

Abstract
Genomic instability at microsatellite loci is a hallmark of many cancers, including breast cancer. However, much of the genomic variation and many of the hereditary components responsible for breast cancer remain undetected. We hypothesized that variation at microsatellites could provide additional genomic markers for breast cancer risk assessment. A total of 1,345 germline and tumor DNA samples from individuals diagnosed with breast cancer, exome sequenced as part of The Cancer Genome Atlas, were analyzed for microsatellite variation. The comparison group for our analysis, representing healthy individuals, consisted of 249 females which were exome sequenced as part of the 1,000 Genomes Project. We applied our microsatellite-based genotyping pipeline to identify 55 microsatellite loci that can distinguish between the germline of individuals diagnosed with breast cancer and healthy individuals with a sensitivity of 88.4 % and a specificity of 77.1 %. Further, we identified additional microsatellite loci that are potentially useful for distinguishing between breast cancer subtypes, revealing a possible fifth subtype. These findings are of clinical interest as possible risk diagnostics and reveal genes that may be of potential therapeutic value, including genes previously not associated with breast cancer.
Electronic supplementary material
The online version of this article (doi:10.1007/s10549-014-2908-8) contains supplementary material, which is available to authorized users.
Keywords: Microsatellite variation, Breast cancer risk assessment, Cancer genomics, The Cancer Genome Atlas
Introduction
The American Cancer Society predicts 232,340 new cases of invasive breast carcinoma (BC) will be diagnosed in 2013 and females have 1 in 8 chance of developing this cancer within their lifetime. An individual’s predisposition, prognosis, and response to therapy of complex diseases such as cancer are mediated to varying degrees by their genomic makeup. Breast cancers have significant known inherited or spontaneous components. However, the accumulated knowledge from extensive studies, many of which have focused on single nucleotide polymorphisms (SNPs), explains less than half of heritable components to date. For example, several dozen variants in the well-studied BRCA1 and BRCA2 genes account for only 5 and 10 % of inherited BC susceptibility, respectively [1–6], and the recent iCOS studies emphasize that there is still a discrepancy between the known BC susceptibility loci and the expected heritable component of BC [7–9]. There is sustained debate between those who believe the missing disease contributions will be explained by rare variants with a large effect or common variants with small effects. However, the truth is probably somewhere in between as it is difficult to explain by large SNP-based Genome Wide Association Studies alone. We hypothesize that much of these significant missing genetic components may be explained by variation in parts of the genome, which have not been explored previously, namely, microsatellite or repetitive DNA loci, notably referred to as “Junk DNA” or more recently “Dark Matter” [10]. Cancer is highly responsive to treatment when diagnosed early; therefore, there is a significant advantage to identifying additional informative, actionable markers that may account for some of the differences between the estimated heritability and the portion of risk that can be explained by known genetic polymorphisms.
Microsatellites are repetitive DNA regions that occur throughout the genome, and variations within microsatellites can affect cellular function through mechanisms including promoting alternative splicing [11], altering protein sequence [12], and affecting gene regulation [13, 14]. Several previous studies on microsatellite variation and its implied instability in cancer focus on variation found between the tumor and somatic genomes of an individual at five mononucleotide “Bethesda” markers [15], which capture a small fraction of variation from the ~1 million microsatellite loci [16]. While variation within the larger set of loci has been generally understudied, two recent technological advances have enabled us to thoroughly characterize microsatellite variation genome-wide: (1) The public release of single-strand, high-throughput next-generation sequence data [17–19]; and (2) The development of algorithms and analytic approaches that enable accurate genotype determination at numerous microsatellite loci [20, 21].
