

Abstract
Recent studies indicate that the pathogenesis of Alzheimer disease may be related to the interaction between prion protein (PrP) and certain oligomeric species of Aβ peptide. However, the mechanism of this interaction remains unclear and controversial. Here we provide direct experimental evidence that, in addition to previously demonstrated binding to Aβ oligomers, PrP also interacts with mature Aβ fibrils. However, contrary to the recent claim that PrP causes fragmentation of Aβ fibrils into oligomeric species, no evidence for such a disassembly could be detected in the present study. In contrast, our data indicate that the addition of PrP to preformed Aβ fibrils results in a lateral association of individual fibrils into larger bundles. These findings have potentially important implications for understanding the mechanism by which PrP might impact Aβ toxicity as well as for the emerging efforts to use PrP-derived compounds as inhibitors of Aβ-induced neurodegeneration.
Keywords: Prion protein, Alzheimer’s disease, Aβ peptide, amyloid fibrils
Alzheimer’s disease (AD) is associated with the accumulation of extracellular amyloid deposits called senile plaques.1 The principal components of these plaques are 40–42 residue amyloid-β (Aβ) peptides derived by proteolytic processing of a transmembrane glycoprotein, the amyloid precursor protein. Even though mature Aβ amyloid fibrils have been shown to be cytotoxic in vitro,2−4 there is a weak correlation between the amyloid burden and progression of cognitive impairment in AD.5,6 The disease symptoms appear to correlate better with the presence of smaller assemblies of Aβ, often referred to as soluble oligomers or Aβ-derived diffusible ligands.7−9 These smaller oligomeric species are also more potent neurotoxins, with their effects including impairment of synaptic plasticity, as indicated by inhibition of long-term potentiation, a neuronal feature believed to be of major importance for learning and memory.7−10 However, the mechanisms by which different types of Aβ assemblies cause synaptic dysfunction and/or neuronal cell death remain unknown.
It was recently shown that the cellular prion protein (PrPC), a glycoprotein tethered to the plasma membrane surface through the GPI anchor, can act as a high affinity receptor for soluble Aβ oligomers.11,12 Several studies also suggested that binding to this receptor may be central to the pathophysiological process in AD, mediating Aβ-induced inhibition of long-term potentiation11,13 as well as neuronal cell death.14 The relevance of prion protein–Aβ interaction to AD pathogenesis has been, however, disputed in some other studies.15,16 This controversy notwithstanding, there appears to be a consensus that both the membrane-anchored PrPC as well as the glycophoshatidylinositol anchor-free recombinant PrP bind with high affinity to at least some oligomeric forms of Aβ.11,15,17,18 The major determinants of this binding have been mapped to PrP segments ∼95–110 and 23–27 within the flexible N-terminal part of the protein. The precise nature of Aβ oligomeric species that bind to PrP is, however, unclear. A recent report suggests that the structures interacting most avidly with PrP are not small globular oligomers but larger protofibrillar assemblies that appear to share some surface features with mature Aβ fibrils.19
The finding of high affinity binding between prion protein and Aβ prompted studies on the aggregation pathway of the peptide in the presence of the recombinant PrP as well as the toxicity of the resulting PrP-Aβ complexes. It was found that the full-length PrP and its N-terminal fragment 23–110 act as strong inhibitors of Aβ assembly into amyloid fibrils.18,20,21 Even more important, it was shown that PrP and its fragments can effectively block neurotoxic effects of soluble oligomers, either by preventing formation of these toxic species or sequestering preformed oligomers in the extracellular space.18,20 Based on these findings, we and others proposed that synthetic analogues/derivatives of prion protein N-terminal fragments may offer a novel approach for pharmacological intervention in AD. However, the mode of PrP-Aβ interaction remains poorly understood and controversial. For example, Younan et al.21 suggested that the prion protein confers Aβ toxicity in AD by disassembling mature Aβ amyloid fibrils into oligomeric species. If PrP indeed has such disaggregating properties, this could impact the strategy for development of PrP-based compounds for treatment of AD. Therefore, here we have revisited the issue of PrP interaction with Aβ fibrils, finding no evidence for fibrils disassembly by PrP. Instead, our data indicate that addition of the recombinant PrP to preformed Aβ fibrils results in a lateral association of these fibrils into thicker bundles.
