

Abstract
Purpose of review
This review highlights recent studies undertaken to further advance the search for successful approaches to eradicate HIV infection.
Recent findings
Small pharmacological compounds such as histone deacetylase inhibitors, inhibitors of bromodomain and extraterminal proteins such as JQ1, and protein kinase C activators such as bryostatin and prostratin are proposed as putative candidates for inducing the expression of latent HIV in a so-called ‘shock and kill’ or ‘kick and kill’ strategy for HIV eradication. However, in order to achieve viral clearance, it is thought likely these compounds will have to be administered in concert with strategies that augment clearance of virus-infected cells in patients that have long been aviremic on successful antiretroviral therapy (ART). Several candidate therapies for this purpose are at hand, such as therapeutic HIV vaccines – recently shown to promote robust cytotoxic T cell responses and blunt viral rebound after ART interruption in clinical studies. HIV-infected patients treated during early infection may be ideal candidates for early studies to test these strategies, as early ART has been shown to limit the establishment of an HIV reservoir.
Summary
HIV latency is multifactorial and thus the eradication of HIV infection may require multiple approaches. Translational efforts employing pharmacological methods to target HIV latency should evaluate in parallel the additional potential benefits of invigorating the immune response of HIV-infected individuals, and limiting the size of the reservoir via early ART.
Keywords: cure, HIV, latency, reactivation, reservoirs
INTRODUCTION
The advent of combinatorial antiretroviral therapy (ART) has significantly decreased HIV morbidity and has made it possible for HIV-infected individuals to live otherwise normal lives. However, in many developing countries nearly 50% of adults and children who are eligible for HIV treatment still lack access to ART and care [1], and HIV continues to kill a significant number of people worldwide [1]. Furthermore, although there has been a decline in the number of individuals that become infected each year, worldwide, there were still 2.5 million new infections in 2011 alone [1]. Thus, given the current number of individuals living with HIV, the number of new annual infections, and potential for long-term ART to induce some toxicities [2], the recent prominence of efforts to develop therapies that could eradicate HIV infection, a ‘cure for AIDS,’ has obvious appeal. To date there have been two documented cases of a potential cure achieved in two different individuals, known in the popular press as the Berlin patient and the Mississippi baby (discussed in more detail below). In the former case, the patient suffered from acute myeloid leukemia and underwent radiation therapy, obliterating his immune cells, followed by engraftment with allogeneic hematopoietic stem cells from a donor homozygous for the delta three-null mutation of the chemokine receptor, CCR5. The patient was thereby engrafted with immune cells refractory to new rounds of HIV infection and in addition graft-versus-host disease very likely contributed to depletion of his reservoir [3].
In the case of the Mississippi baby, full triple combination ART was given to an infant born of an untreated, HIV-infected mother immediately after birth, possibly preventing the establishment of a reservoir [4▪▪]. In both instances a cure was achieved in which the virus has been thus far undetectable. However, the unique circumstances surrounding both cases would make it highly implausible to achieve on a universal scale, and research exploring other modalities of depleting the viral reservoir is currently a high priority.
As efforts to eradicate HIV infection are intensified, it will be important to address the contribution of different immune cell types to the quiescent reservoir. It has recently been reported that gamma/delta T cells harbor silent provirus [5]. However, to date, memory resting CD4+ T cells remained the most well characterized reservoir of quiescent HIV [6-9].
As curative therapies are applied in the clinic, it will also be critical to develop or improve upon pre-existing methods to evaluate the effectiveness of these strategies. The current gold standard for estimating the size of the HIV reservoir is the viral outgrowth assay performed on highly purified resting CD4+ T cells [6,8]. Although this assay measures replicative competent virus, it is costly, labor intensive and requires a large input of cells. Alternative methods to measure the reservoir involve measuring total or integrated DNA from peripheral blood mononuclear cells (PBMCs) or resting CD4 T cells. Given that a large fraction of infected resting CD4 T cells harbor defective provirus [10,11], a disadvantage of PCR-based assays is that they do not distinguish between replication competent and defective virus. In a recent study, Eriksson et al. [12▪▪] compared the viral outgrowth assay to various PCR-based methods to measure the latent reservoir. Using digital droplet PCR to measure total HIV DNA in PBMCs and resting CD4 T cells, the authors observed frequencies of infection that were on average two logs greater than what was measured in the outgrowth assay, possibly reflecting measurement of defective virus by PCR assays. Thus, therapies to eradicate HIV infection will likely need to incorporate assays that reflect depletion of replication competent viruses to measure the efficacy of interventions.
