

Abstract
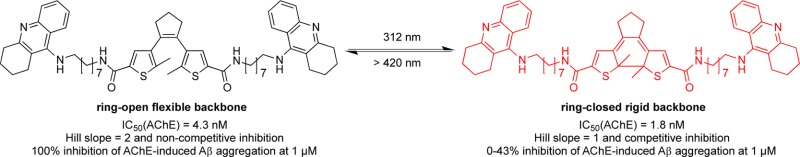
Photochromic cholinesterase inhibitors were obtained from cis-1,2-α-dithienylethene-based compounds by incorporating one or two aminopolymethylene tacrine groups. All target compounds are potent acetyl- (AChE) and butyrylcholinesterase (BChE) inhibitors in the nanomolar concentration range. Compound 11b bearing an octylene linker exhibited interactions with both the catalytic active site (CAS) and the peripheral anionic site (PAS) of AChE. Yet upon irradiation with light, the mechanism of interaction varied from one photochromic form to another, which was investigated by kinetic studies and proved “photoswitchable”. The AChE-induced β-amyloid (Aβ) aggregation assay gave further experimental support to this finding: Aβ1–40 aggregation catalyzed by the PAS of AChE might be inhibited by compound 11b in a concentration-dependent manner and seems to occur only with one photochromic form. Computational docking studies provided potential binding modes of the compound. Docking studies and molecular dynamics (MD) simulations for the ring-open and -closed form indicate a difference in binding. Although both forms can interact with the PAS, more stable interactions are observed for the ring-open form based upon stabilization of a water molecule network within the enzyme, whereas the ring-closed form lacks the required conformational flexibility for an analogous binding mode. The photoswitchable inhibitor identified might serve as valuable molecular tool to investigate the different biological properties of AChE as well as its role in pathogenesis of AD in in vitro assays.
Keywords: Alzheimer’s disease; beta-amyloid aggregation; cholinesterase inhibitors; tacrine; photochromism; 1,2-dithienylethene
Alzheimer’s disease (AD) is the most common cause of dementia and is affecting tens of millions of people (normally aged over 65) worldwide, especially in developed countries due to the higher life expectancy.1 Though research is also focusing on β-secretase 1 (BACE-1) and tau protein hyperphosphorylation, cholinesterase inhibitors (ChEIs), that is, the AD drugs tacrine, rivastigmine, donepezil, and galantamine, are still the only established approved therapy for treatment of AD, apart from the N-methyl-d-aspartate (NMDA) receptor antagonist memantine. Until now, the use of ChEIs is only indicated for symptomatic treatment of AD, since they are not able to prevent the progress of the disease. However, in the last years, ChEIs have again come into the focus of drug development efforts because acetylcholinesterase (AChE) might play a role in the formation of β-amyloid (Aβ) deposits.2,3 Aβ is deposited in extracellular toxic plaques in Alzheimer patients’ brains.3 It is noteworthy that the interaction of AChE with Aβ and its correlation with tau hyperphosphorylation are not related to effects in cholinergic neurotransmission. AChE possesses two binding sites: the catalytic active site (CAS) that is located at the bottom of a 20 Å gorge responsible for neurotransmitter hydrolysis, and the peripheral anionic site (PAS) that is rich in aromatic residues located at the rim of the gorge on the surface of the enzyme.4 The noncholinergic functions of AChE are directly related to the PAS of the enzyme. However, even after many studies, the precise function and pathological mechanism of PAS inhibition still remains unclear, especially its role in the progress of Aβ formation. The PAS may not only stimulate the formation and aggregation of Aβ, but also increase the neurotoxicity of amyloid fibrils. Besides, there may be a positive feedback loop between AChE activity and tau hyperphosphorylation and Aβ formation, respectively.3,5 As a result, proper specific pharmacological testing models are needed as well as suitable tool drugs to decipher more details about inhibition of the PAS and resulting pathophysiological consequences.6
Tacrine, the first anti-AD drug that had been approved by the United States Food and Drug Administration (FDA) two decades ago, still receives a lot of attention despite its dose-dependent hepatotoxicity due to its simple chemical structure and its very high inhibitory activity at both AChE and BChE. Tacrine-based bivalent or hybrid compounds were developed possessing multiple pharmacological actions,7 such as antioxidant,8,9 NO-donor,10 PAS inhibitor,11,12 and so forth. Such versatile compounds were recently reviewed by Tumiatti et al.12 The results are very encouraging, especially from the blockade of the PAS disease-modifying therapeutic strategies for AD might emerge.12−14
Photochromic (or “photoswitchable”) inhibitors can reversibly control biological functions such as the activity of an enzyme. Examples include azobenzene-based ligands and thiophenfulgide derivatives.15 Compounds based on the 1,2-dithienylethene (DTE) scaffold have the advantage of better thermal stability (of both photoisomers) and quantitative rates of photoisomerization compared to azobenzene derivatives.16 DTEs can be reversibly toggled between their photoisomers via a 6-π electrocyclic reaction by UV or visible light (Figure 1).
Figure 1.
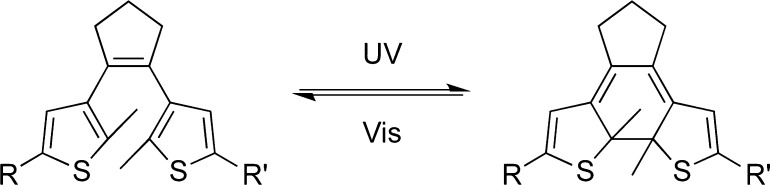
Light-induced switching of a DTE derivative between its ring-open and -closed isomer.
Photochromic compounds are applied as modulators in optoelectronic devices and even as regulators of biological processes.17 König et al. used the DTE scaffold to successfully develop a photoswitchable inhibitor of the enzyme human carbonic anhydrase I, in which the DTE was connected with a 4-amino-sulfonamide, a low-affinity enzyme inhibitor, and an imidazole-binding copper complex.18 Similarly, azo-propofol compounds containing an azobenzene unit were applied at γ-aminobutyric acid A (GABAA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, impressively enabling the control of the neural system in a light-dependent way.19 In another example, the azobenzene motif was used as a spacer to connect chromone structures that inhibit the activation of mast cells in allergy diseases. In the latter case, the target compound not only shows higher potency than the lead compound, but also exhibits photoswitchability during inhibition.20 It has been shown in very recent times that photoswitchable compounds can be successfully used to study biological mechanisms in an innovative manner; the prerequisite is the identification of photoisomers with a pronounced difference in biological activities.19,20
Herein we report the synthesis, physicochemical characterization and several biological studies (ChEs inhibitory activities, enzyme kinetics, and their influence on inhibiting AChE-induced Aβ aggregation, respectively) of molecules constructed from the photochromic diarylethene scaffold combined with aminopolymethylene tacrine moieties. Additionally, computational studies (docking as well as molecular dynamics simulations) were performed to find an explanation on the molecular level for the biological data observed. In this work, we used the photochromic diarylethene scaffold as the photoswitchable component due to its stability of both photoisomers to incorporate it into the well-known bis-tacrines that are able to interact with the two binding sites of AChE, which are both being discussed for their therapeutic value.12 A photoswitchable AChE inhibitor, whose activity can be controlled by light in a spatiotemporal manner, would be a valuable tool to investigate AChE’s roles in the AD disease state and would allow to further explore AChE’s potential for drug development beyond symptomatic treatment via prevention of ACh hydrolysis.
Results and Discussion
Chemistry
Photochromic tacrine-DTE-derivatives were synthesized in a convergent way by amide formation of cis-1,2-α-dithienylcyclopentene carboxylic acid 5 (first path) with 9-aminoalkylamino-1,2,3,4-tetrahydroacridine 9 (second path) using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and 1-hydroxybenzotriazole (HOBt) as coupling reagents (Scheme 1). 1,2-α-Dithienylcyclopentene carboxylic acid 5 for the first path was synthesized according to the method described in the literature (Scheme 1):21 Friedel–Crafts acylation followed by McMurry ring closure reaction gave the dichloro diarylcyclopentene 4, which was converted by chlorine/lithium exchange and reaction with gaseous carbon dioxide to form the corresponding carboxylic acid 5. With regard to the second path, tacrine derivatives 9 were prepared by reacting ethylenediamine or octane-1,8-diamine with 9-chlorotetrahydroacridine 8 using previously described procedures.8,22 The synthesis of bivalent compounds (11a, b) with two tacrine moieties used a 1:2 ratio of DTE dicarboxylic acid and the tacrine derivative, whereas for the synthesis of univalent compounds (10a, b) a 1:1 ratio was applied (Scheme 1). In each case, two different spacer lengths were incorporated. All target compounds were characterized by 1H NMR, 13C NMR, and HRMS, and their purities were determined by HPLC. This also applies to both photoisomeric forms of 11b.
Scheme 1. Synthesis of Photochromic Uni- and Bivalent ChE Inhibitors.

Reagents and conditions: (i) AlCl3, CH3NO2, 0 °C to r.t., 71%; (ii) Zn, TiCl4, dry THF, 40 °C, 58%; (iii) nBuLi, gaseous CO2, dry THF, r.t., 98%; (iv) POCl3, reflux, 60%; (v) ethylenediamine or 1,8-diaminooctane, 1-hexanol, 150 °C, 12 h, 66–73%; (vi) EDCI, HOBt, dry DMF, r.t., 12 h, 22–32%.
Physicochemical Properties
The photochemical properties of all tacrine-DTE-derivatives were investigated by UV/vis spectroscopy. The absorbance spectra of all compounds are presented in the Supporting Information. All target molecules showed reversible photoswitchable behavior. Absorbance spectra were recorded starting with the ring-opened photoisomers and irradiating with 312 nm light in periods of 5 s (e.g., 11b, Figure 2). For all compounds, the absorbances around 270 nm decreased upon UV-irradiation, whereas the absorbance band at 340 nm increased, and a new absorbance band maximum at 525 nm evolved (cf. Supporting Information Figures 1–7) due to the formation of the benzodithiophene conjugation system.16,18 We especially investigated and characterized the structure of the ring-closed form of these compounds, exemplified by compound 11b: the changing of the retention time in HPLC (cf. Supporting Information Figures 8–9) and the shifting of corresponding signals in 1H NMR (cf. Supporting Information Figure 10). At a concentration of 100 μM and after 30 s of irradiation, more than 92% of the ring-closed form is detected by HPLC (cf. Supporting Information Figure 9).
Figure 2.