Results and Discussion
PrP Inhibits Aβ Fibrillization Both in the Nonseeded and Seeded Reactions
Previously we and others found out that the recombinant prion protein inhibits spontaneous (i.e., nonseeded) fibrillization of Aβ1–42,18,20,21 presumably by binding to early oligomeric species and thereby preventing formation of (or neutralizing) the nuclei required for this nucleation-dependent polymerization. In an attempt to gain further insight into the mechanism of PrP-Aβ interaction, here we extended these studies to a seeded fibrillization. Thioflavin T (ThT) fluorescence was used as a tool to follow the progress of the reaction. Under the present experimental conditions, the nonseeded reaction was characterized by a lag phase of approximately 3 h, followed by a rapid growth phase that reached saturation after approximately 8–9 h (Figure 1). Consistent with the previous report,20 addition of PrP (in the present study at the PrP:Aβ1–42 molar ratio of 1:20) strongly inhibited the reaction, with no increase of ThT fluorescence observed up to at least 24 h of incubation. As expected, addition of preformed Aβ1–42 fibrils (fragmented by sonication) as a seed completely eliminated the lag phase observed in the nonseeded reaction, resulting in a rapid increase in ThT fluorescence. However, when seeds were first preincubated in the presence of PrP (at the PrP to fibrillar Aβ molar ratio of 1:2), their ability to initiate fibrillization of Aβ1–42 was greatly diminished, as indicated by much slower increase in ThT fluorescence (Figure 1). Since previous studies consistently failed to detect any significant interaction of PrP with Aβ monomers,15,17,18 this strongly suggests that the prion protein binds to fibrillar seeds, inhibiting their capacity to recruit Aβ substrate to the fibrillar state. This inhibition is, however, only partial, as indicated by a slow, gradual increase in ThT fluorescence over time. Indeed, AFM analysis of the products of seeded reactions both in the absence and presence of PrP indicates the presence of fibrillar aggregates, even though fibrils formed in the presence of PrP appear to be less abundant, shorter and often bundled into larger aggregates (Supporting Information Figure S1).
Figure 1.

PrP inhibits both spontaneous and seeded fibrillization of Aβ1–42. (○) Spontaneous (no seeds added) reaction for Aβ1–42 alone; (▽) Spontaneous reaction for Aβ1–42 in the presence of PrP; (●) Reaction for Aβ1–42 upon seeding with preformed Aβ1–42 fibrils; (▼) Reaction for Aβ1–42 upon seeding with preformed Aβ1–42 fibrils that were preincubated with PrP (Aβ:PrP molar ratio of 2:1). The final concentration of monomeric Aβ1–42, PrP, and Aβ1–42 seeds was 10, 0.5, and 1 μM, respectively. Fibrils used as seeds were presonicated to increase the number of ends.
Direct Evidence for PrP Binding to Aβ fibrils
To further probe whether PrP is indeed able to interact with Aβ fibrils, we performed cosedimentation experiments. To this end, PrP was incubated with intact Aβ1–42 fibrils or fibrils fragmented by sonication (in each case at a PrP to Aβ1–42 molar ratio of 1:25). The fibrillar material was then sedimented by centrifugation, and the supernatants and pellets were analyzed by SDS-PAGE. As shown in Figure 2, essentially all PrP present in samples containing Aβ fibrils cosedimented with fibrillar aggregates, both in the case of intact as well as fragmented fibrils. By contrast, all PrP incubated under identical experimental conditions in the absence of Aβ remained in the supernatant. Thus, these data provide clear experimental evidence that PrP binds to Aβ1–42 fibrils. It should be noted that fibrils fragmented by sonication contain much larger number of ends (per mass unit) as compared to long fibrils. Thus, similar potency of both fibril types to cosediment PrP suggests that the prion protein interacts with Aβ1–42 over the whole length of the fibril rather than binding to fibril ends only.
