopamine (protonated mass of 157) co-elute from the cell extracts (Supporting Information Figure S4a). The mass spectrum for MN9D (Supporting Information Figure S4b, red, upper panel) shows a clear peak at 154, while this peak is hardly detectable for glial cells (Supporting Information Figure S4b, blue, middle panel). The results show that the MN9D cells contain a substantial amount of dopamine (386 ± 18 ng/mg of the dry mass of the cells), while this amount is significantly lower in glial cells (26 ± 13 ng/mg, p < 0.001). Glial cells treated with 10 mM dopamine for 4 h show clear increase in cellular dopamine content (170 ± 74 ng/mg, p < 0.01) (Supporting Information Figure S4b, black, bottom panel). The mass spectrometric results therefore corroborate the image intensity difference observed for these two types of cells. The image intensity ratio is lower than the mass intensity ratio presumably due to the background fluorescence contributed by non-dopamine UV-fluorescent components of the cell. Taken together, these results confirm that two-photon imaging can report the dopamine levels in cultured cells and glia.
We then test if this technique could be extended for imaging dopamine vesicles in live brain slices. Brain slices provide an additional challenge for UV imaging, as the emission signal is expected to be substantially absorbed and scattered by the intervening tissue. We image the Substantia Nigra (SN) region in cultured rat brain slices (coronal sections), which is known to be rich in dopaminergic neurons. Figure 3a shows a typical neuron from SN (summed image of three successive optical sections recorded at 1 μm depth increments). The vesicles/vesicular clusters appear as distinctly bright punctate structures. It is known that somatic monoaminergic vesicles can range in size from <100 nm to >1 μm in some cases.19,24,25 Our resolution (about 300 nm) does not allow us to resolve closely clustered vesicles. Some of the punctate structures we observe are >1 μm and may represent clusters of vesicles, while some of the smaller structures could be individual vesicles. In the rest of this paper, we use the term “vesicle” to collectively denote both lone vesicles and unresolved vesicular clusters. We note that in some cases these bright vesicular structures may also arise from saccules of Golgi, multivesicular bodies and tubulovesicles that are potential sites for intracellular dopamine storage, since they also express the vesicular monoamine transporter-2 (VMAT2).26
Figure 3.
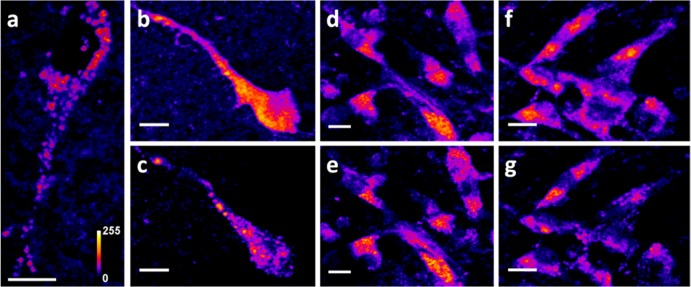
Imaging dopamine and its dynamics in live brain tissue. (a) Substantia Nigra neuron. The bright punctate dopaminergic vesicles/clusters are seen distributed in the cytoplasm and also in the primary neurite. (b, c) Dopaminergic cell before (b) and 15 min after (c) bath application of 50 mM KCl and 8 mM Ca2+. The fluorescence intensity decreases by 25 ± 4%. (d, e) Vehicle treated cells, before (d) and 15 min after (e), show negligible (3 ± 1%) decrease under identical imaging conditions. (f, g) Dopaminergic cells before (f) and 15 min after (g) treatment with 100 μM of PCA. The fluorescence intensity decreases by about 24 ± 2%. Fluorescence intensities are false color coded. Scale bar = 10 μm.
Our images show that the vesicles are distributed throughout the cell body and also in the primary neurite of the neuron. Most of the vesicles are distributed in a perinuclear manner, similar to that observed earlier for serotonin.10,11 Two-photon imaging of the whole brain slice shows that cells containing bright vesicles are mostly localized in the SN region and very few bright cells are observed outside this area, as may be expected (Supporting Information Figure S5). We note that dopaminergic neurons in tissue can be successfully imaged with this configuration without much loss of resolution or signal intensity till a depth of at least 25 μm (Supporting Information Figure S6).
