

Abstract
Opioids remain the mainstay of severe pain management in patients with cancer. The hallmark of pain management is individualization of therapy. Although almost all clinically used drugs act through mu opioid receptors, they display subtle but important differences pharmacologically. Furthermore, not all patients respond equally well to all drugs. Evidence suggests that these variable responses among patients have a biologic basis and are likely to involve both biased agonism and the many mu opioid receptor subtypes that have been cloned.
INTRODUCTION
Cancer pain remains a difficult problem, and its management is an art. The hallmark of pain management in patients with cancer is the need to individualize therapy because of the wide variability of responses among patients to different pain medications and treatments.1–3 One patient may obtain excellent relief with one analgesic, but a different patient may get better results with another. Doses of a specific opioid needed to manage pain can also vary markedly among patients. Although these clinical observations are largely anecdotal, controlled studies find that redheads, who contain a mutated melanocortin 1 receptor, show increased pain tolerance and increased analgesic responses to opioids. Similar results are also seen in mice with a nonfunctional melanocortin 1 receptor.4 Thus, genetics can influence opioid sensitivity, a conclusion that has been validated in other animal studies. The analgesic activity of morphine varies markedly among a series of mouse strains (Fig 1A), and different strains show dramatic differences in the relative potency of opioids. This is well illustrated by the CXBK mouse strain that is insensitive to morphine while maintaining a normal sensitivity toward other mu opioid analgesics (Fig 1B).6–8 Thus, the need to individualize therapy has a biologic basis.9,10
Fig 1.
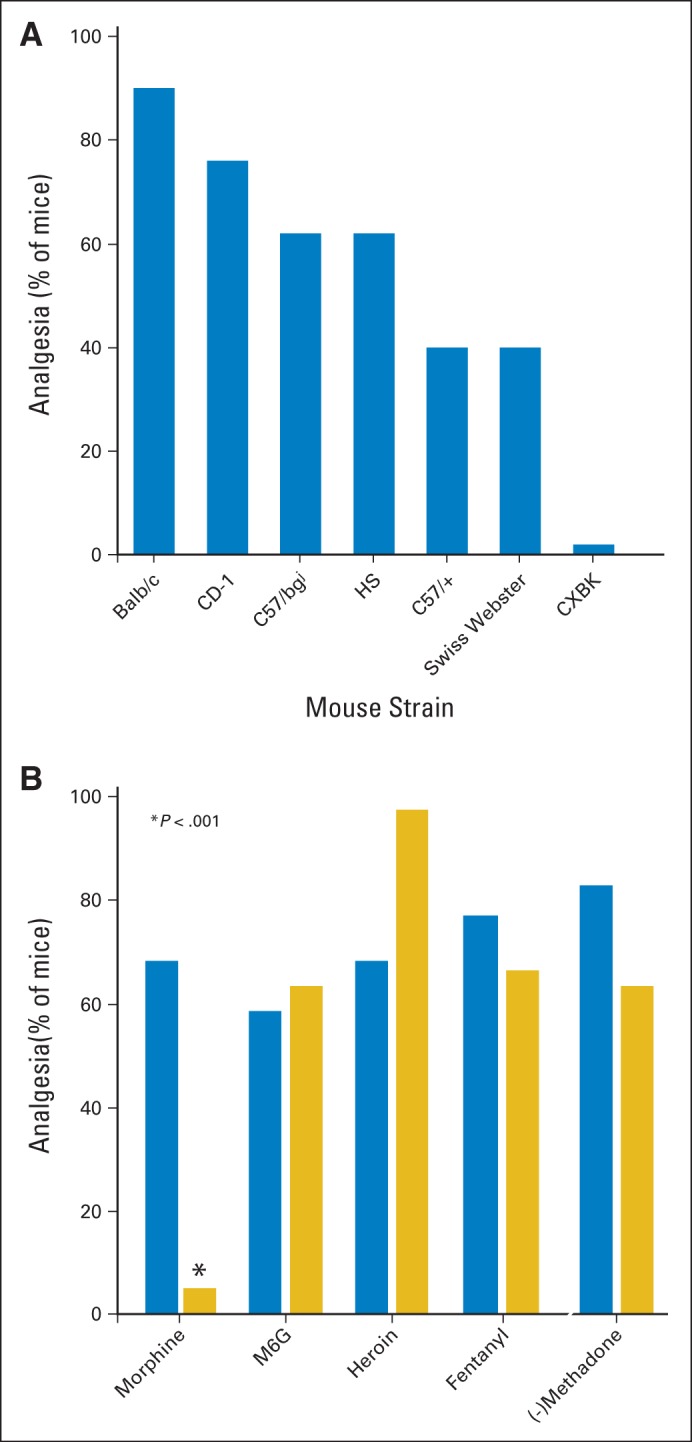
Genetic influence on opioid analgesia. (A) Groups of mice of the indicated strains were tested with morphine (5 mg/kg subcutaneously) in the radiant heat tailflick assay at peak effect. (B) Groups of CD-1 and CXBK mice were tested with equianalgesic doses of the indicated opioid. M6G, morphine-6β-glucuronide. Data adapted.5
Opioids are unique among analgesics in many ways, but what makes them special is their ability to selectively have an impact on the pain of nociceptive stimuli without impairing more objective sensations, such as light touch, temperature, or position sense. However, suffering includes non-nociceptive factors, such as depression and anxiety. To patients, these components may not be separable from nociception itself. Thus, it is important for the physician to distinguish among these various components and recognize that opioids help only the nociceptive component of the suffering, and other therapies may be needed for the other aspects of suffering.
