

Abstract
Idiosyncratic drug-induced liver injury (DILI) is a rare disease that develops independently of drug dose, or route or duration, of administration. Furthermore, idiosyncratic DILI is not a single disease entity, but rather a spectrum of rare diseases with varying clinical, histologic, and laboratory features. The pathogenesis of DILI is not fully understood. Standardization of the DILI nomenclature and methods to assess causality, along with the information provided by the LiverTox website, will harmonize and accelerate DILI research. Studies of new serum biomarkers such as glutamate dehydrogenase, high-mobility group box-1 protein, and microRNA-122 could provide information for use in diagnosis and prognosis, and provide important insights into mechanisms of DILI pathogenesis. Single nucleotide polymorphisms in the HLA region have been associated idiosyncratic hepatotoxicity attributed to flucloxacillin, ximelagatran, lapatanib, and amoxicillin- clavulanate. However, genome-wide association studies of pooled cases have not associated any genetic factors with idiosyncratic DILI. Whole-genome and whole-exome sequencing analyses are underway to study DILI cases attributed to a single medication. Serum proteomic, transcriptome, and metabolome, along with intestinal microbiome, analyses will increase our understanding of the mechanisms of this disorder. Further improvements to in vitro and in vivo test systems should advance our understanding of the causes, risk factors, and mechanisms of idiosyncratic DILI.
Keywords: Acetaminophen, liver failure, side effect, epidemiology
Introduction
There is a growing interest in developing “personalized medicine” wherein a specific drug or treatment is offered to a given patient based upon its predicted efficacy derived from host genomic data and/or tissue specific gene expression (1). Examples of personalized include IL28-B genotyping prior to interferon therapy for hepatitis C virus (HCV) infection and use of CD117 expression in gastrointestinal stromal tumors to guide chemotherapy choices (2-4). Genomic and transcriptomic approaches may also improve patient safety by avoiding potentially hazardous drugs in susceptible individuals. For example, avoidance of abacavir use in HLA-B*5701 positive individuals with HIV infection has reduced the incidence of a potentially severe hypersensitivity reaction from 15% to nearly 0% (5,6). Although severe adverse drug reactions (ADR) like drug induced acute liver failure (ALF) are very uncommon, the inability to reliably identify high-risk individuals has prevented many promising drugs from gaining regulatory approval and led to the removal of other drugs from the marketplace (7). Currently, the role of host genetic, immunologic, and metabolic factors as well as drug and environmental influences in idiosyncratic drug induced liver injury (DILI) pathogenesis are poorly understood. This is, in part, due to the lack of reliable in vitro as well as in vivo test systems to study DILI as well as the difficulty in reliably diagnosing and tracking patients with DILI (8,9). The aim of this review is to summarize recent advances in the epidemiology and diagnosis of idiosyncratic DILI, development of sensitive and specific DILI biomarkers, and insights gleaned from pharmacogenetic studies. As our understanding of the role of the immune system in idiosyncratic DILI evolves, studies of other host factors such as the gut microbiome will hopefully further improve our understanding of the causes and mechanisms of idiosyncratic DILI.
Advances in idiosyncratic DILI Epidemiology
Intrinsic and “idiosyncratic” DILI are commonly thought to arise by different pathophysiologic mechanisms. Intrinsic hepatotoxins such as acetaminophen (APAP) are typically dose dependent and have reproducible animal models that help inform our understanding of the pathways leading to hepatocyte injury (46). In contrast, most instances of DILI seen in clinical practice are termed “idiosyncratic” (i.e. a mixture of characteristics unique to that individual) that are not clearly related to the dose, route, or duration of drug administration (Figure 1). The aim of this review is to provide an update on advances in idiosyncratic DILI research.
Figure 1. Factors implicated in the pathogenesis of “Idiosyncratic” DILI.
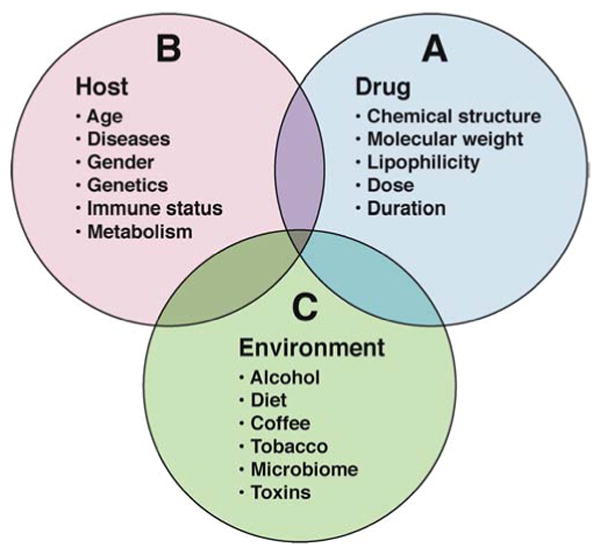
A). Drug factors have not been reliably associated with liver injury in preclinical test systems nor in DILI patients. However, drug-drug or drug-disease interactions could alter the concentration of a drug or reactive metabolite at a cellular level involved in the initiation, maintenance or resolution of liver injury. B) Clinical host risk factors such as age, body weight, and BMI have only rarely been implicated in DILI pathogenesis. However, recent GWA studies have demonstrated consistent associations between various single nucleotide polymorphisms in the HLA region and idiosyncratic DILI susceptibility. C) The micro and macroenvironment vary greatly amongst individuals receiving medications. However, coffee, alcohol consumption and diet have not been identified as bonafide DILI risk factors. The recent development of powerful transcriptomic, metabolomic, and microbiome methods may improve our understanding of environmental factors in DILI pathogenesis using advanced bioinformatics and systems biology approaches.
Overall DILI accounts for < 1% of acute liver injury cases seen by most gastroenterologists in the United States (10, 11). Nonetheless, idiosyncratic DILI is a leading cause of ALF in the US and is likely underdiagnosed due to the need to exclude other more common causes of liver injury and demonstrate improvement following drug discontinuation or “dechallenge” (12). Furthermore, idiosyncratic DILI attributed to a specific drug may present with variable laboratory, clinical and histopathological features making it even more difficult to reliably diagnose and study (Table 1). Until an objective and reliable confirmatory test is developed, idiosyncratic DILI will remain a “clinical diagnosis of exclusion” that requires a high index of suspicion (10).
Table 1. Clinicopathological presentations of Idiosyncratic DILI.
| Phenotype | Histological features (Examples) | Proposed mechanism |
|---|---|---|
| Acute fatty liver with lactic acidosis | Microvesicular hepatic steatosis +/-other tissue involvement (Didianosine, fialuridine) | Severe acute mitochondrial injury |
| Acute hepatic necrosis | Collapse and necrosis of liver parenchyma (Isoniazid, aspirin, niacin) | ? Reactive metabolite +/- immune activation |
| Autoimmune-like hepatitis | Plasma cells & interface hepatitis with detectable autoantibodies (Nitrofurantoin, minocycline) | ? Autoimmuninty |
| Bland cholestasis | Balloon hepatocytes with minimal inflammation (Anabolic steroids) | ? Inhibition of BSEP or other biliary transporters |
| Cholestatic hepatitis | Balloon hepatocytes with inflammation, predominance of serum alk phos elevation (Phenytoin, amoxicllin-clavulanate) | ? Immune mediated +/-reactive metabolite |
| Fibrosis/ cirrhosis | Hepatic collagenization with minimal inflammation (Methotrexate, amiodarone) | ? Stellate cell activation/chronic endothelial cell injury |
| Immunoallergic hepatitis | Skin rash, fever, eosinophilia (Trimethoprim-sulfamethoxasole) | Drug hypersensitivity/allergy |
| Nodular regeneration | Micro or macroscopic liver nodules (Azathioprine, oxaliplatin) | ? Chronic injury to endothelial cells |
| Non-alcoholic fatty liver | Macro and microsteatosis, hepatocyte ballooning and periportal inflammation (Tamoxifen) | ? Chronic mitochondrial injury or altered lipid metabolism |
| Sinusoidal obstruction syndrome | Inflammation with obliteration of central veins (busulfan) | ? Severe, acute endothelial cell injury |
| Vanishing bile duct syndrome | Paucity of interlobular bile ducts (Amoxicllin-clavulanate, sulfonamides) | ? Immune mediated cholangiocyte injury |
Studies of idiosyncratic DILI epidemiology have largely been retrospective case series with highly variable estimates of the incidence and natural history (13-15). The recent adaptation of electronic medical records (EMR) into routine medical practice has created a unique opportunity to track and study various rare ADR's (16,17). Identification of idiosyncratic DILI cases from administrative databases using ICD-9 diagnostic coding has proven to be labor intensive with a low sensitivity and specificity (13,18). However, recent studies that use natural language processing algorithms which can search for key words in a text field such as “hepatotoxicity” or “toxic hepatitis” have demonstrated an improved sensitivity and specificity for DILI (18). In addition, the linking of clinical, laboratory and pathology databases with text searching algorithms may allow for more real time identification of idiosyncratic DILI cases (19, 20).
Idiosyncratic DILI Registries
In 2004, the Drug Induced Liver Injury Network (DILIN) was established by the National Institutes of Health (NIH) to improve our understanding of the causes, mechanisms, and outcomes of idiosyncratic DILI in adults and children (21). Similar multicenter networks have been established in Spain, Iceland, the United Kingdom, Europe, Japan, China and Korea (22-26). These networks are leading efforts to develop standardized nomenclature, grading systems, and causality assessment methods in DILI research (9, 27,28). Harmonization of the approach to DILI phenotyping and causality assessment will hopefully provide an increased number of DILI cases for pooling in genetic association studies (Supplementary Table 1) (27-30). In addition, the NIH in conjunction with the National Library of Medicine has developed a comprehensive, multilayered, and interactive database of the published literature of human drug hepatotoxicity (31). The LiverTox website at http://www.livertox.nih.gov/ has concise overview sections on DILI phenotypes, severity grading, and likelihood scales. In addition, chapters that summarize the clinical and laboratory features of DILI associated with over 650 individual drugs are available along with illustrative cases and annotated references with available hyperlinks to the full PUBMED reference. The LiverTox website has already increased awareness of DILI and will likely prove to be a valuable resource for basic, translational, and clinical research into DILI pathogenesis for years to come.