Materials and methods
The genotypes of microsatellite loci found within 249 ethnically matched healthy female germlines, 656 BC germline exomes, 689 BC tumors (656 matched to the germline samples), and 212 healthy male germlines from exome sequences available through the 1000 Genomes Project (disease-free females and males) or TCGA (BC patients) were computed individually from re-assemblies as described in our previous publications with microsatellite calling accuracy being estimated to be between 94.4 and 96.5 % [20, 21]. We restricted our analysis to those 49,297 microsatellite loci that were genotyped with sufficient coverage (15×) in at least 10 exomes from both the healthy and BC populations, and compared the genotype distribution at each locus for the population. Benjamini–Hochberg False Discovery Rate (FDR) test was applied to the datasets to identify informative loci that distinguish breast cancer from healthy genomes. The sensitivity and specificity of the combined 55 loci to differentiate breast cancer genomes from the healthy genomes were computed using the receiver-operating characteristic (ROC). The genotypes at these loci created a profile used as a risk assessment tool for classifying independent sets of the healthy or BC exomes. Detailed methods are available in the supplemental information.
Results
Many studies attempt to link the presence or absence of specific mutations to a disease state. This has been a successful strategy for discovering disease-associated genes; however, complex disease states are frequently due to additive effects from multiple common variants, as seen, for example, in the multiple SNPs associated with telomere maintenance and BC risk [22]. To uncover this type of interaction, we must employ a methodology that examines the frequency at which alleles are seen across multiple loci in an affected population. However, focusing solely on the frequency at which an allele is represented may result in missing a significant shift in the frequency at which an allele is heterozygous. Therefore, we have performed our analysis on the frequency of genotypes within the examined populations, using an algorithm for genotyping microsatellite loci that we previously designed [20, 21]. We employed this methodology to determine the genotype of all microsatellite loci in exome sequences from the healthy females from the 1000 Genomes Project [18] and in 656 germline exomes from BC patients sequenced as part of TCGA [19] (Suppl. Fig. 1). Comparison of the healthy females from different ethnic backgrounds revealed that variation at some microsatellite loci was correlated with ethnicity. Therefore, we selected 249 individuals of which 87.5 % were of European Ancestry to represent the healthy population (1kGP-EUF) because the microsatellite profile of the BC germline samples was the closest to these exomes (Suppl. Fig. 2), and we did not have information on the ancestry of the BC germline samples at this time.
For each microsatellite locus, the most frequent genotype in the 1kGP-EUF population was identified as the modal genotype and the frequency of alternative genotypes present within both populations was calculated. On average, 29,809 ± 4,688 and 34,849 ± 4,371 microsatellite loci were genotyped per 1kGP-EUF and BC germline sample, with 283 ± 134 and 426 ± 124 nonmodal genotypes, respectively (Suppl. Table 1). We identified 55 loci that each individually showed a statistically significant difference in genotype distribution between 1kGP-EUF and BC germline (two-sided Fisher’s p with adjusted p value ≤0.01 by Benjamini–Hochberg to reduce FDR). A comparison of females from the 1kGP randomly divided into two subgroups did not identify any significant loci using this FDR cut-off, showing that normal variations at loci in two similar populations are not significant using our methods. Figure 1 shows how the genotype distributions for the healthy and cancer populations differ for a sample of the 55 loci, including both those at which there were more nonmodal genotypes present in the healthy population and those at which there were more nonmodal genotypes in the BC population. The genotypes for the BC and 1kGP-EUF exomes can be visualized in Fig. 2, where any genotype that matched the modal genotype identified in the 1kGP-EUF exomes is coded in gray and all nonmodal genotypes are red. 25.1 ± 13.1 and 31.3 ± 9.4 % of the 55 loci were genotyped in the 1kGP-EUF and BC germline exomes, respectively, which is not surprising given that we use very stringent conditions for coverage and alignment, and because Lander–Waterman distributions in random fragment sequencing limits the number of callable loci in each sample [23]. Notably, for the 1kGP-EUF population, the modal genotype at 24 % of the 55 loci is heterozygous, whereas the modal genotype for 36.4 % of the 55 loci in the BC germline exomes is heterozygous. This confirms that we are able to identify loci where the modal genotype is different between the BC and healthy populations. This is important because we are not identifying novel/rare alleles but noting that individuals with BC are more frequently heterozygotic at these loci. Analysis of the genotype distributions at the 55 loci revealed that 80 % (44/55) of the loci are in Hardy–Weinberg equilibrium in the 1kGP-EUF samples, while only 40 % (22/55) are in Hardy–Weinberg equilibrium for the BC germline (Suppl. Table 2), raising the possibility that there is a reduction in selective pressure in BC germline genomes that may result in the increased susceptibility to BC.