The opioids act through the activation of a pain modulating system comprising the endogenous opioid peptides (ie, enkephalins, dynorphins, and β-endorphin) and their receptors.10 The need for a pain modulating system is understandable. Pain serves a valuable role, alerting us to injury. Yet, there are circumstances when pain is detrimental, and the presence of this system has survival advantages. In a study of soldiers during World War II, Beecher11 found that the morphine requirements for wounded soldiers were lower than those for patients undergoing elective surgery back in the United States, implying that the stress of combat was able to turn down the perception of pain. We now know that this involves the activation of endogenous opioid systems.
The many descriptors for pain clearly illustrate the complexity of nociceptive sensations: sharp, dull, shooting, burning, aching, cramping, and so on. Although pain has traditionally been classified as somatic, visceral, or neuropathic, in the clinical setting, it is most often a mixture of pain types and may involve multiple mechanisms, perhaps explaining the utility of combinations of different classes of drugs.1,3,10,12,13 For example, pain from bone metastases can involve bradykinin, endothelins, prostaglandins, proteases, and tyrosine kinase activators.14 The nonsteroidal anti-inflammatory drugs are typically effective for inflammatory pain, and the antidepressant/anticonvulsant drugs are more commonly used for neuropathic pain. Nonsteroidal anti-inflammatory drugs are not as widely used in oncology as they are in the general population because of bleeding issues that can be more problematic in patients with cancer as a result of their treatments and/or their disease. Opioids are effective against all pain but not with the same potency/efficacy. Although they can be used for neuropathic pain, higher doses are often required. From a practical perspective, opioids have no ceiling effect on pain relief, a clear distinction from the other classes of drugs that often have limits on the intensity and nature of pain they can treat. However, because opioid doses are escalated for pain relief, adverse effects can become increasingly problematic and can prevent adequate relief. Medically, the problems of sedation, constipation, respiratory depression, tolerance, and physical dependence need to be continually monitored. It is also becoming important to recognize the endocrine effects of opiates, since recent work has emphasized the lowering of testosterone levels by opioids.15 Methadone may also have an impact on glucose levels, particularly after intravenous administration,16–18 and morphine's effects on prolactin and growth hormone have been known for many years.19 Opiate use also has societal issues, particularly diversion, abuse, and addiction, which require additional care in their use.
All patients chronically on opioids will develop physical dependence, which is a physiological compensation for the chronic exposure to the drug. As long as drug use is maintained, it is rarely an issue. However, if patients stop taking the drug or if they are given an antagonist or a mixed agonist-antagonist such as pentazocine, they may demonstrate the classical signs of opioid withdrawal, ranging from mild dysphoria to going cold turkey, depending on the dose and duration of prior drug use. This is not the same as addiction, which implies drug-seeking behavior in the absence of a medical indication. All patients will develop physical dependence with chronic opioid dosing, but few patients with cancer will become addicted. Nevertheless, all patients being considered for chronic opioid therapy should be carefully screened and monitored for indications of misuse.
MU OPIOIDS
There are three major classes of opioids defined by their receptors: mu, delta, and kappa. Mu receptors are selective for morphine, and most traditional opiate analgesics act through these sites. Delta and kappa1 receptors are selective for the enkephalins and dynorphin, respectively. Although highly selective delta and kappa1 drugs have been generated and extensively studied in preclinical models, none are available clinically, where almost all the opiates used are mu. Although there are some mixed agonists-antagonists with both mu and kappa activity, such as pentazocine, their antagonist actions against mu receptors makes them difficult to use in a patient already being given an opioid since they may precipitate withdrawal.