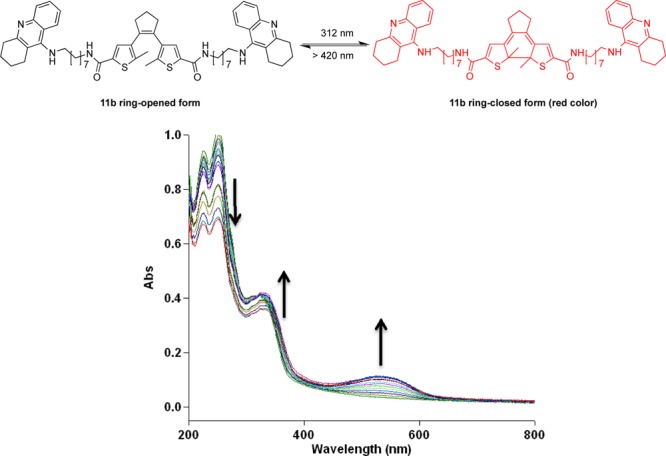
UV–vis absorbance spectra evolution of compound 11b (50 μM in buffer solution with 10% ethanol) by irradiation with 312 nm light. Arrows indicate the changes of the absorption maxima with irradiation periods of 5 s.
We investigated the photostability of the target compounds by successive ring-closing-/ring-opening cycles applying 312 nm and wavelengths ≥ 420 nm, respectively. All target molecules showed a degradation of 20–30% after seven cycles as exemplarily presented for compound 11b in Figure 3. The photodecomposition is also reported for other dithienylcyclopentene systems, but the exact mechanism remains unknown.23 Kept in the dark, the ring-closed photoisomer 11b was stable for several months.18 Photochemical stability was therefore given for the application of the photoisomers under the assay conditions applied and described below, since the ring-opening process demands long exposure (>5 min) and intense light (150 W), whereas during the testing the vial with the testing solution would only be exposed to light for a few seconds.
Figure 3.

Cycle performance of compound 11b, changes in absorption at 525 nm during an alternated irradiation of a solution (50 μM in pH = 8.0 buffer with 10% ethanol) with 312 nm light for 30 s and ≥420 nm light for 5 min. 80% of the compound remained photoactive after seven cycles of on- and off-switching.
Cholinesterase Inhibition
All tacrine-DTE derivatives are very active inhibitors with IC50 values (AChE) ranging from 1.8 nM (bivalent compound 11b) to 153 nM (univalent compound 10a) (note that the terms “univalent” and “bivalent” only refer to the constitution of the compounds with either one or two tacrine moieties, respectively, regardless of the actual binding mode). In most cases, the compounds show similar activities at both AChE and BChE with the ethylene-bridged compounds exhibiting moderately higher inhibitory activities at BChE (Table 1).
Table 1. Inhibition Results of Target Compounds at AChE and BChE and Resulting Selectivities Expressed as the Ratio of IC50 Values.
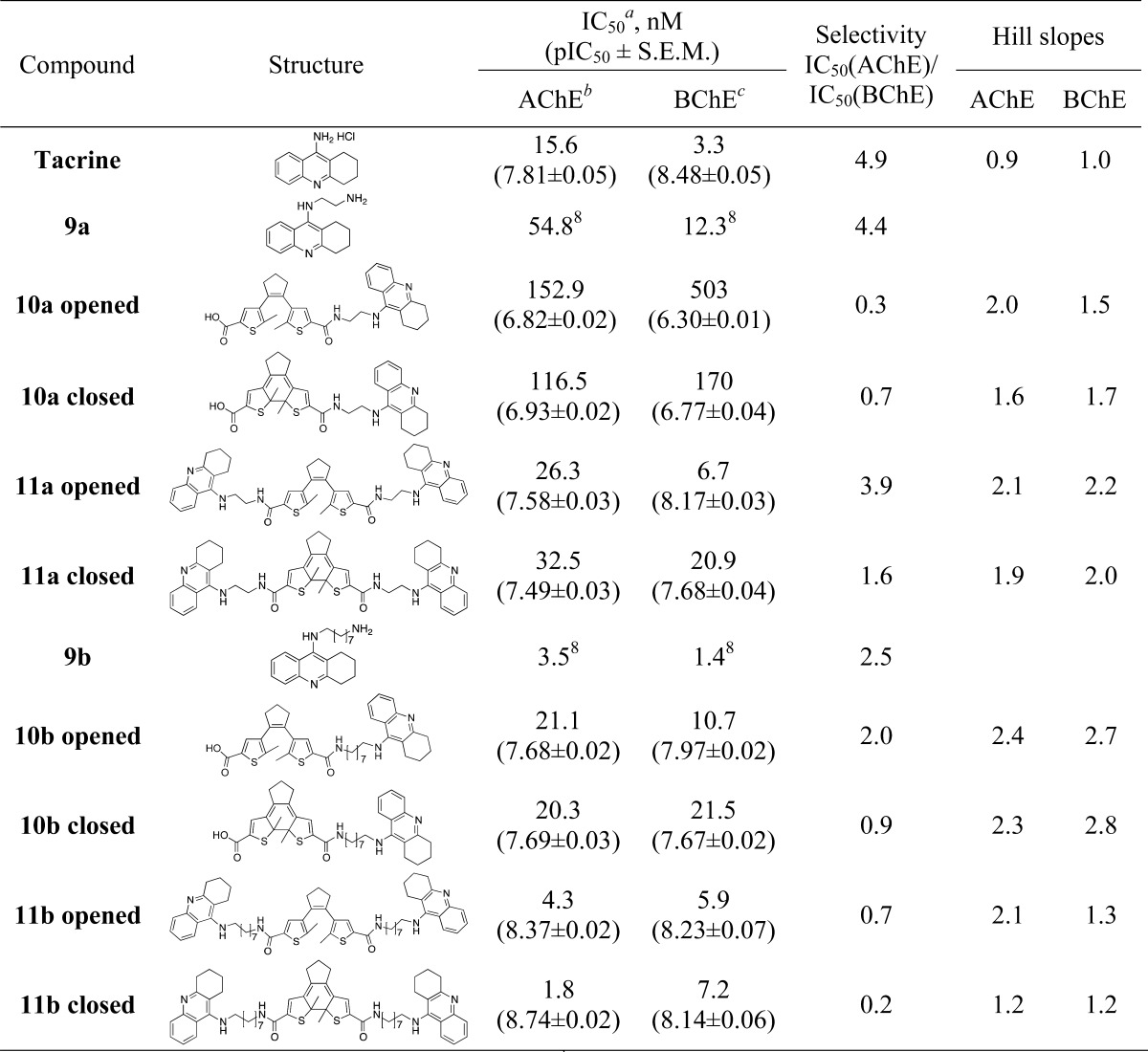
Values are means of at least three determinations, pIC50 = −log IC50.
AChE from electric eel.
BChE from equine serum.
The bivalent inhibitor 11a with a short ethylene unit represents a potent two-digit nanomolar inhibitor in its ring-closed form being only slightly less potent than tacrine, and also the ring-opened form of 11a inhibits AChE in the same concentration range with slightly lower activities. Surprisingly, the octylene-bridged bivalent inhibitor 11b with a sterically demanding DTE group represents (in both the ring-opened and -closed form) a one-digit nanomolar inhibitor being 3- to 4-fold more potent at AChE than even tacrine itself. The respective octylene-bridged univalent inhibitor 10b is also a very active inhibitor (21 nM for ring-opened form, 20 nM for the ring-closed form) when compared to the ethylene-bridged univalent inhibitor 10a with only submicromolar inhibition (153 and 117 nM, respectively). Taken together, both bivalent inhibitors are potent AChE and BChE inhibitors, irrespective of the fact whether the compounds are inhibiting in their ring-opened or -closed forms.
Interestingly, in some cases, the inhibition curves of the ring-opened and -closed forms reveal significant differences between the two photoisomers with regard to their Hill slopes, best exemplified by compound 11b (Table 2, Figure 4). A Hill slope of 1 indicates one site interaction between inhibitor and enzyme, normally with the active center, that is, the CAS of AChE. A steeper Hill slope indicates positive cooperativity, in the case of AChE plausibly due to concomitant interaction with a second binding site.24 For bivalent and hybrid inhibitors, the hydrophobic PAS represents the favored second binding site, the inhibition of which slows down the AChE-induced aggregation of Aβ, a core process in AD pathology.2,6 To confirm this result, we tested the compound 11b further on human AChE, with both usual (4.5 min) and prolonged (1 h) incubation times. The latter was used to ensure that the interaction between the compound and the enzyme has reached equilibrium. The results indicate repeatability of this switch in the Hill slope at AChE from both species at different incubation times (Table 2), with the effect being less pronounced at hAChE.
Table 2. Inhibition of AChE from Electric Eel (eeAChE) and Human (recombinant, hAChE) by Tacrine and Both Photochromic Forms of Compound 11b, with Short and Long Incubation Time.
| 4.5
min incubation |
1 h
incubation |
|||||||
|---|---|---|---|---|---|---|---|---|
| IC50, nM (pIC50 ± SEM) |
Hill
slopes |
IC50, nM (pIC50 ± SEM) |
Hill
slopes |
|||||
| compd | eeAChE | hAChE | eeAChE | hAChE | eeAChE | hAChE | eeAChE | hAChE |
| tacrine | 15.6 | 95.9 | 0.9 | 1.0 | 23.5 | 84.8 | 1.0 | 1.1 |
| (7.81 ± 0.05) | (7.02 ± 0.06) | (7.63 ± 0.01) | (7.07 ± 0.04) | |||||
| 11b opened | 4.3 | 49.6 | 2.1 | 1.6 | 14.0 | 41.4 | 2.1 | 1.6 |
| (8.36 ± 0.03) | (7.30 ± 0.05) | (7.85 ± 0.03) | (7.38 ± 0.06) | |||||
| 11b closed | 1.8 | 19.1 | 1.2 | 1.2 | 12.9 | 14.2 | 1.0 | 1.0 |
| (8.74 ± 0.02) | (7.72 ± 0.06) | (7.89 ± 0.05) | (7.85 ± 0.07) | |||||
Figure 4.
