

Abstract
Background
The legume family (Leguminosae) consists of approx. 17 000 species. A few of these species, including, but not limited to, Phaseolus vulgaris, Cicer arietinum and Cajanus cajan, are important dietary components, providing protein for approx. 300 million people worldwide. Additional species, including soybean (Glycine max) and alfalfa (Medicago sativa), are important crops utilized mainly in animal feed. In addition, legumes are important contributors to biological nitrogen, forming symbiotic relationships with rhizobia to fix atmospheric N2 and providing up to 30 % of available nitrogen for the next season of crops. The application of high-throughput genomic technologies including genome sequencing projects, genome re-sequencing (DNA-seq) and transcriptome sequencing (RNA-seq) by the legume research community has provided major insights into genome evolution, genomic architecture and domestication.
Scope and Conclusions
This review presents an overview of the current state of legume genomics and explores the role that next-generation sequencing technologies play in advancing legume genomics. The adoption of next-generation sequencing and implementation of associated bioinformatic tools has allowed researchers to turn each species of interest into their own model organism. To illustrate the power of next-generation sequencing, an in-depth overview of the transcriptomes of both soybean and white lupin (Lupinus albus) is provided. The soybean transcriptome focuses on analysing seed development in two near-isogenic lines, examining the role of transporters, oil biosynthesis and nitrogen utilization. The white lupin transcriptome analysis examines how phosphate deficiency alters gene expression patterns, inducing the formation of cluster roots. Such studies illustrate the power of next-generation sequencing and bioinformatic analyses in elucidating the gene networks underlying biological processes.
Keywords: Legume genomics, next-generation sequencing, NGS, bioinformatics, Glycine max, soybean, Lupinus albus, white lupin, transcriptome, genome re-sequencing, RNA-seq
INTRODUCTION
With rapidly declining costs and increasing efficiencies, DNA and RNA high-throughput sequencing, via so-called next-generation technologies, are quickly becoming a mainstay in plant genetics, genomics and biochemistry (Chia and Ware, 2011; Ozsolak and Milos, 2011). Rather amazingly, multiple genomes have been sequenced for several species including: arabidopsis (Cao et al., 2011; Gan et al., 2011), rice (Huang et al., 2013), maize (Lai et al., 2010; Jiao et al., 2012), soybean (Kim et al., 2010; Lam et al., 2010), Medicago truncatula (Branca et al., 2011) and chickpea (Varshney et al., 2013). Next-generation sequencing (NGS) of plant genomes is providing new insight into the genomic variation within and between species [single nucleotide polymorphisms (SNPs), presence–absence variations (PAVs), copy number variations (CNVs) and other types of structural variations (SVs)], the introgression of genetic elements such as quantitative trait loci (QTLs), the evolution of organs and pathways, and the status of epigenetic modifications (McMullen et al., 2009; Atwell et al., 2010; Huang et al., 2010; Branca et al., 2011; Schmitz and Zhang, 2011; Schneeberger and Weigel, 2011; Jiao et al., 2012; Tomato Genome Consortium, 2012; Varshney et al., 2013). Likewise, sequencing of plant transcriptomes (RNA-seq) is providing atlases of gene expression, demonstrating alternative splicing events, defining QTL introgressions, characterizing small RNAs and identifying genes and pathways involved in acclimation to biotic and abiotic stresses (Wang et al., 2009; Li et al., 2010; Libault et al., 2010; Severin et al., 2010a, b; Zenoni et al., 2010; Matas et al., 2011; Yang et al., 2011; Meyer et al., 2012; O’Rourke et al., 2013b; Atwood et al., 2014). Next-generation sequencing approaches are replacing chip-based platforms for genomic studies. This change is placing extraordinary pressure on access to bioinformatics, computer speed and storage, and interpretation of data (Fig. 1). The bottleneck to solving long-standing issues in plant biology will be assembling and sifting through terabytes of data from these studies.
Fig. 1.
The genomics bottleneck. Advances in legume genomics have been facilitated by next-generation sequencing technologies. Next-generation sequencing platforms have the power to provide unprecedented insight into biological, genetic, agronomic and academic questions posed by researchers. However, each experiment utilizing next-generation sequencing technology is bound by limitations including data storage, computational power and access to bioinformatic analysis and assistance. With these platforms, the distillation of biologically relevant information (and subsequent interpretation) is the rate-limiting step.
The legume family (Leguminosae) is second only to the grass family in economic and nutritional value (Graham and Vance, 2003). Approximately 17 000 species of legumes are found in nature. Many of these are important grain, pasture and agroforestry species. Grain and forage legumes are grown on some 200 Mha, or about 15 % of the Earth's arable surface. About 33 % of human's dietary nitrogen (N) needs are derived from legumes (maybe up to 60 % in developing countries). Moreover, upwards of 40 % of the worlds cooking oil comes from soybean and peanut (Vance, 2001). In the past decade, legumes have come to rival arabidopsis and maize as model systems for genomic studies. This has occurred not only because of their economic and nutritive value but also due to the fact that they, in symbiosis with soil bacteria collectively known as rhizobia, can provide their individual N needs through symbiotic nitrogen fixation (SNF). The following contribution will focus on addressing issues related to legume biology through the use of NGS. In so doing, references to other species may also be cited.
GENOME SEQUENCING
To date, genome sequence information, either partial or complete, exists for 44 higher plants (41 at http://phytozome.net, accessed 22 January 2014; and three at Legume Information System, www.comparative-legumes.org, accessed 22 January 2014). Six of the currently sequenced genomes are legumes (Table 1); Cajanus cajan, pigeonpea (Varshney et al., 2012); Cicer arietinum, chickpea (Varshney et al., 2013); Glycine max, soybean (Schmutz et al., 2010); Lotus japonicus, trefoil sp. (Sato et al., 2008); Medicago truncatula, barrel medic (Young et al., 2011); and Phaseolus vulgaris, common bean (www.phytozome.net). The estimated high confidence gene calls in these species range from 28 269 to 48 680 genes. The rapid advances in DNA sequencing can be illustrated in that it took some 8 years to sequence and annotate the M. truncatula genome (2003–2010), while the genomes of other species were sequenced in 2 years. Currently, the genomes of alfalfa (Medicago sativa), lupin (Lupinus angustifolius), peanut (Arachis hypogaea) and pea (Pisum sativum) are in the sequencing pipeline. It should be noted that a large collection of expressed sequence tags for a number of plant species are deposited at the Dana Farber Gene Index (http://compbio.dfci.harvard.edu/tgi/plant.html) and these have been invaluable in annotating legume genomes and making gene calls.
Table 1.
Legume sequenced genomes
| Legume | Genome (1N) | No. of genes* | No. of ESTs† | Reference |
|---|---|---|---|---|
| Cajanus cajan (pigeonpea) | 11 | 48 680 | 25 640 | Varshney et al. (2012) |
| Cicer arietinum (chickpea) | 8 | 28 269 | 46 064 | Varshney et al. (2013) |
| Glycine max (soybean) | 20 | 46 430 | 1 530 030 | Schmutz et al. (2010) |
| Lotus japonicus (trefoil) | 6 | 30 799 | 243 067 | Sato et al. (2008) |
| Medicago truncatula (barrel medic) | 8 | 47 845 | 286 175 | Young et al. (2011) |
| Phaseolus vulgaris (common bean) | 11 | 31 600 | 149 769 | In progress (www.phytozome.net) |
*As of January 2014.
†According to the Dana Farber Gene Indices (compbio.dfci.harvard.edu/tgi/plant.html).
A synopsis of the genome sequence data acquired from the six currently sequenced legumes shows that they have both common and unique features. All six sequenced legumes belong to the Papilionoideae, which appears to have undergone a whole-genome duplication (WGD) event some 60 million years ago (MYA), and all appear to be derived from a common ancestor (Supplementary Data Fig. S1; Schmutz et al., 2010; Young et al., 2011; Varshney et al., 2012, 2013]. Soybean underwent a second, probably allopolyploid, WGD event some 13 MYA (Schlueter et al., 2004; Gill et al., 2009; Cannon and Shoemaker, 2012). The WGD event 60 MYA currently appears to be confined to the Papilionoideae, as WGD signatures have not yet been identified in either the Mimosoid or Caesalpinoid subfamilies. A sizable portion (31–59 %) of Papilionoideae genomes are composed of transposable elements. The expansion of transposable elements may be related to the WGD which occurred some 60 MYA, while further expansion of transposable elements in soybean may have occurred at the second WGD event 13 MYA. Comparative analysis within the Papilionoideae subfamily indicates that there is striking synteny among euchromatic regions, with conserved blocks extending as far as a large portion of chromosome arms (Young et al., 2011; Varshney et al., 2012). For example, Mudge et al. (2005) evaluated a region on soybean chromosome 18 which correlated with nematode resistance and was co-linear with medicago for 33 of 35 genes. There appears to be a sizeable component of legume family-specific genes as well as species-specific genes. Varshney et al. (2012) identified from 455 to 3068 legume species-specific genes. A substantial number of the putative legume-specific genes are related to the development of N2-fixing root nodules and to the transduction of biotic and abiotic signalling. Many of these gene families are clustered in the genome and are annotated as nodulins, nodule cysteine-rich (NCR) proteins, leucine-rich repeat (LRR) receptor-like proteins and transporters. Phylogenetic analysis of nodule-specific genes suggests that nodulation arose several times through co-opting of several ancestral genes involved in mycorrhizal colonization (Young et al., 2011).
Some whole-genome sequencing projects were also accompanied by transcriptome analyses via RNA-seq (Young et al., 2011; Varshney et al., 2012, 2013). RNA-seq data allowed for in-depth mining of splice variants, SNPs, simple sequence repeats (SSRs) and small interfering RNA (siRNA) species, the identification of sub-/neo-functionalized genes and transcript expression. Analysis of soybean, which has a high seed oil content, shows an overabundance of genes and transcripts related to lipid biosynthesis compared with other legume sequences (Schmutz et al., 2010). Similarly, in pigeonpea, which thrives in dry arid climates, there is an over-representation of genes related to drought tolerance (Varshney et al., 2012). Interestingly, there are 593 NCRs found in the Medicago genome, with >300 that show enhanced expression in root nodules (Fedorova et al., 2002; Young et al., 2011). The NCRs appear to be required for bacteroid development in mature nodules. A comparable NCR family is not apparent in soybean (Schmutz et al., 2010) or common bean (J.A. O'Rourke and C.P. Vance, unpubl. res.). Lack of NCRs in soybean and common bean suggests that other transcripts in nodules of these species must play a role in bacteroid differentiation and viability.