We then address an unresolved question in dopamine neurotransmission. Extra-cellular measurements have shown that dopamine, along with other monoamines, is involved in volume transmission apart from the conventional synaptic neurotransmission.3,10−15,19,24,25,27−30 Does this arise from specific release-competent pools of somatic vesicles, and if so, where are they located?31 Here we follow the dynamics of the dopaminergic vesicles, after high K+-induced depolarization or after amphetamine administration. Although such treatments may be harsher than physiological stimuli (e.g., action potentials), they serve as robust tests for the ability of our technique to follow dopamine dynamics. Figure 3b and c shows the same cells before and 15 min after the application of 50 mM KCl containing 8 mM Ca2+, respectively. The images consist of a stack of optical slices recorded every 1 μm. Our images clearly show that the somatic dopamine vesicles are mobilized throughout the cell body for exocytosis in response to depolarization. We observe a 25 ± 4% decrease in somatic dopamine reserves in 15 min (n = 17). This decrease reports the net depletion, which is the difference between release and synthesis within this time, under constant depolarization. The control cells (Figure 3d, 0 min; 3e, 15 min) show only a 3 ± 1% decrease (n = 20). This reduction is statistically significant (p < 0.001). Figure 3f and g shows the effect of 100 μM PCA treatment on a collection of cells (Figure 3f, 0 min; Figure 3g, 15 min). We see that dopamine autofluorescence in PCA treated cells decrease by 24 ± 2% (n = 25, p < 0.001) in 15 min and 38 ± 2% in 30 min (n = 16, time point not shown). This amounts to a massive release of dopamine from intracellular stores, equal to or larger than what is caused by chronic depolarization. Using this technique, we are able to precisely quantify the depletion of dopamine reserves upon amphetamine challenge from live tissue. We note that prolonged multiphoton microscopy is known to induce cell-damage through the generation of reactive oxygen species and loss of membrane integrity.32 However, our results show that the photon dosage used in our experiments is low enough to allow the dopaminergic neurons to mount a physiological response to extracellular perturbations even after repeated three-dimensional imaging.
In summary, we describe here a label-free technique by which dopamine dynamics can be imaged in neurons in a relatively benign manner inside live brain tissue. This can be used to probe depolarization induced somatic exocytosis and drug induced efflux of dopamine as a function of time in individual neurons. Our technique also provides a direct opportunity to screen the effectiveness of drugs in modulating dopamine levels and dynamics in live tissue. We note that molecules with similar excitation and emission characteristics may also be amenable to such imaging, though a reliable separation between spectrally similar species will require additional information. We expect the technique developed here to be adaptable to a wider range of problems, which may benefit from the power of mid-UV imaging of native chromophores.
Methods
Imaging
We excite dopamine near its two-photon excitation peak with the 540 nm, ∼100 fs pulse-train output of an optical parametric oscillator (MIRA-OPO, Coherent) (Figure 1). The OPO is synchronously pumped via a mode-locked tunable Ti-Sapphire laser (MIRA, Coherent). The beam is routed through a BIORAD MRC 600 confocal scan-box which is coupled to an inverted microscope (TE 300, Nikon, Japan) equipped with a 60× water immersion objective lens (numerical aperture, NA = 1.2). The laser beam is collimated and the beam diameter reduced to 3 mm by using a two-lens Gaussian telescope to conform to the scanner requirements. At the back of the objective lens, the beam has an average power of 30 mW and fills the back aperture whose diameter is 10 mm. Since the microscope optics does not transmit the dopamine fluorescence (λmax ∼ 314 nm), we collect the emission in the forward (non-epi) direction, with an external photomultiplier tube (PMT, Model: P30A, Electron Tubes, UK) placed directly above the sample in a homemade light-shielded holder. This non-epi detection introduces the additional challenge of separating the weak fluorescence signal from the strong direct laser beam impinging on the detector. A three-photon excitation scheme, similar to that used for serotonin earlier, suffers from the lack of a suitable filter.10−12,15 We use a pair of special glass based absorptive filters (UG11, Schott Glass, Germany; 3 mm thickness each) in front of the PMT which exactly match this stringent requirement. The close proximity of the PMT to the specimen and wide area of detector-face (2 cm × 2 cm) ensures a collection solid angle equivalent to an NA of about 0.6. No separate collection lens is used. Fluorescence obtained from a 2 mM dopamine solution with this setup shows a quadratic dependence on the excitation power, as expected (Supporting Information Figure S1). All imaging is performed at room temperature (25 °C).