Figure 2.
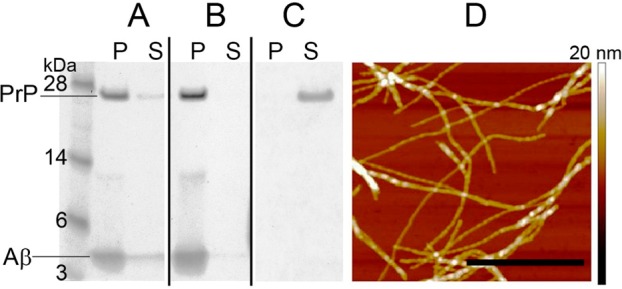
Cosedimentation of PrP with Aβ1–42 fibrils. PrP (2 μM) was incubated in the absence or presence of intact Aβ1–42 fibrils or fibrils fragmented by sonication (50 μM in each case). The samples were subsequently centrifuged under the conditions allowing sedimentation of Aβ fibrils, and the pellets and supernatants were analyzed by SDS-PAGE. (A) PrP preincubated with intact Aβ1–42 fibrils; (B) PrP preincubated with fragmented Aβ1–42 fibrils; (C) PrP alone. Symbols P and S refer to pellet and supernatant, respectively. (D) AFM image of intact Aβ1–42 fibrils used in these experiments is shown to demonstrate that these preparations are indeed highly enriched in fibrillar aggregates. The scale bar represents 0.8 μm.
In our previous study, we probed the interaction between PrP and different forms of Aβ1–42 using electron paramagnetic resonance (EPR) spectroscopy with spin-labeled PrP. While strong binding was observed between PrP and Aβ oligomers, little interaction of the prion protein was detected with Aβ fibrils,17 a result apparently inconsistent with the present sedimentation data. One potential reason for this apparent discrepancy is that the presence of the nitroxide spin label on PrP molecule (at residue 30 in the previous study) could interfere with the interaction with Aβ fibrils. To probe this possibility, we extended EPR experiments to PrP labeled in different region, namely, at residue 113 (113R PrP). EPR spectra for both 30R PrP and 113R PrP in solution show three relatively sharp lines characteristic of a highly mobile spin label (Figure 3A, B). This reflects high flexibility of the entire unstructured N-terminal part of the prion protein molecule. In contrast, the EPR spectrum for 113R PrP in the presence of Aβ1–42 fibrils shows broad features characteristic of a highly immobilized nitroxide (Figure 3D), indicating that under these conditions essentially all PrP molecules are bound to the fibrils. Consistent with the previously published data,17 the spectrum of 30R PrP in the presence of Aβ1–42 fibrils is still dominated by three relatively sharp lines, with the immobilized component being very weak (Figure 3C). Hence, it appears that the modification of the residue 30 with the spin label interferes with PrP binding to the fibrils. An alternative possibility is that 30R PrP still binds to Aβ1–42 fibrils, but this binding does not result in the immobilization of the very N-terminus of the molecule. However, the latter scenario is less likely given that the N-terminal residues 23–27 appear to be important for this interaction (see below). Furthermore, in contrast to wild-type PrP and 113R PrP, little binding to Aβ1–42 fibrils was observed for 30R PrP in the sedimentation experiments (data not shown). In any case, EPR data for 113R PrP provide additional line of evidence that prion protein binds to Aβ1–42 fibrils. Interestingly, strongly immobilized EPR spectra were previously observed for both 30R and 113R PrP in the presence of soluble Aβ1–42 oligomers,17 suggesting substantial differences in the mode of PrP interaction with the oligomers and fibrils.
Figure 3.

Binding of PrP to Aβ1–42 fibrils as probed by EPR spectroscopy. Representative EPR spectra for 30R PrP and 113R PrP (A and B) alone or in the presence of Aβ1–42 fibrils (C and D). The concentration of PrP and Aβ1–42 was 2 and 170 μM, respectively. Note that the EPR spectrum obtained for 113R PrP in the presence of Aβ1–42 fibrils (D) is characteristic of a highly immobilized spin label, whereas the immobilized component in the spectrum of 30R PrP incubated with the fibrils (low-field shoulder in panel C) is very weak.