MULTIPLE GENOMES: RE-SEQUENCING
Re-sequencing the genomes of multiple distinct accessions within a single species can provide greater understanding of evolutionary history and domestication events (Huang et al., 2010; Cao et al., 2011; Jiao et al., 2012). In addition, re-sequencing can lay a foundation for genome-wide association studies (GWAS) and forces contributing to heterosis (Atwell et al., 2010; Lai et al., 2010; Lam et al., 2010; Branca et al., 2011). While the genome of cultivated soybean was sequenced by Schmutz et al. (2010), the whole genome of wild soybean G. soja Sieb and Zucc was sequenced by Kim et al. (2010). Their final G. soja mapped sequence length of 915 Mb covered 97·6 % of the soybean genome to 43× coverage. They found that some 80 % of the G. soja genome was duplicated, reflecting the two WGD events occurring at 60 and 13 MYA (Supplementary Data Fig. S1). As assessed by SNPs and PAVs, the genomes of G. soja differed from that of cultivated soybean by 0·3 %; less than that between arabidopsis accessions (Ossowski et al., 2008). They estimated that while G. max and G. soja diverged approx. 287 000 years ago, soybean was domesticated only 6000–9000 years ago. The authors suggest that some 712 genes representing 32·4 Mb of G. max were partially or completely absent in G. soja, suggesting that cultivated soybean acquired a number of genes during domestication that are not present in the wild species. This was further confirmed by Joshi et al. (2013) who identified 425 genes unique to G. max (completely absent in G. soja) including 12 genes involved in seed development and two involved in lipid metabolism.
Further re-sequencing of soybean was conducted by Lam et al. (2010). They sequenced the genomes of 17 wild (G. soja) and 14 cultivated (G. max) soybean lines. Phylogenetic analysis confirmed that wild and cultivated soybean originated from a common ancestor. The total number of SNPs was much higher in wild soybean as compared with the cultivated lines. A set of 205 614 SNPs was identified which could be useful for QTL mapping and GWAS. Cultivated soybean had a relatively homogeneous genetic background. However, some cultivated lines showed evidence of introgressions from wild soybean. As compared with rice and arabidopsis, both cultivated and wild soybean exhibited higher linkage disequilibrium (LD), decaying at 150 and 75 kb, respectively. Diversity analysis of LD blocks showed that there was a lower SNP ratio in long LD blocks compared with the whole genome. The identification of high LD in soybean along with a high number of SNPs led the authors to conclude that marker-assisted selection (MAS) would be the best approach to breeding for improvement. In addition, since wild and cultivated soybeans are cross-fertile, it is important that wild soybean be protected in order to provide new alleles for cultivated genomes.
Branca et al. (2011) used whole-genome re-sequencing of 26 diverse M. truncatula lines to lay a foundation for GWAS and evaluate LD. They found that M. truncatula was three times more diverse than soybean (some 3 million SNPs). Four clusters of genes had greater replacement site diversity as compared with other gene families: Toll interleukin repeats (TIRs), LRRs, nucleotide binding apoptosis (NB_ARCs) and NCRs. Similar to arabidopsis, LD decayed rapidly, much more so than in soybean. The authors concluded that whole-genome re-sequencing may be more efficient than a tagged SNP approach for GWAS.
Varshney et al. (2013) re-sequenced whole or partial genomes of 90 chickpea genotypes including 60 improved lines, 25 landraces and five wild genera (Cicer) accessions. They identified 4·4 million variants [SNPs and insertions/deletions (Indels)]. The cultivated genotypes displayed limited genetic diversity as compared with the wild landraces. They noted that the breeding for agronomic and phenotypic traits resulted in a loss of genetic diversity through the use of small sets of superior genotypes, which creates genetic bottlenecks. Six genomic regions ranging from 50 to 200 kb carried approx. 122 genes that appear to be candidates for selection in modern breeding efforts. Several genes involved in disease resistance were among those identified as candidates for selection. Extended synteny was detected between chickpea and other sequenced legumes, with the largest number of extended blocks observed for M. truncatula.
Genomic re-sequencing of plants with stable mutant phenotypes provides a powerful method for mapping mutational events in order to link a phenotype with a causal mutation. This type of forward genetics has proven a major genetic tool in determining gene function. In arabidopsis, Belfield et al. (2012) determined that fast neutron (FN) mutagenesis induced smaller genomic deletions and more single base pair substitutions than previously believed, highlighting the limitations of chip-based technologies. In P. vulgaris, we (O’Rourke et al., 2013b) applied NGS technologies to identify three types of SV in an FN population: (1) intercultivar SV, i.e. large sequence segments that are missing from all mutants and the wild-type plant, but are present in the reference genome; (2) intracultivar SV, i.e. sequences exhibiting differences among individuals within the mutant population; and (3) SV specific to a single mutant individual, potentially generated by FN irradiation (Fig. 2). Using this methodology, we identified >17 000 SNPs and 2700 Indels within gene-coding regions potentially impacting the mutant phenotypes. Additionally, large-scale deletions (>1 kb) were identified within gene regions for three of the mutant individuals. The utility of this type of analysis has driven the development of various analysis pipelines designed to identify deletions resulting in mutant phenotypes (Huang et al., 2009; Zuryn et al., 2010; Wang et al., 2011; Leshchiner et al., 2012).
Fig. 2.

Structural variations in Phaseolus vulgaris fast neutron (FN) mutants. DNA-seq data from six Red Hawk individuals aligned to the common bean reference genome (GI19833) available at www.phytozome.net and visualized using IGV (integrative genomics viewer; Robinson et al., 2011). These regions of structural variation (SV) were all identified on chromosome 4 from regions highlighted by filled black boxes on the chromosome at the top of the figure. For each individual, a histogram plot illustrates the read depth, while individual reads are aligned below. Each type of SV is highlighted by a black box. Intercultivar SV: regions present in the reference genome but missing in all Red Hawk individuals, representing differences between common bean cultivars. FN SV: sequences missing in a single individual, but present in all other lines, probably resulting from FN mutagenesis. This type of SV is most probably responsible for the mutant phenotype. Intracultivar SV: regions of genomic heterogeneity within the Red Hawk cultivar. Deletions identified in two or more Red Hawk individuals illustrating the residual heterogeneity in the FN mutant population.
Next-generation technologies are also proving important tools in traditional plant breeding programmes. Combining the power of NGS and QTL maps allows researchers to identify causal genes underlying traits of interest (Edwards et al., 2013; Atwood et al., 2014). Additionally, genotype by sequencing (GBS) provides a rapid and low-cost method for genotyping populations, allowing large-scale genomic selection in breeding programmes (Poland and Rife, 2012). The identification of SNPs and Indels coupled with simultaneous genotyping unique to the breeding lines of interest facilitates high-throughput genotyping of progeny to select for traits of interest and may reduce yield drag by improving progeny selection (Varala et al., 2011). GBS technologies have been successfully applied in soybean and common bean (Hyten et al., 2010; Varala et al., 2011; Sonah et al., 2013).
As the cost of NGS continues to decrease, genome re-sequencing will become even more common. Currently, multiple reference lines of soybean, common bean, lotus and Medicago are being sequenced in addition to WGS analysis of mutant individuals and GBS of mapping populations. These data will provide further insight into evolution, genome architecture, Indels and gene copy number. However, mining useful information from multiple genome sequences will require even greater investment in bioinformatics due to the bottleneck in analysing large data sets (Fig. 1).
LEGUME FUNCTIONAL BIOLOGY THROUGH THE TRANSCRIPTOME
Evolution of plant transcriptomes
Transcriptome analyses of developing tissues and organs, along with that of plants exposed to various biotic and abiotic stresses, can provide insight and understanding of plant genes regulating many processes (Z. Wang et al., 2010a; Mochida and Shinozaki, 2011). Evaluation of plant transcriptomes has undergone a phenomenal expansion in the past 10 years. Several reviews have described the methods and approaches from sequencing individual transcripts [i.e. cDNAs via expressed sequenced tags (ESTs) and serial analyses of gene expression (SAGE)] to massive parallel signature sequencing (MPSS). Upon the sequencing of thousands of ESTs and genome sequencing of many plant species, chip-based technologies involving microarrays emerged as the dominant platform and approach to transcript expression. However, the aforementioned approaches to transcript studies have major limitations including: (1) sample size and normalization; (2) speed; (3) sensitivity; (4) cost; and (5) limited dynamic range (Wang et al., 2009; L. Wang et al., 2010; Ozsolak and Milos, 2011; Schneeberger and Weigel, 2011; Garg and Jain, 2013). Unprecedented advances in high-throughput next-generation RNA-seq have resulted in a paradigm shift in transcriptome studies (Lister et al., 2008; Mochida and Shinozaki, 2011; Garg and Jain, 2013). In addition, the availability of powerful assembly software alleviates the requirement for a sequenced genome (Garg et al., 2011; Kalavacharia et al., 2011; Yang et al., 2011; Zhang et al., 2012; O’Rourke et al., 2013b). RNA-seq has been used in several studies involving plants, including but not limited to: maize leaf development (Li et al., 2010); rice transcriptome complexity (Zhang et al., 2010); rubber plant, Hevea brasiliensis (Xia et al., 2011); panicum development (Meyer et al., 2012); tomato tissue and metabolism (Matas et al., 2011); and grape (Vitis vinifera) berry development (Zenoni et al., 2010).
Although RNA-seq data have been collected for several legume species, a large focus has been on developing genetic markers in chickpea (Garg et al., 2011); lentil, Lens culinaris (Kaur et al., 2011); peanut, A. hypogaea (Zhang et al., 2012); alfalfa, M. sativa (Han et al., 2011); and common bean, P. vulgaris (Kalavacharia et al., 2011). Studies using RNA-seq to understand development, acclimation to stress, composition and N fixation have been limited to soybean (Bolon et al., 2010; Libault et al., 2010; Severin et al., 2010a, b; Atwood et al., 2014); alfalfa, M. sativa (Yang et al., 2011); lupin, L. albus (O’Rourke et al., 2013b); and M. truncatula (Boscari et al., 2013). It is also noteworthy that RNA-seq has been applied to studies of legume microRNAs (miRNAs) in common bean (Peaez et al., 2012), M. truncatula (Lelandais-Briere et al., 2009; Simon et al., 2009) and soybean (Song et al., 2011; Turner et al., 2012). In the sections to follow, we will provide an overview of RNA-seq studies our laboratory has conducted to understand legume biology.