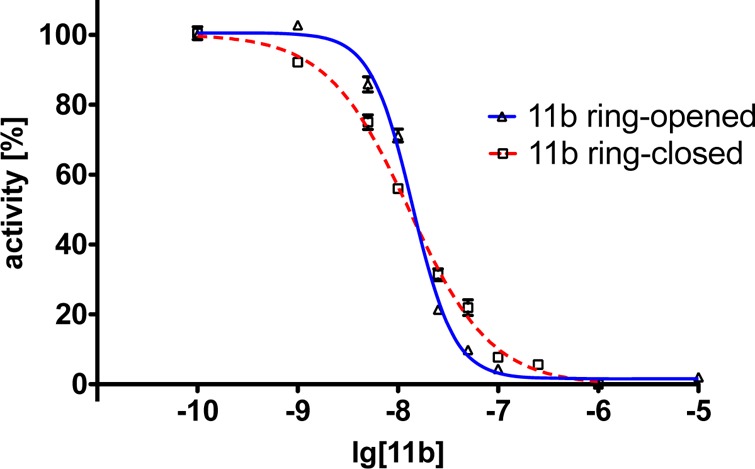
Inhibition curves of compound 11b at eeAChE as both ring-opened (blue solid curve) and -closed (red dashed curve) forms after 1 h incubation. A clear switching of the Hill slope from ring-opened form (nH = 2.1) to ring-closed form (nH = 1.0) after the irradiation with light is observed (for detailed data, cf. Figure 11 in the Supporting Information).
We therefore further investigated the mechanistic details of the mode of inhibition of compound 11b in both its ring-opened and -closed forms by kinetic studies. The mechanism of inhibition was analyzed by recording substrate-velocity curves (Michaelis–Menten) in the presence of different concentrations of compound 11b (cf. Figure 12 in the Supporting Information). Substrate (i.e., acetylthiocholine) concentration was varied between 28.2 and 452 μM. For the ring-opened form, 15, 20, 25, and 30 nM compound 11b were applied, and for the ring-closed form 25, 30, 35, 40, and 50 nM were used. Vmax and Km values were calculated directly from substrate velocity curves using GraphPad Prism. Lineweaver–Burk plots (Figure 5) are shown for better illustration and clearness, that is, reciprocal rates versus reciprocal substrate concentrations for the different inhibitor concentrations, as both ring-opened and -closed forms. For the ring-opened form, when increasing the concentration of 11b, the Vmax value (i.e., the reciprocal of the Y-intercept) decreases, whereas the Km value (i.e., the negative reciprocal of the X-intercept) remains the same. Therefore, compound 11b in its ring-opened form represents a noncompetitive inhibitor of AChE.8,25,26 In contrast, in its ring-closed form, a different Lineweaver–Burk curve illustrates the difference in Km and Vmax values. The Km values differ with different 11b concentrations, while the Vmax values remain essentially unchanged; that is, this photoisomer of 11b is a competitive inhibitor of AChE and therefore presumably might only inhibit the CAS of the enzyme. The Lineweaver–Burk plots for AChE clearly show reversible and noncompetitive inhibition by 11b as ring-opened form, yet competitive inhibition as ring-closed form. These results from the kinetic studies are in accordance with the initial assumptions gained from Hill slopes. Based upon the analyses of the chemical structures of the target compounds as well as of the 3D model of AChE,27 we expect that the more flexible ring-opened form is able to interact with both the CAS and the PAS of AChE, whereas for the ring-closed compound formed after irradiation the rigid backbone of DTE limits this interaction, most likely with the PAS (cf. computational studies described below). The change of interaction keeps the potent inhibition of AChE intact, but binding interaction with the PAS might be different or even lost in 11b’s ring-closed form.
Figure 5.
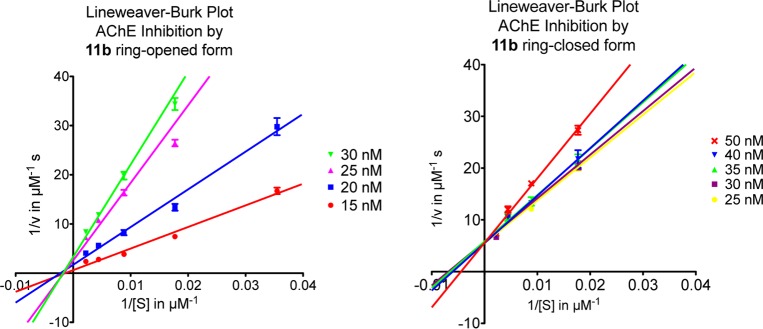
Lineweaver–Burk plots of AChE activity with different substrate concentration (28.2–452 μM) in the presence of suitable concentrations of compound 11b (15–50 nM). The graph on the left was tested with 11b in its ring-opened form, whereas the one on the right was tested in its ring-closed form.
The fact that compound 11b acts as a potent inhibitor of AChE with equal potency for both photoisomers, but is able to interact differently with a second binding site, classifies it as a “photoswitchable” or photochromic PAS-inhibitor. If this is the case, it should also be possible to “switch on or off” AChE’s catalytic effect on Aβ aggregation. Consequently, both photoisomeric forms of compound 11b were incubated together with AChE and their abilities to interfere with the extent of Aβ aggregation as a result of the inhibition of AChE’s PAS were quantified.
AChE Induced β-Amyloid Aggregation
In order to further prove our hypothesis, compound 11b was tested for its influence on AChE-induced β-amyloid aggregation, since the different inhibitory manner of 11b as ring-opened and -closed forms might translate into different abilities to modulate β-amyloid aggregation despite their almost identical IC50 values for AChE inhibition.
An AChE-induced Aβ1–40 aggregation assay was established using the thioflavin T fluorescence method, which was slightly modified from those reported in the literature in order to suit our purpose.2,14,28,29 First, a phosphate buffer containing NaCl was chosen, because of its ability to enhance the ionic strength and consequently accelerate the speed of aggregation.28 Second, the pH value of the buffer was set to 8 where AChE shows the highest activity with regard to substrate hydrolysis.14 Third, only AChE-induced aggregation was presented without considering the influence of self-aggregation due to the fact that under such conditions the rate of self-induced Aβ aggregation should be much slower than the AChE-induced one.2 Lastly, AChE from electric eel instead of the human recombinant one was used to keep the results consistent with the ones obtained in the enzyme inhibition and kinetic studies.
The resulting curves show different inhibitory activities for ring-opened and -closed forms of 11b (Figure 6): As predicted from kinetic studies, all the curves for ring-closed 11b show higher values of aggregation (i.e., lower inhibition) than the curves with corresponding concentrations as ring-opened form. The ring-closed form of 11b does not interfere with the accelerating effect of AChE on aggregation rate resulting in higher fluorescence. As a result, after a certain time, the fluorescence increase (blue lines in Figure 6) should be closer to the blank value (black line in Figure 6). Low concentrations of 11b (0.2 and 1 μM) in the ring-closed form show no or little inhibition of AChE-induced aggregation of Aβ1–40 (Table 3). The effects are above or around the blank value (black line). At a high concentration (5 μM) of 11b in its ring-closed form, a lowered aggregation can be observed which may be due to the inhibitory activity of the compound to prevent self-induced aggregation of Aβ1–40.
Figure 6.
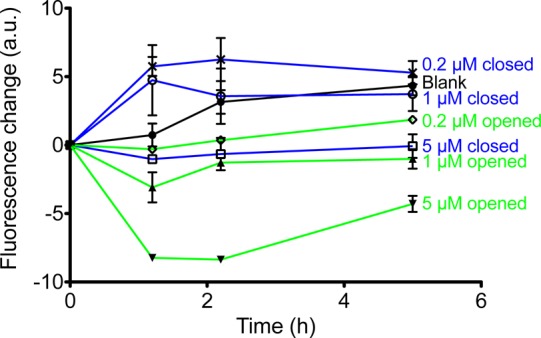
Inhibition curves of AChE-induced Aβ1–40 aggregation by compound 11b in a concentration dependent manner for both its ring-opened and -closed forms (measured in duplicate). The blue lines represent compound 11b in the ring-closed form, whereas the green lines represent the opened form. The quantitative summary of part of the graph is also presented below as a table.
Table 3. Inhibition of AChE-Induced Aβ1-40 Aggregation by Compound 11b in Both Photoforms.
| compound | concentration | percent inhibition with compound after 5 h incubationa |
|---|---|---|
| 11b ring-opened | 1 μM | 100%b |
| 0.2 μM | 53.8–60.2% | |
| 11b ring-closed | 1 μM | 0–43.0% |
| 0.2 μM | 0%c |
All the experiments were performed in duplicate; therefore, the data here are given as a range instead of mean ± SEM.
Experimentally, a slight decrease in fluorescence was observed, due to complete inhibition of Aβ aggregation.
Experimentally, a slight increase in fluorescence was observed compared to the blank value; that is, no standard error of means can be provided due to complete lack of inhibition.
In contrast, the ring-opened form of 11b seems to interfere with the PAS and consequently inhibits the Aβ aggregation in a concentration-dependent manner resulting in lower fluorescence (green lines in Figure 6). At low concentrations of 11b in the ring-opened form (0.2 and 1 μM), fluorescence emission remained in the same range as in the beginning, i.e. the increase is around zero for both concentrations (green lines in Figure 6) reflecting pronounced inhibition of aggregation in contrast to the accelerated Aβ aggregation observed for the blank (Table 3). The reason for an additional decrease of fluorescence emission at a concentration of 5 μM of ring-opened 11b may very well due to the inhibition of self-induced Aβ1–40 aggregation (data not shown). Nevertheless, there is a clear dose-dependent effect on reduction of AChE-induced Aβ1–40 aggregation for the ring-open form of 11b (green lines). The inhibition of Aβ1–40 aggregation by compound 11b in both photoforms after 5 h is also quantitatively summarized in Table 3. At the same concentration, the aggregation of Aβ1–40 inhibited by compound 11b in its ring-opened form is always significantly higher than for the ring-closed form under corresponding conditions (Table 3).
Computational Binding Mode Analysis
Docking studies were carried out to investigate potential binding modes of compound 11b in both its ring-opened and -closed photoforms. Despite the ligands’ large number of conformational degrees of freedom, the docking procedure provided reasonable poses on the top ranks. The partially saturated (cyclohexen-like) ring of the tacrine moiety was generally handled in the twisted half-chair conformation, except for 11b closed, where better results were obtained with a half-chair conformation of the tacrine group located in the PAS (cf. binding-mode description in the following paragraph). Due to the complementarity of interactions, the positions obtained at rank 2 for both 11b open and 11b closed appeared most feasible upon visual inspection. This impression was confirmed by rescoring the docking poses obtained from GOLD with DrugScoreX (DSX):30 With values of −253 (11b opened) and −241 (11b closed) DSX assigned the clearly best score to the poses ranked second in GOLD (see Table 1 in the Supporting Information for the detailed scores of the top five ranks). Accordingly, these poses were selected for further analysis.