HIV latency is multifaceted, and thus it is widely assumed that targeting the HIV reservoir will likely require combinatorial approaches [13,14]. The purpose of this review is to bring to light the many different approaches that have been undertaken in the past year to purge the latent HIV reservoir.
PHARMACOLOGICAL REACTIVATION OF VIRUS AS A CURE STRATEGY
Integration of the HIV genome into the host cellular genes is a critical part of the virus life cycle. As such, once integrated the HIV genome is subject to the controls of the chromatin environment and a variety of different host transcription factors [13-15]. Following initiation of transcription from the HIV long terminal repeat (LTR), the RNA polymerase II (pol II) complex is stalled owing to the actions of the negative elongation factor (NELF) and 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole sensitivity-inducing factor (DSIF) resulting in the production of short abortive transcripts. The viral protein Tat binds to the HIV TAR element and recruits the host positive transcription elongation factor b (PTEF-b) that in turns phosphorylates NELF and DSIF as well as pol II, leading to transcriptional elongation [16,17]. The bromodomain containing 4 (BRD4) protein is a member of the bromodomain extraterminal (BET) family of transcription regulators that binds acetyl lysine residues on histones and nonhistone proteins [18]. Recent studies by several investigators expanded on previous reports that BRD4 competes with Tat for the binding of PTEF-b [19,20▪▪,21▪,22]. It was demonstrated that the small molecule BET inhibitor JQ1 enhanced the association of Tat with PTEF-b and increased HIV production in cell line models of HIV latency. Furthermore, JQ1 was found to synergize with histone deacetylase (HDAC) inhibitors to reactivate HIV from latency [19,20▪▪,21▪,22,23]. However, Boehm et al. [20▪▪] demonstrated that the activity of JQ1 rested on its ability to inhibit another BET family member, BRD2, that paradoxically acts as another suppressor of HIV expression in the absence of HIV Tat. Thus, JQ1 and other BET inhibitors might be additional candidates that can be added to the antilatency arsenal (Fig. 1) [24]. JQ1 was originally developed as an anticancer agent. Although this compound has failed to advance into studies involving human controls, second generation JQ1-like compounds are in development and are likely to be used in clinical trials, therefore making it a very real possibility that such compounds could in turn be used to test the ability of BET inhibitors to disrupt HIV latency in a clinical setting.
FIGURE 1.
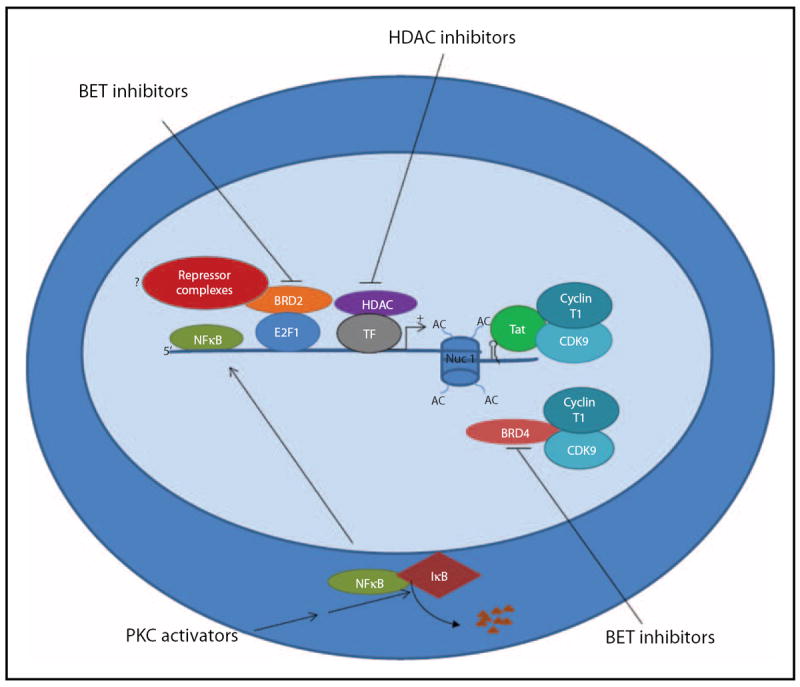
HIV induction by PKC activators and HDAC and BET inhibitors: NFκB is sequestered in the cytoplasm by IκB. PKC activators such as Prostratin and Bryostatin induce HIV from latency through activation of the PKC/NFκB signaling pathway leading to degradation of IκB and translocation of NFκB to the nucleus where it binds to NFκB biding sites at the HIV LTR and activates transcription. HDACs are recruited to the HIV LTR by several different transcription factor complexes (designated here as TF for simplicity) and promote transcriptional repression of the HIV LTR in part by removing acetyl groups from the lysine tails of histones associated with the nucleosome bound provirus, thus increasing the tight interaction of histones with proviral DNA and thereby creating an environment less conducive for transcription. Inhibition of HDAC activities by compounds such as Vorinostat and Panobinostat lead to acetylation (AC) of