The soybean transcriptome
Soybean is a highly valued crop because it accumulates large amounts of oil, protein and seeds, and because, in symbiosis with soil rhizobia bacteria, it forms N2-fixing root nodules. Soybean seed contains approx. 40 % protein and 20 % oil (Ohlrogge and Kuo, 1984; Bolon et al., 2010). Soybean seed protein is a primary food for both livestock and humans. Soybean oil is the most abundant cooking oil on earth (Wilcox, 2004). Symbiotic N2 fixation by soybean greatly reduces the need for added N fertilizer. To understand the genes involved in seed development and root nodule N2 fixation, we evaluated the transcriptomes of seeds and root nodules. To complement these studies, we also analysed the transcriptomes of other soybean tissues (Fig. 3). We isolated total RNA from leaves, flowers, pods, roots, nodules and seven stages of seed development of two near-isogenic lines (NILs). One line segregated for high protein (HiPro; LD04–15154) and the other for low protein (LoPro; LD04–15146) (Seboldt et al., 2000; Nichols et al., 2006; Bolon et al., 2010). These data were used to develop a soybean gene expression atlas (Severin et al., 2010b) and to analyse SNPs between the HiPro and LoPro NILs (Bolon et al., 2010; Bolon and Vance, 2012; Bolon et al., 2013). The close genetic identity of the two NILs was confirmed by identifying only 387 SNPs between the two lines. The 1000 most highly expressed transcripts in each tissue (Combined HiPro and LoPro) are shown in Supplementary Data File S1. It should be noted that Libault et al. (2010) have also developed a soybean gene expression atlas based upon RNA-seq data derived from 14 tissues. They focused primarily on roots and nodules from a single genotype of G. max, Williams 82.
Fig. 3.
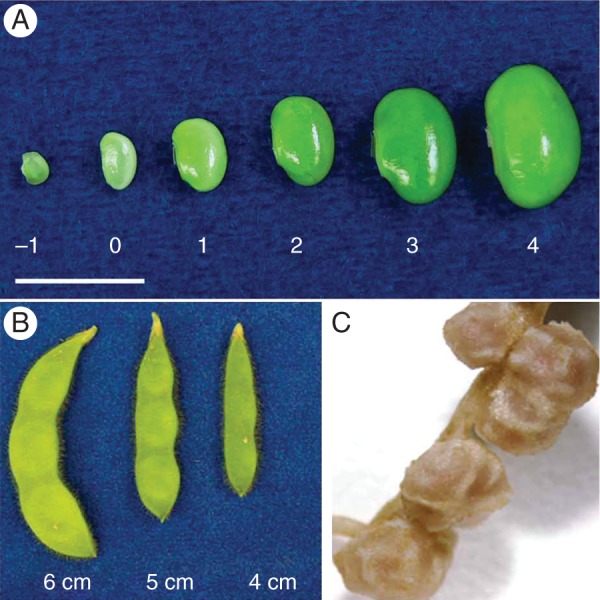
Soybean tissues with RNA-seq transcriptome data. Total RNA was isolated from leaves, flowers, pods, roots, nodules and seven stages of seed development of soybean near-isogenic lines LoPro and HiPro. (A) Seed stages –1 to 4. (B) Pod sizes for corresponding seed stages from stage –1 to stage 2. (C) Nodule tissues attached to soybean roots.
Annotations of the soybean genome (Schmutz et al., 2010) predicted 46 430 high-confidence gene models and 19 780 lower confidence gene models. We found 90·4 % (41 975) high-confidence gene models and 7176 lower confidence gene models to be transcriptionally active. Hierarchical clustering analyses of transcripts between tissues and development suggested three groupings of tissues; underground (roots and nodules), seed related and aerial tissues (flower, leaves and pods) (Severin et al., 2010b). Tissue-specific expression was evaluated by combining counts from each genotype and then selecting transcripts with an RPKM (reads per kilobase per million) ≥1, according to a modified RPKM formula (Severin et al., 2010b). Tissue-specific transcripts in the combined NILs were identified including: seed = 81; leaf = 95; flower = 716; pods and shell pods = 138; root = 727; and nodule = 418 (Supplementary Data File S2).
Due to the capacity of soybean to fix N2 symbiotically, we determined whether genes other than the typical nodulin gene families and leghaemoglobins were highly expressed in nodule tissues. A novel PLAC8 (placenta-specific) transcript was highly expressed. This membrane protein possesses a cysteine-rich region and has been implicated in regulating cell number (Libault and Stacey, 2010). A highly expressed cytochrome P450 was also noted. Annotation for this gene suggests involvement in either flavonoid or abscisic acid metabolism. An embryo-specific protein with a lipoxygenase-related annotation and at least two remorin-like transcripts were abundantly expressed in nodules. Remorins are membrane proteins associated with lipid rafts. Toth et al. (2012) has recently shown that L. japonicus remorin 1 is localized to infected nodule cells, and overexpression of this gene increases nodule numbers. The diverse array of putative functions for soybean nodule-specific transcripts make this data set a valuable addition to root nodule biology.
Because of our interest in transporters, we queried the combined NIL RNA-seq data for transporter function. We found a total of 1733 transporters showing expression in one or more tissues (Supplementary Data File S3). Fifteen of the 20 most highly accumulated transporter gene transcripts in the NIL RNA-seq seed data were found to be aquaporin transporters. The remaining highly expressed transporter genes were annotated as magnesium, copper, nucleotide or sugar-related transporter genes in the seed (Bolon and Vance, 2012). Genes encoding sulfate, ABC, oligopeptide, triose phosphate and sugar transporters were all highly expressed in nodules (Supplementary Data Files S1, S3). The sulfate transporter 1 (SST1) gene has been shown to be critical for effective N2 fixation (Krusell et al., 2005). Sugar import into nodules has been shown to be crucial to maintain effective N2 fixation (Vance and Heichel, 1981). Within the NIL RNA-seq data set, some 5–7 sugar transporters appear to be fairly nodule specific (Supplementary Data File S1). Root tissues, in general, displayed the highest expression of transporter genes, which is consistent with the role of roots in acquiring nutrients necessary for growth.
Seed formation proceeds through three stages: pre-storage, transition and storage maturation (Weber et al., 2005). We performed transcriptome analyses on developing seed stages: –1 seed (<25 mg); +1 seed (approx. 50 mg); +2 seed (approx. 100 mg); +3 seed (approx. 200 mg); and +4 seed (approx. 300 mg) (Fig. 3). These stages correspond to those described by Weber et al. (2005), Bolon et al. (2010) and Severin et al. (2010b). A comparison of –1 seed with the +4 seed gene expression, visualized using MapMan metabolic pathways (Thimm et al., 2004), shows that the greatest changes in transcript abundance occurred as increases in transcript abundance of lipid, primary amino acid metabolism and 1-carbon (C1) metabolism genes (Fig. 4). The greatest decreases in transcript abundance were in cell wall, nucleotide, fermentation, ascorbate and secondary metabolism. These changes in transcript abundance were not unexpected based on metabolic events occurring at each stage of seed development.
Fig. 4.

Overview of soybean seed metabolism throughout seed development. A visualization of soybean metabolism gene transcript abundance changes is shown using MapMan. This map shows a comparison of –1 seed with +4 seed gene expression in general metabolic pathways. The greatest gene transcript abundance increases (blue) are seen in lipid, primary amino acid metabolism and C1-metabolism genes. The largest decreases in transcript abundance (red) are found in cell wall, nucleotide, fermentation, ascorbate and secondary metabolism genes.
Because seed development and filling involves partitioning of N from leaves into pods and seeds, we thought it important to evaluate the expression of N assimilation enzyme gene transcripts from pod formation through seed filling. Transcripts related to 11 gene families were surveyed (Fig. 5; Supplementary Data File S4). Figure 5A shows the gene expression patterns for NADH- and Fd-glutamate synthase (GOGAT). In early pods and seeds, Fd-GOGAT transcripts (Glyma03g28410 and Glyma19g31120) predominate, while later in seed development NADH-GOGAT (Glyma14g32500 and Glyma19g16450) is the dominant form. Expression of Fd-GOGAT in pods and early seed is consistent with the observation that green photosynthesizing tissues utilize Fd-GOGAT for N assimilation while in non-photosynthetic tissues, NADH-GOGAT is more important to N assimilation. Enzymes involved in allantoin metabolism (Fig. 5B; Supplementary Data File S4) appear most highly expressed during early pod set and seed formation, as evidenced by the expression of Glyma13g31430, allantoinase. Expression of genes involved in ammonium transport and ureide degredation (including allantoinase, allantoate amidohydrolase and urease) in early pod set and seed filling reflects the transport and subsequent metabolism of ureides in pods and seeds. Our data support the interpretation that pods and seeds participate in the metabolism of both ammonia and ureides, providing N for protein deposition in seeds.
Fig. 5.
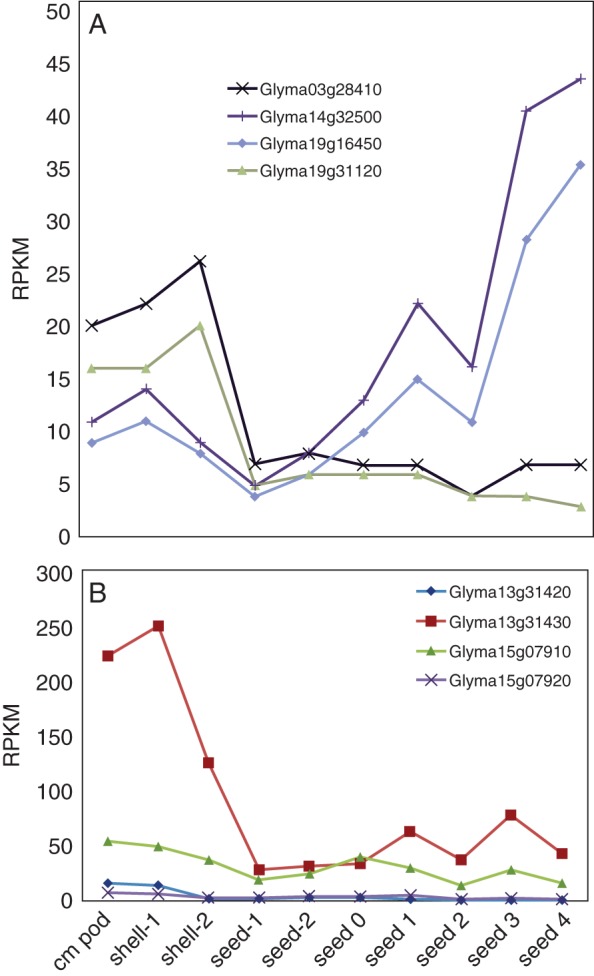
Nitrogen assimilation enzyme gene transcripts from pod formation through to seed filling. (A) Gene transcript abundance patterns for NADH- and Fd-glutamate synthase (GOGAT). In early pods and seeds, Fd-GOGAT transcripts (Glyma03g28410 and Glyma19g31120) are more abundant, while later in seed development NADH-GOGAT (Glyma14g32500 and Glyma19g16450) transcripts are more highly expressed. (B) Allantoin metabolism gene transcript abundance (Supplementary Data File S5) is highest during early pod set and seed formation (Glyma13g31430, allantoinase). RPKM, reads per kilobase per million.