Both photoforms of 11b extend from the CAS to the PAS, reminiscent of the binding mode of the 7-carbon-linker bis-tacrine of structure 2CKM.31 The first tacrine moiety is placed in the CAS (Ser200, Glu327, His440), with its aromatic nitrogen atom in close hydrogen-bonding distance to the backbone oxygen of His440. The ring system is located between Trp84 and Phe330, showing good π-stacking interactions with the Trp84 side chain of the “anionic” site. For both inhibitors, the second tacrine group is sandwiched between Tyr70 and Trp279 of the PAS. The orientation, however, is different (Figure 7): The photochromic units of the two inhibitors are located at the entrance of the binding gorge in an almost perpendicular orientation relative to each other, resulting in different access of the tacrine groups to the PAS. The tacrine moiety of the ring-opened form of 11b enters the PAS from the side of the binding gorge, whereas in the ring-closed 11b the methylene linker wraps around Trp279 to enable access from the more distal site. Presumably, this is required by the restricted conformational freedom of the closed form. In comparison to the 7-carbon-linker bis-tacrine of 2CKM, the much longer dimension of the 11b compounds prevents a direct extension from the CAS to the PAS. Instead, only the alkyl chain between the CAS-tacrine and the photochromic unit is located entirely in the gorge for both photoisoforms, whereas the linker between the photochromic unit and the PAS-tacrine is (necessarily) rather solvent exposed.
Figure 7.
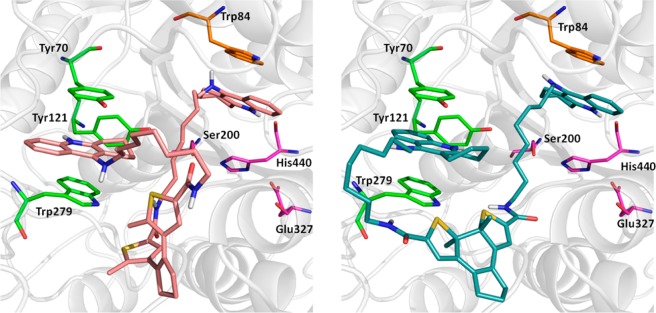
Docked poses of ring-opened 11b (left, colored pink) and ring-closed 11b (right, colored turquoise). The residues of the CAS (Ser200, His440, Glu327) are shown in magenta, Trp84 of the catalytic “anionic” site in orange, and the residues of the PAS (Tyr70, Tyr121, Trp279) in light green.
To investigate whether the docking poses are reliable and form stable complexes with AChE, molecular dynamics (MD) simulations of 18 ns were performed for both complexes. Regarding the interactions in the CAS, the trajectories reveal significantly smaller distances to the His440 backbone for the ring-opened 11b than for the ring-closed 11b, indicating a more stable interaction of the opened form throughout the trajectory (cf. Figure 13 in the Supporting Information). Moreover, the aromatic interaction of the tacrine moiety of 11b (ring-opened) with Trp84 exhibits a smaller average distance of 3.73 Å, compared to 3.94 Å of the ring-closed 11b (statistically significant based on Mann–Whitney U test, p < 10–5). Within the PAS, the most stable π-stacking is observed between ring-opened 11b and Tyr70, underlining the ability of ring-opened 11b to successfully inhibit the PAS. Unlike ring-closed 11b, ring-opened 11b is also further stabilized in the PAS by a complex network of water molecules. This network is formed in a cavity near the PAS as a consequence of a temporary (1 ns) opening of Trp279 toward a water channel. Thereby, the PAS-tacrine moiety of ring-opened 11b is stabilized by hydrogen bonding to water molecules (48% occupancy, Figure 8), whereas similar interactions are only rarely observed for ring-closed 11b (11% occupancy, Figure 8). This stabilization of ring-opened 11b in the PAS is also reflected by representative snapshots obtained from the last conformational family of each trajectory (Figure 9): For ring-opened 11b, each tacrine group shows a virtually identical binding mode as in the docking pose. In contrast, larger shifts (as well as higher fluctuations along the trajectory) are observed for the ring-closed 11b tacrines. These findings indicate that the PAS-tacrine in the ring-closed form is more flexible, compared to the ring-opened form, and might diffuse more easily from the PAS. The PAS interaction is presumably weaker for the ring-closed form compared to the PAS-tacrine of the open form, which is stabilized as part of a hydrogen-bond network. The binding mode of the photoswitchable compound 11b resembles the binding mode of other dual binding AChE inhibitors.10,12,13 However, due to the size and complexity of the compound, the pronounced difference of the photoisomers with regard to PAS inhibition and, therefore, Aβ aggregation is only understandable applying MD simulations. Whereas the ring-opened form is stably fixed in the PAS, the ring-closed form is more loosely bound and retains considerable flexibility. Possibly, this difference translates into the different inhibition behavior of the two isomers with respect to β-amyloid-aggregation. The similar IC50 values, on the other hand, might be due to enthalpy–entropy compensation: While the ring-opened form shows good enthalpic interactions (also with water molecules) at the cost of an entropy loss, the residual flexibility of the ring-closed form reduces the entropy loss, but gains less enthalpy. Obviously, this is only a qualitative interpretation of the observations made in the simulation studies. We are fully aware of their limitations, as well as of the possibility that alternative explanations might exist for the experimental data obtained in this study. A crystallographic determination of the binding mode and calorimetric information regarding enthalpic and entropic contributions would be helpful to support or disprove the model. The nature of the ligands, however, precludes a straightforward access to such experimental data.
Figure 8.

Cumulative occupancies for hydrogen bonds between the PAS-tacrines and water molecules over the entire 18 ns trajectory for ring-closed 11b (left, 11.3%) and ring-opend 11b (right, 47.8%).
Figure 9.

Comparison of the docking pose and a representative MD snapshot for ring-opened 11b (left) and ring-closed 11b (right). The docking poses are colored as in Figure 7, whereas the MD snapshots are shown in dark red (left) and cyan (right). The MD snapshots were obtained in each case from the last conformational family observed in a 2D RMSD analysis of the 18 ns trajectory by averaging all members of this conformational family and selecting the snapshot closest to this average structure as a representative. Figures 7 and 9 were generated with PyMOL (The PyMOL Molecular Graphics System, version 1.5.0.5, Schrödinger, LLC, 2012).
Conclusion
In summary, photochromic uni- and bivalent tacrine-based AChE inhibitors were synthesized and evaluated, whereby the bivalent compounds show potent and almost identical inhibitory activities (with similar or even higher activity than tacrine) as both photoisomers at AChE (and BChE). Surprisingly, even with a sterically demanding 1,2-α-dithienylethene group in the middle of the linker, bivalent compounds still remain nanomolar inhibitors at both ChEs. Computational docking studies suggested that 11b might bind to the CAS and the PAS of AChE in both opened and closed photoforms. However, significant local differences in the binding of the two forms are observed within the 18 ns MD simulation, as only for the opened form a stabilization of the interactions in the CAS and, especially, in the PAS is seen, with the latter one mediated by a water hydrogen-bond network. The mechanism of AChE inhibition by compound 11b might be controlled by light: Upon irradiation with UV–vis light, the binding mode of the inhibitor (CAS and PAS inhibition) can be switched, hinting at the change of the Hill slope in inhibition studies, and proved by kinetic studies. It could also be shown that inhibition of the PAS of AChE might be translated into a reduced Aβ aggregation. The ability to control the mode of AChE inhibition by light may be useful in further investigating the physiological functions of AChE and its role in AD; therefore, these compounds might serve as useful molecular tools to investigate the different biological properties of AChE in in vitro assays.
Methods
Chemistry
General Methods for Synthesis
Melting points are uncorrected and were measured in open capillary tubes, using a Barnstead Electrothermal IA9100 melting point apparatus. 1H NMR and 13C NMR spectral data were obtained from a Bruker Advance spectrometer (300 and 75 MHz, respectively). TLC was performed on silica gel on aluminum foils with fluorescent indicator 254 nm (Fluka) or aluminum oxide on TLC-PET foils with fluorescent indicator 254 nm (Fluka). For detection, iodine vapor or UV light (254 nm), respectively, were used. ESI-MS samples were analyzed using electrospray ionization ion-trap mass spectrometry in nanospray mode using a Thermo Finnigan LCQ Deca. For column chromatography, silica gel 60, 230–400 mesh (Merck) was used. In addition to mass spectrometry and NMR, the purity of the target compound was evaluated by high-resolution mass following HPLC system (confirming purity ≥ 95%).
Analytical HPLC using a VWR HITACHI L-2130 pump coupled to VWR HITACHI column oven L-2350 and L-2455 diode array detector: The solvents were as follows: (A) water +0.05% trifluoroacetic acid and (B) acetonitrile +0.05% trifluoroacetic acid; flow, 0.4 mL/min. Column Hibra 125-4 Purospher STAR RP-18e (3 μm) at 20 °C, detecting at 249 nm; solvent A from 90 to 10% for 30 min, then 10% for 15 min, from 10% to 90% for 10 min, 90% for 5 min.