Table 1.
Many of the genes associated with our 55 signature microsatellite loci are known to be associated with cancer generally, specifically with BC, or are involved in other cellular pathways associated with cancer
| Cancer | NUFIP1, KDM1A, SPHK2, STC1, PIAS2, MLL, TLN2, CUL1, POP4, PDGFRA, NCOR1, MME, RASA1, ANAPC7, HSP90AA1, FANCI, WRN, TBP, DNAH3, MT1X, PTPN22, NUP54, ADAM2, KIF1B, CORIN, ADAMTSL3, CPOX, ACRC, NXF1, RDX, CDS2, SLC13A1 |
| Breast cancer | NUFIP1, KDM1A, SPHK2, STC1, PIAS2, MLL, TLN2, CUL1, POP4, PDGFRA, NCOR1, MME, RASA1, ANAPC7, HSP90AA1, FANCI, WRN, TBP |
| Cell cycle | CUL1, PTPN22, KIF1B, DNAH3, PDGFA, CCDC46, WRN, MICALL1, ANAPC7 |
| Apoptosis | CUL1, SPHK2, ADAM2, PDGFRA, PDCD6IP |
Fig. 1.

Individual microsatellite loci vary significantly between breast cancer and healthy genomes. Genotype distributions for a representative subset of our 55 signature loci are shown. Gray bars represent genotypes present in the healthy population, and red bars represent genotypes in the BC samples
Fig. 2.

Modal and nonmodal genotypes present in germline exomes of BC and healthy individuals. Individuals with BC show a distinct genotype pattern compared with the healthy females. Gray modal, red nonmodal. luminal A [LA], luminal B [LB], ERBB2/HER2+ [HER2], and basal-like [BL], UNKNOWN = no indicated subtype, BC germline IND [independent set of BC germline exomes of mixed ethnicity], BC germline IND-2 [independent set of BC germline exomes of “white” ethnicity], 1kGP-EUF, IND [independent set of healthy females, aka 1kGP-EUF IND]
Thirty-two of the genes associated with the 55 microsatellite loci have previously been associated with cancer, and 18 are specifically linked to BC (Table 1). 49 of the same loci are located in introns, of which 24 are within 50 nt of an exon/intron boundary; three additional loci are intergenic. Notably, four are in the 3′UTRs of PIAS2, WWC3, MT1X, and TBP, and one is exonic (a CAG repeat in FAM157A; see Suppl. Fig. 3 for detailed analysis of this variant).
The genotypic differences at these 55 loci appear to have two effects on the likelihood of BC. At 30 of the 55 loci, the presence of a nonmodal genotype is potentially protective against BC (relative risk of <0.6; Suppl. Table 2), whereas at 25 of the loci, a nonmodal genotype appears to promote BC (relative risk >1.3). Gene ontology enrichment analysis showed that genes involved in notch signaling were enriched among those potential BC-promoting loci, while the set that potentially protects against BC includes proteins known to be involved in maintaining genomic stability (e.g., WRN, FANCI, HSP90) and programmed cell death (e.g., PDCD6IP). Supplementary Fig. 4 highlights some of the genes involved in signaling pathways including p53, integrin, and MAPKK pathways.
We performed a similar analysis on the 508 BC germline exomes that were classified as “white” once the information on the ancestry of the BC samples was made available. This analysis identified a set of 52 microsatellite loci, of which 42 overlapped with the original 55 loci set. Of the loci that “fell out”, 9 loci fell below our statistical cut-offs (had adjusted p-values of 0.05–0.01). In addition, gene ontology analysis of the 52 loci was similar to that of the original 55 loci. This analysis not only gives us confidence in our original loci set as robust, but also shows that there are likely additional informative loci that can be identified as more BC exomes are available.