As mentioned previously, soybean seed value is determined by the oil and protein composition. Using tissues from ten different stages of reproductive development, transcripts encoding enzymes related to oil biosynthesis were evaluated. Our analysis of this RNA-seq data set provides a comprehensive overview of oil metabolism throughout soybean seed development and a foundation for modelling gene networks involved in seed oil deposition. Transcripts encoding enzymes of 11 gene families involved in oil biosyntheses were identified, including 15 members of the fatty acid desaturase gene family (Supplementary Data File S5). Three patterns of lipid gene expression were noted based on the stage of seed development: (1) high expression in early pod and seed; (2) uniform expression throughout development; and (3) high expression during seed filling. Early pod seed expression was exemplified by glycerol-3-phosphate transferase and phospholipid diacylglycerol acyltransferase gene expression. Examples of genes with transcripts expressed throughout seed development include triacylglycerol lipase and diacylglycerol acyltransferase.
Unexpectedly, our data show that FAD2–2B and FAD2–2C are expressed early in seed formation. In contrast, and as expected, transcripts for FAD2–1B and FAD2–1A were overabundant during later seed development as oil bodies are deposited. In a previous study of seed filling, Severin et al. (2010b) noted 143 genes annotated by gene ontology (GO) analysis as nutrient reservoir. Included among these 143 genes are oleosins, sucrose-binding proteins, glycinins and lipoxygenases (Supplementary Data Files S1, S2), consistent with seed transcriptome analysis in the soybean NIL pair (Bolon et al., 2010; Bolon and Vance, 2012). Proteomic analyses on the developing soybean seed (Hajduch et al., 2005) also provide support for the presence of gene transcripts associated with metabolism, protein destination and storage.
During seed filling of dicot plants, particularly soybeans, the embryo enlarges as proteins, oil and carbohydrates are stored in the cotyledons (Le et al., 2007; Agrawal et al., 2008; Verdier and Thompson, 2008; Verdier et al., 2008; Hajduch et al., 2010). Several transcription factors act as major regulators of these processes (Verdier et al., 2008; Le et al., 2010), while an additional 900–1300 transcription factors have been associated with seed development (Benedito et al., 2008; Le et al., 2010). Our data set allowed us to profile transcription factors throughout seed formation and filling. Some 54 transcripts related to 12 transcription factor families were evaluated (Z. Wang et al., 2010b; Supplementary Data File S6). Transcripts encoding AP2, VAL2, GL2, PKL, FIE and DOF4, were most highly expressed in early pod and embryo development. PKL is a crucial activator of embryo development while FIE suppresses premature development of the endosperm (Mosquna et al., 2004; Li et al., 2005). The transition from embryo to seedling development is regulated by VAL1 and 2 (Wang and Perry, 2013). Seed filling development is regulated by ABI3, FUS3 and WRI1. These transcription factors appear to affect oil, carbohydrate and protein deposition during seed fill. The interaction of transcription factors throughout embryo and seed development is quite complex, with multiple transcription factors interacting at each stage.
The soybean NIL RNA-seq data set from 14 soybean tissues has significantly enhanced the options for understanding the soybean whole transcriptome. With this data set, along with those of Libault et al. (2010), legume researchers now have a stronger foundation for analysing gene networks in soybean. The data serve as a valuable resource for the legume community.
White lupin transcriptome: acclimation to phosphorus deficiency
Phosphorus (Pi) is a critical nutrient required for plant growth and development (Bieleski, 1973). While the Pi content is high in many soils, availability is often limited due to slow diffusion and high fixation rates. Moreover, in developing countries, the aged and weathered soils render Pi essentially unavailable (Bieleski, 1973; Vance et al., 2003; Lambers et al., 2008). Additionally, Pi fertilizer production is not sustainable, with inexpensive world supplies becoming limited by 2050–2080 (Cordell et al., 2009; Gilbert, 2009). Thus, efforts to improve Pi acquisition and utilization by plants are imperative.
White lupin (L. albus) forms cluster roots (Fig. 6) in response to Pi deficiency (Neumann and Romheld, 1999; Vance et al., 2003; Cheng et al., 2011; O’Rourke et al., 2013b). In Pi-deficient plants, upwards of 70 % of the root mass may occur as cluster roots. Cluster root formation is accompanied by striking biochemical changes including the release of enzymes and proteins, exudation of protons and organic acids, modified metabolism and altered hormone balance. The biochemical and molecular changes that occur during cluster root formation have made white lupin a model for studies of plant acclimation to Pi deficiency stress.
Fig. 6.

White lupin cluster root phenotype. White lupin (Lupinus albus; inset image) cluster root formed when grown in Pi-deficient hydroponics for 14 d. A bracket spanning the region of closely spaced tertiary lateral rootlets delineates the cluster root region.
In an effort to better understand plant acclimation to Pi deficiency and the genes involved, we conducted a whole-genome transcript expression study in white lupin plants grown under Pi-sufficient and Pi-deficient conditions (O’Rourke et al., 2013b). The Pi-deficient root samples were composed primarily of cluster roots. The transcripts were assembled de novo and used to build the first white lupin gene index (LAGI 1·0), which is available on the white lupin species information page at http://comparative-legumes.org. We project 50 734 transcriptionally active sequences representing 7·8 % of the genome. Considering all fully sequenced legumes, approx. 6 % of the genome is predicted to encode proteins (M. truncatula, 8·3 %; L. japonicus, 4·4 %; pigeonpea, 5·6 %; soybean, 5 %). The 1000 most highly expressed transcripts are provided in Supplementary Data File S7.
We detected 2128 transcripts that are differentially expressed due to Pi deficiency (Fig. 7; Supplementary Data File S8). In leaves, 1342 transcripts were differentially expressed due to Pi deficiency while 903 transcripts were differentially expressed in roots (some transcripts were differentially expressed in both leaves and root). Several functional classes of transcripts having increased expression under Pi-deficient conditions were over-represented in the transcripts of Pi-deficient cluster roots including glycolysis, C1-metabolism, glyoxylate related, cell wall, lipid metabolism, metal handling and transport (Fig. 8). Similar classes of transcripts were over-represented in Pi-deficient leaves. In comparison, classes of transcripts over-represented in Pi-sufficient leaf samples included photosynthesis, ATP synthesis and stress. Previous studies from our laboratory (Johnson et al., 1994, 1996) as well as others (Neumann and Romheld, 1999; Plaxon and Tran, 2011) have shown that Pi deficiency enhances glycolysis and organic acid metabolism. The lupin transcriptome reveals that other carbon pathways are also altered (Supplementary Figs S2, S3). Specifically, C1-metabolism, especially formate, glyoxylate and methionine metabolism, was significantly upregulated in Pi-deficient cluster roots. Acetyl-CoA metabolism was also upregulated in Pi-deficient cluster roots. Each of the above-mentioned new carbon metabolism pathways activated due to Pi deficiency contributes to carbon cycling through organic acids which can be exuded into the rhizosphere to increase the solubility of bound Pi (Supplementary Figure S3).
Fig. 7.
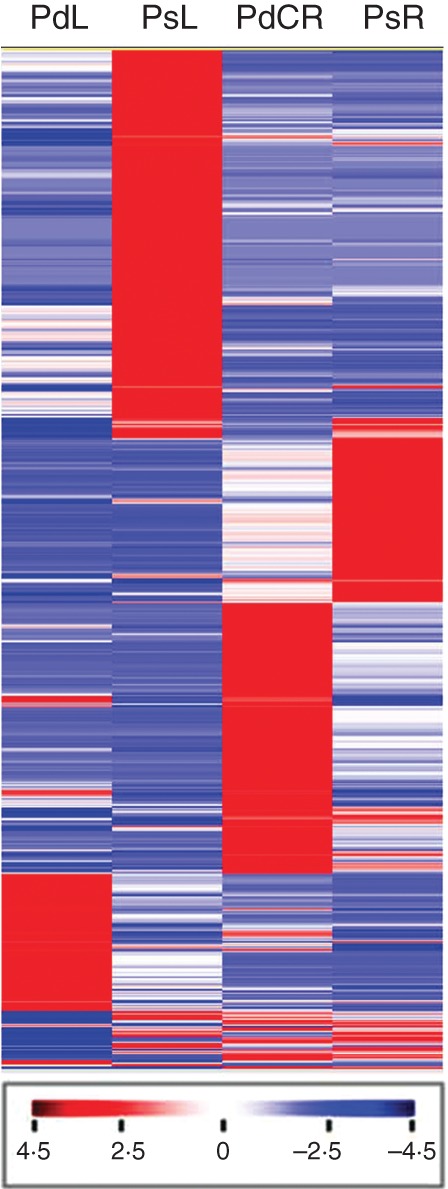
Heatmap of genes differentially expressed due to Pi deficiency. A heatmap of 2128 transcripts in the LAGI 1·0 assembly differentially expressed due to Pi deficiency. To be considered differentially expressed, the transcript must have an RPKM ≥3 in at least one tissue, ≥2-fold change between tissues and P ≤ 0·05. A total of 1342 transcripts were differentially expressed between leaf samples and 903 between root samples due to Pi deficiency (117 transcripts were differentially expressed in both leaves and roots). Expression profiles are presented as Z-scores. Red indicates high expression, white indicates intermediate expression and blue indicates low expression. PdL, phosphate-deficient leaves; PsL, phosphate-sufficient leaves; PdCR, phosphate-deficient cluster roots; PsR, phosphate-sufficient normal roots.
Fig. 8.