4,4′-(Cyclopent-1-ene-1,2-diyl)bis(5-methyl-N-(2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)thiophene-2-carboxamide) (11a)
To the solution of dithiophene carboxylic acid 5 (150 mg, 0.43 mmol) in tetrahydrofuran (10 mL) was added 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDCI, 182 mg, 0.95 mmol) and 1-hydroxybenzotriazole (HOBt, 15 mg, 0.1 mmol), and the resulting solution was stirred at room temperature for 1 h. The solution of aminoalkyl tacrine 9a (230 mg, 0.95 mmol) in tetrahydrofuran (5 mL) was added to the above solution, and agitation continued overnight. The target compound precipitated from the solution and was collected by filtration as yellow solid (50 mg, 22%). MP 192–195 °C. ESI-MS 398.2 m/z [M+2H]2+, 795.3 m/z [M+H]+. 1H NMR (MeOH-d4, 300 MHz) δ: 1.79–1.88 (m, 14H, 2 × CH2(CH2)2CH2 (tacrine), 2 × CH3), 2.02–2.07 (m, 2H, CH2CH2CH2 (cyclopentene)), 2.67–2.79 (m, 8H, 2 × CH2(CH2)2CH2C=N (tacrine), CH2CH2CH2 (cyclopentene)), 2.92–2.94 (m, 4H, 2 × CH2(CH2)2CH2C=N (tacrine)), 3.64–3.68 (t, 4H, 2 × CONHCH2CH2NH, J = 4.97 Hz), 4.09–4.12 (t, 4H, 2 × CONHCH2CH2NH, J = 5.31 Hz), 7.34 (s, 2H, 2 × CH (thiophene)), 7.49–7.53 (t, 2H, arom, J = 8.23 Hz), 7.66–7.79 (m, 4H, arom), 8.39–8.41 (d, 2H, arom, J = 8.59 Hz) ppm. 13C NMR (MeOH-d4, 75 MHz) δ: 15.42 (CH3), 22.05 (CH2CH2CH2 (cyclopentene)), 23.21 (CH2(CH2)2CH2 (tacrine)), 25.24 (CH2(CH2)3C=N (tacrine)), 29.60 (CH2)3CH2C=N (tacrine)), 39.79 (CH2CH2CH2 (cyclopentene)), 41.11 (NHCH2CH2NHCO), 51.17 (NHCH2CH2NHCO), 113.36 (arom), 117.36 (arom), 120.42 (CH (tacrine)), 126.64 (CH (tacrine)), 126.66 (CH (tacrine)), 131.81 (CH (tacrine)), 134.15 (CH (thiophene)), 134.91 (arom), 136.37 (arom), 138.18 (arom), 139.97 (arom), 142.49 (arom), 151.71 (arom), 158.24 (arom), 166.00 (CONH) ppm. ESI-HRMS: C47H52N6O2S2 calcd 398.1791 m/z [M+2H]2+, 795.3509 m/z [M+H]+, anal. 398.1797 m/z [M+2H]2+, 795.3509 m/z [M+H]+. HPLC purity 95.5%, retention time 23 min.
5-Methyl-4-(2-(2-methyl-5-((2-((1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)carbamoyl)thiophen-3-yl)cyclopent-1-en-1-yl)thiophene-2-carboxylic Acid (10a)
The mixture of dithiophene carboxylic acid 5 (100 mg, 0.29 mmol), EDCI (56 mg, 0.29), and HOBt (15 mg, 0.1 mmol) in DMF (5 mL) was agitated for 30 min at room temperature. The solution of aminoalkyl tacrine 9a (69 mg, 0.29 mmol) in DMF (3 mL) was added to the above solution, and agitation was continued overnight. Water (30 mL) was added to the reaction solution, and the aqueous phase was extracted with dichloromethane/methanol mixture (10:1, 3 × 30 mL). The combined organic phases were washed with brine (2 × 20 mL), dried over anhydrous sodium sulfate, and concentrated under vacuum. The residue was purified via column chromatography with dichloromethane/:methanol/ammonia 5:1:0.1 as eluent system. The target compound was obtained as yellow solid (40 mg, 24%). MP > 300 °C (decomp.). ESI-MS 482.2 m/z [M+2H]2+, 963.6 m/z [M+H]+. 1H NMR (MeOH-d4 + CDCl3, 300 MHz) δ: 1.87–1.98 (m, 10H, CH2(CH2)2CH2 (tacrine), 2 × CH3), 2.01–2.06 (m, 2H, CH2CH2CH2 (cyclopentene)), 2.72–2.96 (m, 8H, CH2(CH2)2CH2 (tacrine), CH2CH2CH2 (cyclopentene), 3.68–3.72 (t, 2H, J = 5.48 Hz, CONHCH2CH2NH), 4.14–4.17 (t, 2H, J = 5.35 Hz, CONHCH2CH2NH), 7.22 (s, 1H, CH (thiophene)), 7.26 (s, 1H, CH (thiophene)), 7.52–7.57 (m, 1H, arom), 7.73–7.80 (m, 2H, arom), 7.95 (s, 1H, CONH), 8.36–8.39 (d, 1H, J = 8.66 Hz, arom) ppm. 13C NMR (MeOH-d4, 75 MHz) δ: 15.16 (CH3), 15.35 (CH3), 21.95 (CH2CH2CH2 (cyclopentene)), 23.12, 23.97 (CH2(CH2)2CH2 (tacrine)), 25.14 (CH2(CH2)3C=N (tacrine)), 29.45 (CH2)3CH2C=N (tacrine)), 39.54, 39.64 (CH2CH2CH2 (cyclopentene)), 41.09 (NHCH2CH2NHCO), 51.15 (NHCH2CH2NHCO), 113.34 (arom), 117.30 (arom), 120.24 (CH (tacrine)), 126.59 (CH (tacrine)), 126.65 (CH (tacrine)), 131.53 (CH (tacrine)), 134.19 (CH (tacrine)), 134.93 (CH (tacrine)), 135.87 (CH (thiophene)), 136.27 (arom), 138.07 (arom), 138.14 (arom), 142.57 (arom), 144.21 (arom), 158.46 (arom), 159.42 (COOH), 165.96 (CONH) ppm. ESI-HRMS: C32H34N3O3S2 calcd 572.2036 m/z [M+H]+, anal. 572.2037 m/z [M+H]+. HPLC purity 95.0%, retention time 25 min.
4,4′-(Cyclopent-1-ene-1,2-diyl)bis(5-methyl-N-(8-((1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)thiophene-2-carboxamide) (11b Ring-Opened Form)
This compound was synthesized analogously as 11a, using compound 5 (91 mg, 0.26 mmol), aminoalkyl tacrine 9b (170 mg, 0.53 mmol), EDCI (105 mg, 0.55 mmol), triethylamine (77 μL, 0.55 mmol), and a catalytic amount of HOBt (10 mg, 0.07 mmol) in DMF (10 mL). The crude compound was purified via column chromatography with dichloromethane/methanol/ammonia 10:1:0.1 as eluent system. The target compound was obtained as yellow solid (80 mg, 32%). MP 145–148 °C. ESI-MS 482.2 m/z [M+2H]2+, 963.6 m/z [M+H]+. ESI-HRMS: 482.2735 m/z [M+2H]2+, 965.5377 m/z [M+H]+. 1H NMR (MeOH-d4, 300 MHz) δ: 1.28–1.35 (m, 16H, 2 × NHCH2CH2(CH2)4CH2CH2NHCO), 1.50–1.54 (m, 4H, 2 × NH(CH2)6CH2CH2NHCO), 1.67–1.74 (m, 4H, 2 × NHCH2CH2(CH2)6NHCO), 1.86–1.92 (m, 14H, 2 × CH2(CH2)2CH2 (tacrine), 2 × CH3), 2.03–2.08 (m, 2H, CH2CH2CH2 (cyclopentene)), 2.66–2.69 (m, 4H, 2 × CH2(CH2)2CH2C=N (tacrine)), 2.77–2.82 (t, 4H, CH2CH2CH2 (cyclopentene), J = 7.48 Hz), 2.96–2.98 (m, 4H, 2 × CH2(CH2)2CH2C=N (tacrine)), 3.23–3.28 (t, 4H, 2 × CONHCH2(CH2)7NH, J = 7.00 Hz), 3.71–3.76 (t, 4H, 2 × CONH(CH2)7CH2NH, J = 7.26 Hz), 7.42–7.48 (m, 4H, arom (tacrine), 2 × CH (thiophene)), 7.65–7.75 (m, 4H, arom), 8.21–8.24 (d, 2H, arom, J = 8.67 Hz) ppm. 13C NMR (MeOH-d4, 75 MHz) δ: 14.76 (CH3), 22.66 (CH2CH2CH2 (cycloheptene)), 23.50 (CH2(CH2)2CH2 (tacrine)), 23.92, 25.44, 27.72, 27.87, 30.21, 30.53, 31.35, 31.82 (CH2(CH2)3C=N (tacrine), (CH2)3CH2C=N (tacrine), NHCH2(CH2)6CH2NHCO), 39.52 (CH2CH2CH2 (cyclopentene)), 40.78 (NHCH2(CH2)6CH2NHCO), 49.43 (NHCH2(CH2)6CH2NHCO), 114.44 (arom), 118.80 (arom), 123.36 (CH (tacrine)), 125.69 (2 × CH (tacrine)), 130.63 (CH (tacrine)), 132.35 (CH (thiophene)), 136.24 (arom), 137.90 (arom), 141.39 (arom), 143.13 (arom), 154.69 (arom), 156.07 (arom), 164.18 (CONH) ppm. ESI-HRMS: C59H75N6O2S2 calcd 482.2730 m/z [M+2H]2+, 963.5387 m/z [M+H]+, anal. 482.2735 m/z [M+2H]2+, 963.5377 m/z [M+H]+. HPLC purity 95.4%, retention time 21.26 min.
9a,9b-Dimethyl-N2,N8-bis(8-((1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)-5,6,9a,9b-tetrahydro-4H-indeno[5,4-b:6,7-b′]dithiophene-2,8-dicarboxamide (11b Ring-Closed form)
ESI-MS 482.2 m/z [M+2H]2+, 963.6 m/z [M+H]+. 1H NMR (MeOH-d4, 400 MHz) δ: 1.29–1.44 (m, 16H, 2 × NHCH2CH2(CH2)4CH2CH2NHCO), 1.51–1.58 (m, 4H, 2 × NH(CH2)6CH2CH2NHCO), 1.80–1.88 (m, 6H, 2 × NHCH2CH2(CH2)6NHCO, CH2CH2CH2 (cyclopentene)), 1.94 (s, 6H, 2 × CH3), 1.95–1.99 (m, 8H, 2 × CH2(CH2)2CH2 (tacrine)), 2.44–2.48 (t, 4H, CH2CH2CH2 (cyclopentene), J = 2.46 Hz), 2.69–2.72 (m, 4H, 2 × CH2(CH2)2CH2C=N (tacrine)), 3.01–3.03 (m, 4H, 2 × CH2(CH2)2CH2C=N (tacrine)), 3.23–3.27 (t, 4H, 2 × CONHCH2(CH2)7NH, J = 3.25 Hz), 3.94–3.97 (t, 4H, 2 × CONH(CH2)7CH2NH, J = 3.95 Hz), 6.63 (s, 2H, 2 × CH (thiophene)), 7.57–7.61 (m, 4H, arom (tacrine)), 7.75–7.87 (m, 4H, arom), 8.38–8.41 (d, 2H, arom, J = 8.39 Hz) ppm. HPLC purity 95.4%, retention time 22.22 min.