Opiates have a wide range of structures despite their common affinity for mu opiate receptors. Morphine and codeine are natural products of opium, as is thebaine, a precursor used in the synthesis of several drugs, including oxycodone, naloxone, naltrexone, and buprenorphine. Methadone, meperidine, and the fentanyl series are all fully synthetic and are not dependent on opium. Although they are all potent analgesics, not all patients respond equally well to each mu opioid, presumably in large part because of genetic factors as shown in preclinical studies (Fig 1B). In addition to variations in the relative analgesic potency among patients, adverse effects also may differ. There is no way to predict the optimal drug for a specific patient. The choice is empirical, and several drugs may need to be tried before finding the most appropriate one.
Structurally, the antagonists naloxone and naltexone are similar to oxymorphone, differing only in the replacement of the N-methyl group with an N-allyl 1 (naloxone) or N-methylcyclopropyl group (naltrexone). Naloxone reverses opioid actions. Its onset of action following intravenous administration is rapid, often within less than 1 minute. However, care must be taken when using it to reverse the longer-acting analgesics since naloxone has a relatively short duration of action that requires repeated dosing to prevent the reappearance of the overdose as the naloxone effects wear off. Both naloxone and naltrexone are subject to a large first-pass effect, making their oral use difficult. However, this feature of their metabolism has been used to advantage by including a low dose with oral preparations of analgesics to minimize abuse potential. When taken orally, the antagonists have little effect because of their hepatic inactivation. However, if they are injected, the antagonist dose is sufficient to precipitate withdrawal in people who are dependent.
OPIOID ANALGESIA
Opiates act in several places within the neuraxis.20–23 Supraspinally, several structures play a role in morphine analgesia, including the locus ceruleus, nucleus raphe magnus, periaqueductal gray, medial thalamus, and limbic structures. At the spinal level, opioid action involves the dorsal horn where the receptors are both pre- and postsynaptic. The presynaptic ones are associated with the nerves entering the dorsal horn from the dorsal root ganglia and from descending pathways. The dorsal root ganglia neurons also extend into the periphery where the presynaptic receptors on the nerve modulate peripheral opioid analgesia.
Given systemically, opioids act through all sites simultaneously with the supraspinal systems being the most sensitive, which explains why peripherally restricted antagonists such as methylnaltrexone can be given to ameliorate constipation without having a major effect on analgesic activity. Their regional interactions are important in understanding opioid action. Studies by Yeung and Rudy24 clearly show profound synergy when morphine is administered both spinally and supraspinally, and microinjection studies demonstrate synergy among the brainstem nuclei.25 Studies also reveal synergy between spinal and peripheral sites and between spinal and systemic drug (Table 1).26,27 Small doses of spinal morphine dramatically potentiate the activity of a systemic drug. This is relevant clinically. Intrathecal and epidural opioids are widely used. Epidurally, the opioid diffuses into the intrathecal space to produce concentrations at the spinal cord far greater than would be seen with systemic administration. However, the epidural space has a dense network of veins (Batson's plexus) through which opioids and other drugs are absorbed, leading to significant systemic blood levels of drug.28 Indeed, the serum level of morphine (approximately 50 ng/mL) following epidural dosing (10 mg) is not very different from that seen after systemic administration of the same morphine dose (approximately 80 ng/mL). Thus, epidural morphine resembles the paradigm in which spinal morphine dramatically potentiates systemic drug, thus shifting the dose-response curve several-fold to the left.
Table 1.
Synergy Between Spinal and Systemic Morphine
| Drug and Method of Administration | Systemic Morphine ED50 | Shift |
|---|---|---|
| Systemic morphine alone | 3.1 mg/kg subcutaneously | |
| Systemic morphine + 25 ng intrathecal morphine* | 0.5 mg/kg subcutaneously | More than six-fold |
NOTE. The analgesic ED50 in the radiant heat tailflick assay of systemic morphine given subcutaneously was determined in groups of CD-1 mice alone and following an intrathecal injection of morphine (50 ng), a dose less than 10% of morphine's intrathecal ED50. Higher intrathecal morphine doses shifted the systemic ED50 as much as 50-fold. Isobolographic analysis of the doses reveal synergy between the intrathecal and the systemic routes. Data adapted.27
Morphine intrathecal ED50 is 305 ng.