Several reports of the etiologies and outcomes of idiosyncratic DILI from around the world have recently been published (Table 2) (23, 24,26, 32, 33). The laboratory profile of DILI at presentation is defined by the ratio of serum alanine aminotransferase (ALT) to alkaline phosphatase levels (i.e. R value = (ALT/ ULN)/ (Alk phos/ ULN)) and can be categorized as hepatocellular (R > 5), mixed (R = 2 to 5) or cholestatic (R < 2). Similarities across the DILI cohorts include the proportion of female patients (49% to 65%), median subject age (48 to 55 years), and proportion with acute hepatocellular injury at presentation (42% to 58%). Although the DILIN and Spanish networks tend to enroll sicker patients that are more likely to be hospitalized, the proportion of patients that have died or required liver transplantation is remarkably similar. Antibiotics are the most commonly identified drug class leading to idiosyncratic DILI but the specific implicated agents differ substantially. In addition, DILIN has reported a significant proportion of cases attributed to herbal and dietary supplements (HDS) that has increased over the past 10 years from 7% in ‘04/’05 to 20% in ‘10/’11 (34). The most frequently implicated HDS products in DILIN are body building supplements and weight loss products that contain green tea extract (35, 36).
Table 2. Presenting clinical features and outcomes in Prospective studies of DILI.
| Feature | DILIN US N=300 (ref 32) |
Spain N=446 (ref 23) |
Japan N=1676 (ref 26) |
Iceland N=96 (ref 24) |
France N=34 (ref 33) |
|---|---|---|---|---|---|
|
| |||||
| Study Design | Multicenter (8 sites) ‘04 to ‘08 | Multicenter (32 sites) ’94-‘05 | Multicenter ’97-‘06 | Population based ’10-‘12 | Population based ’97-‘01 |
|
| |||||
| Causality method | Expert opinion | RUCAM | RUCAM | RUCAM | CIOMS |
|
| |||||
| F/U duration (mon) | 6 to 24 | 3 | NA | 3 | 3 |
|
| |||||
| Mean age (yrs) | 48 | 53 | 55 | 55 | 55 |
|
| |||||
| % Female | 60% | 49% | 57% | 56% | 65% |
|
| |||||
| Race | |||||
| % Caucasian | 79% | 100% | 0% | 100% | 100% |
| % African Amer | 11% | 0% | |||
| % Asian | 4% | 100% | |||
| % Other | 6% | 0% | |||
|
| |||||
| Liver injury type | |||||
| % Hepatocellular | 57% | 58% | 59% | 42% | 53% |
| % Mixed/Cholestatic | 20%/23% | 22%/20% | 20%/21% | 26%/32% | 26%/21% |
|
| |||||
| % Jaundice | 69% | 71% | NA | 27% | 29% |
|
| |||||
| % Liver biopsy | 50% | 25% | NA | 11% | NA |
|
| |||||
| % Hospitalized | 60% | 53% | NA | 23% | 12% |
|
| |||||
| % Died or transplanted | 10% | 7% | 3.7% | 11% | 6% |
|
| |||||
| % Chronic | 14% | 10% | 8.4% | 7% | NA |
|
| |||||
| Suspect drugs | |||||
| % Antibiotics | 45% | 32% | 14% | 37% | 25% |
| % Psychotropic | 15% | 17% | 10% | 7% | 22% |
| % HDS products | 9% | 0% | 17.1% | 16% | 0% |
| % Hypolipidemic | 3% | 3% | 0% | 3% | 13% |
NA= Not available
DILIN is prospectively following all study subjects for a minimum of 6 months after enrollment (21,32). A competing cause of liver injury has been identified in up to 15% of DILIN patients including previously unsuspected acute HCV and hepatitis E virus (HEV) infection (37-39). DILIN has also confirmed the association of high serum aminotransferase levels at presentation in jaundiced DILI patients with a greater likelihood of early death/ transplant (i.e. Hy's law = serum ALT > 5X ULN with elevated total bilirubin) (40-42). However, the observation that nearly 50% of the deaths in DILIN patients were attributed to an underlying non-hepatic medical condition such as malignancy or heart failure is also an important observation (40). In addition, DILIN and other groups have confirmed the greater likelihood of patients presenting with a severe cholestatic liver injury to develop chronic liver injury during follow-up (41,42). These findings highlight the need for careful clinical and laboratory monitoring of DILI patients and also provide the rationale for a clinical trial to improve patient outcomes (43-45).
Idiosyncratic DILI epidemiology- Mechanistic insights
Although drugs undergo extensive safety testing in in vitro test systems and various animal species prior to clinical development, this testing frequently fails to identify potentially hepatotoxic drugs. In addition, the low overall incidence of idiosyncratic DILI with most available drugs of only 1 in 10,000 to 1 in 100,000 patient years prevents most hepatotoxic drugs from being identified in clinical trials. Recent studies have suggested that drugs which are administered at a daily dose > 50 to 100 mg/day with greater lipophilicity are more prone to cause DILI compared to agents given at a lower daily dose with less lipophilicity (47, 48). Possible explanations for these simple but intriguing observations include the fact that drugs given in high daily doses may lead to higher intrahepatic levels of the parent drug or a metabolite involved in DILI pathogenesis. In addition, lipophilic drugs require greater metabolism to be eliminated from the body which may increase their likelihood of causing liver damage. It is also possible that extensively metabolized drugs may induce covalently- bound haptens which can illicit an adverse adaptive immune response in a genetically susceptible individual (49, 50). However, the daily dose of a medication, its lipophilicity, and extent of hepatic metabolism are inadequate features to reliably predict DILI risk from individual drugs. In addition, studies of chemoinformatics have failed to identify chemical moieties in drugs that are more prone to lead to idiosyncratic hepatotoxicity (51, 52).
In western DILI patients, antibacterial antibiotics and psychoactive drugs are the most frequently implicated therapeutic drug classes (Table 2). Although antibacterial antibiotics are used by millions of Americans each day, they are usually only taken for a few days or weeks. This suggests that an active or recent infection may increase susceptibility to an ADR via aberrant innate or adaptive immune pathways or alterations in the gut microbiome in susceptible patients (46). In support of the “danger hypothesis” (Figure 2) administration of tumor necrosis factor (TNF) to hepatocyte cultures as well as in animal models of DILI can potentiate the hepatotoxicity of multiple drugs (53). In addition, the greater frequency of DILI in immunosuppressed liver transplant recipients (1 in 100 patient years) compared to the general population (1 in 10,000 patient years) suggests that specific patient populations may be at increased risk of idiosyncratic DILI (20). However, a plausible explanation as to why the majority of patients with an acute or chronic illness who receive multiple medications including antibiotics do not develop DILI remains unclear.
Figure 2. Potential mechanism(s) involved in Idiosyncratic DILI pathogenesis.
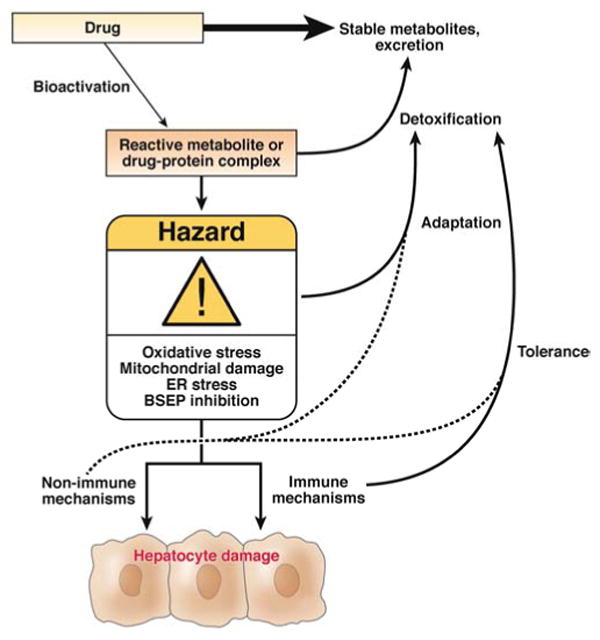
It is plausible that several cellular mechanisms may be involved in DILI pathogenesis. One major hypothesis of idiosyncratic DILI pathogenesis is the inadvertent generation of a reactive metabolite or parent drug-protein complex that can directly or indirectly mediate damage to intracellular proteins and/or organelles resulting in the initiation of “Danger” signals. DILI patients may be uniquely susceptible to develop liver injury from reduced detoxification, adaptation, or tolerance pathways that would normally rescue damaged hepatocytes or an increased likelihood of “Bioactivation” pathways. In the hapten hypothesis, the drug-protein or metabolite-protein adduct leads to inadvertent activation of the adaptive immune system. Alternatively, non-immune mechanisms wherein damage associated molecular pathway (DAMP's) proteins such as HMGB1, heat shock proteins, or cellular DNA released from necrotic cells lead to the recruitment of localized tissue injury. Cytokines, chemokines, and co-stimulatory molecules may play an integral role in macrophage activation and magnification of the DAMP response via their impact on drug metabolizing enzyme activity, the density of HLA molecules on antigen presenting cells, the ability of the presented antigens to activate T-Cells, and the ability of activated T-cells to cause hepatocyte death.
A recent population based study from Iceland provides the best estimates of the incidence of idiosyncratic DILI in a western country (24). In this study, all suspected DILI episodes regardless of severity occurring amongst the 250,000 inhabitants of Iceland over a 2 year period were reviewed. The crude overall annual incidence of idiosyncratic DILI was 19.1 cases per 100,000 population which is remarkably similar to the rate of 13.9 cases per 100,000 reported from France (33). These rates are likely higher than prior estimates due to the prospective nature of these studies, inclusion of subclinical cases, and the ability to systematically canvas an entire population. Although no differences in gender were noted, there was a strong association of DILI risk with patient age that varied from 9 per 100,000 (age 15 to 29) to 41 per 100,000 (age > 70). Although amoxicillin-clavunate was the most frequently implicated drug, the estimate of DILI risk was 1 per 2350 users and substantially lower than that observed with azathioprine at 1 per 133 users and infliximab at 1 per 148 users. These data lend support to the notion that some drugs used in clinical practice are intrinsically more hepatotoxic than others. Although DILIN is not a population based study, amoxicillin/clavulante is also the most commonly implicated agent (> 120 DILIN cases) but is prescribed to more than 70 million Americans each year. In contrast, isoniazid (> 50 DILIN cases and < 200,000 prescriptions per year) and nitrofurantoin (> 50 DILIN cases and < 500,000 prescriptions each year) appear to be over-represented in the DILIN as well as the Acute Liver Failure Study Group (ALFSG) databases (43). Similarly, the infrequent reporting of statins, beta-blockers, and calcium channel blockers in DILI registries despite their widespread use suggests that these drug classes are probably not as intrinsically hepatotoxic as previously suggested (54-55). The LiverTox website has developed a categorization of the likelihood of DILI from a specific agent based upon the frequency of bonafide cases in the world's literature that varies from Category A (“Well known with > 50 cases described” like amoxicillin/clavulanate and phenytoin) to Category E (“Despite extensive use, no evidence of liver injury” like felodipine or propranolol).