5-Methyl-4-(2-(2-methyl-5-((8-((1,2,3,4-tetrahydroacridin-9-yl)amino)octyl)carbamoyl)thiophen-3-yl)cyclopent-1-en-1-yl)thiophene-2-carboxylic Acid (10b)
This compound was synthesized analogously as 11a, using compound 5 (64 mg, 0.18 mmol), aminoalkyl tacrine 9b (60 mg, 0.18 mmol), EDCI (35 mg, 0.18 mmol), triethylamine (28 μL, 0.2 mmol) and a catalytic amount of HOBt (10 mg, 0.07 mmol) in DMF (10 mL). The crude compound was purified via column chromatography with dichloromethane/methanol/ammonia 10:1:0.1 as eluent system. The target compound was obtained as yellow solid (30 mg, 25%). MP 195–200 °C. ESI-MS: 655.3 m/z [M+H]+. ESI-HRMS: 328.1612 m/z [M+2H]2+, 655.3147 m/z [M+H]+. 1H NMR (MeOH-d4, 300 MHz) δ: 1.15–1.20 (m, 8H, NHCH2CH2(CH2)4CH2CH2NHCO), 1.46–1.56 (m, 2H, NH(CH2)6CH2CH2NHCO), 1.65–1.70 (m, 2H, NHCH2CH2(CH2)6NHCO), 1.89–1.93 (m, 10H, CH2(CH2)2CH2 (tacrine), 2 × CH3), 2.03–2.13 (m, 2H, CH2CH2CH2 (cyclopentene)), 2.72–2.85 (m, 6H, CH2(CH2)2CH2C=N (tacrine)), CH2CH2CH2 (cyclopentene)), 2.97–2.99 (m, 2H, CH2(CH2)2CH2C=N (tacrine)), 3.23–3.28 (t, 2H, NH(CH2)7CH2NHCO, J = 7.09 Hz), 3.63–3.69 (t, 2H, NHCH2(CH2)7NHCO, J = 7.22 Hz), 7.40–7.47 (m, 3H, arom (tacrine), 2 × CH (thiophene)), 7.61–7.66 (m, 1H, arom (tacrine)), 7.73–7.76 (m, 1H, arom (tacrine)), 8.17–8.20 (d, 1H, arom (tacrine), J = 8.77 Hz) ppm. 13C NMR (MeOH-d4, 75 MHz) δ: 14.65 (CH3), 14.68 (CH3), 21.80, 22.99 (CH2(CH2)2CH2 (tacrine)), 23.87 (CH2CH2CH2 (cyclopentene)), 24.83 (CH2(CH2)2CH2C=N (tacrine)), 27.54, 27.76, 29.29, 30.07, 30.08, 30.45, 31.41 (NHCH2(CH2)6CH2NHCO, CH2(CH2)2CH2C=N (tacrine)), 39.41, 39.46 (CH2CH2CH2 (cyclopentene)), 40.66 (NHCH2(CH2)6CH2NHCO), 49.62 (NHCH2(CH2)6CH2NHCO), 112.87 (arom), 117.08 (arom), 120.05 (CH (tacrine)), 126.34 (CH (tacrine)), 126.54 (CH (tacrine)), 130.55 (CH (thiophene)), 131.77 (CH (thiophene)), 134.14 (CH (tacrine)), 135.63 (arom), 136.14 (arom), 136.20 (arom), 136.25 (arom), 137.86 (arom), 137.97 (arom), 139.80 (arom), 141.42 (arom), 142.19 (arom), 158.09 (arom), 164.20 (COOH), 166.43 (CONH) ppm. ESI-HRMS: C38H47N4O2S2 calcd 655.3135 m/z [M+H]+, anal. 328.1612 m/z [M+2H]2+, 655.3147 m/z [M+H]+. HPLC purity 95.1%, retention time 26 min.
Experimental Procedures for the Pharmacological Investigations
Cholinesterase Inhibition and Kinetic Studies
AChE (E.C.3.1.1.7, Type VI-S, from electric eel), hAChE (recombinant, expressed in HEK 293 cells), and BChE (E.C.3.1.1.8, from equine serum) were purchased from Sigma-Aldrich (Germany). 5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB, Ellman’s reagent), acetylthiocholine (ATC), and butyrylthiocholine (BTC) iodides were also obtained from Sigma-Aldrich (Germany). UV measurements were performed on Varian Cary 50 Scan UV–vis spectrometer, and 4 mL disposable cuvettes were used for the testing. As reference for ChE inhibition, the established drug tacrine hydrochloride was used, and purchased from Sigma-Aldrich (UK).
Ellman’s assay was performed as described in the following procedure: stock solutions of the test compounds were prepared in ethanol, 100 μL of which gave a final concentration of 10–3 M when diluted to the final volume of 3.32 mL. The highest concentration of the test compounds applied in the assay was 10–4 M (10% EtOH in the stock solution did not influence enzyme activity). The rest of the testing concentrations were diluted from stock solution using the buffer below. In order to obtain an inhibition curve, at least five different concentrations (normally 10–4–10–9 M) of the test compound were measured at 20 °C and 412 nm, each concentration in triplicate.9,22
For buffer preparation, 1.36 g of potassium dihydrogen phosphate (10 mmol) was dissolved in 100 mL of water and adjusted with NaOH to pH = 8.0 ± 0.1. Enzyme solutions were prepared to give 2.5 units/mL in 1.4 mL aliquots. Furthermore, 0.01 M DTNB solution, 0.075 M ATC solution, and 0.075 M BTC solution were used. A cuvette containing 3.0 mL of phosphate buffer, 100 μL of the respective enzyme, 100 μL of DTNB, and 100 μL of the test compound solution was allowed to stand at 25 °C for 4.5 min (or 1 h in the case of prolonged incubation). The reaction was started by addition of 20 μL of the substrate solution (ATC/BTC). The solution was mixed immediately, and exactly 2.5 min after substrate addition the absorption was measured. For the reference value, 100 μL of water replaced the test compound solution. For determining the blank value, additionally 100 μL of water replaced the enzyme solution. The inhibition curve was obtained by plotting the percentage enzyme activity (100% for the reference) versus the logarithm of test compound concentration using GraphPad Prism software (version 5.0).
For human AChE, the reaction time after the addition of ATC was slightly prolonged to 5 min.
For the kinetic measurements the following substrate concentrations were used: 28.2, 56.5, 113, 226, and 452 μM. In contrast to the above-described affinity measurements, the reaction time was extended to 4 min before measurement of the absorption. Vmax and Km values of the Michaelis–Menten kinetics were calculated by nonlinear regression out of the substrate velocity curves. Linear regression (double reciprocal) was used for calculating the Lineweaver–Burk plots for reasons of illustration. All kinetic studies were also processed using GraphPad Prism 5 software (version 5.0).32
AChE-Induced β-Amyloid Aggregation Testing
Thioflavin T (Basic Yellow 1) was purchased from Acros (Belgium). AChE (from electric eel), 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), and absolute DMSO (BioReagent) were purchased from Sigma-Aldrich (Germany). Deionized water was used to prepare the buffer solution. Amyloid β-protein (1–40) trifluoroacetate was purchased from Bachem AG (Burbendorf, Switzerland). Buffers and other chemicals were of analytical grade. The stock solutions of thioflavin T and AChE were prepared using the same buffer solution (25 mM NaH2PO4, 100 mM NaCl and final pH 8.0) to reach final concentration at 15 μM and 2.5 U/mL, respectively.
Aβ1–40 was suspended in HFIP and left to stay at room temperature for 4 h until a clear solution (20 mg/mL) was formed. The solvent was removed under a stream of nitrogen until a clear film remained, following with drying under high vacuum for 2 h. The residue was then dissolved in DMSO to form a 2 mM stock solution, which was subsequently stored frozen at −20 °C. The testing compound 11b was dissolved in DMSO to form 500 μM stock solution and subsequently diluted to 100, 20, and 4 μM solutions in DMSO (corresponding to 5, 1, and 0.2 μM in final testing), which were stored in the freezer at −20 °C.
For the thioflavin T-based fluorometric assay, analyses were performed with a Varian Cary Eclipse fluorescence spectrophotometer using 96-well fluorescence microtiter plates (Nunc, Denmark). Fluorescence was monitored at 450 nm (λexc) and 482 nm (λem) with excitation and emission slits of 5 nm bandwidth using Cary Eclipse Kinetics program. The fluorescence emission spectrum was recorded as a scan of the average intensity values around 5 min after subtracting the background fluorescence from thioflavin T (2.5 μM) and AChE (1 U/mL) solution with or without the corresponding inhibitor. Each assay was run in duplicate.
To the mixed solution of AChE (40 μL, final concentration 1U/mL), buffer (50 μL), and inhibitor (5 μL as either ring-opened or closed form, final concentration 5, 1, and 0.2 μM) was added the solution of Aβ1–40 (5 μL, final concentration 100 μM). The resulting solution (100 μL volume) was incubated at 25 °C for 1, 2, and 5 h with or without the presence of inhibitor. Thioflavin T solution (20 μL, final concentration 2.5 μM) was added to the corresponding well at the specific time spot, that is, 1, 2, and 5 h, and the fluorescence was measured according to the method described above.
All the data were processed on GraphPad Prism 5 software (version 5.0) to generate the aggregation curves.
Docking and Molecular Dynamics Simulations
To reveal potential binding modes of compound 11b, docking studies were carried out with the open and the closed form of the inhibitor. The AChE crystal structure with PDB code 2CKM from the RCSB Protein Data Bank was selected for this purpose.31,33 It corresponds to the structure of Torpedo californica AChE (TcAChE) in complex with a bis-tacrine linked by a seven-carbon spacer. The selection was based on the good resolution of the structure (2.15 Å) and the similarity of the bound ligand with the compounds investigated here. Moreover, TcAChE shows a high sequence similarity (58% identity and 73% similarity) and a conserved binding site with respect to Electrophorus electricus AChE (eeAChE) used for the biochemical/pharmacological studies. The identity and similarity calculations were performed by the pairwise sequence alignment of TcAChE (2CKM) and EeAChE (1C2O) with the Needle program, using the EBLOSUM62 matrix, of EMBOSS v.6.3.1.34
Docking was carried out with GOLD 5.1.35 The protein was set up for docking by removing all water molecules and nonprotein atoms and protonating the amino acids with MOE 2011.10 using Protonate 3D at pH 7.36 The search region was focused on the binding area comprising the CAS, the binding gorge, and the PAS. The binding site was defined by the following residues: Tyr70, Asp72, Trp84, Asn85, Gly117, Gly118, Gly119, Tyr121, Glu199, Ser200, Trp279, Phe288, Phe290, Glu327, Phe330, Phe331, Tyr334, Trp432, Met436, Ile439, His440, and Tyr442. The ligand structures were either built in MOE (compounds 11b opened and closed) or extracted from the crystal structure (ligands for redocking experiments, cf. below). The protonation was set according to the expected ionization state at pH 7; for 11b, this implied protonation at the tacrine nitrogen atoms, consistent with their pKa values between 8.7 and 9.6, as calculated with MoKa.37 The structures were energy minimized in MOE using the MMFF94x force field and an rms-gradient of 0.001 kcal/(mol·Å).