lysine residues on histones tails resulting in a relaxation of the interaction of histones and DNA, facilitating the binding of positive transcription factors that induce HIV transcription initiation and elongation. Finally, inhibition of BET protein family members BRD2 and BRD4 by BET inhibitors such as JQ1 leads to HIV promoter activation through separate modes of action. BRD4 competes with Tat for the binding of PTEF-b (composed of CDK9 and cyclin T1). Inhibition of BRD4 allows enhanced Tat binding to PTEF-b and transactivation that is required for productive transcription elongation. Although less clear, it is hypothesized that inhibition of BRD2 may block recruitment of repressive transcription complexes (in exchange for activator complexes), thus promoting HIV transcription. BET, bromodomain extraterminal; BRD, bromodomain containing; HDAC, histone deacetylase; LTR, long terminal repeat; PKC, protein kinase C; PTEF-b, positive transcription elongation factor b.
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is a ubiquitous regulatory protein involved in promoting transcription of many genes. NF-κB is maintained in an in active state in the cytoplasm via its association with IκB proteins. Phosphorylation of IκB proteins leads to their degradation and the release of NF-κB which in turn translocates to the nucleus and binds to NF-κB response elements and promotes transcription by recruiting other coactivators. As NF-κB activation has been shown to induce LTR transcription, a recent study by Fernandez et al. [25▪] sought to delineate the roles of the different IκB isoforms in regulating NF-κB activation and subsequently expression of HIV. The authors found that siRNA knockdown of IκBα and IκBε leads to increased transcription at the LTR. Interestingly, IκBε knockdown produced the most expression of virus. Ironically, IκBε is expressed predominantly in T cells of the thymus, spleen and lymph nodes [26], sites that represent potential HIV reservoirs. This suggests that selective targeting of IκBε may prove to be an effective mechanism to deplete persistent HIV infection.
One of the many proposed mechanisms to deplete the HIV reservoirs involves pharmacological modulation of signaling pathways involved in HIV replication. Protein kinase C (PKC) is one such pathway. The medicinal potential of the PKC activator prostratin was first recognized in the 1990s when it was discovered that it was the active component of the traditional medicinal plant, Homalanthus nutans [27]. Prostratin has been shown to reactivate HIV from latency via PKC-mediated phosphorylation of IκB leading to the activation of NFκB (Fig. 1). Activated NF-κB translocates to the nucleus where it binds NF-κB binding sites at the HIV promoter, promoting transcription [28]. Prostratin is also believed to downregulate CD4 and CXCR4, and in some cases CCR5, on the surface of cells [29,30], suggesting that this candidate for antilatency therapy may reactivate virus and at the same time prevent de-novo infection of bystander cells. Until 2008, the difficulty of synthesizing prostratin from plant- based sources and the accompanying low yield had mostly limited its usage in preclinical trials. Extending on previous work that successfully increased the yield of plant-based prostratin, the Wender laboratory has recently described a high yield synthesis of prostratin analogues. These analogues were shown to be 100-fold more potent than prostratin in reactivating HIV from cell line models of latency and also from the resting CD4+ T cells of HIV-infected individuals [31▪▪].