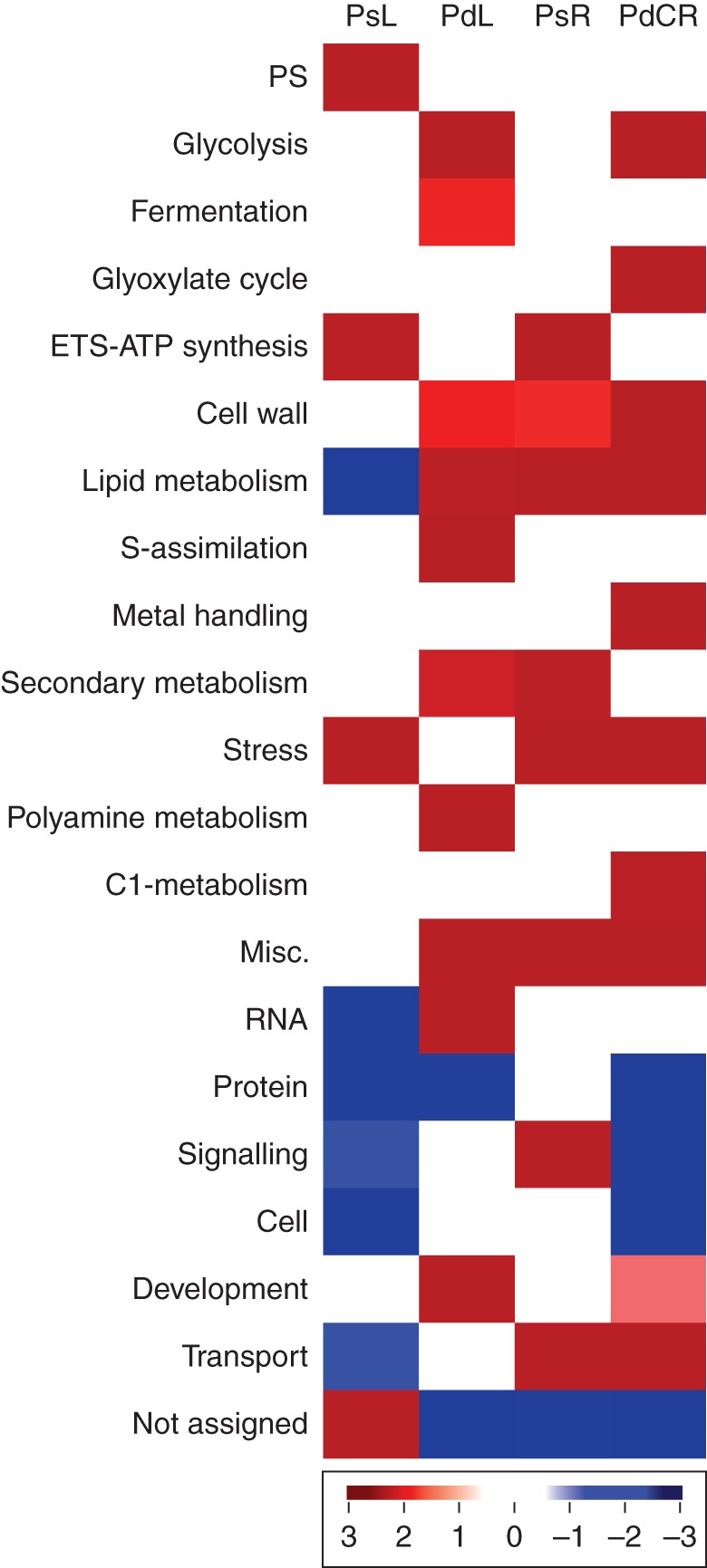
Gene categories with altered expression due to Pi deficiency in white lupin. The identification of over-represented MapMan categories (Thim et al., 2004) among genes differentially expressed due to Pi deficiency relative to the LAGI 1·0 transcriptome (Fisher test and Benjamin–Hochberg correction, Z-value cut-off = 1). MapMan categories over- (red) or under- (blue) represented in Pi-sufficient leaves (PsL), Pi-deficient leaves (PdL), Pi-sufficient roots (PsR) and Pi-deficient cluster roots (PdCR).
The increase in C1-metabolism in Pi-deficient roots also appears to contribute to the Yang cycle and ethylene biosynthesis (O’Rourke et al., 2013b). Pi deficiency is known to increase ethylene synthesis in roots (Gilbert et al., 2000; Ma et al., 2003; Zhang et al., 2003; Nagarajan and Smith, 2012; Niu et al., 2013). Ethylene interacts with auxin to alter root hair and lateral root development, therefore impacting white lupin root architecture when subjected to Pi-deficient conditions.
The noticed effect of Pi deficiency on ethylene-related transcripts in cluster roots stimulated us to assess transcripts involved in the metabolism of additional plant hormones, since interactions between hormone synthesis and degradation are known to regulate root architecture and development. Plant hormones are also implicated in nutrient sensing and signalling (Torrey, 1976; Lopez-Bucio et al., 2002; Rubio et al., 2009; Chiou and Lin, 2011; Niu et al., 2013). Transcripts involved in the biosynthesis and degradation of auxin, gibberellin (GA) and cytokinin all exhibited differential expression in Pi-deficient cluster roots (Supplementary Data File S9).
Auxin is a well known effector of lateral root development and density (Torrey, 1976; Casimiro et al., 2001). Previous findings from our lab (Gilbert et al., 2000; Cheng et al., 2011) showed an important role for auxin in signalling cluster root development under Pi deficiency. The addition of exogenous auxin stimulated cluster root development, even under Pi-sufficient conditions. Furthermore, the inhibition of auxin transport blocked cluster root formation. The RNA-seq data further support a role for auxin homeostasis in cluster root formation. We found transcripts related to auxin homeostasis to be more highly expressed in Pi deficiency-induced cluster roots than in other treatments (Supplementary Data File S9). Pi deficiency results in enhanced sensitivity to auxin and increased auxin concentration in Pi-deficient roots (Lopez-Bucio et al., 2002; Al-Ghazi et al., 2003; Cheng et al., 2011).
Cytokinin applications have been reported to inhibit root growth and suppress lateral root formation (Li et al., 2006; Werner et al., 2010; Cui et al., 2011). Moreover, Pi deficiency is known to reduce cytokinin activity in roots (Lo et al., 2008; Werner et al., 2010). Additional evidence for a role for cytokinin in lateral root formation has been demonstrated by the overexpression of cytokinin oxidase which results in reduced cytokinin levels and increased lateral root formation (Lo et al., 2008; Werner et al., 2010). Our RNA-seq data revealed that Pi deficiency increases the expression of transcripts encoding cytokinin oxidase, a gene regulating cytokinin degradation. We treated Pi-deficient plants with a synthetic cytokinin, benzyadenine (BA), which resulted in an inhibition of cluster root formation in Pi-deficient plants. Thus, the increased expression of cytokinin oxidase in Pi-deficient cluster roots may be related to maintaining cytokinin homeostasis at levels that promote cluster root development during Pi deficiency.
Similarly, we noted that Pi deficiency appeared to stimulate the expression of transcripts involved in GA biosynthesis. To test the effect of GA on cluster root formation we added either GA3 or paclobutrazol, an inhibitor of GA biosynthesis, to white lupin plants. Altering GA availability produced striking phenotypes. Additional GA3 reduced the density of lateral rootlets within the cluster root zone of Pi-deficient plants, while the application of paclobutrazol greatly stimulated cluster root formation in both Pi-sufficient and Pi-deficient plants (O’Rourke et al., 2013b). Similarly, silencing GA oxidase in Populus increased root GA concentration and decreased lateral root numbers, while in arabidopsis exogenous GA application had little effect on lateral root formation (Jiang et al., 2007). The lupin transcriptome analysis also revealed that the GA receptor, GID16, and phytochrome-interacting factor (PIF1) transcripts, which modulate GA signalling through DELLA proteins, had increased transcript abundance under Pi-deficient conditions. It appears that GA plays an important role in cluster root formation as evidenced by GA transcript expression and chemical modulation of GA availability. Our data suggest that Pi deficiency-induced cluster root formation is modulated by either increased GA production or increased sensitivity of roots to GA.
The expression of transporter genes is required for acclimation to nutrient deficiencies. Of the 2128 transcripts differentially expressed in white lupin in response to Pi deficiency, 110 were annotated as transporters (Supplementary Data File S10). In Pi-deficient cluster roots, 11 transporter families representing 42 transcripts had enhanced expression when compared with Pi-sufficient roots. The most abundant transporter families were found to be phosphate, membrane H+, ATPases, divalent cation and multidrug and toxin efflux (MATE) transporters. In comparison, eight transporter families, represented by 22 transcripts, exhibited decreased expression in Pi-deficient cluster roots, including lipid, sugar and sulfate transporters. Few transporter families in leaves were impacted by low Pi. Increased transporter gene expression during nutrient-deficient conditions has been noted for arabidopsis (Misson et al., 2005), common bean (Hernandez et al., 2007), tomato (Liu et al., 1998) and rice (Li et al., 2010). Wang et al. (2002) reported an interaction among Pi, K and Fe transporters in tomatoes exposed to various nutrient stresses, suggesting that multiple transporters are co-ordinately regulated irrespective of the particular nutrient deficiency. Our RNA-seq data support such an interpretation.
Previous studies have shown miRNAs (miR399 and miR395) directly regulate sulfate and phosphate deficiency responses (Sunkar and Zhu, 2004; Sunkar, 2010). Additional studies have investigated the post-transcriptional regulation by miRNAs in response to nutritional, biotic and abiotic stresses (Jones-Rhoades and Bartel, 2004; Sunkar and Zhu, 2004; Fujii et al., 2005; Bari et al., 2006; Chiou et al., 2006; Hsieh et al., 2009; Zhu et al., 2009; Sunkar, 2010; Valdes-Lopez et al., 2010). However, sequencing of miRNAs requires special library construction and was thus beyond the scope of our study.
A major goal for crop production is to develop lines with improved acclimation to Pi deficiency. Identification of genes shared in common between species that respond to Pi deficiency may be useful for MAS. We identified 12 genes commonly expressed due to Pi deficiency in three species; white lupin (Uhde-Stone et al., 2003; O’Rourke et al., 2013b), potato (Hammond et al., 2011) and arabidopsis (Misson et al., 2005; Morcuende et al., 2007; Thibaud et al., 2010) (Table 2). These genes, among others reported by Hammond et al. (2011), may serve as molecular markers for crop improvement of Pi stress acclimation. In recent years, genes regulating Pi acclimation QTLs have been identified. In arabidopsis, the LOW PHOSPHATE ROOTS (LPR) QTL was identified as a multicopper oxidase (Svistoonoff et al., 2007). Similarly, the PUP1 QTL in rice was shown to be a protein kinase and designated as phosphate starvation tolerance 1 (PSTOL1). Shi et al. (2013) identified monogalactosyl diacyl glycerol synthase (MGDG) synthase and a glucose-6-phosphate transporter (G-6-PT) as controlling Pi acclimation in Brassica napus. We found LPR1, MGDG synthase and G-6-PT all highly upregulated in Pi-deficient cluster roots and designated them as important candidates for MAS.
Table 2.
Candidate genes for phosphorus acclimation marker-assisted selection
| LAGI 1.0 ID | PsR | PdCR | PsL | PdL | Annotation |
|---|---|---|---|---|---|
| LAGI01_23220 | 6·3 | 16·6 | 1·3 | 7·5 | MGDG synthase |
| LAGI01_41314 | 5·0 | 14·0 | 3·6 | 8·0 | SPX domain |
| LAGI01_24353 | 2·6 | 13·6 | 0 | 0 | Phosphate transporter |
| LAGI01_3168 | 1·3 | 9·6 | 0 | 4·5 | Phospholipase |
| LAGI01_53773 | 2·0 | 7·0 | 0 | 5·0 | MGDG synthase |
| LAGI01_47701 | 0 | 5·6 | 1·6 | 7·0 | SPX domain |
| LAGI01_62170 | 0 | 4·3 | 0 | 1·0 | SPX domain |
| LAGI01_31197 | 0 | 10·0 | 0 | 19·5 | Pyrophosphatase |
| LAGI01_79787 | 2·0 | 11·6 | 0 | 14·0 | Purple acid phosphatase |
| LAGI01_35670 | 3·3 | 14·3 | 5·6 | 16·5 | Glycosyltransferase |
| LAGI01_57598 | 6·0 | 60·0 | 2·6 | 21·5 | GPX-PDE8 |
| LAGI01_81721/74210/75025/63491 | 4·3 | 22·3 | 27·4 | 80·0 | Ferritin |
Expression patterns (as RPKM) for LAGI 1·0 transcripts identified as potential candidates for phosphorus deficiency marker-assisted selection.