INCOMPLETE CROSS-TOLERANCE AND OPIOID ROTATION
All opioids produce tolerance with chronic dosing, and many patients require dose escalation to accommodate their decreased sensitivity toward the drugs. It is important to note that this is not addiction, and physicians must assess patients carefully. One cannot simply assume that the request for increasing doses of drug represents drug-seeking behavior. Once the pain is controlled, highly tolerant patients with cancer often can be maintained on a fixed opioid dose for long periods of time, suggesting that tolerance is not a continually progressive process. Rather, it is possible to achieve an equilibrium between tolerance and analgesic activity. Indeed, the need to escalate drug dose in patients who have been stable on their opioid dose for prolonged periods of time may be an early indication of progression of disease.3,29
Tolerance to all opioid actions does not develop at the same rate, and adverse effects become increasingly problematic with dose escalation as the therapeutic index decreases. In this situation, many physicians will switch the patient to a different opioid, an approach termed “opioid rotation.” In opioid rotation, a patient is switched to a second drug because of problematic adverse effects or inadequate pain relief with the first drug.30,31 The effectiveness of the second drug is most likely a result of incomplete cross-tolerance in which the degree of tolerance to the second drug is less than that for the first. Although the phenomenon has long been known clinically, we have only recently gained insights into the mechanisms for incomplete cross-tolerance, which likely involves differing activities at subtypes of mu opioid receptors or biased agonism. The choice of the second drug is empirical, and more than one drug may need to be tried. There are several tables showing the relative potency of opioids. It is important to note that these are equianalgesic tables, not conversion tables. The values in these tables were determined in opiod-naive patients, and the ratios in tolerant patients can vary dramatically. Thus, rotation must be carefully carried out. In practice, the dose of the second drug is typically calculated by using the equianalgesic chart and then reduced by 50% to 75% to obtain the conversion dose to avoid overdose. The emphasis is on safety. Thus, it is not uncommon to find that after the reduction, the dose of the second drug must be carefully titrated upward to achieve pain relief. Morphine-to-methadone conversions are particularly difficult, and when making that conversion, the calculated dose should be decreased by 75%. Patients tolerant to morphine are not as tolerant to methadone, meaning that methadone will be far more potent in the morphine-tolerant patient than anticipated. Special care also must be taken with methadone and other long-acting drugs, since it may take days for a fixed dose of these drugs to achieve steady-state levels. Escalating too rapidly can present serious problems, even overdose, and should be done over many days.
MOLECULAR BIOLOGY AND PHARMACOLOGY OF MULTIPLE MU RECEPTOR SUBTYPES
Opioid pharmacology is unusual in that our clinical experience for a vast array of analgesics preceded the discovery of their receptors and their mechanisms of action. Indeed, clinical experience molded the direction of basic research which, in turn, has provided insights into why the clinically proven approaches used today are effective. The hallmark of pain management is individualization of care, taking into account the differing pharmacology of mu opioids and various sensitivity of patients to the drugs. In this era of evidence-based medicine, an understanding of the clinical approaches and why they are effective is important.
The opiate receptor was first demonstrated in 1973 in receptor-binding studies. The initial assumption of a single mu receptor soon was challenged by detailed ligand-binding studies and the actions of naloxonazine and naloxazone, antagonists that dissociated morphine analgesia from respiratory depression, constipation, and most signs of physical dependence.10,32–34 Evidence is accumulating that the complexity of mu opioid pharmacology results from a combination of the extensive splicing of the receptor and biased agonism.
The mu receptor is a member of the G-protein coupled receptor (GPCR) family. This is a broad family of more than 300 receptors that account for a large proportion of drugs currently used in medicine. Structurally, they all contain seven-transmembrane (7-TM) domains with an extracellular N-terminus and an intracellular C-terminus (Fig 2). Classically, once an agonist binds, the receptor activates a G-protein, which dissociates into its α and its β and γ subunits which, in turn, signal through downstream transduction pathways to modulate enzyme activity, such as adenylyl cyclase, or ion channels. However, GPCRs can also signal through alternative pathways, such as β-arrestin.35–39 What makes these alternative pathways important is that the relative activation of these pathways may differ from one drug to another, with one drug activating primarily one pathway while another drug activates a different pathway. Because the different pathways yield different pharmacologic responses, the pharmacologic profile of drugs can vary even if they label a single receptor.10,40 This is referred to as biased agonism.
Fig 2.

Schematic of the opioid receptors. The exon composition of the MOR-1 gene (Oprm1; top) and its splice patterns (bottom) are shown schematically. Intron distances are not shown to scale. The full-length variants contain seven-transmembrane (7TM) domains encoded by exons 1, 2, and 3, with 3′ splicing downstream of exon 3. The 6TM domain receptor variants lack exon 1 but contain exon 11. Exon 11 does not traverse the membrane and remains intracellular, resulting in the loss of the first transmembrane domain. The single transmembrane domain variants contain exon 1, with 3′ downstream splicing. aa, amino acid.