Risk classifier
We used the frequency of modal or nonmodal genotypes at each of the original 55 informative loci within the BC population relative to the 1kGP-EUF population to create a breast cancer profile, i.e., we assigned a “modal” or “nonmodal” designation for each of the loci depending on the overall consensus for the BC germline samples in relation to the 1kGP-EUF. We then determined for each individual sample whether it matched the BC cancer profile at each locus at which it was genotyped. Figure 3 shows the distribution of exomes based on the number of genotypes at the 55 signature loci that match the cancer profile. Using the false positive and false negative rates within the training set, we were able determine the ROC for the 55 loci. By means of maximizing the area under the ROC curve, we determined the optimal cut-off for a classifier as having 76 % of the 55 BC loci matching the breast cancer genotype profile (Suppl. Fig. 5). We were then able to classify the BC germline exomes as “cancer-like” (≥76 %) or the healthy (<76 %) with a sensitivity of 88.4 % and a specificity of 77.1 % (Fig. 3). A similar risk classifier analysis done using the 52 loci from the “white” BC exomes had the same high sensitivity (88.4 %), but the specificity was only 59 % (Suppl. Fig. 5B). Using these risk classifiers on a set of BC tumor samples, we identified 88.1 % of the BC tumor exomes as cancer-like using the 55 loci set from all BC exomes and 88 % of the “white” BC tumor exomes as “cancer-like” using the 52 loci from the “white” BC exomes, a difference that was not statistically significant from the number of germline BC samples that were cancer-like in either case (Fig. 3; Suppl. Fig. 6). This is in contrast to the 1kGP-EUF samples, of which 77.1 % were normal, and only 22.9 % were cancer-like (Fig. 3; Table 2). In addition, two independent sets of BC germline samples (BC IND with 60 samples of mixed ethnicity and BC IND-2 with 137 samples that were all “white”) showed a similar frequency of exomes classified as “cancer-like”, whereas the other healthy individuals, including males and nonEuropean females, and an independent set of 52 European females are more similar to the 1kGP-EUF exomes (Table 2; Suppl. Fig. 7).
Fig. 3.

BC exomes have a higher average percentage of loci matching the breast cancer profile. Distributions of exomes based on their genotypes at the 55 BC-associated microsatellite loci. We classify genomes having ≥76 % of callable genotypes as cancer-like and those having <76 % as similar to the healthy population
Table 2.
Classification of exome sets using our BC risk classifier
| Sample set | Number of exomes | % Healthy | % Cancer-like |
|---|---|---|---|
| 1kGP-EUF | 249 | 77.1 | 22.9 |
| 1kGP-EUF IND | 52 | 61.5 | 38.5 |
| BC germline | 656 | 11.6 | 88.4 |
| BC IND | 60 | 15.0 | 85.0 |
| BC IND-2 | 137 | 14.6 | 85.4 |
The 55 signature loci were derived from the analysis of BC germline exomes regardless of BC subtype. We divided the BC samples into their subtypes and a set of samples where a subtype was not specified (unknown) to determine if we are able to classify exomes according to subset. Surprisingly, the BC “unknown” exome samples appeared to have a distinct profile within the 55 informative loci, distinguishing them from established BC subtypes (Fig. 2). Based on the “unknown” classification, we do not know if these samples constitute individuals with BC which would be consistent with a known subtype but were simply not classified or if these samples are unidentifiable using traditional subtype classification methods. We suggest that the latter explanation is more consistent with their distinct genotype profile within our loci set. The two independent sets of BC germline samples had similar genotype profiles to those BC germlines for which there was a subtype specified as opposed to the 1kGP-EUF samples or the unknown BC germline samples (Fig. 2), whereas the independent set of healthy European females (IND) was more similar to the 1kGP-EUF. We re-analyzed all microsatellites for each subtype with respect to the 1kGP-EUF to identify additional loci that are associated with each or multiple subtypes. We found additional informative loci that distinguish the LA and “unknown” subtypes in addition to the 55 that distinguish all BC from the healthy genomes (Fig. 4). For the LA subtype, we identified four informative loci, two of which were unique to the LA subtype. For the “unknown” subtype, there were 41 informative loci identified, 22 of which were unique and included loci in genes involved in cell–cycle control, chromatin remodeling and programmed cell death. That there are loci unique to specific BC subtypes indicates that our method may be useful for distinguishing between BC subtypes. No loci passed our rigorous statistical requirements for the luminal B (LB), ERBB2/HER2+ (HER2), or basal-like/triple negative (BL) subtypes, likely because of the smaller number of exomes available for these BC subtypes.