Another potent activator of the PKC pathway is the macrolide bryostatin. Like prostratin, bryostatin has been shown to reactivate HIV in cell line models of HIV latency and is considered to be a key candidate to purge the HIV reservoir, especially given its prior usage as an investigational anticancer therapeutic in human clinical trials, thus making it more likely that the compound could easily be tested in humans for its ability to deplete HIV in vivo [32,33]. However, studies involving bryostatin have been limited thus far because of the cost associated with its synthesis, which is restricted by its structural complexity and low abundance of bryozoan from which it is synthesized [34]. Again, the Wender group recently described the synthesis of bryostatin analogues, the aptly named bryologs, that were shown to effectively reactivate HIV from latency at greater potency than bryostatin [35▪]. In addition, the bryologs were 1000-fold more potent than prostratin in activating the LTR, suggesting that the bryologs may even be more potent than the recently synthesized prostratin analogues described above. Whether or not the bryologs or the prostratin analogs will prove to be effective at depleting HIV in vivo remains to be determined.
Histone deacetylases (HDACs) maintain HIV in a transcriptionally silent state [36] (Fig. 1). Indeed, the redundancy with which these enzymes are recruited to the HIV promoter perhaps highlights their importance in maintaining latency [36]. To date, HDAC inhibition by small pharmacological molecules to induce transcription at the HIV LTR is the most well characterized strategy to purge latent HIV. Our group recently demonstrated that a single dose of vorinosat, a class I HDAC, can disrupt latency in humans [37]. Currently, the effect of panobinostat, a class I HDAC inhibitor more potent than vorinostat that is in phase III clinical trials for Hodgkin’s lymphoma and cutaneous T-cell lymphoma, is being analyzed for its ability to perturb latency in human controls (clinical trial #NCT01680094).
IMMUNE STRATEGIES FOR ERADICATION
Recent studies by Shan et al. [38] suggest that the reversal of latency by drugs such as HDAC inhibitors may not be sufficient to induce viral cytopathic effects and death of the infected cell. Such cells can be cleared by antigen-specific cytotoxic T cells (CTLs), and so perhaps approaches to disrupt latency using small molecules will also require reinvigoration of an anergic immune system. Furthermore, several studies have shown that the ability of elite suppressors to control their virus infection in the absence of ART is in part due to robust anti-HIV immune responses, thus underscoring the potential of an augmented immune response to clear virus [39,40]. To that end, several studies that seek to improve the function of the immune system in conjunction with ART are ongoing. In a double blind study involving 17 patients, administration of an HIV DNA vaccine coding for clade B gag, Pol and Nef and Env from clades A, B and C, followed by a replication-deficient adenovirus boost (encoding all the above antigens, except Nef) resulted in a broadening of HIV-specific CTL repertoire with increased polyfunctionality [41▪▪]. However, the authors did not observe a change in the frequency of infection of latently infected CD4+ T cells.
In another recent clinical trial, a therapeutic vaccine designated AGS-004 was demonstrated to significantly augment the HIV-specific immune response and reduce the viral load set point after ART interruption post-vaccine therapy. This vaccine, a dendritic cell-based RNA vaccine with Gag, Nef, Rev and VPR as the patient-specific RNA antigen payload, was evaluated in 19 patients; in 25% of the patients a significant decline in integrated HIV DNA was also observed [42]. However, given a recent publication showing that HIV DNA measurements might be over-estimating the true size of the reservoir as it may be measuring defective virus [12▪▪], it remains to be determined whether declines observed above are translated into true decline of nondefective virus using more accurate measurements of replicative competent virus such as viral outgrowth assays [12▪▪].
As studies using pharmacological approaches to disrupt latency advance into clinical testing, there is concern that the compounds studied in vivo thus far may not perturb latency sufficiently to allow subsequent clearance of productively infected cells by the immune system [38]. A recent investigation by Graf et al. [43▪] demonstrated that CTL from elite controllers can recognize and clear infected cells producing Gag in the absence of virion production [43▪], supporting the hypothesis that enhancing the CTL response in HIV-infected individuals concomitantly with the administration of antilatency pharmacological therapeutics will be critical in depleting the HIV reservoir (Fig. 2).