The mu opioid receptor MOR-1 has been cloned41–44 and crystallized, and its structure has been established.45 The TM domains form a circular structure with the opioid-binding pocket located deep within the TM region of the protein. Following the cloning of MOR-1, a vast array of splice variants were identified involving both 3′ and 5′ splicing, with similar patterns seen across a wide range of species, including humans, rats, and mice (Fig 2).10 Three major categories of variants have been identified. Most are classical full-length variants with all 7-TM domains in which 3′ splicing leads to changes in only the tip of the intracellular tail. Because the remainder of these full-length variants are the same, including all 7-TM domains, they have identical binding pockets. However, the 3′ splicing yields unique C-terminal amino acid sequences for each. These differences are important because of the concept of biased agonism. There is a G-protein coupled receptor kinase–induced phosphorylation “bar code” at the C-terminus of GPCRs that regulates β-arrestin functions.46 The 3′ splicing of MOR-1 leads to a wide range of potential phosphorylation sites in the C-tail, the region displaying the bar code, thereby offering the possibility of vastly differing levels of biased agonism among the variants.
A second set of variants are truncated, containing only 6-TM domains because of the absence of exon 1 and the first TM domain that it encodes (Fig 2). These truncated 6-TM variants are pharmacologically important and play a major role in the actions of several established opioid analgesics, providing a target for new agents with unique pharmacologic properties.
The last set comprises variants that contain only a single TM domain (Fig 2). Although these variants do not bind opioids directly, they help modulate opioid analgesia by increasing expression of the full-length 7-TM variants through a chaperone-like action.47
FUNCTIONAL CHARACTERIZATION OF THE MOR-1 SPLICE VARIANTS
The various MOR-1 knockout models have provided important insights into mu opioid action.10 Two models in particular have proven valuable. One MOR-1 knockout targeting exon 1 still expresses the 6-TM truncated variants.48 Conversely, an exon 11 knockout mouse lacks these 6-TM variants without appreciable changes in the levels of the full-length variants.49 These two knockout models reveal several different profiles of activity (Table 2). Morphine is the prototype of the group that is totally dependent on the full-length variants but unaffected by loss of the 6-TM variants. Disruption of exon 1 eliminates all the full-length variants and morphine activity. Buprenorphine is an example of the second group, which depends on both the full-length and the truncated 6TM variants. Loss of either set of variants lowers buprenorphine activity. Heroin and morphine-6β-glucuronide (M6G) also fall within this group. The potency of both heroin and M6G are slightly reduced in the exon 1 knockout, but higher doses still elicit a full analgesic response, unlike morphine which is totally inactive (Fig 3). Unlike morphine, the potency of heroin and M6G are significantly reduced in the exon 11 knockout mouse.
Table 2.
MOR-1 Knockout Models and Mu Opioid Analgesia
| Variable | Exon 1 KO | Exon 11 KO |
|---|---|---|
| Variant | ||
| Full length 7-TM | Lost | Retained |
| Truncated 6-TM | Retained | Lost |
| Truncated 1-TM | Lost | Retained |
| Analgesia | ||
| Morphine | Lost | Retained |
| Buprenorphine | Lost | Lost |
| IBNtxA | Retained | Lost |
NOTE. Two MOR-1 knockout (KO) models have proven valuable in the characterization of mu opioid analgesics. In one model, exon 1 is deleted without having an impact on the expression of the exon 11-associated six-transmembrane (6-TM) splice variants.48 The other knockout model has a disruption in exon 11, eliminating all the 6-TM variants without affecting the full-length variants.49
Abbreviation: IBNtxA, 3-iodobenzoyl-6β-naltrexamide.
Fig 3.

Differentiating morphine from heroin analgesia. Knockout (KO) mice that lacked exon 1 but still expressed exon 11 and the truncated six-transmembrane variants were tested in the radiant heat tailflick assay for analgesia and compared with wild-type controls. Drugs included morphine, heroin, and the morphine metabolite morphine-6β-glucuronide (M6G). Data adapted.47
The third group may be the most interesting. The experimental drug 3-iodobenzoyl-6β-naltrexamide (IBNtxA) provides a window into their possibilities.50 Unlike morphine, IBNtxA actions are unaffected by the loss of the full-length MOR-1 splice variants or delta or kappa1 receptors, but loss of the exon 11 variants eliminates its activity. IBNtxA is a potent analgesic with an unusual pharmacologic profile. Like traditional opioids, it is quite potent, with an activity potency 10-fold greater than that of morphine.50,51 However, IBNtxA is active in neuropathic/inflammatory pain models, which is unusual for opioids.10 This is particularly important in cancer pain since much of it is neuropathic in nature. Its adverse effect profile is intriguing. IBNtxA shows no respiratory depression, and it does not produce physical dependence with chronic administration. Its inhibition of GI transit, a measure of constipation, is quite limited and it displays no reward behavior in the conditioned place preference paradigm, raising the possibility that it may be possible to dissociate abuse liability from analgesia. Targeting these 6-TM variants may yield important new analgesics in the future.