The present findings regarding PrP binding to Aβ1–42 fibrils contrast with the inability to detect this binding in previous studies using surface plasmon resonance (SPR).15,17,18 The reason for this apparent discrepancy is not clear. However, it should be noted that SPR is less than ideally suited for this type of studies due to very “sticky” nature of Aβ fibrils. It is possible that fibrils suspended in the flow buffer became nonspecifically absorbed to the large surface of tubing and the flow cell of the SPR system before reaching the sensor with the attached PrP. The present conclusion that PrP does interact with Aβ1–42 fibrils is supported not only by the cosedimentation and EPR data but also by morphological studies using atomic force microscopy (AFM) and transmission electron microscopy (TEM) as described below. Furthermore, this conclusion is consistent with an earlier report that, at least under certain conditions, Aβ1–42 fibrils can promote conversion of monomeric PrP into a protease-resistant form.22
The Role of the N-Terminal PrP Region in the Interaction with Aβ Fibrils
To identify a region within the PrP molecule responsible for the interaction with Aβ1–42 fibrils we performed cosedimentation experiments similar to those described above employing a number of truncated PrP variants. As shown in Figure 4, under the present experimental conditions (Aβ1–42:PrP molar ratio of 10:1), more than 90% of the full-length PrP (PrP23–231) cosedimented with the fibrils. By contrast, the PrP variant lacking the N-terminal region 23–121 (PrP122–231) did not cosediment with the fibrils at all, indicating the lack of any measurable interaction. Removal of the shorter N-terminal fragment 23–89 (PrP90–231) resulted in an approximately 2-fold reduction in the binding capacity as compared to the full-length protein. A similar reduction in binding capacity was observed for the PrP variant with a deletion of only five N-terminal residues 23–27 (PrP28–231), pointing to an important role of this cluster of basic amino acids. Altogether, these data indicate that PrP residues involved in the interaction with Aβ1–42 fibrils map entirely to the largely unstructured 23–121 region of the protein.
Figure 4.

Cosedimentation of PrP and its fragments with Aβ1–42 fibrils. (A) Schematic diagram of PrP fragments used. Full-length PrP or its fragments (2 μM) were preincubated in the absence (B) or presence of Aβ1–42 fibrils (20 μM) (C). Samples were then centrifuged, and pellets and supernatants were analyzed by SDS-PAGE. Symbols P and S refer to pellet and supernatant, respectively. (D) Percent fraction of PrP cosedimented with fibrils was determined by densitometric analysis of the gels.
Previous studies revealed that PrP regions of critical importance for binding to soluble Aβ oligomers map to PrP sequence ∼95–110 and a cluster of basic N-terminal residues 23–27.17 These two regions appear also to govern the interaction of PrP with mature Aβ fibrils. However, it should be noted that while the physiologically generated N1 fragment of PrP (residues 23–110) was reported to retain full ability to interact with Aβ1–42 oligomers,18 our present cosedimentation data indicate reduced binding of this fragment to Aβ1–42 fibrils as compared with that of the full-length PrP (Figure 4). This suggests that PrP residues 111–121 may also be involved in PrP interaction with fibrillar aggregates of Aβ. Another difference in the mechanism of PrP interaction with Aβ oligomers and fibrils is suggested by EPR experiments. While for oligomers this interaction could be observed both for PrP spin-labeled at position 30 and 113,17 binding to fibrils was inhibited by the presence of spin label at position 30, that is, in close proximity to the basic cluster of residues 23–27.