FIGURE 2.
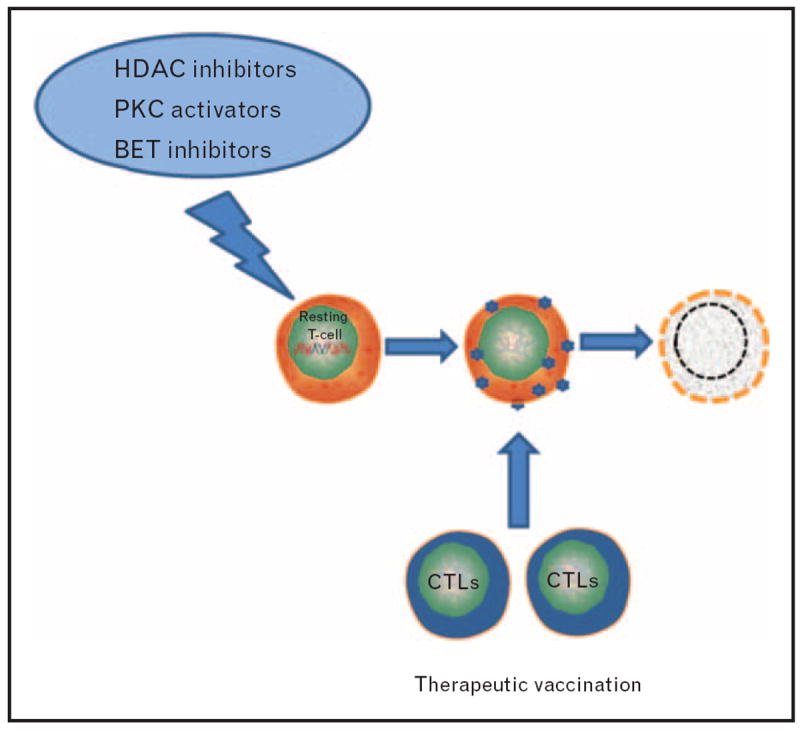
A method to deplete the latent HIV reservoir: latently infected cells are induced to reveal themselves to the immune system by the administration of pharmaceutical small molecules that disrupt latency. In parallel, therapeutic vaccination enhance the immune response for effective viral clearance. BET, bromodomain extraterminal; CTL, cytotoxic T cell; HDAC, histone deacetylase; PKC, protein kinase C.
ANTIRETROVIRAL THERAPY AS CURE THERAPY
The Virological and Immunological Studies in Controllers after Treatment Interruption (VISCONTI) group reported that 14 HIV-infected patients who initiated ART during primary HIV infection controlled their virus for a median of 7.5 years after the interruption of prolonged ART [44▪▪]. This study has revived interest in ART with the goal of achieving ‘posttreatment control,’ a kind of functional cure. VISCONTI patients also had a very small reservoir as measured by HIV DNA, suggesting that by limiting the pool of infected cells early ART may decrease the pool of latently infected cells ready to reseed new rounds of infection in the absence of therapy. However, a small reservoir alone does not explain why these patients did not experience viral rebound after the cessation of ART as re-emergence of viremia in patients treated early and who have small reservoirs has been reported [45]. One interesting feature of the VISCONTI post-treatment controllers is that their long-lived central memory T cells appeared to contribute marginally to the HIV reservoir as opposed to most patients where central memory T cells are the major contributors of the HIV reservoir. This suggests that perhaps the nature of the viral reservoir may play an important role in controlling infection in the absence of ART.
A recent report of the possible achievement of a functional cure in a child has further supported the idea that early therapeutic interventions may be promising in certain patients as an approach to eradicate HIV infection, or at the very least a remission of HIV in the absence of therapy. A child who was born to an HIV-positive mother not on ART had detectable HIV DNA and 19 812 copies of HIV RNA/ml at 30 h of age [4▪▪]. Three-drug ART was initiated immediately and HIV RNA subsequently declined to less than 48 copies/ml when the child was 29 days old. At 18 months of age, ART was terminated because of noncompliance on the part of the mother of the child. Interestingly, even in the absence of therapy, viral load was undetectable at 23 months of age when the child was returned to care. Although more sensitive assays revealed low levels of RNA (2 copies/ml), virus could not be recovered from a quantitative viral outgrowth assay performed with 22 million resting CD4+ T cells. Whether or not this case can be repeated in other patients remains to be determined.