Inferences of DILI Pathogenesis from Liver Histology
DILI is a well-known imitator of most forms of acute and chronic liver injury (Table 1). Furthermore, some drugs may cause differing patterns of liver injury in individual patients like valproate that can vary from hepatic steatosis to massive necrosis (56). A careful review of liver histopathology in 249 DILIN patients indicates that 5 liver injury patterns accounted for 83% of the cases (57). In addition, poorer outcomes were associated with higher degrees of necrosis, microvesicular steatosis and a ductular reaction while subjects with intra-hepatic eosinophils and/or granulomas tended to fare better. The prognostic utility of these histologic features is consistent with prior reports and suggests that peripheral or intrahepatic eosinophilia is protective (58, 59).
Other studies have suggested potential histopathological differences in patients with idiopathic and drug-induced autoimmune hepatitis (60). Similarly, studies of lymphocyte subsets in liver tissue samples have demonstrated that DILI subjects are less likely to have Natural killer cells, CD4+ T-Helper, and B-cells compared to patients with acute viral hepatitis (61). The predominance of CD8 + cytotoxic T-cells in the livers of idiosyncratic DILI patients is consistent with the hypothesis that the intrahepatic generation of neo-antigens from the drug or its metabolite may lead to the recruitment and activation of T-lymphocytes that can initiate or perpetuate the liver injury (62). In contrast, neutrophil and macrophage infiltration into the liver is felt to be a late event in patients with APAP overdose and other drugs that directly injure peri-central hepatocytes (63). Similarly, patients with bland cholestasis due to anabolic steroids may develop liver injury through direct toxicity to bile salt or other drug transporters (64).
Drugs that disrupt mitochondrial function via depletion of mitochondrial DNA or triggering of outer mitochondrial membrane permeabilization can lead to variable patterns of liver tissue injury. Acute impairment of mitochondrial function is a distinctive and frequently dramatic clinical syndrome characterized by small droplets of fat (microvesicular) that accumulate in the hepatocyte cytosol from impaired B-oxidation of fatty acids as reported with tetracycline and valproic acid (56, 65). In contrast, drugs that lead to partial but chronic depletion of mitochondrial function can lead to accumulation of large fat droplets (macrovesicular) that are often eccentrically located in the cell. Drugs associated with the latter sub-acute or chronic phenotype include tamoxifen and various dideoxynucleoside analogues used to treat HIV infection (66, 67). Other drugs like oxaliplatin can damage liver endothelial cells and lead to nodular regenerative hyperplasia and portal hypertension in some patients (68, 69).
In vitro test systems to study DILI mechanisms
Extensive in vitro and animal toxicology testing is required in drug development but these approaches have consistently failed to predict the development of various ADR's including DILI. In 2007, the US National Academy of Sciences suggested that greater use of human biological test systems be employed to improve detection and provide mechanistic insight into human ADR's (70). In addition, the need for pathways analysis of toxicological mechanisms was emphasized using metabolomic, transcriptomic, and genomic approaches. Currently, potential drug-drug interactions can be reliably predicted from in vitro inhibition of known phase 1 and 2 polymorphisms in cultured human hepatocytes. Recently, several chimeric mice strains expressing highly differentiated human hepatocytes that can be used in cell culture experiments as well as human hepatoma cell lines with highly differentiated cellular function have been developed (71,72). In addition, the discovery that human and mouse fibroblasts and somatic cells can be preprogrammed into inducible pleuripotent stem (iPS) cells has generated a great deal of interest in using this novel technology to study drug hepatotoxicity, liver regeneration and various genetic diseases (73-75). Derivation of hepatocytes from iPS cells of patients who develop DILI could allow for a long-term and renewable source of cells to study idiosyncratic DILI mechanisms in affected patients compared to treated controls (76).
Advances in the development of in vitro culture methods of T-cells implicated in various hypersensitivity drug reactions have also recently been reported including techniques to expand the number of available T-cells derived from peripheral blood of patients with allergic drug reactions (77). Since antigens are processed and presented on human leukocyte antigen (HLA) molecules of antigen presenting cells to the T-cell receptor on T-cells, additional studies of T-cell physiology can now be conducted (Figure 3). Patients with known hypersensitivity to piperacillin which by itself is unlikely to stimulate an immune response have demonstrated alterations in the binding site of albumin that can lead to highly immunogenic drug metabolite-albumin conjugates (78, 79). In addition, modifications of the Lymphocyte proliferation assay have been developed with readouts of T-cell cytokine expression and microarrays rather than pure cellular proliferation (80). Whether these modified lymphocyte transformation assays will improve drug specific diagnoses or provide mechanistic insights into DILI pathogenesis remains to be determined.
Figure 3. Proposed role of abberant immunity in DILI pathogenesis.

Drugs are small molecules capable of binding to serum proteins under normal physiological circumstances for transport, metabolism, and elimination. In most instances, a drug-protein conjugate will not illicit a host immune response. However, a minority of individuals with specific class II HLA alleles that are ubiquitously expressed may be uniquely predisposed to have the native drug-protein or drug metabolite-protein conjugate activate an antigen presenting cell such as a dendritic cell or macrophage. The processing and handling of drug- protein conjugates in these individuals can then inadvertently activate T-Cell receptors which may proliferate and mediate end-organ damage.
Biomarkers of DILI pathogenesis and outcomes
Biomarkers are analytes from blood, urine, or other biological samples that may provide insight into the severity, cause, or outcome of a DILI episode. In addition, biomarkers may improve the speed or accuracy of making a diagnosis of DILI (81). Ideally, direct examination of liver tissue would provide the greatest insight into the pathophysiological steps involved in idiosyncratic DILI but is impractical for obvious reasons. The serum biomarkers most commonly used to detect and manage most forms of acute and chronic liver injury are serum ALT, alkaline phosphatase and total bilirubin levels. Serum ALT is more liver-specific than AST but it is not etiology specific and can also be elevated in subjects with extensive hepatic glycogen, hepatocyte autophagy, and hepatic steatosis (82, 83). Furthermore, monitoring for serum ALT elevations in subjects receiving a potentially hepatotoxic drug like isoniazid has consistently failed to identify subjects at risk of developing DILI compared to the larger group of patients who “adapt” with ALT normalization during continued treatment (84, 85). Similarly, serum alkaline phosphatase levels are not liver-specific and may be spuriously elevated in other disease states (81). Total bilirubin levels are an insensitive marker for most forms of liver disease increasing only when there has been extensive liver damage or via direct inhibition of biliary transporters. Fractionation of total bilirubin levels can help exclude benign elevations due to intravascular hemolysis and genetic polymorphisms in uridine glucuronyltransferase activity (Gilberts’) which are present in 1 to 5% of the general population.
New Serum biomarkers
Ongoing efforts to identify new DILI biomarkers include the large scale initiative of the Safer and Faster Evidence-based Translation (SAFE-T) Consortium in Europe (86). Proposed biomarker candidates include serum markers of liver injury (sorbitol dehydrogenase (SDH), glutathione s-transferase, GSTα) and mitochondrial dysfunction from disrupted hepatocytes (glutatmate dehydrogenase or GLDH) (Table 3). In addition, the discovery of circulating microRNA's in the serum has demonstrated the novel tissue specificity of miR-122 for liver injury. Assessment of full length keratin-18 (K-18) and high mobility group box protein-1 (HMGB-1) in the serum has been shown to be a sensitive biomarker for necrotic cell death while caspase cleaved K-18 fragments are noted in subjects with ongoing apoptosis. However, neither of these markers are liver disease specific. M-30 is a serum protein which selectively recognizes caspase cleaved neoepitopes of K-18 released from hepatocytes undergoing apoptotic death while serum M-65 reflects total hepatocyte death (apoptosis and necrosis)(87, 88). An index of serum M-30 levels in combination with other lab parameters was recently shown to be superior to the King's College criteria and MELD score in predicting spontaneous survival in ALF (89, 90).
Table 3. Proposed DILI biomarkers.
| Analyte | Source and significance | Test performance to date |
|---|---|---|
| Liver injury markers | ||
| Sorbitol dehydrogenase (SDH) | Hepatocyte specific injury | Earlier marker of acute liver injury |
| Glutathione S-transferase alpha (GSTα) | Centrilobular liver damage and kidney damage | Early marker of acute liver (serum) and kidney (urine) injury |
| Bile acids | Synthesized in the liver; disruption of hepatic excretion | More sensitive than bilirubin for excretory abnormalities |
| Glutamate dehydrogenase (GLDH) | Mitochondrial disruption | Increased in some patients with chronic liver disease |
| Serum cytokine profiles | ||
| Micro RNA's | ||
| miR-122 miR-192 |
Liver specific release from damaged hepatocytes | Released into plasma with acute and chronic injury; validation ongoing |
| Mechanistic biomarkers | ||
| HMGB1 | Necrosis marker | Not liver specific |
| Acetylated HMGB1 | Innate immune activation marker | Acetylation requires mass spec for detection |
| Cytokeratin 18 fragments | Marker of caspase cleaved proteins in apoptotic cell death | Not liver specific |
| M-30 | Apoptosis marker | |
| M-65 | Total apoptosis and necrosis marker | |
| Serum Cys-APAP adducts | Marker of APAP overdose | Ability to distinguish therapeutic dosing from drug overdose being tested. |
| Metabolomics | ||
| Urine or serum metabolome | Amount and type of endogenous substances | Exploratory with substantial drug, dietary, environmental, and microbiome influences; bioinformatics for data reduction ongoing |
Heparins are a widely used class of biological agents that are frequently associated with mild and non-progressive serum ALT elevations but rarely if ever lead to clinically significant liver injury (91). A recent study of 48 healthy men given a heparin formulation for 5 days demonstrated asymptomatic elevations in serum AST and ALT levels in 90% of the treated patients (92). In addition, significant elevations in the levels of serum SDH, GLDH, miR-122, and HMGB1 in both its native and acetylated form were noted (93). However, serum K-18 fragment levels indicative of apoptosis remained unchanged. These data suggest that heparins cause a self-limited and mild hepatocyte necrosis with secondary activation of the innate immune system. HMGB1, a damage associated molecular pattern (DAMP) protein, can bind to toll-like receptor 4 and initiate an innate immune response at its site of release in the liver (94). The detection of acetylated HMGB1 is suggestive of activated innate immune cells which may be linked to tissue repair. However, the reason for a lack of progressive liver injury despite continued dosing with heparins remains unclear (95).