PrP Induces Lateral Association of Preformed Aβ Fibrils
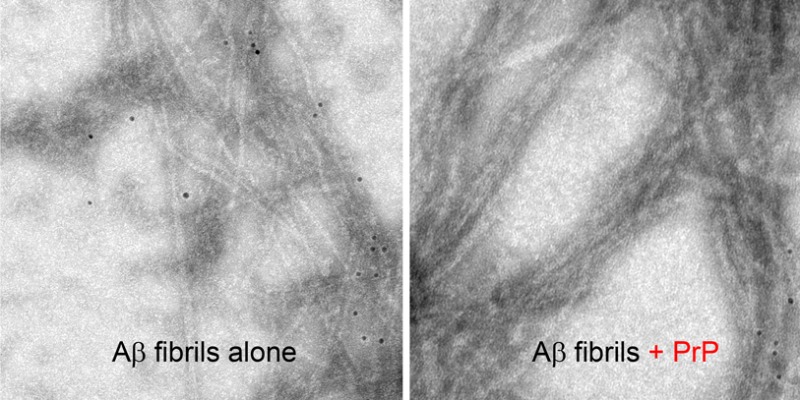
Recently Younan et al.21 reported that the addition of the recombinant prion protein to preformed Aβ fibrils resulted in the disassembly of these fibrils into smaller oligomeric structures. Here we assessed the effect of PrP on the morphology of Aβ1–42 fibrils by AFM and TEM. However, in numerous experiments we could not detect any disassembly or fragmentation of these fibrillar structures in the presence of PrP. By contrast, we found that prion protein induces massive lateral association of individual fibrils into much thicker bundles. This bundling is evident both in the height and amplitude mode of AFM images (Figure 5) as well as TEM micrographs (Supporting Information Figure S2A). Furthermore, direct association of PrP with mature Aβ1–42 fibrils can be shown by means of immuno-gold TEM (Supporting Information Figure S2B). Clearly, upon addition of PrP, most nanogold particles associated with antibodies against His-tagged PrP become concentrated around Aβ1–42 fibrils. They appear to decorate the entire length of fibrillar bundles, indicating that PrP interacts not only with fibril ends but with the entire surface of the fibrillar structures.
Figure 5.

Lateral association of mature Aβ fibrils by PrP as evidenced by AFM. The images show Aβ1–42 or Aβ1–40 fibrils alone and upon addition of PrP. The concentration of both Aβ and PrP was 14 μM. Left and right panels show AFM images in height and amplitude modes, respectively. The color coded bars at left panels illustrate the height scale in the range between 0 nm (darkest color) and 50 nm (white color).
The lack of fibril dissociation into soluble oligomers in the presence of PrP was further verified by sedimentation analysis. In these experiments, fibrillar preparations of Aβ1–42 were preincubated with PrP and subjected to low-speed centrifugation to sediment fibrils. The pellets and supernatants were then spotted on nitrocellulose and analyzed using mAb BAM-10 against Aβ. Over the entire range of PrP: Aβ1–42 molar ratios tested (1:25 to 1:1), vast majority of Aβ was found in the pellet (Supporting Information Figure S3), clearly indicating that even at PrP:Aβ molar ratio as high as 1:1 there is no dissociation of Aβ fibrils into small, soluble oligomers.
Since the study of Younan et al.21 was performed using Aβ1–40 peptide, we repeated some of our key experiments using the latter peptide. Again, no evidence for any disassembly of long Aβ1–40 fibrils in the presence of PrP could be detected in the present study. Instead, as in the case of Aβ1–42, prion protein was found to induce lateral association of individual Aβ1–40 fibrils into much thicker bundles (Figure 5). Furthermore, the binding of PrP to Aβ1–40 fibrils (similar to that described above for Aβ1–42 fibrils) was independently confirmed by sedimentation experiments (Supporting Information Figure S4).