MN9D Cell Culture
MN9D cells are a fusion of rostral mesencephalic neurons from E14 C57/BL/6J mice with N18TG2 neuroblastoma cells, obtained from a cancer of the sympathetic nervous system.20 These cells are known to produce measurable amount of dopamine.21 The cells were a kind gift from Prof. Ruth G. Perez, University of Pittsburgh. MN9D cells are grown in DMEM media with high glucose (Sigma) supplemented with Fetal clone III (Hyclone), containing penicillin and streptomycin (Penstrep, Gibco) as antibiotics. Cells are incubated in poly-l-Lysine (1 mg/mL, Sigma) coated T-75 tissue culture flasks in an environment maintained at 37 °C and 5% CO2. For dopamine imaging, the cells are grown in coverslip bottomed Petri dishes (poly-l-lysine coated) for 1 day. Imaging is performed in Thomson’s buffer (TB) (146 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, 0.4 mM KH2PO4, 0.3 mM Na2HPO4, 10 mM d-glucose, and 20 mM Na-HEPES; pH adjusted to 7.4). All the buffer salts were purchased from Sigma.
Glial Cell Culture
Neonatal female Wister rat P0–P3 pups are anesthetized and the brain is dissected out. All animal handling procedures are approved by the Animal Ethics Committee of the institute. The meninges are removed with the help of fine forceps under a stereomicroscope placed inside a standard laminar hood, without damaging the tissue. A small part of cortex is cut and incubated with 1× trypsin (Gibco) in TB for 20 min at 37 °C. The tissue is then triturated and grown in T-75 tissue culture flasks as described for MN9D cells. After 2 days, it is split and transferred to coverslip bottomed Petri dishes. Since glial cells outnumber the neurons by far in the cortex and only glia can divide and proliferate, this protocol ensures that glial cells predominate in the culture. Imaging is performed in TB after 1 day.
Tissue Sample Preparation
In order to visualize dopamine in live neurons, neonatal P2–P4 pups are anesthetized to dissect out the entire brain. Meninges are removed with a pair of forceps without damaging the tissue. Coronal slices (200 μm thick) are cut using a tissue chopper. Slices containing the Substantia Nigra (SN) are identified by the geometry of the adjacent hippocampus and are placed on culture plate inserts (Millipore), which goes into standard six-well plate chambers. These are maintained in DMEM (high glucose, Gibco) based media supplemented with fetal calf serum (Gibco), B27, and glutamine (Gibco) for 2 days in the incubator at 37 °C and 5% CO2. The method was adapted from Seibt et al.33 and modified as required. Imaging is performed after 2 days by placing the tissue in TB in a coverslip bottomed Petri dish. We note that the slice thins down considerably (to about 50 μm) during those 2 days.
Following Dopamine Dynamics
For drug induced dopamine release, 200 μM para-chloroamphetamine (PCA) solution in TB is added in equal volume to the dish containing cells/tissue slice in TB to yield a final concentration of 100 μM of PCA. The cells/slices are kept in this bath for 15 (or 30) min before being imaged again. For inducing depolarization in the brain slices, we use a modified TB, replacing a part of the Na+ ions with K+ ions and keeping the ionic strength and osmolarity constant. This yields a concentration of 100 mM K+ and 16 mM Ca2+ in the exocytosing solution. Equal volume of this solution is added to the existing TB in the bath, resulting in a final concentration of 50 mM K+ and 8 mM Ca2+, which induces depolarization. The slices are kept in this bath for 15 min before being imaged again. Before imaging glial cells incubated with dopamine (10 mM), they were washed with TB (3 times). The analysis was performed by selecting the cytoplasms of individual cells as the regions of interest (ROI) and computing the average brightness of the ROIs using ImageJ software (open source, available from the Web site: http://rsbweb.nih.gov/ij/). This was repeated for multiple cells under each experimental condition and the average of these values along with the standard error of the mean (SEM) are reported in the text. Student’s t test was performed in each case to check the statistical significance of the differences.