CONCLUSION
HIV latency is established early during infection and is a complex phenomenon. HIV eradication will likely require multiple approaches. HDAC inhibitors have shown promise at disrupting latency in humans. However, they will likely have to be used concomitantly with immunotherapy to obtain viral clearance of infected cells and therefore depletion of the reservoir. It is also plausible that drugs that target other pathways involved in HIV latency such as activation of NF-κB through the PKC pathway or increasing the availability of PTEF-b at the HIV promoter may augment the effect of HDAC inhibitors in vivo, but such compounds are not yet ready to enter clinical trials. Meanwhile, it is apparent that early ART is instrumental in minimizing the pool of latently infected cells and most likely will be exploited further as a means to a functional cure. Finally, whatever combination of antilatency therapy that is undertaken, it must be of such nature that it can become scalable, affordable and accessible to make a significant impact on the HIV pandemic.
KEY POINTS.
Targeting pathways involved in maintaining HIV quiescence with pharmacological compounds such as PKC activators, HDAC and BET protein inhibitors may be important in the ‘shock and kill’ strategy to deplete the HIV reservoir.
Immune augmentation strategies will be critical for achieving viral clearance in antilatency therapies.
Early ART limits the establishment of an HIV reservoir and is likely to be exploited as a means to achieve either eradication or a functional cure.
Acknowledgments
This work was supported by the National Institutes of Health grant DA030156 to D.M.M.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
-
▪
of special interest
-
▪▪
of outstanding interest
- 1.UNAIDS world AIDS day 2012 report. 2012 Nov 20; Press Release. http://www.unaids.org/en/resources/publications/2012/name,76120,en.asp.
- 2.Vidal F, Gutierrez F, Gutierrez M, et al. Pharmacogenetics of adverse effects due to antiretroviral drugs. AIDS Rev. 2010;12:15–30. [PubMed] [Google Scholar]
- 3.Allers K, Hutter G, Hofmann J, et al. Evidence for the cure of HIV infection by ccr5delta32/delta32 stem cell transplantation. Blood. 2011;117:2791–2799. doi: 10.1182/blood-2010-09-309591. [DOI] [PubMed] [Google Scholar]
- 4▪▪.Persaud D, Gay H, Ziemniak C, et al. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N Engl J Med. 2013;23:23. doi: 10.1056/NEJMoa1302976. This study describes a functional cure achieved in a child for the first time. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soriano-Sarabia N, Archin N, Margolis D. Study of transitional memory CD4+ T cells and G/D T cells as latent reservoirs for replication competent HIV-1. Conference on Retroviruses and Opportunistic Infections; March 2013; Atlanta, GA. [abstract] [Google Scholar]
- 6.Chun TW, Finzi D, Margolick J, et al. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med. 1995;1:1284–1290. doi: 10.1038/nm1295-1284. [DOI] [PubMed] [Google Scholar]
- 7.Chun TW, Stuyver L, Mizell SB, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 1997;94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finzi D, Hermankova M, Pierson T, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 9.Wong JK, Hezareh M, Gunthard HF, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez G, Xu X, Chermann JC, Hirsch I. Accumulation of defective viral genomes in peripheral blood mononuclear cells of human immunodeficiency virus type 1-infected individuals. J Virol. 1997;71:2233–2240. doi: 10.1128/jvi.71.3.2233-2240.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kieffer TL, Kwon P, Nettles RE, et al. G→A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J Virol. 2005;79:1975–1980. doi: 10.1128/JVI.79.3.1975-1980.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12▪▪.Eriksson S, Graf EH, Dahl V, et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 2013;9:e1003174. doi: 10.1371/journal.ppat.1003174. The authors of the above reference performed a detailed comparison of the different methods currently used to measure HIV reservoirs and showed that DNA measurements by PCR methods may overestimate the size of the reservoir as they do not discriminate against defective proviruses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barton KM, Burch BD, Soriano-Sarabia N, Margolis DM. Prospects for treatment of latent HIV. Clin Pharmacol Ther. 2013;93:46–56. doi: 10.1038/clpt.