In conclusion, opiates are effective in the management of cancer pain. However, their use is not always straightforward because of differences in their overall effectiveness and in the adverse effects among patients. Treatment needs to be individualized for each patient. The variability in response to the opioids among patients has not always been appreciated. Too often, clinicians will conclude that opioids are ineffective after trying a single agent or will misinterpret a request for additional medication from a patient who is not responding at doses normally effective for other patients. Many of these differences are biologically based, as shown by the preclinical studies, and we cannot predict which patient will respond best to which drug or dose. Recognizing the need to try different drugs and to escalate dose to effectiveness goes a long way toward improving patient care.
The molecular mechanisms of opioid action are complex, but they offer insights into why many of the approaches used in pain medicine are effective, such as trying more than one drug to enhance efficacy and minimize adverse effects and by using opioid rotation when faced with mounting problematic tolerance. The recognition of biased agonism helps explain how two drugs working through a single receptor can elicit different pharmacologic profiles. The identification of multiple splice variants expands these possibilities exponentially since each variant is subject to biased agonism. There have been few new opioids in recent years. The discovery of the truncated variants provides a new target for potent analgesics lacking many of the problematic adverse effects seen with current drugs. Several available opioids appear to work, in part, through this target, including buprenorphine, butorphanol, nalbuphine, and levorphanol, supporting the potential of this approach. The future holds much promise in the effective management of cancer pain.
Footnotes
Supported in part by Grants No. DA02615, DA06241, and DA07242 from the National Institute on Drug Abuse, National Institutes of Health (G.W.P.) and by Core Grant No. CA08748 from the National Cancer Institute.
Author's disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: Gavril W. Pasternak, QRxPharma (C), Sarentis Therapeutics (C), Purdue Pharma (C), Cubist Pharmaceuticals (C), Cytogel (C) Stock Ownership: None Honoraria: None Research Funding: None Expert Testimony: None Patents, Royalties, and Licenses: None Other Remuneration: None
REFERENCES
- 1.Payne R. Pathophysiology of cancer pain. In: McDonald N, Doyle D, Hanks G, editors. Oxford Textbook of Medicine. London, United Kingdom: Oxford University Press; 1997. [Google Scholar]
- 2.Payne R, Pasternak GW. Pain. In: Johnston MV, Macdonald RL, Young AB, editors. Principles of Drug Therapy in Neurology. Philadelphia, PA: F.A. Davis; 1992. pp. 268–301. [Google Scholar]
- 3.Foley KM. Controlling the pain of cancer. Sci Am. 1996;275:164–165. doi: 10.1038/scientificamerican0996-164. [DOI] [PubMed] [Google Scholar]
- 4.Mogil JS, Ritchie J, Smith SB, et al. Melanocortin-1 receptor gene variants affect pain and mu-opioid analgesia in mice and humans. J Med Genet. 2005;42:583–587. doi: 10.1136/jmg.2004.027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rossi GC, Brown GP, Leventhal L, et al. Novel receptor mechanisms for heroin and morphine-6 beta-glucuronide analgesia. Neurosci Lett. 1996;216:1–4. doi: 10.1016/0304-3940(96)12976-1. [DOI] [PubMed] [Google Scholar]
- 6.Chang A, Emmel DW, Rossi GC, et al. Methadone analgesia in morphine-insensitive CXBK mice. Eur J Pharmacol. 1998;351:189–191. doi: 10.1016/s0014-2999(98)00366-5. [DOI] [PubMed] [Google Scholar]
- 7.Neilan CL, King MA, Rossi G, et al. Differential sensitivities of mouse strains to morphine and [Dmt1]DALDA analgesia. Brain Res. 2003;974:254–257. doi: 10.1016/s0006-8993(03)02590-3. [DOI] [PubMed] [Google Scholar]
- 8.Baran A, Shuster L, Eleftheriou BE, et al. Opiate receptors in mice: Genetic differences. Life Sci. 1975;17:633–640. doi: 10.1016/0024-3205(75)90101-0. [DOI] [PubMed] [Google Scholar]
- 9.Pasternak GW. Incomplete cross tolerance and multiple mu opioid peptide receptors. Trends Pharmacol Sci. 2001;22:67–70. doi: 10.1016/s0165-6147(00)01616-3. [DOI] [PubMed] [Google Scholar]