PsR, phosphate-sufficient normal roots; PdCR, phosphate-deficient cluster roots; PsL, phosphate-sufficient leaves; PdL, phosphate-deficient leaves.
The multiple transcripts (LAGI01_81721, LAGI01_74210, LAGI01_75025 and LAGI01_63491) all share the same expression patterns and all annotate as ferritin, suggesting that all the transcripts encode the same gene.
Overall, our white lupin transcriptome analyses have provided novel insight and detail into how plants acclimate to Pi deficiency. We have shown previously unknown genes and processes to be major effectors for Pi deficiency tolerance. The lupin RNA-seq data also provided a foundation for gene calling and identification of splice variants upon sequencing of the L. angustifolius genome.
CONCLUSIONS
Studies using high-throughput DNA and RNA sequencing have resulted in profound paradigm shifts in plant genetics and biochemistry. Our fundamental understanding of genome architecture (Jiao et al., 2012), species evolution (Lam et al., 2010), adaptation (O’Rourke et al., 2013b), transcript variants (Libault et al., 2010), gene networks (Tomato Genome Consortium, 2012) and biochemical pathways (Li et al., 2012; O’Rourke et al., 2013b) has been expanded. Divergent plant lines such as ecotype collections (Branca et al., 2011; Varshney et al., 2013), mutant populations (Bolon et al., 2011; Fu et al., 2013; O’Rourke et al., 2013a) and nested association lines (Fu et al., 2013) can now be readily sequenced at both RNA and DNA levels. Such studies allow the identification of variants for almost any trait at the nucleotide level (Severin et al., 2010a; Peiffer et al., 2012). Access to genome-wide transcripts facilitates the identification of transcript variants (Libault et al., 2010; Severin et al., 2010b), miRNAs and their target RNAs (Sunkar, 2010), cell- and tissue-specific genes (Libault et al., 2010; Severin et al., 2010b) and epigenetic regulation (Weigel and Colot, 2012). The power of both forward and reverse genetics can be more fully realized through rapid, low-cost sequencing. The broad umbrella of genomics extended by high-throughput sequencing now impacts the disciplines of agronomy, biochemistry, physiology and microbiology. However, to translate the vast array to improved plants still lies through plant breeding and genetics (Supplementary Fig. S4). Multidisciplinary collaboration among plant biologists will be essential to feeding the worlds 7·5 billion inhabitants.
SUPPLEMENTARY DATA
ACKNOWLEDGEMENTS
The analyses were carried out in part using resources provided by the University of Minnesota Supercomputing Institute. This work was supported by the US Department of Agriculture-Agricultural Research Service [CRIS number 3640–21000–028–00D] and the United Soybean Board [projects 1240 and 2240].
LITERATURE CITED
- Agrawal GK, Hajduch M, Graham K, Thelen JJ. In-depth investigation of the soybean seed-filling proteome and comparison with a parallel study of rapeseed. Plant Physiology. 2008;148:504–518. doi: 10.1104/pp.108.119222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ghazi Y, Muller B, Pinloche S, et al. Temporal responses of Arabidopsis root architecture to phosphate starvation: evidence for the involvement of auxin signalling. Plant, Cell and Environment. 2003;26:1053–1066. [Google Scholar]
- Atwell S, Huang YS, Vihjalmsson BJ, et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010;465:627–631. doi: 10.1038/nature08800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood SE, O'Rourke JA, Peiffer GA, et al. Replication protein A subunit 3 and the iron efficiency response in soybean. Plant, Cell and Environment, 2014;37:213–234. doi: 10.1111/pce.12147. [DOI] [PubMed] [Google Scholar]
- Bari R, Pant BD, Stitt M, Scheible W-R. PHO2, microRNA399 and PHR1 define a phosphate signaling pathway in plants. Plant Physiology. 2006;141:988–999. doi: 10.1104/pp.106.079707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfield EJ, Gan X, Mithani A, et al. Genome-wide analysis of mutations in mutant lineages selected following fast-neutron irradiation mutagenesis of Arabidopsis thaliana. Genome Research. 2012;22:1306–1315. doi: 10.1101/gr.131474.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedito VA, Torres-Jerez I, Murray JD, et al. A gene expression atlas of the model legume Medicago truncatula. The Plant Journal. 2008;55:504–513. doi: 10.1111/j.1365-313X.2008.03519.x. [DOI] [PubMed] [Google Scholar]
- Bieleski RL. Phosphate pools, phosphate transport, and phosphate availability. Annual Review of Plant Physiology. 1973;24:225–252. [Google Scholar]
- Bolon Y-T, Vance CP. Characterization of the linkage group I seed protein QTL in soybean. In: Wilson RF, editor. Designing soybeans for the 21st century. AOCS Press; 2012. [Google Scholar]
- Bolon Y-T, Joseph B, Cannon SB, et al. Complementary genetic and genomic approaches help characterize the linkage group I seed protein QTL in soybean. BMC Plant Biology. 2010;3:10–41. doi: 10.1186/1471-2229-10-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon Y-T, Haun WJ, Xu WW, et al. Phenotypic and genomic analyses of a fast neutron mutant population resource in soybean. Plant Physiology. 2011;156:240–253. doi: 10.1104/pp.110.170811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon Y-T, Hyten DL, Orf JH, Vance CP, Muehlbauer GJ. eQTL networks reveal complex genetic architecture in the immature soybean seed. Plant Genome. 2013;7 10.3835/plantgenome2013.08.0027. [Google Scholar]
- Boscari A, Del Giudice J, Ferrarini A, et al. Expression dynamics of the Medicago truncatula transcriptome during the symbiotic interaction with Sinorhizobium mellioti: which role for nitric oxide? Plant Physiology. 2013;161:425–439. doi: 10.1104/pp.112.208538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branca A, Paape TD, Zhou P, et al. Whole-genome nucleotide diversity, recombination, and linkage disequilibrium in the model legume Medicago truncatula. Proceedings of the National Academy of Sciences USA; 2011. pp. E864–E870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SB, Shoemaker RC. Evolutionary and comparative analysis of the soybean genome. Breeding Science. 2012;61:437–444. doi: 10.1270/jsbbs.61.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, et al. Whole-genome sequencing of mulitple Arabidopsis thaliana populations. Nature Genetics. 2011;43:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- Casimiro I, Marchant A, Bhalerao RP, et al. Auxin transport promotes Arabidopsis lateral root initiation. The Plant Cell. 2001;13:843–852. doi: 10.1105/tpc.13.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Bucciarelli B, Liu J, et al. White lupin cluster root acclimation to phosphorus deficiency and root hair development involve unique glycerophosphodiester phosphodiesterases. Plant Physiology. 2011;156:1131–1148. doi: 10.1104/pp.111.173724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia JM, Ware D. Sequencing for the cream of the crop. Nature Biotechnology. 2011;29:138–139. doi: 10.1038/nbt.1756. [DOI] [PubMed] [Google Scholar]
- Chiou T-J, Aung K, Lin S-I, Wu C-C, Chiang S-F, Su C-L. Regulation of phosphate homeostais by microRNA in Arabidopsis. The Plant Cell. 2006;18:412–421. doi: 10.1105/tpc.105.038943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou T-J, Lin S-I. Signaling network in sensing phosphate availability in plants. Annual Review of Plant Biology. 2011;62:185–206. doi: 10.1146/annurev-arplant-042110-103849. [DOI] [PubMed] [Google Scholar]
- Cordell D, Drangert J, White S. The story of phosphorus: global food security and food for thought. Global Environmental Change. 2009;19:292–305. [Google Scholar]
- Cui H, Hao Y, Kovtun M, et al. Genome-wide direct target analysis reveals a role for SHORT-ROOT in root vascular patterning through cytokinin homeostasis. Plant Physiology. 2011;157:1221–1231. doi: 10.1104/pp.111.183178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards D, Batley J, Snowdon RJ. Accessing complex crop genomes with next-generation sequencing. Theoretical and Applied Genetics. 2013;126:1–11. doi: 10.1007/s00122-012-1964-x. [DOI] [PubMed] [Google Scholar]
- Fedorova M, van de Mortel J, Matsumoto PA, Cho J, Town CD, VandenBosch KA. Genome-wide identification of nodule specific transcripts in the model legume Medicago truncatula. Plant Physiology. 2002;130:519–537. doi: 10.1104/pp.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Cheng Y, Linghu J, et al. RNA sequencing reveals the complex regulatory network in the maize kernel. Nature Communications. 2013;4:2832. doi: 10.1038/ncomms3832. [DOI] [PubMed] [Google Scholar]
- Fujii H, Chiou T-J, Lin S-I, Aung K, Zhu J-K. A miRNA involved in phosphate-starvation response in Arabidopsis. Current Biology. 2005;15:2038–2043. doi: 10.1016/j.cub.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Gan X, Stegle O, Behr J, et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature. 2011;477:419–423. doi: 10.1038/nature10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg R, Jain M. RNA-Seq for transcriptome analysis in non-model plants. Methods in Molecular Biology. 2013;1069:43–58. doi: 10.1007/978-1-62703-613-9_4. [DOI] [PubMed] [Google Scholar]
- Garg R, Patel RK, Tyagi AK, Jain M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Research. 2011;18:53–63. doi: 10.1093/dnares/dsq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert GA, Knight JD, Vance CP, Allan DL. Proteoid root development of phosphorus deficient lupin is mimicked by auxin and phosphonate. Annals of Botany. 2000;85:921–928. [Google Scholar]
- Gilbert N. Environment: the disappearing nutrient. Nature. 2009;461:716–718. doi: 10.1038/461716a. [DOI] [PubMed] [Google Scholar]
- Gill N, Findley S, Walling JG, et al. Molecular and chromosomal evidence for allopolyploidy in soybean. Plant Physiology. 2009;151:1167–1174. doi: 10.1104/pp.109.137935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham PH, Vance CP. Legumes: importance and constraints to greater use. Plant Physiology. 2003;131:872–877. doi: 10.1104/pp.017004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduch M, Ganapathy A, Stein JW, Thelen JJ. A systematic proteomic study of seed filling in soybean. Establishment of high-resolution two-dimensional reference maps, expression profiles, and an interactive proteome database. Plant Physiology. 2005;137:1397–1419. doi: 10.1104/pp.104.056614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduch M, Hearne LB, Miernyk JA, et al. Systems analysis of seed filling in Arabidopsis: using general lineral modeling to assess concordance of transcript and protein expression. Plant Physiology. 2010;152:2078–2087. doi: 10.1104/pp.109.152413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JP, Broadley MR, Bowen HC, Spracklen WP, Hayden RM, White PJ. Gene expression changes in phosphorus deficient potato (Solanum tuberosum L.) leaves and the potential for diagnostic gene expression markers. PLoS One. 2011;6:e24606. doi: 10.1371/journal.pone.0024606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Kang Y., I. Torres-Jerez FC, et al. Genome-wide SNP discovery in tetraploid alfalfa using 454 sequencing and high resolution melting analysis. BMC Genomics. 2011;12:1–11. doi: 10.1186/1471-2164-12-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez G, Ramirez M, Valdes-Lopez O, et al. Phosphorus stress in common bean: root transcript and metabolic responses. Plant Physiology. 2007;144:752–767. doi: 10.1104/pp.107.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh L-C, Lin S-I, Shih ACC, et al. Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiology. 2009;151:2120–2132. doi: 10.1104/pp.109.147280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Feng Q, Qian Q, et al. High-throughput genotyping by whole-genome resequencing. Genome Research. 2009;19:1068–1076. doi: 10.1101/gr.089516.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wei X, Sang T, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Genetics. 2010;42:961–967. doi: 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- Huang X, Lu T, Han B. Resequencing rice genomes: an emerging new era of rice genomics. Trends in Genetics. 2013;29:225–232. doi: 10.1016/j.tig.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Hyten DL, Song Q, Fickus EW, et al. High-throughput SNP discovery and assay development in common bean. BMC Genomics. 2010;11:475–485. doi: 10.1186/1471-2164-11-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Gao X, Liao L, Harberd NP, Fu X. Phosphate starvation, root architecture and anthocyanin accumulation responses are modulated by the gibberellin–DELLA signaling pathway in Arabidopsis. Plant Physiology. 2007;145:1460–1470. doi: 10.1104/pp.107.103788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Zhao H, Ren R, et al. Genome wide genetic changes during modern breeding of maize. Nature Genetics. 2012;44:812–815. doi: 10.1038/ng.2312. [DOI] [PubMed] [Google Scholar]