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Lint C, Bouchat S, Marcello A. HIV-1 transcription and latency: an update. Retrovirology. 2013;10:1742–4690. doi: 10.1186/1742-4690-10-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherrill-Mix S, Lewinski MK, Famiglietti M, et al. HIV latency and integration site placement in five cell-based models. Retrovirology. 2013;10:90. doi: 10.1186/1742-4690-10-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karn J. The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit. Curr Opin HIV AIDS. 2011;6:4–11. doi: 10.1097/COH.0b013e328340ffbb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ott M, Geyer M, Zhou Q. The control of HIV transcription: keeping RNA polymerase II on track. Cell Host Microbe. 2011;10:426–435. doi: 10.1016/j.chom.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boehm D, Conrad RJ, Ott M. Bromodomain proteins in HIV infection. Viruses. 2013;5:1571–1586. doi: 10.3390/v5061571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartholomeeusen K, Xiang Y, Fujinaga K, Peterlin BM. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J Biol Chem. 2012;287:36609–36616. doi: 10.1074/jbc.M112.410746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20▪▪.Boehm D, Calvanese V, Dar RD, et al. Bet bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle. 2013;12:452–462. doi: 10.4161/cc.23309. A very detailed study on inhibition of BET bromodomain and reactivation of HIV from latency that argues against the inhibition of BRD4 being the primary mechanism for BET inhibition leading to HIV reactivation, rather that the related protein BRD2 plays a significant role in maintaining HIV latency in the absence of Tat. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21▪.Zhu J, Gaiha GD, John SP, et al. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012;2:807–816. doi: 10.1016/j.celrep.2012.09.008. The authors of this reference showed that JQ1 treatment, in addition to inhibiting BRD4, enhancesTat association with PTEF-b leading to proviral transcription. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z, Guo J, Wu Y, Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing BRD4 inhibition of Tat-transactivation. Nucl Acids Res. 2013;41:277–287. doi: 10.1093/nar/gks976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banerjee C, Archin N, Michaels D, et al. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J Leukocyte Biol. 2012;92:1147–1154. doi: 10.1189/jlb.0312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karn J. A new BET on the control of HIV latency. Cell Cycle. 2013;12:545–546. doi: 10.4161/cc.23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25▪.Fernandez G, Zaikos TD, Khan SZ, et al. Targeting IkappaB proteins for HIV latency activation: the role of individual IkappaB and NF-kappaB proteins. J Virol. 2013;87:3966–3978. doi: 10.1128/JVI.03251-12. This work shows that IκBε may play a prior unrecognized role in suppression of HIV transcription. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Memet S, Laouini D, Epinat JC, et al. IkappaBepsilon-deficient mice: reduction of one T cell precursor subspecies and enhanced Ig isotype switching and cytokine synthesis. J Immunol. 1999;163:5994–6005. [PubMed] [Google Scholar]
- 27.Gustafson KR, Cardellina JH, 2nd, McMahon JB, et al. A nonpromoting phorbol from the samoan medicinal plant Homalanthus nutans inhibits cell killing by HIV-1. J Med Chem. 1992;35:1978–1986. doi: 10.1021/jm00089a006. [DOI] [PubMed] [Google Scholar]
- 28.Trushin SA, Bren GD, Asin S, et al. Human immunodeficiency virus reactivation by phorbol esters or T-cell receptor ligation requires both PKCalpha and PKCtheta. J Virol. 2005;79:9821–9830. doi: 10.1128/JVI.79.15.9821-9830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulkosky J, Culnan DM, Roman J, et al. Prostratin: activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood. 2001;98:3006–3015. doi: 10.1182/blood.v98.10.3006. [DOI] [PubMed] [Google Scholar]
- 30.Rullas J, Bermejo M, Garcia-Perez J, et al. Prostratin induces HIV activation and downregulates HIV receptors in peripheral blood lymphocytes. Antivir Ther. 2004;9:545–554. [PubMed] [Google Scholar]