Fig. 4.
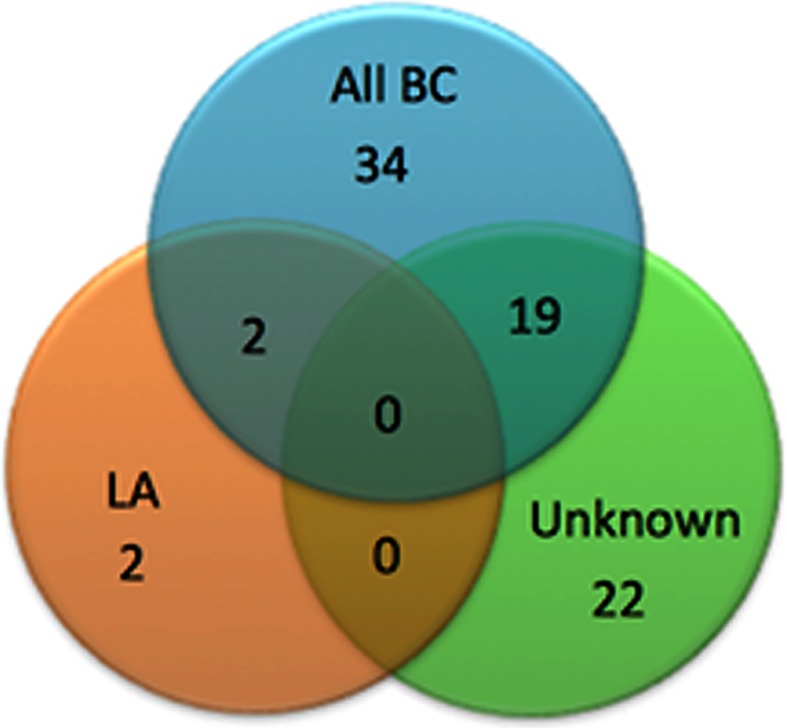
Overlap of informative loci distinguishing BC subtypes
Breast cancer tumor versus germline exomes
595 of the BC germline exome samples had matched tumor/germline exome data available (Suppl. Table 1). Supplementary Fig. 8 shows the genotypic concordance between the BC germline and tumor samples. For the 496 matched samples where we could genotype at least 10 of the 55 loci in the germline and tumor, 75.2 % were cases where the tumor and germline were cancer-like, 8.9 % the tumor was cancer-like, while the germline was not; and 12.1 % the germline was cancer-like, while the tumor was not (Suppl. Table 1). There were only 3.8 % of cases where neither the germline nor the matched tumor was cancer-like. It is important to note that no exome was sequenced with >15× coverage at all 55 loci (Fig. 2), and so in instances where only one of the matched germline and tumor exomes was classified as cancer-like, the difference may be due to differences in which loci could be genotyped for a given sample. Comparison of the tumor and matched germline exomes with our analytic pipeline did not reveal additional loci that were statistically different. This is not unexpected given that microsatellite instability associated with tumors could re-distribute genotypes nonuniformly across a population or even within a single individual. This analysis highlights the strength of our methodology for identifying cancer-like exomes from germline sequencing data.