The conclusion of Younan et al.21 that PrP induces disassembly of Aβ1–40 fibrils was partly based on the observation that upon PrP addition to preformed fibrils there is a rapid reduction of ThT fluorescence intensity. However, such a loss of ThT signal could equally well result from PrP-induced clumping of individual fibrils into larger aggregates as observed in the present study (e.g., due to precipitation of these aggregates). Indeed, a drop in ThT fluorescence upon fibril clumping has been previously observed in the studies with other amyloid-forming proteins.23 A second line of evidence in the study of Younan et al.21 for PrP-induced fibril disassembly was based on TEM images. It should be noted, however, that the structures shown for fibrils treated with PrP appear to be much wider (∼60 nm) than the width of typical Aβ fibrils alone. Thus, it is difficult to understand how these structures could be created by fibril fragmentation as proposed by the authors. Furthermore, at the mechanistic level it is not clear how PrP in the absence of other cofactors could act as a fibril-disaggregating agent. Such a disaggregating activity is a known property of HSP100 family of chaperones.24 However, these chaperones are large oligomeric proteins that utilize energy derived from ATP hydrolysis for remodeling of their substrates.
Conclusions and Implications
While recent studies point to a role of PrP-Aβ interaction in the pathogenesis of AD, the mechanism of this interaction and molecular nature of Aβ assemblies that bind to PrP remain unclear and controversial. Even though some of the previous studies concluded that PrP binds exclusively to some types of soluble Aβ oligomers and/or protofibrils, our present data clearly show that the protein also interacts with mature Aβ fibrils. However, in contrast to the recent report that PrP interaction with Aβ fibrils results in their disassembly into smaller oligomeric species,21 no evidence for such a disassembly could be detected in the present study. In contrast, our data consistently indicate that the addition of PrP to preformed Aβ1–42 or Aβ1–40 fibrils results in a lateral self-association of these fibrils into larger aggregates.
The present findings have two potentially important implications. First, it was previously proposed that PrP-induced disassembly of mature Aβ fibrils into smaller oligomers provides the mechanism by which prion protein confers Aβ toxicity in AD, as oligomers are believed to be the major toxic form of Aβ.21 However, given the present data, such a scenario appears to be highly unlikely. If PrPC indeed plays a major role in AD pathogenesis, this would have to be through the mechanism involving direct interaction with transient Aβ oligomeric species that are formed early in the aggregation process (i.e., before but not after formation of mature fibrils). Second, an increasing body of evidence suggests that recombinant PrP and its N-terminal fragments act as strong inhibitors of Aβ oligomers neurotoxicity and thus may offer a novel approach for pharmacological intervention in AD. The previously postulated ability of PrP to disaggregate mature Aβ fibrils into smaller, presumably more toxic oligomers would obviously complicate efforts to develop PrP-based compounds against AD. However, the present data indicate that such a complication is unlikely.
Methods
Preparation of PrP and Aβ Peptides
Full-length recombinant human PrP (PrP23–231), its fragments and Cys mutants were expressed and purified as previously described.17,25 Spin labeling of PrP with engineered Cys residues was performed using a previously published procedure.17 Human Aβ1–42 and Aβ1–40 peptides were purchased from American Peptide Co., Sunnyvale, CA. Before use, the peptides were disaggregated as described previously.20
ThT Fluorescence Assay
The kinetics of fibril formation by Aβ1–42 in the absence and presence of PrP was monitored by ThT assay as described previously.20 The peptide (final concentration of 10 μM) was added to wells of a 96-well plate containing 50 mM sodium phosphate, pH 7.4, and 10 μM ThT. The plate was placed in Bio Tek FLx800 plate reader and fluorescence intensity was measured every 20 min at 485 nm upon excitation at 440 nm. Before each reading, the plate was subjected to shaking for 10 s.
Preparation of Aβ Fibrils
Fibrils for cosedimentation, EPR, and morphological studies were prepared by incubating disaggregated Aβ peptides (100 μM) in 7.5 mM sodium phosphate, pH 7.4, at 25 °C for at least four days. Fibril formation was confirmed by AFM. To separate fibrillar aggregates from residual oligomers and/or monomers, the preparations were subjected to low-speed centrifugation (16 000g; 20 min).26 The pellet containing mature fibrils was washed with deionized water and subsequently resuspended in water. To prepare Aβ1–42 seeds, the fibrils were fragmented by sonication (four cycles of 25 s, 550 Sonic Dismembrator).