- 31▪▪.Beans EJ, Fournogerakis D, Gauntlett C, et al. Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo. Proc Natl Acad Sci U S A. 2013;110:11698–11703. doi: 10.1073/pnas.1302634110. The author of this reference describes the synthesis of prostratin anlogs that are 100 times more potent than prostratin at reactivating HIV from latency in cell line models of latency as well as in resting CD4+ T cells isolated from HIV-positive patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams SA, Chen LF, Kwon H, et al. Prostratin antagonizes HIV latency by activating NF-kappaB. J Biol Chem. 2004;279:42008–42017. doi: 10.1074/jbc.M402124200. [DOI] [PubMed] [Google Scholar]
- 33.Barr PM, Lazarus HM, Cooper BW, et al. Phase II study of bryostatin 1 and vincristine for aggressive nonHodgkin lymphoma relapsing after an autologous stem cell transplant. Am J Hematol. 2009;84:484–487. doi: 10.1002/ajh.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaufelberger DE, Koleck MP, Beutler JA, et al. The large-scale isolation of bryostatin 1 from Bugula neritina following current good manufacturing practices. J Nat Prod. 1991;54:1265–1270. doi: 10.1021/np50077a004. [DOI] [PubMed] [Google Scholar]
- 35▪.DeChristopher BA, Loy BA, Marsden MD, et al. Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat Chem. 2012;4:705–710. doi: 10.1038/nchem.1395. The structural complexity of the structure of byostatin and its low abundance from its natural source has often made its cost prohibitive to use. The authors of this reference described for the first time the successful economical synthesis of bryostatin analogs that were 1000 times more potent than prostratin at inducing HIV expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choudhary SK, Margolis DM. Curing HIV: pharmacologic approaches to target HIV-1 latency. Annu Rev Pharmacol Toxicol. 2011;51:397–418. doi: 10.1146/annurev-pharmtox-010510-100237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Archin NM, Liberty AL, Kashuba AD, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shan L, Deng K, Shroff NS, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012;36:491–501. doi: 10.1016/j.immuni.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porichis F, Kaufmann DE. HIV-specific CD4 T cells and immune control of viral replication. Curr Opin HIV AIDS. 2011;6:174–180. doi: 10.1097/COH.0b013e3283454058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Theze J, Chakrabarti LA, Vingert B, et al. HIV controllers: a multifactorial phenotype of spontaneous viral suppression. Clin Immunol. 2011;141:15–30. doi: 10.1016/j.clim.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41▪▪.Casazza JP, Bowman KA, Adzaku S, et al. Therapeutic vaccination expands and improves the function of the HIV-specific memory T-cell repertoire. J Infect Dis. 2013;207:1829–1840. doi: 10.1093/infdis/jit098. The authors of this reference showed that a therapeutic DNA vaccine with clade B Gag, Pol and Nef and clade A, B, and C Env as the antigen payload induced a strong anti-HIV immune response, including an augmentation of the antigen- specific CTL response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicolette C. Design and development considerations for immune augmentation to assist in virus eradication. Towards an HIV Cure: IAS 2013 Symposium; Kuala Lumpur, Malaysia. 2013. [abstract] [Google Scholar]
- 43▪.Graf EH, Pace MJ, Peterson BA, et al. Gag-positive reservoir cells are susceptible to HIV-specific cytotoxic T lymphocyte mediated clearance. PLoS One. 2013;8:e71879. doi: 10.1371/journal.pone.0071879. Gag production by CD4+ T cells is sufficient to induce clearance of these cells by CD8+ T cells of elite controllers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44▪▪.Saez-Cirion A, Bacchus C, Hocqueloux L, et al. Posttreatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy: ANRS VISCONTI study. PLoS Pathog. 2013;9:e1003211. doi: 10.1371/journal.ppat.1003211. The VISCONTI patients who began ART during primary HIV infection controlled their viremia for a median of 7.5 years after treatment interruption, suggesting that in some patients early ART may lead to HIV remission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chun TW, Justement JS, Murray D, et al. Rebound of plasma viremia following cessation of antiretroviral therapy despite profoundly low levels of HIV reservoir: implications for eradication. AIDS. 2010;24:2803–2808. doi: 10.1097/QAD.0b013e328340a239. [DOI] [PMC free article] [PubMed] [Google Scholar]