Biomarkers in Acetaminophen hepatotoxicity
Another study that used therapeutic doses of APAP for 5 days in healthy volunteers showed that nearly 30% of the treated patients experienced an increase in serum ALT levels (96). There was also an increase in serum SDH, GLDH, and HMGB1 levels (96, 97). A panel of these early DILI biomarkers were recently tested in patients presenting to the hospital following APAP overdose with initially normal serum aminotransferase levels (98). A combination of miR-122, HMGB1, and K-18 levels identified the development of liver injury with a higher degree of accuracy than the initial serum ALT, INR, and plasma APAP level. In addition, acetylated HMGB1 levels reflective of immune activation were noted later in the course of APAP overdose and associated with a poorer outcome in an independent cohort of 78 patients (99).
Several studies have demonstrated that detection of acetaminophen-protein adducts in serum can confirm a diagnosis of APAP hepatotoxicity. The premise of this test is based on the formation of covalent adducts between intrahepatocyte proteins and the major reactive metabolite of APAP formed in the liver, NAPQI. Since serum acetaminophen-protein adducts have a longer half-life in serum compared to the parent drug or its metabolites, this test may be particularly informative in patients presenting late after an unintentional APAP overdose (100). In addition, 15% of patients with indeterminate ALF also had detectable acetaminophen-protein adducts (101). However, low levels of APAP-protein adducts have also been detected in the blood of patients taking therapeutic doses of APAP over 5 days (102). The development of tests to detect circulating drug-protein adducts in other patients with idiosyncratic DILI would be desirable. However, idiosyncratic DILI due to most drugs occurs at lower daily doses and toxic metabolites implicated in DILI pathogenesis are believed to represent a smaller fraction of the total testable drug in the serum compared to APAP making it technically difficult to identify and quantify drug-protein adducts (103).
Serum Proteomics
Proteomic studies that simultaneously identify and quantify thousands of proteins utilize high-pressure liquid chromatography (HPLC) and tandem mass spectroscopy. With advanced bioinformatics software, the protein(s) and/ or pathways involved in DILI pathogenesis can then be studied. The serum proteomic profile of 74 DILIN patients who had a baseline sample collected within 2 weeks of DILI onset and 40 healthy controls were recently analyzed using a label-free, mass spectrometry quantitative approach (104). Several proteins were expressed at a higher level in subjects with hepatocellular versus cholestatic DILI including fructose-bisphosphate and aldolase B. This hepatic enzyme correlated with serum ALT and AST levels at baseline and returned to normal during follow-up. Of note, autoantibodies to this protein have previously been reported in patients with troglitazone hepatotoxicity (105). In addition, elevations in apolipoprotein E, an abundant lipoprotein of triglyceride rich chylomicrons, had the greatest ability to distinguish DILI patients from controls. The longitudinal analysis of 21 patients with baseline and 6 month samples demonstrated that expression of 53 priority 1 proteins either increased or decreased over time including components of inflammatory, immune system activation, and several hepatotoxicity-specific pathways. A proteomics platform in combination with metabolomics was recently used to distinguish patients that developed ximelagatran hepatotoxicity from unaffected controls (106). In that study, biomarkers that predicted patients at risk of ALT elevation included apolipoproteins A-II, A-IV, and E. These provocative data suggest that further proteomic studies are indicated particularly in samples obtained prior to or shortly after DILI onset.
Serum Cytokines and chemokines
Serum cytokine and chemokine levels may also prove to be useful diagnostic or prognostic DILI biomarkers (107). DILIN recently completed an analysis of 27 immune analytes in 78 subjects who were enrolled within 2 weeks of DILI onset and 6 months after enrollment (108). Disparate patterns of immune responses were evident and low values of IL-9, IL-17, PDGG-bb, and RANTES combined with serum albumin were predictive of early death. Lower levels of the cytokines associated with innate immunity (i.e. low IL-9 and IL-17) were associated with a poorer prognosis suggesting a role for these biomarkers and immune pathways in DILI pathogenesis. These data are consistent with recent studies demonstrating a role of the TH17 adaptive immunity pathway in idiosyncratic DILI pathogenesis (109). However, a study from the ALFSG failed to demonstrate a difference in circulating IL-17 levels of patients with ALF due to idiosyncratic DILI compared to those with APAP overdose (110).
Transcriptomics and metabolomics
Transcriptomics represents the detection and quantification of transcribed genes or messenger RNA in the serum and other fluids. In contrast, metabolomics represents the study of endogenous small molecules and metabolites in the serum and urine which reflect normal physiological and diseased states. Transcriptomic studies require the collection of RNA from specialized tubes and then detection of relative gene expression levels using oligo array chips and quantitation of mRNA expression using PCR. In metabolomic studies, samples are analyzed using NMR spectroscopy or mass spectral techniques to simultaneously detect and quantify thousands of endogenous metabolites.
A recent study of 8 hepatotoxicants administered to rats demonstrated that transcriptomes present in peripheral blood correlated with direct measures of these genes in the liver (111). In fact these studies suggested the peripheral blood transcriptomic data might be more sensitive to liver injury than traditional liver injury tests such as serum ALT and that unique signatures for individual drugs including APAP hepatotoxicity could be determined (112). Furthermore, human whole blood transcriptome data from patients with overt APAP overdose could differentiate patients with toxicity from those without (112). However, marked down-regulation of genes involved in oxidative phosphorylation and mitochondrial function as well as metabolomic changes in the blood and urine were observed in patients receiving therapeutic dose of APAP compared to those with overt APAP hepatotoxicity (113, 114).
Advantages of metabolomic and transcriptomic studies in biomarker discovery include the large amount of data that can be generated and quantified from small sample aliquots. Furthermore, pathways analysis can be conducted using bioinformatics platforms. Finally, the relationship between changes in gene expression from the transcriptomic platforms can be integrated with the physiological changes detected from metabolomics. However, the transcriptome in whole blood may chiefly reflect the transcriptional activity of lymphocytes and it appears that these approaches may not be able to distinguish the pharmacological effect of a drug like APAP from a toxic dose (108). In addition, serum and urine metabolomes in patients can be influenced by dietary and environmental factors including the gut microbiome (115). Therefore, further studies of the drivers of inter-individual differences in the metabolome and transcriptome are needed as part of a systems biology approach to DILI susceptibility (116, 117).
MicroRNA's
MicroRNA's are small regulatory, non-coding RNA's that are 18 to 25 nucleotides in length and can be detected in microvesicles in the serum. Although a number of miRNA's are widely expressed, certain miRNA's appear to be tissue specific. Liver specific miRNA's include miR-122, miR-21, and miR-192 and these molecules are believed to repress a set of target messenger RNA (mRNA) and thereby regulate specific cellular proteins and cell phenotype. Liver derived miRNA's may be a highly sensitive and specific biomarker of APAP hepatotoxicity that parallel ALT levels but increase earlier in the course of liver injury (118). In addition, they appear to have prognostic significance for APAP overdose patients in need of transplant versus those more likely to survive (119, 120). However, serum miR-122 levels have not been directly correlated against hepatic expression in humans. Nonetheless, their short half-life, liver tissue specificity, and quantifiability make them attractive biomarkers for severe acute liver injury.
Pharmacogenetic studies of idiosyncratic DILI
Due to its low incidence in the general population, genetic variation in host receptors, immune response, and metabolic pathways have been implicated in idiosyncratic DILI pathogenesis. Previous genetic association studies have largely focused on candidate genes involved in the uptake, metabolism, transport, or detoxification of a drug that can be used to predict drug pharmacokinetic and pharmacodynamic parameters. For example, reduced activity in the NAT2 gene and increased activity in CYP2E1 mediated oxidative metabolism have been implicated in isoniazid hepatotoxicity (121-123). However, these hypothesis-driven, biologically plausible approaches have yielded only weak associations which are frequently not replicated in independent cohorts (124).
An alternative approach to identify genetic associations is to scan the entire human genome in affected cases and population controls without a specific a priori hypothesis. In most genome wide association (GWA) studies, the frequency of single nucleotide polymorphisms (SNP) is at least 1 to 5% in the general population. This “discovery” platform allows for the unbiased detection of up to 1 million SNP's that may associate with the disease trait of interest. The first successful GWA study in DILI identified a very strong association between flucloxacillin-induced liver injury and the HLA-B*5701 allele on chromosome 6 (125). Other GWA studies have identified additional HLA alleles associated with lumiracoxcib, ximelagatran, and lapatanib hepatotoxicity (Supplementary Table 2) (126-128).
Genetic studies of Flucloxacillin hepatotoxicity
Flucloxacillin is a parenterally administered B-lactam antibiotic that is widely used in Europe and other countries to treat staphylococcus and streptococcal infections. Flucloxacillin is associated with a rare but potentially severe cholestatic hepatitis that is more common in females, the elderly, and after prolonged courses of treatment (129). Daly et al performed a GWA study in 51 patients with bonafide flucloxacillin cholestasis compared to 64 ethnically matched treated controls as well as population controls (125). In the initial and validation cohorts, possession of the HLA-B*5701 allele was associated with an 80-fold increased risk of developing DILI (p=9 × 10-19). This HLA allele has also been associated with abacavir-hypersensitivity in patients with HIV infection (5). Although this is one of the strongest genetic associations ever reported from a GWA study, the high frequency of the HLA-B*5701 in the general population (6 to 8%) coupled with the low incidence of DILI in treated patients (1 in 10,000) leads to a low positive predictive value (0.12%) for identifying patients at risk for flucloxacillin cholestasis but a high negative predictive value (99.99%) (130). Therefore, testing for HLA-B*5701 may help diagnose flucloxacillin cholestasis in exposed patients that develop jaundice but use of the drug should not be withheld in HLA-B*5701 positive individuals.