- 10.Pasternak GW, Pan YX. Mu opioids and their receptors: Evolution of a concept. Pharmacol Rev. 2013;65:1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beecher HK. Pain in men wounded in battle. Ann Surg. 1946;123:96–105. [PMC free article] [PubMed] [Google Scholar]
- 12.Portenoy RK, Lesage P. Management of cancer pain. Lancet. 1999;353:1695–1700. doi: 10.1016/S0140-6736(99)01310-0. [DOI] [PubMed] [Google Scholar]
- 13.Reisine T, Pasternak GW. Opioid analgesics and antagonists. In: Hardman JG, Limbird LE, editors. Goodman & Gilman's: The Pharmacological Basis of Therapeutics (ed 9) New York, NY: McGraw-Hill; 1996. pp. 521–556. [Google Scholar]
- 14.Mantyh P. Bone cancer pain: Causes, consequences, and therapeutic opportunities. Pain. doi: 10.1016/j.pain.2013.07.044. [epub ahead of print on July 31, 2013] [DOI] [PubMed] [Google Scholar]
- 15.Elliott JA, Horton E, Fibuch EE. The endocrine effects of long-term oral opioid therapy: A case report and review of the literature. J Opioid Manag. 2011;7:145–154. doi: 10.5055/jom.2011.0057. [DOI] [PubMed] [Google Scholar]
- 16.Faskowitz AJ, Kramskiy VN, Pasternak GW. Methadone-induced hypoglycemia. Cell Mol Neurobiol. 2013;33:537–542. doi: 10.1007/s10571-013-9919-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maingi S, Moryl N, Andrew F. Symptomatic hypoglycemia due to escalating doses of intravenous methadone. J Pain. 2008;9:37. [Google Scholar]
- 18.Wiederholt IC, Genco M, Foley JM. Recurrent episodes of hypoglycemia induced by propoxyphene. Neurology. 1967;17:703–706. doi: 10.1212/wnl.17.7.703. [DOI] [PubMed] [Google Scholar]
- 19.Spiegel K, Kourides IA, Pasternak GW. Prolactin and growth hormone release by morphine in the rat: Different receptor mechanisms. Science. 1982;217:745–747. doi: 10.1126/science.6285470. [DOI] [PubMed] [Google Scholar]
- 20.Wei E, Loh HH, Way EL. Neuroanatomical correlates of morphine dependence. Science. 1972;177:616–617. doi: 10.1126/science.177.4049.616. [DOI] [PubMed] [Google Scholar]
- 21.Pert A, Yaksh T. Sites of morphine induced analgesia in primate brain: Relation to pain pathways. Brain Res. 1974;80:135–140. doi: 10.1016/0006-8993(74)90731-8. [DOI] [PubMed] [Google Scholar]
- 22.Fields HL, Martin JB. Pain: pathophysiology and management. In: Braunwald E, Hauser SL, Fauci AS, et al., editors. Harrison's Principles of Internal Medicine (ed 15) New York, NY: McGraw-Hill; 2001. pp. 55–60. [Google Scholar]
- 23.Basbaum AI, Fields HL. Endogenous pain control systems: Brainstem spinal pathways and endorphin circuitry. Annu Rev Neurosci. 1984;7:309–338. doi: 10.1146/annurev.ne.07.030184.001521. [DOI] [PubMed] [Google Scholar]
- 24.Yeung JC, Rudy TA. Multiplicative interaction between narcotic agonisms expressed at spinal and supraspinal sites of antinociceptive action as revealed by concurrent intrathecal and intracerebroventricular injections of morphine. J Pharmacol Exp Ther. 1980;215:633–642. [PubMed] [Google Scholar]
- 25.Rossi GC, Pasternak GW, Bodnar RJ. Synergistic brainstem interactions for morphine analgesia. Brain Res. 1993;624:171–180. doi: 10.1016/0006-8993(93)90075-x. [DOI] [PubMed] [Google Scholar]
- 26.Kolesnikov Y, Pasternak GW. Topical opioids in mice: Analgesia and reversal of tolerance by a topical N-methyl-D-aspartate antagonist. J Pharmacol Exp Ther. 1999;290:247–252. [PubMed] [Google Scholar]
- 27.Kolesnikov YA, Jain S, Wilson R, et al. Peripheral morphine analgesia: Synergy with central sites and a target of morphine tolerance. J Pharmacol Exp Ther. 1996;279:502–506. [PubMed] [Google Scholar]
- 28.Weddel SJ, Ritter RR. Serum levels following epidural administration of morphine and correlation with relief of postsurgical pain. Anesthesiology. 1981;54:210–214. doi: 10.1097/00000542-198103000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Foley KM. Management of cancer pain in cancer. In: DeVita VT, Hellman S, Rosenberg SA, editors. Principles and Practice of Oncology. New York, NY: Lippincott; 1985. pp. 1940–1961. [Google Scholar]
- 30.Cherny N, Ripamonti C, Pereira J, et al. Strategies to manage the adverse effects of oral morphine: An evidence-based report. J Clin Oncol. 2001;19:2542–2554. doi: 10.1200/JCO.2001.19.9.2542. [DOI] [PubMed] [Google Scholar]