Mass Spectrometric Analysis of Cellular Dopamine
We estimate the relative amount of dopamine in MN9D or glial cells by lysing the cells and analyzing the dopamine content in the cell lysate by high performance liquid chromatography (HPLC) coupled to mass spectrometry (MS). MN9D (or glia) cells grown in T-75 dishes are washed twice with water followed by addition of 3–4 mL of perchloric acid (0.17 N). For estimating dopamine uptake by glia, 10 mM dopamine was added to the cell culture media. After 4 h of incubation, the media was removed and the cells were washed with water to remove extracellular dopamine. The washing was repeated till no detectable fluorescence of dopamine (performed in a steady-state fluorimeter, Fluoromax3, Horiba Scientific, Japan) was observed in the cell-washings. This was followed by addition of 4 mL of perchloric acid. Cells are brought in suspension by freeze–thaw and repeated flushing of perchloric acid. We lyse the cells by probe sonication under ice-cold conditions followed by centrifugation. The precipitates are lyophillized (Freeze Drier CS110-4pro, Labogene Aps, Denmark) to obtain the dry mass of the cells. Isotopically labeled dopamine (all three ring protons are deutarated, protonated mass of 157, Cambridge Isotope Laboratories) is added to the cell lysate at a final concentration of 10 μM as an external standard. The cell lysate (100 μL) is loaded into the HPLC-MS system (model: Prominence, Shimadzu Corporation, Kyoto, Japan) through a C18 analytical column (Kromasil, Eka Chemicals AB, Bohus, Sweden). A gradient of water and acetonitrile (both containing 0.1% formic acid, v/v) is used as the eluent, running at a flow rate of 0.5 mL/min for elution. The time-normalized mass intensities corresponding to m/z = 154 (for dopamine, Supporting Information Figure S4a, black) and 157 (for isotopically labeled dopamine, Supporting Information Figure S4a, red) for 22.5–24.5 min of elution (where dopamine and isotopically labeled dopamine co-elute, shaded gray in Supporting Information Figure S4a) are background subtracted (for 0–22.5 min of elution) and used for ratiometric quantification of dopamine concentration in the cell lysate. This concentration is normalized with respect to the total volume of the cell lysate and the dry cell mass to obtain the absolute dopamine content (in ng/mg of dry cell mass). Student’s t test was performed for each pair to verify the statistical significance of the results.
Acknowledgments
This work is supported in part by grant no. BT/53/NE/TBP/2010 from the Dept. of Biotechnology, India to S.M. and a CSIR Network Project grant, miND (BSC0115) to S.S. We thank Ullas Kolthur, Shubha Tole, Vidita Vaidya, Ankona Datta, Sujit Ghosh, and Senthil Arumugam for their technical help in various phases of this project, and Ruth Perez for the MN9D cell line.
Supporting Information Available
Determination of order of excitation, determination of the sensitivity of detection, absorption and emission spectra of dopamine and related UV-chromophores, details of estimation of cellular dopamine content by HPLC-coupled mass spectrometry, imaging the localization of dopaminergic cells in different regions of the brain, and three-dimensional imaging in thick brain tissue, Figures S1–S6. This material is available free of charge via the Internet at http://pubs.acs.org/.
Author Contributions
§ B.S. and A. B.: Equal contribution.
The authors declare no competing financial interest.