- Johnson JF, Allan DL, Vance CP. Phosphorus stress-induced proteoid roots show altered metabolism in Lupinus albus. Plant Physiology. 1994;104:657–665. doi: 10.1104/pp.104.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J, Allan D, Vance CP, Weiblen G. Root carbon dioxide fixation by phosphorus-deficient Lupinus albus (contribution to organic acid exudation by proteoid roots) Plant Physiology. 1996;112:19–30. doi: 10.1104/pp.112.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including stress-induced miRNA. Molecular Cell. 2004;14:787–799. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- Joshi T, Valliyodan B, Wu J-H, Lee S-H, Xu D, Nguyen HT. Genomic differences between cultivated soybean, G. max and its wild relative G. soja. BMC Genomics. 2013;14(Suppl):55. doi: 10.1186/1471-2164-14-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalavacharia V, Liu Z, Meyers BC, Thimmapuram J, Melmaiee K. Identification and analysis of common bean (Phaseolus vulgaris L.) transcriptomes by massively parallel pyrosequencing. BMC Plant Biology. 2011;11:135–107. doi: 10.1186/1471-2229-11-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Cogan NO, Pembleton LW, et al. Transcriptome sequencing of lentil based on second-generation technology permits large-scale unigene assembly and SSR marker discovery. BMC Genomics. 2011;12:265. doi: 10.1186/1471-2164-12-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Lee S, Van K, et al. Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proceedings of the National Academy of Sciences, USA; 2010. pp. 22032–22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krusell L, Krause K, Ott T, et al. The sulfate transporter SST1 is crucial for symbiotic nitrogen fixation in Lotus japonicus root nodules. The Plant Cell, 2005;17:1625–1636. doi: 10.1105/tpc.104.030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Li R, Xu X, et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nature Genetics. 2010;42:1027–1031. doi: 10.1038/ng.684. [DOI] [PubMed] [Google Scholar]
- Lam H-M, Xu X, Lui X, et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nature Genetics. 2010;42:1053–1059. doi: 10.1038/ng.715. [DOI] [PubMed] [Google Scholar]
- Lambers H, Raven JA, Shaver GR, Smith SE. Plant nutrient-acquisition strategies change with soil age. Trends in Ecology and Evolution. 2008;23:95–103. doi: 10.1016/j.tree.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Le BH, Wagmaister JA, Bui AQ, Harada JJ, Goldberg RB. Using genomics to study legume seed development. Plant Physiology. 2007;144:562–574. doi: 10.1104/pp.107.100362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le BH, Cheng C, Bui AQ, et al. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proceedings of the National Academy of Sciences, USA; 2010. pp. 8063–8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelandais-Briere C, Naya L, Sallet E, et al. Genome-wide Medicago truncatula small RNA analysis revealed novel microRNAs and isoforms differentially regulated in roots and nodules. The Plant Cell. 2009;21:2780–2796. doi: 10.1105/tpc.109.068130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshchiner I, Alexa K, Kelsey P, et al. Mutation mapping and identification by whole-genome sequencing. Genome Research. 2012;22:1541–1548. doi: 10.1101/gr.135541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Peng Z, Yang X, et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nature Genetics. 2012;45:43–50. doi: 10.1038/ng.2484. [DOI] [PubMed] [Google Scholar]
- Li HC, Chuang K, Henderson JT, et al. PICKLE acts during germination to repress expression of embryonic traits. The Plant Journal. 2005;44:1010–1022. doi: 10.1111/j.1365-313X.2005.02602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Liu C, Lian X. Gene expression profiles in rice roots under low phosphorus stress. Plant Molecular Biology. 2010;72:423–432. doi: 10.1007/s11103-009-9580-0. [DOI] [PubMed] [Google Scholar]
- Li X, Mo X, Shou H, Wu P. Cytokinin-mediated cell cycling arrest of pericycle founder cells in lateral root initiation of Arabidopsis. Plant and Cell Physiology. 2006;47:1112–1123. doi: 10.1093/pcp/pcj082. [DOI] [PubMed] [Google Scholar]
- Libault M, Stacey G. Evolution of FW2.2-like (FWL) and PLAC8 genes in eukaryotes. Plant Signaling and Behavior. 2010;5:1226–1228. doi: 10.4161/psb.5.10.12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libault M, Farmer A, Joshi T, et al. An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. The Plant Journal. 2010;63:86–99. doi: 10.1111/j.1365-313X.2010.04222.x. [DOI] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti-Filippini J, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CM, Muchhal US, Uthappa M, Kononowicz AK, Raghothama KG. Tomato phosphate transporter genes are differentially regulated in plant tissues by phosphorus. Plant Physiology. 1998;116:91–99. doi: 10.1104/pp.116.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo S-F, Yang S-Y, Chen K-T, et al. A novel class of gibberellin 2-oxidases control semidwarfism, tillering, and root development in rice. The Plant Cell. 2008;20:2603–2618. doi: 10.1105/tpc.108.060913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bucio J, Hernandez-Abreu E, Sanchez-Calderon L, Nieto-Jacobo MF, Simpson J, Herrera-Estrella L. Phosphate availability alters architecture and causes changes in hormone sensitivity in the Arabidopsis root system. Plant Physiology. 2002;129:244–256. doi: 10.1104/pp.010934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Baskin TI, Brown KM, Lynch JP. Regulation of root elongation under phosphorus stress involves changes in ethylene responsiveness. Plant Physiology. 2003;131:1381–1390. doi: 10.1104/pp.012161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matas AJ, Yeats TH, Buda GJ, et al. Tissue and cell-type specific transcriptome profiling of expanding tomato fruit provides insights into metabolic and regulatory specialization and cuticle formation. The Plant Cell. 2011;23:3893–3910. doi: 10.1105/tpc.111.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen MD, Kreshovich S, Villeda HS, et al. Genetic properties of the maize nested association mapping population. Science. 2009;7:737–740. doi: 10.1126/science.1174320. [DOI] [PubMed] [Google Scholar]
- Meyer K, Stecca KL, Ewell-Hicks K, Allen SM, Everard JD. Oil and protein accumulation in developing seeds is influenced by the expression of a cytosolic pyrophosphatase in Arabidopsis. Plant Physiology. 2012;159:1221–1234. doi: 10.1104/pp.112.198309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misson J, Raghothama K, Jain A, et al. A genome-wide transcriptional analysis using Arabidopsis thaliana Affymetrix gene chips determined plant responses to phosphate deprivation. Proceedings of the National Academy of Sciences, USA; 2005. pp. 11934–11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida K, Shinozaki K. Advances in omics and bioinformatics tools for systems analyses of plant functions. Plant and Cell Physiology. 2011;52:2017–2038. doi: 10.1093/pcp/pcr153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcuende R, Bari R, Gibon Y, et al. Genome-wide reprogramming of metabolism and regulatory networks of Arabidopsis in response to phosphorus. Plant, Cell and Environment. 2007;30:85–112. doi: 10.1111/j.1365-3040.2006.01608.x. [DOI] [PubMed] [Google Scholar]
- Mosquna A, Katz A, Shochat S, Grafi G, Ohad N. Interaction of FIE, a polycomb protein with pRb: a possible mechanism regulating endosperm development. Molecular Genetics and Genomics. 2004;271:651–657. doi: 10.1007/s00438-004-1024-6. [DOI] [PubMed] [Google Scholar]
- Mudge J, Cannon SB, Kalo P, et al. Highly syntenic regions in the genomes of soybean, Medicago truncatula, and Arabidopsis thaliana. BMC Plant Biology. 2005;5:15. doi: 10.1186/1471-2229-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan VK, Smith AP. Ethylene's role in phosphate starvation signaling: more than just a root growth regulator. Plant and Cell Physiology. 2012;52:277–286. doi: 10.1093/pcp/pcr186. [DOI] [PubMed] [Google Scholar]
- Neumann G, Romheld V. Root excretion of carboxylic acids and protons in phosphorus-deficient plants. Plant and Soil. 1999;211:121–130. [Google Scholar]
- Nichols DM, Glover KD, Carlson SR, Specht E, Diers BW. Fine mapping of a seed protein QTL on soybean linkage group I and its correlated effects on agronomic traits. Crop Science. 2006;46:834–839. [Google Scholar]
- Niu YF, Chai RS, Jin GL, Wang H, Tang CX, Zhang YS. Responses of root architecture development to low phosphorus availability: a review. Annals of Botany. 2013;112:391–408. doi: 10.1093/aob/mcs285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlrogge JB, Kuo T-M. Control of lipid synthesis during soybean seed development: enzymic and immunochemical assay of acyl carrier protein. Plant Physiology. 1984;74:622–625. doi: 10.1104/pp.74.3.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke JA, Iniguez LP, Bucciarelli B, et al. A re-sequencing based assessment of genomic heterogeneity and fast neutron-induced deletions in a common bean cultivar. Frontiers in Plant Science. 2013a;4:210. doi: 10.3389/fpls.2013.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke JA, Yang SS, Miller SS, et al. An RNA-Seq transcriptome analysis of orthophosphate-deficient white lupin reveals novel insights into phosphorus acclimation in plants. Plant Physiology. 2013b;161:705–724. doi: 10.1104/pp.112.209254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Clark RM, Lanz C, Warthmann N, Weigel D. Sequencing of natural strains of Arabidopis thaliana with short reads. Genome Research. 2008;18:2024–2033. doi: 10.1101/gr.080200.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nature Reviews Genetics. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peaez P, Trejo MS, Iniguez LP, et al. Identification and characterization of microRNAs in Phaseolus vulgaris by high-throughput sequencing. BMC Genomics. 2012;13:83. doi: 10.1186/1471-2164-13-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer GA, King KE, Severin AJ, et al. Identification of candidate genes underlying an iron efficiency quantitative trait locus in soybean. Plant Physiology. 2012;158:1745–1754. doi: 10.1104/pp.111.189860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaxon WC, Tran HT. Metabolic adaptations of phosphate-starved plants. Plant Physiology. 2011;156:1006–1015. doi: 10.1104/pp.111.175281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland JA, Rife TW. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome. 2012;5:92–102. [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nature Biotechnology. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio V, Bustos R, Irigoyen ML, Cardona-Lopez X, Rojas-Triana M, Paz-Ares J. Plant hormones and nutrient signaling. Plant Molecular Biology. 2009;69:361–373. doi: 10.1007/s11103-008-9380-y. [DOI] [PubMed] [Google Scholar]