- 31.Fine PG, Portenoy RK. Ad Hoc Expert Panel on Evidence Review and Guidelines for Opioid Rotation: Establishing “best practices” for opioid rotation: Conclusions of an expert panel. J Pain Symptom Manage. 2009;38:418–425. doi: 10.1016/j.jpainsymman.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ling GS, Spiegel K, Lockhart SH, et al. Separation of opioid analgesia from respiratory depression: Evidence for different receptor mechanisms. J Pharmacol Exp Ther. 1985;232:149–155. [PubMed] [Google Scholar]
- 33.Ling GS, MacLeod JM, Lee S, et al. Separation of morphine analgesia from physical dependence. Science. 1984;226:462–464. doi: 10.1126/science.6541807. [DOI] [PubMed] [Google Scholar]
- 34.Ling GS, Spiegel K, Nishimura SL, et al. Dissociation of morphine's analgesic and respiratory depressant actions. Eur J Pharmacol. 1983;86:487–488. doi: 10.1016/0014-2999(83)90203-0. [DOI] [PubMed] [Google Scholar]
- 35.Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Groer CE, Schmid CL, Jaeger AM, et al. Agonist-directed interactions with specific beta-arrestins determine mu-opioid receptor trafficking, ubiquitination, and dephosphorylation. J Biol Chem. 2011;286:31731–31741. doi: 10.1074/jbc.M111.248310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raehal KM, Schmid CL, Groer CE, et al. Functional selectivity at the μ-opioid receptor: Implications for understanding opioid analgesia and tolerance. Pharmacol Rev. 2011;63:1001–1019. doi: 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly E. Efficacy and ligand bias at the μ-opioid receptor. Br J Pharmacol. 2013;169:1430–1446. doi: 10.1111/bph.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tidgewell K, Groer CE, Harding WW, et al. Herkinorin analogues with differential beta-arrestin-2 interactions. J Med Chem. 2008;51:2421–2431. doi: 10.1021/jm701162g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeWire SM, Yamashita DS, Rominger DH, et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, Mestek A, Liu J, et al. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol Pharmacol. 1993;44:8–12. [PubMed] [Google Scholar]
- 42.Eppler CM, Hulmes JD, Wang JB, et al. Purification and partial amino acid sequence of a mu opioid receptor from rat brain. J Biol Chem. 1993;268:26447–26451. [PubMed] [Google Scholar]
- 43.Thompson RC, Mansour A, Akil H, et al. Cloning and pharmacological characterization of a rat mu opioid receptor. Neuron. 1993;11:903–913. doi: 10.1016/0896-6273(93)90120-g. [DOI] [PubMed] [Google Scholar]
- 44.Wang JB, Imai Y, Eppler CM, et al. mu opiate receptor: cDNA cloning and expression. Proc Natl Acad Sci U S A. 1993;90:10230–10234. doi: 10.1073/pnas.90.21.10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manglik A, Kruse AC, Kobilka TS, et al. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reiter E, Ahn S, Shukla AK, et al. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu J, Xu M, Brown T, et al. Stabilization of the μ-opioid receptor by truncated single transmembrane splice variants through a chaperone-like action. J Biol Chem. 2013;288:21211–21227. doi: 10.1074/jbc.M113.458687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schuller AG, King MA, Zhang J, et al. Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat Neurosci. 1999;2:151–156. doi: 10.1038/5706. [DOI] [PubMed] [Google Scholar]
- 49.Pan YX, Xu J, Xu M, et al. Involvement of exon 11-associated variants of the mu opioid receptor MOR-1 in heroin, but not morphine, actions. Proc Natl Acad Sci U S A. 2009;106:4917–4922. doi: 10.1073/pnas.0811586106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Majumdar S, Grinnell S, Le Rouzic V, et al. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc Natl Acad Sci U S A. 2011;108:19776–19783. doi: 10.1073/pnas.1115231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Majumdar S, Subrath J, Le Rouzic V, et al. Synthesis and evaluation of aryl-naloxamide opiate analgesics targeting truncated exon 11-associated μ opioid receptor (MOR-1) splice variants. J Med Chem. 2012;55:6352–6362. doi: 10.1021/jm300305c. [DOI] [PMC free article] [PubMed] [Google Scholar]