Flucloxacillin is also an agonist of the human pregnane X receptor (PXR) and further studies have shown that patients with flucloxacillin cholestasis are more likely to have polymorphisms in the PXR promoter region (131). Other studies demonstrate an important role for the adaptive immune system with T-cell clones of afflicted individuals and previously unexposed HLA-B*5701 positive patients showing increased reactivity to flucloxacillin-albumin conjugates in a dose dependent manner (132). These studies also showed that the reactive T-cell clones from afflicted patients had cell surface markers that are associated with hepatic localization. These data confirm that flucloxacillin-protein binding is critical for the formation of functional T-cell antigens. This is in contrast to recent mechanistic studies of abacavir and carbamazepine hypersensitivity reactions wherein the native drug itself has been shown to bind in the antigen binding cleft of an HLA molecule and directly stimulate an immune response (133). Additional studies to further understand how flucloxacillin mediates liver damage and the role of hapten formation versus direct stimulation of T-cells are ongoing (Figure 3) (134).
Genetic studies in pooled idiosyncratic DILI patients
DILIN and other groups have been collecting DNA from idiosyncratic DILI patients for pooled pharmacogenetic studies (22). Hypothesizing that DILI susceptibility may be shared across multiple drugs, a GWA was recently undertaken in 783 Caucasian individuals who experienced DILI from over 200 implicated agents (135, 136). Unfortunately, there were no genome wide significant associations noted and further stratification of cases according to clinical phenotypes such as injury pattern, latency, severity, drug class, and patient age did not reveal any significant associations. The lack of GWA findings in the pooled DILIN cases supports the notion that genetic determinants of idiosyncratic DILI risk may be largely drug-specific or due to rarer genetic variants not assessed on the GWA chip. Going forward, newer techniques including exome arrays that can assess for functional genetic variants present in 1 in 1000 to 1 in 5,000 individuals at over 250,000 loci are being undertaken. In addition, improvements in the speed, accuracy, and costs of whole exome and whole genome sequencing now allow for a more in-depth search of causal variants from smaller samples of well phenotyped, high causality cases attributed to a single drug (137). The role of DNA methylation, copy number variants and epigenetics in most forms of acute liver injury is largely unknown but also worthy of further study (138).
Mechanistic Inferences into DILI pathogenesis
The strong and consistent association of DILI susceptibility with various SNP's in the HLA region suggests that the host immune response plays a key role in DILI pathogenesis. Human leukocyte antigens are highly polymorphic proteins that are designed to initiate immunity by presenting pathogen derived peptides to T-Cells. Polymorphisms in HLA genes mostly map to the antigen-binding cleft which allows diversification of the repertoire of self-derived and pathogen derived peptide antigens to be presented to T-Cells (139). A growing number of other immunologically based ADR's including dermatological reactions and idiosyncratic DILI are also associating with various HLA alleles (140). In most of these instances, the implicated drug does not directly bind to the antigen binding cleft of the HLA- molecule. Rather, a series of drug-protein modification steps or conversion of the drug to an intermediate or reactive metabolite is required to form an immunogenic hapten. However, it is likely that other host genetic or intracellular pathways may also be required for an ADR to develop. Furthermore, since many susceptible individuals with a given HLA haplotype do not develop DILI or other ADR's upon drug exposure, the role of other intracellular “bioactivation” and “detoxification” pathways that may allow for adaptation to occur need to be evaluated (Figure 2). In addition, since HLA polymorphisms are ethnically restricted, the absence of a genetic association of DILI susceptibility in one patient population will not preclude a positive association in another group as recently noted in differing HLA susceptibility alleles in Han Chinese and Europeans to carbamazepine hypersensitivity reactions (141, 142).
The potential for the gut microbiome to impact susceptibility and outcome to DILI is another intriguing hypothesis worthy of further study. Early studies have demonstrated interesting alterations in the gut microbiome of patients with obesity compared to non-obese controls and in animal models of fatty liver (143, 144). Recent studies of complex biliary tract disorders also demonstrates that variance in microbiome content may exceed that explained by genomic variation (145). However, there have been no studies of the gut microbiome in idiosyncratic DILI patients but there are interesting animal data demonstrating a significant impact of the gut microbiome in mediating melamine related kidney injury (146, 147).
Future directions in DILI research
Over the next 5 to 10 years, additional studies of host genetic polymorphisms in idiosyncratic DILI susceptibility attributed to individual agents will be completed using next generation sequencing. To conduct such studies, DNA samples collected from bonafide DILI cases wherein competing viral, immunological, and metabolic causes of liver disease have been definitively excluded are needed as well as validation samples from independent cohorts. It is hoped that EMR mining with natural language processing algorithms will improve the speed and accuracy of DILI case acquisition and inform pharmacoepidemiological studies regarding the causes of DILI in a given population. In addition, continued efforts from multicenter research networks like DILIN will help provide biological samples for mechanistic studies. Improved causality assessment tools, case definitions, and further development of a web-based portal of human hepatotoxicity such as LiverTox will also be essential. Finally, the integration of data from divergent research platforms (i.e.proteomics, transcriptomics, metabolomics, genomics) using a systems biology approach as well as data derived from improved in vitro and in vivo test systems may provide an unprecedented opportunity to study human drug metabolism and idiosyncratic DILI.
Supplementary Material
Supplementary Table 1. Causality assessment methods used in DILI studies
Supplementary Table 2. Genome wide association studies of DILI susceptibility
Acknowledgments
Dr. Fontana is a NIH funded investigator with research support as a principal investigator in and the Drug Induced Liver Injury Network (2U01-DK065184-06) and the Acute Liver Failure Study Group (DK-U-01-58369)
Disclosures: Dr. Fontana has received research support from Vertex Pharmaceuticals and Gilead Sciences and served as a consultant to GlaxoSmithkline and Tibotec.
Abbreviations
- APAP
Acetaminophen
- ADR
Adverse drug reaction
- ALF
Acute liver failure
- ALFSG
Acute Liver Failure Study Group
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- DAMP
Damage associated molecular pattern
- DILI
Drug induced liver injury
- DILIN
Drug induced liver injury network
- EMR
Electronic medical records
- GLDH
glutatmate dehydrogenase
- GSTα
glutathione S-transferase alpha
- GWA
Genome wide association
- HCV
Hepatitis C virus
- HDS
Herbal and dietary supplement
- HEV
Hepatitis E virus
- HLA
Human leukocyte antigen
- HMGB1
high-mobility group box-1 protein
- iPS
inducible pleuripotent stem cells
- K-18
Keratin-18
- NIH
National Institutes of Health
- RUCAM
Roussel-Uclaf causality assessment method
- SDH
Sorbitol dehydrogenase
- SNP
Single nucleotide polymorphism
- TNF
Tumor necrosis factor
- ULN
upper limit of normal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guttmacher AE, Collins FS. Realizing the promise of genomics in biomedical research. JAMA. 2005;294:1399–1402. doi: 10.1001/jama.294.11.1399. [DOI] [PubMed] [Google Scholar]
- 2.Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 3.Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology. 2010;139:120–9 e18. doi: 10.1053/j.gastro.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. NEJM. 2002;347:472. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 5.Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359:727–732. doi: 10.1016/s0140-6736(02)07873-x. [DOI] [PubMed] [Google Scholar]
- 6.Zucman D, deTruchis P, Majerholc C, et al. Prospective screening for human leukocyte antigen-B*5701 avoids abacavir hypersensitivity reaction in the ethnically mixed French HIV population. J AIDS. 2007;45:1–3. doi: 10.1097/QAI.0b013e318046ea31. [DOI] [PubMed] [Google Scholar]
- 7.Watkins PB. Drug safety sciences and the bottleneck in drug development. Clin Pharmacol Ther. 2011;89:788–790. doi: 10.1038/clpt.2011.63. [DOI] [PubMed] [Google Scholar]
- 8.Watkins P, Seeff LB. Drug-induced liver injury: summary of a single topic clinical research conference. Hepatology. 2006;43:618–631. doi: 10.1002/hep.21095. [DOI] [PubMed] [Google Scholar]
- 9.Fontana RJ, Seeff LB, Andrade RJ, et al. Standardization of Nomenclature and causality assessment in drug-induced liver injury: Summary of a Clinical Research Workshop. Hepatology. 2010;52:730–742. doi: 10.1002/hep.23696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fontana RJ. Approaches to the Study of Drug-Induced Liver Injury. Clin Pharm Ther. 2010:1–4. doi: 10.1038/clpt.2010.100. [DOI] [PubMed] [Google Scholar]
- 11.Galan MV, Potts JA, Silverman AL, et al. The burden of acute non-fulminant drug-induced hepatitis in a United States tertiary care referral center. J Clin Gastroenterol. 2005;39:64–67. [PubMed] [Google Scholar]