Thirty-three germline exome sequenced samples had known mutations in TP53 [19]; of these, 28 were identified by our method as cancer-like. In addition, 15 samples were identified as having a potential mutation in BRCA1 or BRCA2 of which 14 are identified by our method as cancer-like (Suppl. Fig. 9). That the majority of exomes with BRCA/TP53 mutations are also classified by our method as cancer-like is not surprising since these genes are important for maintaining genomic stability. Our measure is not restricted to identifying individuals carrying these known high-risk markers allowing us to identify 541 individuals who did not carry these disease–predisposing mutations as cancer-like.
RNAseq data were available for 636 of the BC tumors and 87 of the BC germline samples that were in our BC exome sets. We performed genotype prediction from the RNAseq data for 18,148 exonic microsatellite loci that were potentially callable in the matched RNAseq genotypes and the respective genotypes in the germline and tumor samples. At 99.98 % of loci, the predicted genotype from RNAseq was consistent with the genotype determined from the matched exome sequencing. Those loci that were genotyped differently between the matched exome and RNASeq data were located at 72 loci,1 none of which is in genes associated with our 55 loci. However, genes associated with loci that differ between BC germline and RNAseq data are enriched for the VEGF signaling pathway, which influences vascular growth and angiogenesis. These loci may be additional biomarkers for alternatively spliced transcripts that may contribute to BC.
Gene set enrichment analysis (GSEA) [24, 25] indicated that the 55 informative loci and those loci that were identified in the individual subtypes were enriched for association with genes expression of which positively correlates with BRCA1. We analyzed the RNAseq data to identify additional potential shifts in gene expression that might correlate with BC. We were able to analyze the expression level for 52 of the genes in the BC tumor exomes but only 46 genes in the BC germline samples because gene expression data were provided for 304 tumor samples but only 39 germline samples from the TCGA [19]. No expression information was available for FAM157A or TRG. Of the signature loci, 48 had previously been shown to have some levels of expression in breast tissue (Suppl. Table 2; [26]). Comparing all germline and tumor samples, analysis of the expression levels of the genes associated with the 55 informative microsatellite loci revealed that seven of these showed >2× increased expression in tumors, while four showed decreased expression (Suppl. Table 3). One gene in the germline set (CRISP1) and one gene in the tumor set (ABHD12B) showed >2× difference in expression between individuals who had a genotype matching the BC profile, and those who did not. In both cases, the individuals with a genotype that matched the BC profile showed a higher expression level.
Microsatellite variation at intronic loci may result in alternatively spliced transcripts [11] that have the potential to contribute to oncogenesis, with estimates that ~95 % of multi-exon genes exhibit alternative splicing [27]. In addition, 49.0 % of the intronic loci were within 50 nt of an exon/intron junction, a higher frequency than expected given that only 3.4 % of all intronic microsatellites that were genotyped in at least one exome sample were within this boundary. This led us to hypothesize that RNA splicing is affected. We used Cufflinks [28] to identify possible alternative splicing events in transcripts from genes containing the signature loci. For those loci at which we had data about both the transcript splicing and genotype data, we found that, for the germline and tumor sets respectively, 84.9 and 81.5 % of the transcripts from loci genotype of which matched the BC profile showed possible alternative splicing compared with 77.4 and 79.8 % of those transcripts from loci genotype of which did not match the profile.
Ten of the genes associated with the 55 loci are targets of, or affected by, pharmaceuticals several of which are prescribed or in clinical trials for BC (Suppl. Table 4). This is ~1.2× greater than expected given the drug–target interactions within the CancerResource database [29]. Thus, our analysis may provide novel drug–targets or drug re-positioning opportunities for additional or combinatorial BC treatment plans.