Supplementary Material
References
- Schultz W. (2007) Multiple dopamine functions at different time courses. Annu. Rev. Neurosci. 30, 259–88. [DOI] [PubMed] [Google Scholar]
- Boileau I.; Dagher A.; Leyton M.; Welfeld K.; Booij L.; Diksic M.; Benkelfat C. (2007) Conditioned dopamine release in humans: a positron emission tomography [11C]raclopride study with amphetamine. J. Neurosci. 27, 3998–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe E. H.; Marty A.; Schulte A.; Chow R. H. (1998) Extrasynaptic vesicular transmitter release from the somata of substantia nigra neurons in rat midbrain slices. J. Neurosci. 18, 3548–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubernator N. G.; Zhang H.; Staal R. G.; Mosharov E. V.; Pereira D. B.; Yue M.; Balsanek V.; Vadola P. A.; Mukherjee B.; Edwards R. H.; Sulzer D.; Sames D. (2009) Fluorescent false neurotransmitters visualize dopamine release from individual presynaptic terminals. Science 324, 1441–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez P. C.; Pereira D. B.; Borgkvist A.; Wong M. Y.; Barnard C.; Sonders M. S.; Zhang H.; Sames D.; Sulzer D. (2013) Fluorescent dopamine tracer resolves individual dopaminergic synapses and their activity in the brain. Proc. Natl. Acad. Sci. U.S.A. 110, 870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G.; Henke A.; Karpowicz R. J. Jr.; Sonders M. S.; Farrimond F.; Edwards R.; Sulzer D.; Sames D. (2013) New fluorescent substrate enables quantitative and high-throughput examination of vesicular monoamine transporter 2 (VMAT2). ACS Chem. Biol. 8, 1947–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro M. G.; Westmeyer G. G.; Romero P. A.; Szablowski J. O.; Kuster B.; Shah A.; Otey C. R.; Langer R.; Arnold F. H.; Jasanoff A. (2010) Directed evolution of a magnetic resonance imaging contrast agent for noninvasive imaging of dopamine. Nat. Biotechnol. 28, 264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti S.; Shear J. B.; Williams R. M.; Zipfel W. R.; Webb W. W. (1997) Measuring serotonin distribution in live cells with three-photon excitation. Science 275, 530–532. [DOI] [PubMed] [Google Scholar]
- Shear J. B.; Xu C.; Webb W. W. (1997) Multiphoton-excited visible emission by serotonin solutions. Photochem. Photobiol. 65, 931–936. [DOI] [PubMed] [Google Scholar]
- Balaji J.; Desai R.; Kaushalya S. K.; Eaton M. J.; Maiti S. (2005) Quantitative measurement of serotonin synthesis and sequestration in individual live neuronal cells. J. Neurochem. 95, 1217–1226. [DOI] [PubMed] [Google Scholar]
- Kaushalya S. K.; Desai R.; Arumugam S.; Ghosh H.; Balaji J.; Maiti S. (2008) Three-photon microscopy shows that somatic release can be a quantitatively significant component of serotonergic neurotransmission in the mammalian brain. J. Neurosci. Res. 86, 3469–3480. [DOI] [PubMed] [Google Scholar]
- Kaushalya S. K.; Nag S.; Ghosh H.; Arumugam S.; Maiti S. (2008) A high-resolution large area serotonin map of a live rat brain section. NeuroReport 19, 717–721. [DOI] [PubMed] [Google Scholar]
- Kumar M.; Kaushalya S. K.; Gressens P.; Maiti S.; Mani S. (2009) Optimized derivation and functional characterization of 5-HT neurons from human embryonic stem cells. Stem Cells Dev. 18, 615–627. [DOI] [PubMed] [Google Scholar]
- Colgan L. A.; Putzier I.; Levitan E. S. (2009) Activity-dependent vesicular monoamine transporter-mediated depletion of the nucleus supports somatic release by serotonin neurons. J. Neurosci. 29, 15878–15887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar B.; Das A. K.; Arumugam S.; Kaushalya S. K.; Bandyopadhyay A.; Balaji J.; Maiti S. (2012) The dynamics of somatic exocytosis in monoaminergic neurons. Front. Physiol. 3, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Pastila R. K.; Pitsillides C.; Runnels J. M.; Puoris’haag M.; Cote D.; Lin C. P. (2010) Imaging leukocyte trafficking in vivo with two-photon-excited endogenous tryptophan fluorescence. Opt. Express 18, 988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeskind B. J.; Jordan C. D.; Timp W.; Trapani L.; Waller G.; Horodincu V.; Ehrlich D. J.; Matsudaira P. (2007) Nucleic acid and protein mass mapping by live-cell deep-ultraviolet microscopy. Nat. Methods 4, 567–569. [DOI] [PubMed] [Google Scholar]