- 12.Ostapowicz G, Fontana RJ, Schiodt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Int Med. 2002;137:947–954. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 13.Vuppalanchi R, Liangpunsakul S, Chalasani N. Etiology of new-onset jaundice: how often is it caused by idiosyncratic DILI in the United States? Am J Gastroenterol. 2006;101:1–5. doi: 10.1111/j.1572-0241.2006.01019.x. [DOI] [PubMed] [Google Scholar]
- 14.Meier Y, Cavallaro M, Roos M, et al. Incidence of drug-induced liver injury in medical inpatients. Eur J Clini Pharmacol. 2005;61:135–153. doi: 10.1007/s00228-004-0888-z. [DOI] [PubMed] [Google Scholar]
- 15.Duh MS, Walker AM, Kronlund KH. Descriptive epidemiology of acute liver enzyme abnormalities in the general population of central Massachusettes. Pharmacoepid Drug Saf. 1999;8:275–283. doi: 10.1002/(SICI)1099-1557(199907)8:4<275::AID-PDS427>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 16.Bielinski SJ, Chai HS, Pathak J, et al. Mayo Genome Consortia: A genotype-phenotype resource for genome-wide association studies with an application to the analysis of circulating bilirubin levels. Mayo Clin Proc. 2011;86:606–614. doi: 10.4065/mcp.2011.0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarty CA, Chisholm RL, Chute CG, et al. The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genomics. 2011;4:13. doi: 10.1186/1755-8794-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jinjuvadia K, Kwan W, Fontana RJ. Searching for a needle in a haystack: use of ICD-9-CM codes in drug induced liver injury. Am J Gastroenterol. 2007;102:2437–2443. doi: 10.1111/j.1572-0241.2007.01456.x. [DOI] [PubMed] [Google Scholar]
- 19.Overby CL, Pathak J, Gotteman O, et al. A collaborative approach to developing an electronic health record phenotyping algorithim for drug induced liver injury. J Am Med Informatics. 2013 doi: 10.1136/amiajnl-2013-001930. (accepted)-eprint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sembera S, Lammert C, Talwalkar JA, et al. Frequency, clinical presentation, and outcomes of drug-induced liver injury after liver transplantation. Liver Transpl. 2012;18:803–810. doi: 10.1002/lt.23424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fontana RJ, Watkins PB, Bonkovsky HL, et al. Rationale, design, and conduct of the Drug induced Liver Injury Network Prospective study. Drug Safety. 2009;32:55–68. doi: 10.2165/00002018-200932010-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fontana RJ. In: Drug-Induced Liver Injury Networks. 3rd. Kaplowitz, DeLeve, editors. Elsevier; 2013. pp. 713–723. [Google Scholar]
- 23.Andrade RJ, Lucena MJ, Fernandez MC, et al. Drug induced liver injury: an analysis of 461 incidences submitted to the Spanish registry over a 10 year period. Gastroenterology. 2005;129:512–521. doi: 10.1016/j.gastro.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Bjornsson ES, Bergmann OM, Bjornsson HK, et al. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the General population of Iceland. Gastroenterology. 2013;144:1419–1425. doi: 10.1053/j.gastro.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Molookhia M, McKeigue P. EUDRAGENE: European collaboration to establish a case-control DNA collection for studying the genetic basis of adverse drug reactions. Pharmacogenomics. 2006;7:633–8. doi: 10.2217/14622416.7.4.633. [DOI] [PubMed] [Google Scholar]
- 26.Takikawa H, Murata Y, Horiike N, et al. Drug-induced liver injury in Japan: an analysis of 1676 cases between 1997 and 2006. Hepatol Res. 2009;39:427–431. doi: 10.1111/j.1872-034X.2008.00486.x. [DOI] [PubMed] [Google Scholar]
- 27.Aithal GP, Watkins PB, Andrade RJ, et al. Case definition and phenotype standardization in drug-induced liver injury. Clin Pharm Ther. 2011;89:1–20. doi: 10.1038/clpt.2011.58. [DOI] [PubMed] [Google Scholar]
- 28.Agarwal V, McHutchison JG, Hoofnagle JH. Important elements for the diagnosis of drug-induced liver injury. Clin Gastro Hep. 2010;8:463–470. doi: 10.1016/j.cgh.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rockey DC, Seeff LB, Rochon J, et al. Causality assessment in drug-induced liver injury using a structured expert opinion process: comparison to the Roussel-Uclaf Assessment method. Hepatology. 2010;51:2117–2126. doi: 10.1002/hep.23577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pirmohamed M, Aithal GP, Behr E, et al. The phenotype standardization Project: Improving pharmacogenetic studies of serious adverse drug reactions. Clin Pharm Ther. 2011;89:784–785. doi: 10.1038/clpt.2011.30. [DOI] [PubMed] [Google Scholar]
- 31.Hoofnagle JH, Serrano J, Knoben JE, Navarro VJ. LiverTox: A website on Drug-Induced. Liver Injury Hepatology. 2013:873–874. doi: 10.1002/hep.26175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chalasani N, Fontana RJ, Bonkovsky HL, et al. Causes, clinical features, and outcomes from a prospective study of drug induced liver injury in the United States. Gastroenterology. 2008;135:1924–1934. doi: 10.1053/j.gastro.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sgro C, Clinard F, Quazir K, et al. Incidence of drug-induced hepatic injuries: A French Population-based study. Hepatology. 2002;36:451–455. doi: 10.1053/jhep.2002.34857. [DOI] [PubMed] [Google Scholar]
- 34.Navarro VJ, Barnhart HX, Bonkovsky HL, et al. The rising burden of Herbal and Dietary Supplement induced hepatotoxicity in the USA (Abstract) Hepatology. 2013 Submitted. [Google Scholar]
- 35.Galati G, Lin A, Sultan AM, O'Brien PJ. Cellular and in vivo hepatotoxicity caused by green tea phenolic acids and catechins. Free Radic Biol Med. 2006;40:570–80. doi: 10.1016/j.freeradbiomed.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 36.Navarro VJ, Barnhart HX, Bonkovsky HL, Hwang S. Green tea extract containing herbal and dieteary supplements are frequently mislabelled in the US: The US DILIN experience (Abstract) Hepatology. 2011;54 #336. [Google Scholar]
- 37.Davern TJ, Chalasani N, Fontana RJ, et al. Role of acute hepatitis E in suspected drug-induced liver injury. Gastroenterology. 2011;141:1665–1672. doi: 10.1053/j.gastro.2011.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dalton HR, Fellows HJ, Stableforth W, et al. The role of hepatitis E virus testing in drug-induced liver Injury. Aliment Pharmacol Ther. 2007;26:1429–1435. doi: 10.1111/j.1365-2036.2007.03504.x. [DOI] [PubMed] [Google Scholar]
- 39.Hoofnagle JH, Nelson KE, Purcell RH, Hepatitis E. NEJM. 2012;367:1237–1244. doi: 10.1056/NEJMra1204512. [DOI] [PubMed] [Google Scholar]
- 40.Fontana RJ, Gu J, Barnhart H, et al. Subject race and liver biochemical profile are strongly associated with clinical outcomes and chronic liver injury: Results from the DILIN Prospective Study (Abstract) Hepatology. 2012;56:600A. [Google Scholar]
- 41.Andrade RJ, Lucena MI, Kaplowitz N, et al. Outcome of acute idiosyncratic drug-induced liver injury: Long-term follow-up in a hepatotoxicity registry. Hepatology. 2006;44:1581–1588. doi: 10.1002/hep.21424. [DOI] [PubMed] [Google Scholar]
- 42.Bjornsson ES, Davidsdottir L. The long-term follow-up after idiosyncratic drug-induced liver injury with jaundice. J Hepatology. 2009;50:511–517. doi: 10.1016/j.jhep.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 43.Reuben A, Koch DG, Lee WM Acute Liver Failure Study Group. Drug induced acute liver Study Group: results of a US Multicenter, prospective study. Hepatology. 2010;52:2065–2076. doi: 10.1002/hep.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anstee QM, Day CP. S-adenosylmethionine (SAMe) therapy in liver disease: A review of current evidence and clinical utility. J Hepatology. 2012;57:1097–1109. doi: 10.1016/j.jhep.2012.04.041. [DOI] [PubMed] [Google Scholar]
- 45.Patel SJ, Milwid JM, King KR, et al. Gap junction inhibition prevents drug-induced liver toxicity and fulminant hepatic failure. Nature Biotechnology. 2012:1–5. doi: 10.1038/nbt.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roth RA, Ganey PE. Intrinsic versus idiosyncratic drug-induced hepatotoxicity- Two villains or one? J Pharm Exp Ther. 2010;332:692–7. doi: 10.1124/jpet.109.162651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lammert C, Einarsson S, Saha C, et al. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: search for signals. Hepatology. 2009;47(6):2003–9. doi: 10.1002/hep.22272. [DOI] [PubMed] [Google Scholar]
- 48.Chen M, Borlak J, Tong W. High lipophilicity and high daily dose of oral medication are associated with significant risk for drug-induced liver injury. Hepatology. 2013;58:388–396. doi: 10.1002/hep.26208. [DOI] [PubMed] [Google Scholar]
- 49.Kaplowitz N. Avoiding idiosyncratic DILI: Two is better than one. Hepatology. 2013;58:15–17. doi: 10.1002/hep.26295. [DOI] [PubMed] [Google Scholar]
- 50.Lammert C, Bjornsson E, Niklasson A, Chalasani N. Oral medications with significant hepatic metabolism at higher risk for hepatic adverse events. Hepatology. 2010;51:615–620. doi: 10.1002/hep.23317. [DOI] [PubMed] [Google Scholar]
- 51.Low Y, Uehera T, Minowa Y, et al. Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem Res in Toxicology. 2011;24:1251–1262. doi: 10.1021/tx200148a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodgers AD, Zhu H, Fourches D, et al. Modeling liver-related adverse effects of drugs using nearest neighbor quantitative structure-activity relationship methods. Chem Res Toxicol. 2010;23:724–732. doi: 10.1021/tx900451r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaw PJ, Ganey PE, Roth RA. Idiosyncractic drug-induced liver injury and the role of inflammatory stress with an emphasis on an animal model of trovofloxacin hepatotoxicity. Toxicological Sciences. 2010;118:7–18. doi: 10.1093/toxsci/kfq168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lewis JH, Mortensen ME, Zweig S, et al. Efficacy and safety of high-dose pravastatin in hypercholesterolemic patients with well compensated chronic liver disease: Results of a prospective, randomized, double-blind placebo-controlled multicenter trial. Hepatology. 2007;46:1453–1463. doi: 10.1002/hep.21848. [DOI] [PubMed] [Google Scholar]
- 55.FDA Drug Safety Communication: Important Safety label changes to Cholesterol lowering statin drugs. 2013 Apr 13th; http://www.fda.gov/Drugs/DrugSafety/ucm293101.htm.