Cosedimentation Experiments
Aβ1–42 fibrils (untreated or fragmented by sonication) were incubated with full-length PrP or its fragments in 50 mM sodium phosphate, pH 7.4, at 25 °C for 5 min. As a control, the same PrP variants were incubated under identical conditions without fibrils. The samples were then centrifuged (16 000g; 10 min) and pellets (resuspended in the initial volume of the sample) as well as supernatants were analyzed by SDS-PAGE on 12% precast gels.
EPR Spectroscopy
Samples of spin-labeled PrP (2 μM) alone or preincubated with Aβ1–42 fibrils (170 μM) in 50 mM sodium phosphate, pH 7.4, were transferred into glass capillaries. EPR spectra were obtained using a Bruker EMX spectrometer as described previously.17
Atomic Force Microscopy
Samples of Aβ1–40 or Aβ1–42 fibrils alone or fibrils (14 μM) preincubated for 0.5 h, at 25 °C, in 50 mM sodium phosphate, pH 7.4, with full-length PrP (14 μM) were placed on freshly cleaved mica for 3 min. After rinsing with deionized water and drying, the samples were imaged in a tapping mode using a MultiMode atomic force microscope equipped with NanoScope IV controller (Digital Instruments, Santa Barbara, CA).
Transmission Electron Microscopy
Aβ1–42 fibrils (14 μM in 50 mM sodium phosphate, pH 7.4) were preincubated with or without full-length PrP (14 μM) containing N-terminal 6 × His tag. For immuno-gold staining, the samples were incubated for 1 h with 100-fold diluted mouse mAb against His-tag (Millipore) and subsequently for 1 h with 10-fold diluted gold-conjugated goat antibodies (20 nm colloidal gold particles) against mouse IgG. Preparations were spotted on mesh 400 copper grids covered with collodion and carbon, negatively stained with 2% uranium acetate, and analyzed in a JEM 1400 electron microscope equipped with a digital camera.
Glossary
Abbreviations
- Aβ
amyloid-β
- AD
Alzheimer’s disease
- AFM
atomic force microscopy
- EPR
electron paramagnetic resonance
- PrP
prion protein
- SPR
surface plasmon resonance
- ThT
thioflavin T
- TEM
transmission electron microscopy
Supporting Information Available
Figure S1 showing AFM images of Aβ1–42 fibrils generated in the presence or absence of PrP in the seeded reaction. Figure S2 showing TEM micrographs of Aβ1–42 fibrils alone and upon incubation with PrP. Figure S3 showing sedimentation of fibrillar Aβ1–42 in the presence of PrP. Figure S4 showing cosedimentation of PrP with Aβ1–40 fibrils. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
K.N and W.K.S. designed research; K.N., K.S., S.C. performed research; K.N. and W.K.S. analyzed data and wrote the paper.
This work was supported by the Spitz Brain Health Innovation Pilot Grant program and National Institutes of Health Grants NS044158 and NS074317.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Selkoe D. J. (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766. [DOI] [PubMed] [Google Scholar]
- Yankner B. A.; Duffy L. K.; Kirschner D. A. (1990) Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science 250, 279–282. [DOI] [PubMed] [Google Scholar]
- Pike C. J.; Walencewicz A. J.; Glabe C. G.; Cotman C. W. (1991) Aggregation-related toxicity of synthetic beta-amyloid protein in hippocampal cultures. Eur. J. Pharmacol. 207, 367–368. [DOI] [PubMed] [Google Scholar]
- Lorenzo A.; Yankner B. A. (1994) Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc. Natl. Acad. Sci. U.S.A. 91, 12243–12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann D. M.; Jones D.; South P. W.; Snowden J. S.; Neary D. (1992) Deposition of amyloid beta protein in non-Alzheimer dementias: evidence for a neuronal origin of parenchymal deposits of beta protein in neurodegenerative disease. Acta Neuropathol. 83, 415–419. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P.; Herrmann F. R.; Bussiere T.; Bouras C.; Kovari E.; Perl D. P.; Morrison J. H.; Gold G.; Hof P. R. (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Selkoe D. J. (2007) Abeta oligomers - a decade of discovery. J. Neurochem. 101, 1172–1184. [DOI] [PubMed] [Google Scholar]