- Sato S, Nakamura Y, Kaneko T, et al. Genome structure of the legume, Lotus japonicus. DNA Research. 2008;15:227–239. doi: 10.1093/dnares/dsn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlueter JA, Dixon P, Granger C, et al. Mining EST databases to resolve evolutionary events in major crop species. Genome. 2004;47:868–876. doi: 10.1139/g04-047. [DOI] [PubMed] [Google Scholar]
- Schmitz RJ, Zhang X. High-throughput approaches for studying plant epigenomics. Current Opinion in Plant Biology. 2011;14:130–136. doi: 10.1016/j.pbi.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz J, Cannon SB, Schlueter J, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;14:178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- Schneeberger K, Weigel D. Fast-forward genetics enabled by new sequencing technologies. Trends in Plant Science. 2011;16:282–288. doi: 10.1016/j.tplants.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Seboldt AM, Shoemaker RC, Diers BW. Analysis of a quantitative trait locus allele from wild soybean that increases seed protein concentration in soybean. Crop Science. 2000;40:1438–1444. [Google Scholar]
- Severin AJ, Peiffer GA, Xu WW, et al. An integrative approach to genomic introgression mapping. Plant Physiology. 2010a;154:3–12. doi: 10.1104/pp.110.158949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severin AJ, Woody JL, Bolon Y-T, et al. RNA-Seq atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biology. 2010b;10:160. doi: 10.1186/1471-2229-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Li R, Zhao Z, et al. QTL for yield traits and their association with functional genes in response to phosphorus deficiency in Brassica napus. PLoS One. 2013;8:e54559. doi: 10.1371/journal.pone.0054559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon SA, Meyers BC, Sherrier DJ. MicroRNAs in the rhizobia legume symbiosis. Plant Physiology. 2009;151:1002–1008. doi: 10.1104/pp.109.144345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonah H, Bastien M, Iquira E, et al. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS One. 2013;8:e54603. doi: 10.1371/journal.pone.0054603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song QX, Liu YF, Hu XY, et al. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biology. 2011;10:5. doi: 10.1186/1471-2229-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar R. MicroRNAs with macro-effects on plant stress responses. Seminars in Cell and Developmental Biology. 2010;21:805–811. doi: 10.1016/j.semcdb.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Sunkar R, Zhu J-K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. The Plant Cell. 2004;16:2001–2019. doi: 10.1105/tpc.104.022830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svistoonoff S, Creff A, Reymond M, et al. Root tip contact with low-phosphate media reprograms plant root architecture. Nature Genetics. 2007;39:792–796. doi: 10.1038/ng2041. [DOI] [PubMed] [Google Scholar]
- Thibaud M-C, Arrighi J-F, Bayle V, et al. Dissection of local and systemic transcriptional responses to phosphate starvation in Arabidopsis. The Plant Journal. 2010;64:775–789. doi: 10.1111/j.1365-313X.2010.04375.x. [DOI] [PubMed] [Google Scholar]
- Thimm O, Blasing O, Gibon Y, et al. MAPMAN: a user driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. The Plant Journal. 2004;37:914–939. doi: 10.1111/j.1365-313x.2004.02016.x. [DOI] [PubMed] [Google Scholar]
- Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrey JG. Root hormones and plant growth. Annual Review of Plant Physiology. 1976;27:435–459. [Google Scholar]
- Toth K, Stratil TF, Madsen EB, et al. Functional domain analysis of the remorin protein LjSYMREM1 in Lotus japonicus. PLoS One. 2012;7:e30817. doi: 10.1371/journal.pone.0030817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner M, Yu O, Subramanian S. Genome organization and characteristics of soybean microRNAs. BMC Genomics. 2012;13:169. doi: 10.1186/1471-2164-13-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhde-Stone C, Zinn KE, Ramirez-Yanez M, Li A, Vance CP, Allan DL. Nylon filter arrays reveal differential gene expression in proteoid roots of white lupin in response to phosphorus deficiency. Plant Physiology. 2003;131:1064–1079. doi: 10.1104/pp.102.016881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes-Lopez O, Yang SS, Aparicio-Fabre R, et al. MicroRNA expression profile in common bean (Phaseolus vulgaris) under nutrient deficiency stresses and manganese toxicity. New Phytologist. 2010;187:805–818. doi: 10.1111/j.1469-8137.2010.03320.x. [DOI] [PubMed] [Google Scholar]
- Vance CP. Symbiotic nitrogen fixation and phosphorus acquisition. Plant nutrition in a world of declining renewable resources. Plant Physiology. 2001;127:390–397. [PMC free article] [PubMed] [Google Scholar]
- Vance CP, Heichel GH. Nitrate assimilation during vegetative regrowth of alfalfa. Plant Physiology. 1981;68:1052–1057. doi: 10.1104/pp.68.5.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance CP, Uhde-Stone C, Allan DLP. Phosphorus acquisition and use: critical adaptations by plants for securing a nonrenewable resource. New Phytologist, 2003;157:423–447. doi: 10.1046/j.1469-8137.2003.00695.x. [DOI] [PubMed] [Google Scholar]
- Varala K, Swaminathan K, Li Y, Hudson ME. Rapid genotyping of soybean cultivars using high throughput sequencing. PLoS One. 2011;6:e24811. doi: 10.1371/journal.pone.0024811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney RK, Song C, Saxena RK, et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nature Biotechnology, 2013;31:240–246. doi: 10.1038/nbt.2491. [DOI] [PubMed] [Google Scholar]
- Varshney RK, Chen W, Li Y, et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol. 2012;30:83–9. doi: 10.1038/nbt.2022. [DOI] [PubMed] [Google Scholar]
- Verdier J, Kaker K., Gallardo K., et al. Gene expression profiling of M. truncatula transcription factors identifies putative regulators of grain legume seed filling. Plant Molecular Biology. 2008;67:567–580. doi: 10.1007/s11103-008-9320-x. [DOI] [PubMed] [Google Scholar]
- Verdier J, Thompson RD. Transcriptional regulation of storage protein synthesis during dicotyledon seed filling. Plant and Cell Physiology. 2008;49:1263–1271. doi: 10.1093/pcp/pcn116. [DOI] [PubMed] [Google Scholar]
- Wang F, Perry SE. Identification of direct targets of FUSCA3, a key regulator of Arabidopsis seed development. Plant Physiology. 2013;161:1251–1264. doi: 10.1104/pp.112.212282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Mullighan CG, Easton J, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nature Methods. 2011;8:652–654. doi: 10.1038/nmeth.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li P, Brutnell TP. Exploring plant transcriptomes using ultra high-throughput sequencing. Briefings in Functional Genomics. 2010;9:118–128. doi: 10.1093/bfgp/elp057. [DOI] [PubMed] [Google Scholar]
- Wang Y-H, Garin DF, Kochian LV. Rapid induction of regulatory and transporter genes in response to phosphorus, potassium, and iron deficiencies in tomato roots. Evidence for cross talk and root/rhizosphere-mediated signals. Plant Physiology. 2002;130:1261–1370. doi: 10.1104/pp.008854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Fang B, Chen J, et al. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas) BMC Genomics. 2010a;11:726. doi: 10.1186/1471-2164-11-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Libault M, Joshi T, et al. SoyDB: a knowledge database of soybean transcription factors. BMC Plant Biology. 2010b;10:14. doi: 10.1186/1471-2229-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber H, Borisjuk L, Wobus U. Molecular physiology of legume seed development. Annual Review of Plant Biology. 2005;56:253–279. doi: 10.1146/annurev.arplant.56.032604.144201. [DOI] [PubMed] [Google Scholar]
- Weigel D, Colot V. Epialleles in plant evolution. Genome Biology. 2012;13:249. doi: 10.1186/gb-2012-13-10-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner T, Nehnevajova E, Kollmer I, et al. Root-specific reduction of cytokinin causes enhanced root growth, drought tolerance, and leaf mineral enrichment in Arabidopsis and tobacco. The Plant Cell. 2010;22:3905–3920. doi: 10.1105/tpc.109.072694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox JR. World distribution and trade of soybean. In: Boerma HR, Specht JE, editors. Soybean: improvement, production, and uses. Madison, WI: American Society of Agronomy; 2004. pp. 1–14. [Google Scholar]
- Xia Z, Xu H, Zhai J, et al. RNA-Seq analysis and de novo transcriptome assembly of Hevea brasiliensis. Plant Molecular Biology. 2011;77:299–308. doi: 10.1007/s11103-011-9811-z. [DOI] [PubMed] [Google Scholar]
- Yang SS, Tu JZ, Foo C, et al. Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genomics. 2011;12:199. doi: 10.1186/1471-2164-12-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ND, Debelle F, Oldroyd GE, et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011;480:520–524. doi: 10.1038/nature10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenoni S, Ferraini A, Giacomelli E, et al. Characterization of transcriptonal complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiology. 2010;152:1787–1795. doi: 10.1104/pp.109.149716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Guo G, Hu X, et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Research. 2010;20:646–654. doi: 10.1101/gr.100677.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liang S, Duan J, et al. De novo assembly and characterization of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.) BMC Genomics. 2012;12:13–90. doi: 10.1186/1471-2164-13-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YL, Lynch JP, Brown KM. Ethylene and phosphorus availability have interacting yet distinct effects on root hair development. Journal of Experimental Botany. 2003;54:2351–2361. doi: 10.1093/jxb/erg250. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Zeng H, Dong C, Yin X, Shen Q, Yang Z. MicroRNA expression profiles associated with phosphorus deficiency in white lupin (Lupinus albus L.) Plant Science. 2009;178:23–29. [Google Scholar]
- Zuryn S, Le Gras S, Jamet K, Jarriault S. A strategy for direct mapping and identification of mutations by whole-genome sequencing. Genetics. 2010;186:427–430. doi: 10.1534/genetics.110.119230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.