- 56.Stewart JD, Horvath R, Baruffini E, et al. Polymerase G gene, POLG, determines the risk of sodium-valproate-induced liver toxicity. Hepatology. 2010;52:1791–1796. doi: 10.1002/hep.23891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kleiner D, Chalasani N, Lee WML, et al. Hepatic histological findings in suspected drug-induced liver injury: Systematic evaluation and clinical associations. Hepatology. 2013 doi: 10.1002/hep.26709. (Accepted Aug 2013). DOI xxxx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bjornsson E, Kalaitzakis E, Olsson R. The impact of eosinophilia and hepatic necrosis on prognosis in patients with drug-induced liver injury. Alim Pharm Ther. 2007;25:1411–1421. doi: 10.1111/j.1365-2036.2007.03330.x. [DOI] [PubMed] [Google Scholar]
- 59.Katoonizadeh A, Nevens F, Verslype C, et al. Liver regeneration in acute severe liver impairment: a clincopathological correlation study. Liver Int. 2006;26:1225–1233. doi: 10.1111/j.1478-3231.2006.01377.x. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki A, Brunt EM, Kleiner DE, et al. The use of liver biopsy evaluation in the discrimination of idiopathic autoimmune hepatitis versus drug-induced liver injury. Hepatology. 2011;54:931–939. doi: 10.1002/hep.24481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Foureau D, Walling T, Maddukuri V, et al. Comparative analysis of portal hepatic infiltrating leukocytes in acute drug-induced liver injury, idiopathic autoimmune and viral hepatitis. Liver International. 2013 doi: 10.1111/cei.12558. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaplowitz N. Drug induced Liver Injury. Clin Infect Dis. 2004;38(Suppl 2):S44–48. doi: 10.1086/381446. [DOI] [PubMed] [Google Scholar]
- 63.Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen0induced liver injury. J Leuk Biol. 2008;84:14, 10–21. doi: 10.1189/jlb.0308173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krishnan P, Feng ZZ, Gordon S. Prolonged intrahepatic cholestasis and renal failure secondary to anabolic androgenic steroid-enriched dietary supplements. A Review of the Literature. J Clin Gastro. 2009;43:672–675. doi: 10.1097/MCG.0b013e318188be6d. [DOI] [PubMed] [Google Scholar]
- 65.Robinson MJ, Rywlin AM. Tetracycline-associated fatty liver in the male. Report of an autopsied case. Am J Dig Dis. 1970;15:857–862. doi: 10.1007/BF02236049. [DOI] [PubMed] [Google Scholar]
- 66.Bleeker-Rovers C, Kadir S, van Leusen R, Richter C. Hepatic steatosis and lactic acidosis caused by stavudine in an HIV-infected patient. Neth J Med. 2000;57:190–193. doi: 10.1016/s0300-2977(00)00064-4. [DOI] [PubMed] [Google Scholar]
- 67.Larosche I, Letteron P, Fromenty B, et al. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J Pharmacol Exp ther. 2007;321:526–535. doi: 10.1124/jpet.106.114546. [DOI] [PubMed] [Google Scholar]
- 68.Narita M, Oussoultzoglou E, Chenard MP, et al. Liver injury due to chemotherapy-induced sinusoidal obstruction syndrome is associated with sinusoidal capillarization. Ann Surg Oncol. 2012;19:2230–2237. doi: 10.1245/s10434-011-2112-6. [DOI] [PubMed] [Google Scholar]
- 69.Vieto NO, George BJ. Oxaliplatin-induced hepatocellular injury and ototoxicity: a review of the literature and report of unusual side effects of a commonly used chemotherapeutic agent. J Oncol Pharm Pract. 2012;18:355–359. doi: 10.1177/1078155212437901. [DOI] [PubMed] [Google Scholar]
- 70.Toxicity testing in the 21st Century: A Vision and a Strategy. National Academies Press; Washington, DC: 2007. Committee on Toxicity Testing and Assessment of Environmental Agents, National Research Council. [Google Scholar]
- 71.Grompe M, Strom S. Mice with Human livers. Gastroenterology. 2013 Sep 13; doi: 10.0153/j.gastro2013.09.009. Pii:S0016-5085 (13)01308-5. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 72.McGill MR, Yan HU, Ramachandran A, et al. HepaRG cells: A human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology. 2011;53:974–982. doi: 10.1002/hep.24132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Si-Tayeb K, Fallon KN, Nagaoka M, et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Song Z, Cai J, Liu Y, et al. Efficient generation of hepatocyte-like cells from human induced pleuripotent stem cells. Cell Res. 2009;19:1233–42. doi: 10.1038/cr.2009.107. [DOI] [PubMed] [Google Scholar]
- 75.Guguen-Guillouzo C, Corlu A, Guillouzo A. Stem cell-derived hepatocytes and their use in toxicology. Toxicology. 2010;270:3–9. doi: 10.1016/j.tox.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 76.Jozefczuk W, Prigione A, Chavez L, et al. Comparative analysis of human embryonic stem cell and induced pluripotent stem-cell derived hepatocyte like cells reveals current drawbacks and possible strategies for improved differentiation. Stem Cells Dev. 2011 Jul;20(7):1259–75. doi: 10.1089/scd.2010.0361. [DOI] [PubMed] [Google Scholar]
- 77.Faulkner L, Martinsson K, Santoyo-Castelazo A, et al. The development of In Vitro culture methods to characterize primary T-cell responses to drugs. Tox Sciences. 2012;127:150–158. doi: 10.1093/toxsci/kfs080. [DOI] [PubMed] [Google Scholar]
- 78.El-Chaiesh S, Monshi MM, Whitaker P, et al. Characterization of the antigen specificity of T-cell clones from piperacillin-hypersensitive patients with cystic fibrosis. J Pharm Exp Ther. 2012;341:597–610. doi: 10.1124/jpet.111.190900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chessman D, Kostenko L, Lethborg T, et al. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity. 2008;28:822–832. doi: 10.1016/j.immuni.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 80.Porebski G, Gschwend-azwodniak A, Pichler WJ. In vitro diagnosis of T cell-mediate drug allergy. Clin Exp Allergy. 2011;4:461–470. doi: 10.1111/j.1365-2222.2011.03701.x. [DOI] [PubMed] [Google Scholar]
- 81.Watkins PB. Biomarkers for the Diagnosis and Management of Drug Induced Liver Injury. Sem Liv Dis. 2009;29:394–400. doi: 10.1055/s-0029-1240008. [DOI] [PubMed] [Google Scholar]
- 82.Green RM, Flamm S. AGA technical review on the evaluation of liver chemistry tests. Gastroenterology. 2002;123:1367–1384. doi: 10.1053/gast.2002.36061. [DOI] [PubMed] [Google Scholar]
- 83.Sayuk GS, Elwing JE, Lisker-Melman M. Hepatic glycogenosis: an underrecognized source of abnormal liver function tests? Dig Dis Sci. 2007;52:936–938. doi: 10.1007/s10620-006-9430-8. [DOI] [PubMed] [Google Scholar]
- 84.Senior JR. Monitoring for hepatotoxicity: what is the predictive value of liver “function” tests? Clin Pharmacol Ther. 2009;85:331–334. doi: 10.1038/clpt.2008.262. [DOI] [PubMed] [Google Scholar]
- 85.Mitchell JR, Long MW, Thorgeisson UP, et al. Acetylation rates and monthly liver function tests during one year of isoniazid preventive therapy. Chest. 1975;68:181–190. doi: 10.1378/chest.68.2.181. [DOI] [PubMed] [Google Scholar]
- 86.M’Kada H, Perazzo H, Munteanu M, et al. Real time identification of drug-induced liver injury (DILI) through daily screening of ALT results: A prospective, pilot cohort Study. PLOS one. 2012;7:1–7. doi: 10.1371/journal.pone.0042418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rutherford AE, Hynan LS, Borges CB, et al. Serum apoptosis markers in acute liver failure: a pilot study. Clin Gastroenterol Hepatol. 2007;5(12):1477–83. doi: 10.1016/j.cgh.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 88.Bantel H, Lugering A, Hidemnan J, et al. Detection of apoptotic caspase activation in sera from patients with chronic HCV infection is associated with fibrotic liver injury. Hepatology. 2004;40:1078–87. doi: 10.1002/hep.20411. [DOI] [PubMed] [Google Scholar]
- 89.Rutherford A, King LY, Hynan LS, et al. Development of an accurate index for predicting outcomes of patients with acute liver failure. Gastroenterology. 2012;143(5):1237–43. doi: 10.1053/j.gastro.2012.07.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chung RT, Stravitz T, Fontana RJ, et al. Pathogenesis of Liver Injury in Acute Liver Failure. Gastroenterology. 2012;143:e1–e7. doi: 10.1053/j.gastro.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Monreal M, Lafoz E, Salvador R, et al. Adverse effects of three different forms of heparin therapy: thrombocytopenia, increased transaminases, and hyperkalemia. Eur J Clin Pharmaol. 1989;37:415–418. doi: 10.1007/BF00558513. [DOI] [PubMed] [Google Scholar]
- 92.Harrill AH, Roach J, Fier I, et al. The effects of heparins on the liver: Application of mechanistic serum biomarkers in a randomized study in healthy volunteers. Clin Pharm Ther. 2012;92:214–220. doi: 10.1038/clpt.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pisetsky DS, Jiang W. Roll of Toll-like receptors in HGMB1 release from macrophages. Ann NY Acad Sci. 2007;1109:58–65. doi: 10.1196/annals.1398.008. [DOI] [PubMed] [Google Scholar]
- 94.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 95.Uetrecht J. Mechanisms of Immune-mediated liver Injury. Toxicol Sci. 2010;115:307–321. doi: 10.1093/toxsci/kfq009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized, controlled trial. JAMA. 2006;296:87–93. doi: 10.1001/jama.296.1.87. [DOI] [PubMed] [Google Scholar]
- 97.Wetmore BA, Brees DJ, Singh R, et al. Quantitative analyses and transcriptomic profiling of circulating messenger RNAs as biomarkers of rat liver injury. Hepatology. 2010;51:2127–2139. doi: 10.1002/hep.23574. [DOI] [PubMed] [Google Scholar]
- 98.Antoine D, Dear JW, Lewis PS, et al. Mechanistic biomarkers provide early and sensitive detection of acetaminophen-induced acute liver injury at first presentation to Hospital. Hepatology. 2013 doi: 10.1002/hep.26294. on line. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Antoine DJ, Jenkins RE, Dear JW, et al. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatology. 2012;56:1070–1079. doi: 10.1016/j.jhep.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 100.Davern TJ, James LP, Hinson JA, et al. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology. 2006;130:687–694. doi: 10.1053/j.gastro.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 101.Khandelwal N, James LP, Sanders C, et al. Unrecognized acetaminophen toxicity as a cause of indeterminate acute Liver failure. Hepatology. 2011;53:567–576. doi: 10.1002/hep.24060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.James LP, Simpson P, Russo M, et al. Detection of acetaminophen-protein adducts in serum during therapeutic exposure to acetaminophen in healthy volunteers (Abstract) Hepatology. 2007;46(Suppl 1):812A. [Google Scholar]