Using default settings in GOLD, the best-suited scoring function was selected based on redocking experiments with the seven-carbon spacer bis-tacrine of crystal structure 2CKM. Fifty docking runs carried out with each of the four scoring functions available in GOLD (ASP, CHEMPLP, ChemScore, GoldScore) showed that best results are obtained with the Astex Statistical Potential (ASP),38 with good clustering on the top ranks and rmsd values of 1.07 Å (rank 1) and 0.74 Å (rank 2) of the docked pose with respect to the native pose in the crystal structure. Similar results were obtained in redocking experiments with the five-carbon spacer bis-tacrine to crystal structure 2CMF.31
Accordingly, docking of the 11b inhibitors was carried out with the ASP function. Due to the higher conformational flexibility of the inhibitors in comparison to the bis-tacrines of 2CKM and 2CMF, the number of operations in the GOLD GA settings was increased to 4 000 000. Furthermore, to improve the convergence, a scaffold match constraint was applied to place one tacrine moiety in the CAS, using the corresponding moiety of the bis-tacrine crystal structure 2CMF as reference for the least-squares fit (with a weight set to 5.0 in GOLD).
Molecular dynamics (MD) simulations were carried out to analyze the stability and plausibility of the binding modes suggested by docking. The ligands were setup for simulation with modules of Amber 12.39 RESP charges were calculated for all ligands based on HF/6-31G electrostatic potentials obtained with Gaussian 03.40,41 The General Amber Force Field (GAFF) was used for the ligands, assigning atom and bond types with antechamber and missing force field parameters with parmchk.42,43 For the protein, the Amber ff99SB force field was used. All simulations were based on protein structure 2CKM. The missing amino acids (His486, Ser487, Gln488, and Asn489) between Pro485 and Ser490 were inserted using Modeller 9.11 with rigid protein during model refinement.44 Three disulfide bonds were identified and retained in the MD setup (between cysteines 67–94, 254–265, and 402–521, respectively). All histidines were protonated at the ε-nitrogen, except His398 and His440, which were both protonated at the δ-nitrogen to ensure a proper hydrogen-bonding network. Water molecules present in the crystal structure within a radius of 4 Å of the seven pocket residues (Tyr70, Trp84, Tyr121, Ser200, Trp279, Glu327, and His440) were included in the setup.
To relieve initial strains, very short energy minimizations (30 steps) of the docked ligands within the rigid proteins were performed using a generalized Born implicit solvent model as implemented in the Amber module sander.45,46 Subsequently, the entire complexes were minimized for 200 steps. After minimization, the complexes were solvated with tleap in rectangular TIP3P water boxes,47 resulting in box sizes of approximately 85 Å × 83 Å × 82 Å. For equilibration, water molecules were allowed to move freely (keeping protein and ligand rigid) in the constant-volume box while increasing the temperature from 100 to 300 K over 20 ps, followed by cooling to 100 K over 5 ps. The temperature was adjusted by means of the Berendsen weak coupling algorithm with a time constant of 0.5 ps. Subsequently, the complete systems were treated without constraints and gradually heated to 300 K over a time period of 25 ps. The systems thus obtained were used as input for MD production runs with NAMD.48 Using a time step of 2 fs, trajectories of 18 ns were generated for each system. The systems were simulated under NPT periodic boundary conditions. Constant pressure was assured by the Nosé-Hoover Langevin piston pressure control,49,50 while constant temperature was achieved by the use of Langevin dynamics. A van der Waals interaction cutoff of 12 Å was used, whereas the particle mesh Ewald methodology was applied for electrostatic interactions.51 All analyses of the trajectories were performed with VMD52 and the statistical framework R.53,54
Acknowledgments
The authors thank Michaela Prinz for her kind help on establishing the Aβ aggregation assay.
Glossary
Abbreviations
- Aβ
β-amyloid protein
- AChE
acetylcholinesterase
- AD
Alzheimer’s disease
- BACE-1
beta-site amyloid precursor protein cleaving enzyme 1, or beta-secretase 1
- BChE
butyrylcholinesterase
- CAS
catalytic active site
- ChE(I)
cholinesterase (inhibitor)
- DTE
1,2-α-dithienylethene
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- HFIP
1,1,1,3,3,3-hexafluoro-2-propanol
- HOBt
hydroxybenzotriazole
- NMDA
N-methyl-d-aspartate
- PAS
peripheral anionic site
- SARs
structure–activity relationships
- TFA
trifluoroacetic acid
Supporting Information Available
Spectral data of target compounds; HPLC chromatography for compound 11b; figures showing results of pharmacological tests; figures showing results of computational studies. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
M.D. was responsible for the supervision and development of the whole project in collaboration with B.K. N.K. under the supervision of B.K. performed the synthesis of compound 5 and the testing of physicochemical properties of the target compounds. X.C. performed the syntheses of the target compounds, enzyme inhibition, and kinetic studies as well as AChE-induced β-amyloid inhibition testing with the help from U.H. S.W. conducted the computaional docking studies and B.M. helped with the molecular dynamics study, both under the supervision of C.A.S.
Part of this work was funded by the German Research Foundation (Deutsche Forschungsgemeinschaft under DFG DE1546/6-1).
The authors declare no competing financial interest.
Supplementary Material
References
- Ballard C.; Gauthier S.; Corbett A.; Brayne C.; Aarsland D.; Jones E. (2011) Alzheimer’s disease. Lancet 377, 1019–1931. [DOI] [PubMed] [Google Scholar]
- Bartolini M.; Bertucci C.; Cavrini V.; Andrisano V. (2003) β-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem. Pharmacol. 65, 407–416. [DOI] [PubMed] [Google Scholar]
- García-Allón M.-S.; Small D. H.; Avila J.; Sáez-Valero J. (2011) Revisiting the role of acetylcholinesterase in Alzheimer’s disease: cross-talk with P-tau and β-amyloid. Front. Mol. Neurosci. 4:22, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal F. J.; Inestrosa N. C. (2011) Interactions of AChE with Aβ aggregates in Alzheimer’s brain; therapeutic relevance of IDN 5706. Front. Mol. Neurosci. 4:19, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveyra M.-X.; García-Ayllón M.-S.; de Barreda E. G.; Small D. H.; Martínez S.; Avila J.; Sáez-Valero J. (2012) Altered expression of brain acetylcholinestease in FTDP-17 human tau transgenic mice. Neurobiol. Aging 33, 624.e23–34. [DOI] [PubMed] [Google Scholar]
- Piazzi L.; Rampa A.; Bisi A.; Gobbi S.; Belluti F.; Cavalli A.; Bartolini M.; Andrisano V.; Valenti P.; Recanatini M. (2003) 3-(4-{[Benzyl(methyl)amino]methyl}phenyl)-6,7-dimethoxy-2H-2-chromenone (AP2238) inhibits both acetylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation: A dual function lead for Alzheimer’s disease therapy. J. Med. Chem. 46, 2279–2282. [DOI] [PubMed] [Google Scholar]
- León R.; Garcia A. G.; Marco-Contelles J. (2013) Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 33, 139–189. [DOI] [PubMed] [Google Scholar]
- Fang L.; Kraus B.; Lehmann J.; Heilmann J.; Zhang Y.; Decker M. (2008) Design and synthesis of tacrine-ferulic acid hybrids as multi-potent anti-Alzheimer drug candidates. Bioorg. Med. Chem. Lett. 18, 2905–2909. [DOI] [PubMed] [Google Scholar]
- Chen X.; Zenger K.; Lupp A.; Kling B.; Heilmann J.; Fleck C.; Kraus B.; Decker M. (2012) Tacrine-silibinin codrug shows neuro- and hepatoprotective effects in vitro and pro-cognitive and hepatoprotective effects in vivo. J. Med. Chem. 55, 5231–5242. [DOI] [PubMed] [Google Scholar]
- Fang L.; Appenroth D.; Decker M.; Kiehntopf M.; Lupp A.; Peng S.; Fleck C.; Zhang Y.; Lehmann J. (2008) NO-donating tacrine hybrid compounds improve scopolamine-induced cognition impairment and show less hepatotoxicity. J. Med. Chem. 51, 7666–7669. [DOI] [PubMed] [Google Scholar]
- a Bourne Y.; Radic Z.; Kolb H. C.; Sharpless B.; Taylor P.; Marchot P. (2005) Structural insights into conformational flexibility at the peripheral site and within the active center gorge of AChE. Chem.-Biol. Interact. 157, 159–165. [DOI] [PubMed] [Google Scholar]; b Bourne Y.; Radic Z.; Taylor P.; Marchot P. (2010) Conformational remodeling of femtomolar inhibitor – acetylcholinesterase complexes in the crystalline state. J. Am. Chem. Soc. 132, 18292–18300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumiatti V.; Miniarini A.; Bolognesi M. L.; Milelli A.; Rosini M.; Melchiorre C. (2010) Tacrine derivatives and Alzheimer’s disease. Curr. Med. Chem. 17, 1825–1838. [DOI] [PubMed] [Google Scholar]
- Muñoz-Torrero D. (2008) Acetylcholinesterase inhibitors as disease-modifying therapies for Alzheimer’s disease. Curr. Med. Chem. 15, 2433–2455. [DOI] [PubMed] [Google Scholar]
- Galdeano C.; Viayna E.; Sola I.; Formosa X.; Camps P.; Badia A.; Clos M. V.; Relat J.; Ratia M.; Bartolini M.; Mancini F.; Andrisano V.; Salmona M.; Minguillón C.; González-Muñoz G. C.; Rodríguez-Franco M. I.; Bidon-Chanal A.; Luque F. J.; Muñoz-Torrero D. (2012) Huprine-tacrine heterodimers as anti-amyloidogenic compounds of potential interest against Alzheimer’s and prion diseases. J. Med. Chem. 55, 661–669. [DOI] [PubMed] [Google Scholar]