- Klein W. L.; Krafft G. A.; Finch C. E. (2001) Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum?. Trends Neurosci. 24, 219–224. [DOI] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. [DOI] [PubMed] [Google Scholar]
- Dahlgren K. N.; Manelli A. M.; Stine W. B. Jr.; Baker L. K.; Krafft G. A.; LaDu M. J. (2002) Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053. [DOI] [PubMed] [Google Scholar]
- Lauren J.; Gimbel D. A.; Nygaard H. B.; Gilbert J. W.; Strittmatter S. M. (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimbel D. A.; Nygaard H. B.; Coffey E. E.; Gunther E. C.; Lauren J.; Gimbel Z. A.; Strittmatter S. M. (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 30, 6367–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freir D. B.; Nicoll A. J.; Klyubin I.; Panico S.; Mc Donald J. M.; Risse E.; Asante E. A.; Farrow M. A.; Sessions R. B.; Saibil H. R.; Clarke A. R.; Rowan M. J.; Walsh D. M.; Collinge J. (2011) Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo W.; Lee H. P.; Zou W. Q.; Wang X.; Perry G.; Zhu X.; Smith M. A.; Petersen R. B.; Lee H. G. (2012) Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum. Mol. Genet. 21, 1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balducci C.; Beeg M.; Stravalaci M.; Bastone A.; Sclip A.; Biasini E.; Tapella L.; Colombo L.; Manzoni C.; Borsello T.; Chiesa R.; Gobbi M.; Salmona M.; Forloni G. (2010) Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calella A. M.; Farinelli M.; Nuvolone M.; Mirante O.; Moos R.; Falsig J.; Mansuy I. M.; Aguzzi A. (2010) Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol. Med. 2, 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Yadav S. P.; Surewicz W. K. (2010) Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J. Biol. Chem. 285, 26377–26383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluharty B. R.; Biasini E.; Stravalaci M.; Sclip A.; Diomede L.; Balducci C.; La Vitola P.; Messa M.; Colombo L.; Forloni G.; Borsello T.; Gobbi M.; Harris D. A. (2013) An N-terminal fragment of the prion protein binds to amyloid-β oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 288, 7857–7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll A. J.; Panico S.; Freir D. B.; Wright D.; Terry C.; Risse E.; Herron C. E.; O’Malley T.; Wadsworth J. D.; Farrow M. A.; Walsh D. M.; Saibil H. R.; Collinge J. (2013) Amyloid-β nanotubes are associated with prion protein-dependent synaptotoxicity. Nat. Commun. 4, 2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieznanski K.; Choi J. K.; Chen S.; Surewicz K.; Surewicz W. K. (2012) Soluble prion protein inhibits amyloid-β (Aβ) fibrillization and toxicity. J. Biol. Chem. 287, 33104–33108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younan N. D.; Sarell C. J.; Davies P.; Brown D. R.; Viles J. H. (2013) The cellular prion protein traps Alzheimer’s Aβ in an oligomeric form and disassembles amyloid fibers. FASEB J. 27, 1847–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R.; Estrada L. D.; Diaz-Espinoza R.; Morales-Scheihing D.; Jara M. C.; Castilla J.; Soto C. (2010) Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J. Neurosci. 30, 4528–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanik D. L.; Surewicz K. A.; Surewicz W. K. (2004) Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol. Cell 14, 139–145. [DOI] [PubMed] [Google Scholar]
- Zolkiewski M.; Zhang T.; Nagy M. (2012) Aggregate reactivation mediated by the Hsp100 chaperones. Arch. Biochem. Biophys. 520, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillas M.; Swietnicki W.; Gambetti P.; Surewicz W. K. (1999) Membrane environment alters the conformational structure of the recombinant human prion protein. J. Biol. Chem. 274, 36859–36865. [DOI] [PubMed] [Google Scholar]
- Stine W. B. Jr.; Dahlgren K. N.; Krafft G. A.; LaDu M. J. (2003) In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.