- 103.Antoine DJ, William DP, Parke BK. Understanding the role of reactive metabolites in drug-induced hepatotoxicity. State of the science. Exp Drug Met Tox. 2008;4:1415–1427. doi: 10.1517/17425255.4.11.1415. [DOI] [PubMed] [Google Scholar]
- 104.Bell LN, Vuppalanchi R, Watkins PB, et al. Serum proteomic profiling in patients with drug-induced liver injury APT. 2012;35:600–612. doi: 10.1111/j.1365-2036.2011.04982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maniratanachote R, Shibata A, Kaneko S, et al. Detection of autoantibody to aldolase B in sera from patients with troglitazone-induced liver dysfunction. Toxicology. 2005;216:15–23. doi: 10.1016/j.tox.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 106.Andersson U, Lindberg J, Wang S, et al. A systems biology approach to understanding elevated serum alanine transaminase levels in a clinical trial with ximelagatran. Biomarkers. 2009;14:572–586. doi: 10.3109/13547500903261354. [DOI] [PubMed] [Google Scholar]
- 107.Shi Q, Hong H, Senior J, et al. Biomarkers for drug-induced liver injury. Exp rev Gastroenterol Hepatol. 2010;4:225–34. doi: 10.1586/egh.10.8. [DOI] [PubMed] [Google Scholar]
- 108.Steuerwald NM, Foureau DM, Norton HJ, et al. Profiles of serum cytokines in acute drug-induced Liver Injury and their prognostic significance. doi: 10.1371/journal.pone.0081974. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hammerich L, Heymann F, Tacke F. Role of IL-17 and Th17 cells in liver diseases. Clin Deve Immunol. 2011;345:803. doi: 10.1155/2011/345803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li J, Zhu X, Liu F, et al. Cytokine and autoantibody patterns in acute liver failure. J Immunotoxicol. 2010;7:157–164. doi: 10.3109/15476910903501748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lobenhofer EK, Auman JT, Blackshear PE, et al. Gene expression response in target organ and whole blood varies as a function of target organ injury phenotype. Genome Biol. 2008;9:R100. doi: 10.1186/gb-2008-9-6-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bushel PR, Heinloth AN, Li J, et al. Blood gene expression signatures predict exposure levels. PNAS. 2007;104:18211–18216. doi: 10.1073/pnas.0706987104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fannin RD, Russo M, O’Connell TM, et al. Acetaminophen dosing of humans results in blood Transcriptome and Metabolome changes consistent with impaired oxidative phosphorylation. Hepatology. 2010;51:227–236. doi: 10.1002/hep.23330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Winnike JH, Li Z, Wright FA, et al. Use of pharmco-metabonomics for early prediction of acetaminophen-induced hepatotoxicity in humans. Clin Pharm & Ther. 2010;88:45–50. doi: 10.1038/clpt.2009.240. [DOI] [PubMed] [Google Scholar]
- 115.Clayton TA, Baker D, Lindon JC, et al. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. PNAS. 2009;106:14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.O’Connell TM, Watkins PB. The application of metabonomics to predict Drug-induced Liver Injury. Clin Pharm Ther. 2010;88:384–399. doi: 10.1038/clpt.2010.151. [DOI] [PubMed] [Google Scholar]
- 117.Robertson DG, Watkins PB, Reily MD. Metabolomics in Toxicology: Preclinical and clinical applications. Tox Sciences. 2011;120:S146–S170. doi: 10.1093/toxsci/kfq358. [DOI] [PubMed] [Google Scholar]
- 118.Wang K, Zhang S, Marzolf B, et al. Circulating microRNA's, potential biomarkers for DILI. PNAS. 2009;106:4402–4407. doi: 10.1073/pnas.0813371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McGill MR, Sharpe MR, Williams CD, et al. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 2012;122:1574–1583. doi: 10.1172/JCI59755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Starkey LPJ, Dear J, Platt V, et al. Circulating microRNA's as potential markers of human drug-induced liver injury. Hepatology. 2011;54:1767–1776. doi: 10.1002/hep.24538. [DOI] [PubMed] [Google Scholar]
- 121.Huang YS, Chern HD, Su WJ, et al. Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatitis. Hepatology. 2002;35:883–9. doi: 10.1053/jhep.2002.32102. [DOI] [PubMed] [Google Scholar]
- 122.Huang YS, Chern HD, Su WJ, et al. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug-induced hepatitis. Hepatology. 2003;37:924–930. doi: 10.1053/jhep.2003.50144. [DOI] [PubMed] [Google Scholar]
- 123.Hautekeete ML, Horsmans Y, Van Waeyenberge C, et al. HLA association of amoxicillin clavuanate- induced hepatitis. Gastroenterology. 1999;117:1181–6. doi: 10.1016/s0016-5085(99)70404-x. [DOI] [PubMed] [Google Scholar]
- 124.Daly AK, Day CP. Genetic association studies in Drug-Induced Liver Injury. Sem Liv Dis. 2009;29:400–411. doi: 10.1055/s-0029-1240009. [DOI] [PubMed] [Google Scholar]
- 125.Daly AK, Donaldson PT, Bhatnagar P, et al. DILIGEN Study; International SAE Consortium HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41:816–9. doi: 10.1038/ng.379. [DOI] [PubMed] [Google Scholar]
- 126.Kindmark A, Jawaid A, Harbron CG, et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J. 2008;8:186–95. doi: 10.1038/sj.tpj.6500458. [DOI] [PubMed] [Google Scholar]
- 127.Singer JB, Lewitzky S, Leroy E, et al. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat Genet. 2010;42(8):711–4. doi: 10.1038/ng.632. [DOI] [PubMed] [Google Scholar]
- 128.Spraggs CF, Budde LR, Briley LP, et al. HLA-DQA1*01:01 is a major risk factor for lapatanib-induced hepatotoxicity in women with advanced breast cancer. J Clin Onc. 2011;29:667–673. doi: 10.1200/JCO.2010.31.3197. [DOI] [PubMed] [Google Scholar]
- 129.Russmann S, Kaye JA, Jick SS, et al. Risk of cholestatic liver disease associated with flucloxacillin and flucloxacillin prescribing habits in the UK: cohort study using data from the UK General Practice Research Database. Br J Clin Pharmacol. 2005;60:76–82. doi: 10.1111/j.1365-2125.2005.02370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Phillips EJ, Mallal SA. HLA-B*5701 and flucloxacillin associated drug-induced liver disease. AIDS. 2013;27:1–492. doi: 10.1097/QAD.0b013e32835ca9d5. [DOI] [PubMed] [Google Scholar]
- 131.Andrews E, Armstrong M, Tugwood J, et al. A role for the pregnane X receptor in Flucloxacillin-induced liver injury. Hepatology. 2010;51:1656–1664. doi: 10.1002/hep.23549. [DOI] [PubMed] [Google Scholar]
- 132.Monshi M, Faulkner L, Gibson A, et al. Human leukocyte antigen (HLA)-B*57:01-restricted activation of drug-specific T-cells provides the immunological basis for flucloxacillin-induced liver injury. Hepatology. 2013;57:727–739. doi: 10.1002/hep.26077. [DOI] [PubMed] [Google Scholar]
- 133.Illing PT, Vivain JP, Dudek NL, et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature. 2012;486:554–558. doi: 10.1038/nature11147. [DOI] [PubMed] [Google Scholar]
- 134.Wuillemin N, Adam J, Fontana S, et al. HLA haplotype determines hapten or p-I T cell reactivity to flucloxacillin. J Immunology. 2013;190:4956–4964. doi: 10.4049/jimmunol.1202949. [DOI] [PubMed] [Google Scholar]
- 135.Urban TJ, Shen Y, Stolz A, et al. Limited contribution of common genetic variants to risk of liver injury due to a variety of drugs. Pharmacogenetic and Genomics. 2012;22:784–795. doi: 10.1097/FPC.0b013e3283589a76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lucena MI, Molokhia M, Shen Y, Urban TJ, Aithal GP, Andrade RJ, et al. Susceptibility to Amoxicillin-Clavulanate-Induced Liver Injury is Influenced by Multiple HLA Class I and II Alleles. Gastroenterology. 2011;131:338–347. doi: 10.1053/j.gastro.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Urban TJ, Goldstein DB, Watkins PB. Genetic basis of susceptibility to drug-induced liver injury: what have we learned and where do we go from here? Pharmacogenomics. 2012;13:1–4. doi: 10.2217/pgs.12.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ventham NT, Kennedy NA, Nimmo ER, et al. Beyond gene discovery in inflammatory bowel disease: The emerging role of epigenetics. Gastroenterology. 2013;145:293–308. doi: 10.1053/j.gastro.2013.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Parham P, Onta T. Population biology of antigen presentation by MHC class I molecules. Science. 1996;272:67–74. doi: 10.1126/science.272.5258.67. [DOI] [PubMed] [Google Scholar]
- 140.Bharadwaj M, Iling P, Theodossis A, et al. Drug hypersensitivity and human leukocyte antigens of the major histocompatablity complex. Annu Rev Pharmacol Toxicol. 2012;52:401–431. doi: 10.1146/annurev-pharmtox-010611-134701. [DOI] [PubMed] [Google Scholar]
- 141.Chen P, Lin JJ, Lu CS, et al. Carbamazepine-induced toxic effects and HLA-B*5802 screening in Taiwan. NEJM. 2011;364:1126–1133. doi: 10.1056/NEJMoa1009717. [DOI] [PubMed] [Google Scholar]
- 142.McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. NEJM. 2011;364:1134–1143. doi: 10.1056/NEJMoa1013297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Markle JG, Frank DN, Mortin-Toth S, et al. Sex differences in the gut microbiome drive hormone dependent regulation of autoimmunity. Science. 2013;339:1084–1088. doi: 10.1126/science.1233521. [DOI] [PubMed] [Google Scholar]
- 144.Henao-Mejia J, Elinav E, Jin C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hirschfield GM, Chapman RW, Karlsen TH, et al. The Genetics of complex cholestatic disorders. Gastroenterology. 2013;144:1357–1374. doi: 10.1053/j.gastro.2013.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Holmes E, Li JV, Marchesi JR<, et al. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Met. 2012;16:559–564. doi: 10.1016/j.cmet.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 147.Zheng X, Zhao A, Xie G, et al. Melamine-induced renal toxicity is mediated by the gut microbiota. Sci Trans Med. 2013;5:172. doi: 10.1126/scitranslmed.3005114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Causality assessment methods used in DILI studies
Supplementary Table 2. Genome wide association studies of DILI susceptibility