- Balaji J.; Reddy C. S.; Kaushalya S. K.; Maiti S. (2004) Microfluorometric detection of catecholamines with multiphoton-excited fluorescence. Appl. Opt. 43, 2412–2417. [DOI] [PubMed] [Google Scholar]
- Puopolo M.; Hochstetler S. E.; Gustincich S.; Wightman R. M.; Raviola E. (2001) Extrasynaptic release of dopamine in a retinal neuron: activity dependence and transmitter modulation. Neuron 30, 211–225. [DOI] [PubMed] [Google Scholar]
- Choi H. K.; Won L. A.; Kontur P. J.; Hammond D. N.; Fox A. P.; Wainer B. H.; Hoffmann P. C.; Heller A. (1991) Immortalization of embryonic mesencephalic dopaminergic neurons by somatic cell fusion. Brain Res. 552, 67–76. [DOI] [PubMed] [Google Scholar]
- Choi H. K.; Won L.; Roback J. D.; Wainer B. H.; Heller A. (1992) Specific modulation of dopamine expression in neuronal hybrid cells by primary cells from different brain regions. Proc. Natl. Acad. Sci. U.S.A. 89, 8943–8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. P.; Huang X. M.; Oberlender R.; Nash J. F.; Nichols D. E. (1990) Behavioral, biochemical and neurotoxicological actions of the α-ethyl homologue of p-chloroamphetamine. Eur. J. Pharmacol. 191, 1–10. [DOI] [PubMed] [Google Scholar]
- Engel K.; Zhou M.; Wang J. (2004) Identification and characterization of a novel monoamine transporter in the human brain. J. Biol. Chem. 279, 50042–50049. [DOI] [PubMed] [Google Scholar]
- Trueta C.; Mendez B.; De-Miguel F. F. (2003) Somatic exocytosis of serotonin mediated by L-type calcium channels in cultured leech neurones. J. Physiol. 547, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns D.; Riedel D.; Klingauf J.; Jahn R. (2000) Quantal release of serotonin. Neuron 28, 205–220. [DOI] [PubMed] [Google Scholar]
- Nirenberg M. J.; Chan J.; Liu Y.; Edwards R. H.; Pickel V. M. (1996) Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J. Neurosci. 16, 4135–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descarries L.; Beaudet A.; Watkins K. C. (1975) Serotonin nerve terminals in adult rat neocortex. Brain Res. 100, 563–588. [DOI] [PubMed] [Google Scholar]
- Suetake K.; Kojima H.; Inanaga K.; Koketsu K. (1981) Catecholamine is released from non-synaptic cell-soma membrane: histochemical evidence in bullfrog sympathetic ganglion cells. Brain Res. 205, 436–440. [DOI] [PubMed] [Google Scholar]
- Chen G.; Gavin P. F.; Luo G.; Ewing A. G. (1995) Observation and quantitation of exocytosis from the cell body of a fully developed neuron in Planorbis corneus. J. Neurosci. 15, 7747–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Zhang X. Y.; Luo P. F.; Huang W.; Zhu F. P.; Liu T.; Du Y. R.; Wu Q. H.; Lu J.; Xiu Y.; Liu L. N.; Huang H. P.; Guo S.; Zheng H.; Zhang C. X.; Zhou Z. (2012) Action potential-triggered somatic exocytosis in mesencephalic trigeminal nucleus neurons in rat brain slices. J. Physiol. 590, 753–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueta C.; De-Miguel F. F. (2012) Extrasynaptic exocytosis and its mechanisms: a source of molecules mediating volume transmission in the nervous system. Front. Physiol. 3, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirlapur U. K.; Konig K.; Peuckert C.; Krieg R.; Halbhuber K. J. (2001) Femtosecond near-infrared laser pulses elicit generation of reactive oxygen species in mammalian cells leading to apoptosis-like death. Exp. Cell Res. 263, 88–97. [DOI] [PubMed] [Google Scholar]
- Seibt J.; Schuurmans C.; Gradwhol G.; Dehay C.; Vanderhaeghen P.; Guillemot F.; Polleux F. (2003) Neurogenin2 specifies the connectivity of thalamic neurons by controlling axon responsiveness to intermediate target cues. Neuron 39, 439–452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.