- a Harvey J. H.; Trauner D. (2008) Regulating enzymatic activity with a photoswitchable affinity label. ChemBioChem 9, 191–193. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fehrentz T.; Schönberger M.; Trauner D. (2011) Optochemical genetics. Angew. Chem., Int. Ed. 50, 12156–12182. [DOI] [PubMed] [Google Scholar]; c Fehrentz T.; Kuttruff C. A.; Huber F. M. E.; Kienzler M. A.; Mayer P.; Trauner D. (2012) Exploring the pharmacology and action spectra of photochromic open-channel blockers. ChemBioChem 13, 1746–1749. [DOI] [PubMed] [Google Scholar]
- Irie M. (2000) Diarylethenes for memories and switches. Chem. Rev. 100, 1685–1716. [DOI] [PubMed] [Google Scholar]
- Mayer G.; Heckel A. (2006) Biologically active molecules with a “light switch”. Angew. Chem., Int. Ed. 45, 4900–4921. [DOI] [PubMed] [Google Scholar]
- a Vomasta D.; Högner C.; Branda N. R.; König B. (2008) Regulation of human carbonic anhydrase I (hCAI) activity by using a photochromic inhibitor. Angew. Chem., Int. Ed. 47, 7644–7647. [DOI] [PubMed] [Google Scholar]; b Vomasta D.; Innocenti A.; König B.; Supuran C. T. (2009) Carbonic anhydrase inhibitors: two-prong versus mono-prong inhibitors of isomers I, II, IX, and XII exemplified by photochromic cis-1,2-α-dithienylethene derivatives. Bioorg. Med. Chem. Lett. 19, 1283–1286. [DOI] [PubMed] [Google Scholar]
- a Stawski P.; Sumser M.; Trauner D. (2012) A photochromic agonist of AMPA receptors. Angew. Chem., Int. Ed. 51, 5748–5751. [DOI] [PubMed] [Google Scholar]; b Stein M.; Middendorp S. J.; Carta V.; Pejo E.; Raines D. E.; Forman S. A.; Sigel E.; Trauner D. (2012) Azo-propofols: photochromic potentiators of GABAA receptors. Angew. Chem., Int. Ed. 51, 10500–10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a For review, see:Velema W. A.; Szymanski W.; Feringa B. L. (2014) Photopharmacology: beyond proof of principle. J. Am. Chem. Soc. 136, 2178–2191. [DOI] [PubMed] [Google Scholar]; b Velema W. A.; Stuart M. C.; Szymanski W.; Feringa B. L. (2013) Light-triggered self-assembly of a dichromonyl compound in water. Chem. Commun. 49, 5001–5003. [DOI] [PubMed] [Google Scholar]; c Velema W. A.; Van der Toorn M.; Szymanski W.; Feringa B. L. (2013) Design, synthesis, and inhibitory activity of potent, photoswitchable mast cell activation inhibitors. J. Med. Chem. 56, 4456–4464. [DOI] [PubMed] [Google Scholar]
- Lucas L. N.; de Jong J. J. D.; van Esch J. H.; Kellogg R. M.; Feringa B. L. (2003) Syntheses of dithienylcyclopentene optical molecular switches. Eur. J. Org. Chem. 1, 155–166. [Google Scholar]
- a Hu M.-K.; Lu C.-F. (2000) A facile synthesis of bis-tacrine isosteres. Tetrahedron Lett. 41, 1815–1818. [Google Scholar]; b Frideling A.; Faure R.; Galy J.-P.; Kenz A.; Alkorta I.; Elguero J. (2004) Tetrahydroacridin-9-ones, 9-chlorotetrahydroacridines, 9-amino-tetrahydroacridines and 9-(pyrazol-1-yl)-tetrahydroacridines derived from chiral cyclanones. Eur. J. Med. Chem. 39, 37–48. [DOI] [PubMed] [Google Scholar]
- Van der Molen S. J.; Liao J.; Kudernac T.; Agustsson J. S.; Bernard L.; Calame M.; van Wees B. J.; Feringa B. L.; Schönenberger C. (2009) Light-controlled conductance switching of ordered metal-molecule-metal devices. Nano Lett. 9, 76–80. [DOI] [PubMed] [Google Scholar]
- Kühhorn J.; Hübner H.; Gmeiner P. (2011) Bivalent dopamine D2 receptor ligands: synthesis and binding properties. J. Med. Chem. 54, 4896–4903. [DOI] [PubMed] [Google Scholar]
- Rosini M.; Andrisano V.; Bartolini M.; Bolognesi M. L.; Tarozzi A.; Melchiorre C. (2005) Rational approach to discover multipotent anti-Alzheimer drugs. J. Med. Chem. 48, 360–363. [DOI] [PubMed] [Google Scholar]
- Viayna E.; Gómez T.; Galdeano C.; Ramírez L.; Ratia M.; Badia A.; Clos M. V.; Verdaguer E.; Junyent F.; Camins A.; Pallàs M.; Bartolini M.; Mancini F.; Andrisano V.; Arce M. P.; Rodríguez-Franco M. I.; Bidon-Chanal A.; Luque F. J.; Camps P.; Muñoz-Torrero D. (2010) Novel huprine derivatives with inhibitory activity toward β-amyloid aggregation and formation as disease-modifying anti-Alzheimer drug candidates. ChemMedChem 5, 1855–1870. [DOI] [PubMed] [Google Scholar]
- Chen X.; Tikhonova I. G.; Decker M. (2011) Probing the mid-gorge of cholinesterases with spacer-modified bivalent quinazolinimines leads to highly potent and selective butyrylcholinesterase inhibitors. Bioorg. Med. Chem. 19, 1222–1235. [DOI] [PubMed] [Google Scholar]
- Bartolini M.; Bertucci C.; Bolognesi M. L.; Cavalli A.; Melchiorre C.; Andrisano V. (2007) Insight into the kinetic of amyloid β (1–42) peptide self-aggregation: Elucidation of inhibitors’ mechanism of action. ChemBioChem 8, 2152–2161. [DOI] [PubMed] [Google Scholar]
- a Alptüzün V.; Prinz M.; Hörr V.; Scheiber J.; Radacki K.; Fallarero A.; Vuorela P.; Engels B.; Braunschweig H.; Erciyas E.; Holzgrabe (2010) Interaction of (benzyliden-hydrazono)-1,4-dihydropyridines with β-amyloid, acetylcholine, and butyrylcholine esterases. Bioorg. Med. Chem. 18, 2049–2059. [DOI] [PubMed] [Google Scholar]; b Prinz M.; Parlar S.; Bayraktar G.; Alptüzün V.; Erciyas E.; Fallarero A.; Karlsson D.; Vuorela P.; Burek M.; Förster C.; Turunc E.; Armagan G.; Yalcin A.; Schiller C.; Leuner K.; Krug M.; Sotriffer C.; Holzgrabe U. (2013) 1,4-Substituted 4-(1H)-pyridylene-hydrazone-type inhibitors of AChE, BuChE, and amyloid-β aggregation crossing the blood-brain barrier. Eur. J. Pharm. Sci. 49, 603–613. [DOI] [PubMed] [Google Scholar]
- Neudert G.; Klebe G. (2011) DSX: A Knowledge-Based Scoring Function for the Assessment of Protein–Ligand Complexes. J. Chem. Inf. Model. 51, 2731–2745. [DOI] [PubMed] [Google Scholar]
- Rydberg E. H.; Brumshtein B.; Greenblatt H. M.; Wong D. M.; Shaya D.; Williams L. D.; Carlier P. R.; Pang Y.-P.; Silman I.; Sussman J. L. (2006) Complexes of Alkylene-Linked Tacrine Dimers with Torpedo californica Acetylcholinesterase: Binding of Bis(5)-tacrine Produces a Dramatic Rearrangement in the Active-Site Gorge. J. Med. Chem. 49, 5491–5500. [DOI] [PubMed] [Google Scholar]
- Decker M.; Krauth F.; Lehmann J. (2006) Novel tricyclic quinazolinimines and related tetracyclic nitrogen bridgehead compounds as cholinesterase inhibitors with selectivity towards butyrylcholinesterase. Bioorg. Med. Chem. 14, 1966–1977. [DOI] [PubMed] [Google Scholar]
- Bernstein F. C.; Koetzle T. F.; Williams G. J.; Meyer E. E. Jr.; Brice M. D.; Rodgers J. R.; Kennard O.; Shimanouchi T.; Tasumi M. (1997) The Protein Data Bank: A Computer-based Archival File For Macromolecular Structures. J. Mol. Biol. 112, 535–542. [DOI] [PubMed] [Google Scholar]
- Rice P.; Longden I.; Bleasby A. (2000) EMBOSS: the European molecular biology open software suite. Trends Genet. 16, 276–277. [DOI] [PubMed] [Google Scholar]
- a GOLDSUITE 5.1, CCDC Software, www.ccdc.cam.ac.uk.; b Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. (2003) Improved Protein-Ligand Docking Using GOLD. Proteins 52, 609–623. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE), 2011.10, Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2011. [Google Scholar]
- Milletti F.; Storchi L.; Sforna G.; Cruciani G. (2007) New and Original pKa Prediction Method Using Grid Molecular Interaction Fields. J. Chem. Inf. Model. 47, 2172–2181. [DOI] [PubMed] [Google Scholar]
- Mooij W. T. M.; Verdonk M. L. (2005) General and Targeted Statistical Potentials for Protein–Ligand Interactions. Proteins 61, 272–287. [DOI] [PubMed] [Google Scholar]
- Case D. A., Darden T. A., Cheatham T. E. III, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., Goetz A. W., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Wolf R. M., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M.-J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., and Kollman P. A. (2012) AMBER 12, University of California, San Francisco.
- Bayly C. I.; Cieplak P.; Cornell W.; Kollman P. A. (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10269–10280. [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J. A. Jr., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., and Pople J. A. (2004) Gaussian 03, Revision D.01, Gaussian, Inc., Wallingford, CT. [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Model. 25, 247–260. [DOI] [PubMed] [Google Scholar]
- Šali A.; Blundell T. L. (1993) Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Still W. C.; Tempczyk A.; Hawley R. C.; Hendrickson T. (1990) Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 112, 6127–6129. [Google Scholar]
- Srinivasan J.; Trevathan M. W.; Case D. (1999) Application of a pairwise generalized Born model to proteins and nucleic acids: inclusion of salt effects. Theor. Chem. Acc. 101, 426–434. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L. (1994) Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189. [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. (1995) Constant-pressure molecular-dynamics simulation-the Langevin piston method. J. Chem. Phys. 103, 4613–4621. [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle mesh Ewald: an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- R Core Team (2013) R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, ISBN 3-900051-07-0, http://www.R-project.org/. [Google Scholar]
- Sarkar D. (2008) Lattice: Multivariate Data Visualization with R, Springer, New York, ISBN 978-0-387-75968-5. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.