Discussion
In summary, the comparison of “healthy” and breast cancer patient exomes at microsatellite loci revealed variations in nonmodal genotype frequency, while comparably, only a small number of variations were seen between matched breast cancer germline and tumor exomes. We applied our microsatellite genotyping pipeline to nearly 50,000 microsatellite loci from BC and disease-free females and identified 55 loci at which the frequency of nonmodal genotypes was statistically significantly different between the two populations, of which 30 showed a risk ratio below 0.6, while 25 had a risk ratio greater than 1.3. Importantly, the presence or the absence of nonmodal genotypes at the 55 loci was used to create a “BC profile” that can be used as a risk classifier. The overwhelming majority of exomes classified as cancer-like did not carry any known BC-associated mutation. If the analysis is confirmed in independent matched cohorts, then an assay consisting of these 55 loci might be clinically informative with a sensitivity of 88.4 %, which exceeds current test performance, while the specificity is about two fold which would be expected, given that 12 % of the “healthy” female population will be future BC patients. Many of the 55 loci are within genes implicated in breast cancer, while several represent potential new drug–targets with protein variants resulting from alternative splicing or, in one case, an exonic variation.
Such surveys of large cohorts of the microsatellite genomes of the affected individuals and the matched “healthy” populations could be a platform for identifying clinically actionable risk diagnostics, companion diagnostics, and drug–targets when applied to complex multigenic diseases for which disease severity, therapy response, and other metadata are known.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This study was funded by the Virginia Bioinformatics Institute Medical Informatics Systems Division director’s funds, the National Institute of Health, National Human Genome Research Institute, 1000 Genomes Project Dataset Analysis Grant (T-55818-363-1), and high performance computing was supported by a Grant from the National Science Foundation (OCI-1124123). This project was made possible through data provided by the 1000 Genomes Project and The Cancer Genome Atlas Project. The authors thank the system administrators in the VBI computational core (Michael Snow, Jeremy Johnson, Dominik Borkowski, David Bynum, Douglas McMaster, and Vedavyas Duggirala) for technical support. The authors would like to thank Hunt Willard for comments during the preparation of the manuscript. All the authors contributed to the experimental design, data analysis and the writing of this manuscript. In addition, L.J.M. designed, wrote, and ran all code, N.C.F. and E.K. provided biological interpretations, N.C.F. wrote the paper, and H.R.G. conceived and directed the study.
Footnotes
Large data file, content available upon request.
References
- 1.Langston AA, Malone KE, Thompson JD, Daling JR, Ostrander EA. BRCA1 mutations in a population-based sample of young women with breast cancer. New Engl J Med. 1996;334(3):137–142. doi: 10.1056/NEJM199601183340301. [DOI] [PubMed] [Google Scholar]
- 2.Newman B, Mu H, Butler LM, Millikan RC, Moorman PG, King MC. Frequency of breast cancer attributable to BRCA1 in a population-based series of American women. JAMA J Am Med Assoc. 1998;279(12):915–921. doi: 10.1001/jama.279.12.915. [DOI] [PubMed] [Google Scholar]
- 3.Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, Easton DF, Evans C, et al. Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst. 1999;91(11):943–949. doi: 10.1093/jnci/91.11.943. [DOI] [PubMed] [Google Scholar]
- 4.Malone KE, Daling JR, Neal C, Suter NM, O’Brien C, Cushing-Haugen K, Jonasdottir TJ, Thompson JD, et al. Frequency of BRCA1/BRCA2 mutations in a population-based sample of young breast carcinoma cases. Cancer. 2000;88(6):1393–1402. doi: 10.1002/(SICI)1097-0142(20000315)88:6<1393::AID-CNCR17>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 5.Johnson N, Fletcher O, Palles C, Rudd M, Webb E, Sellick G, dos Santos Silva I, McCormack V, et al. Counting potentially functional variants in BRCA1, BRCA2 and ATM predicts breast cancer susceptibility. Human Mol Genet. 2007;16(9):1051–1057. doi: 10.1093/hmg/ddm050. [DOI] [PubMed] [Google Scholar]
- 6.Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro AN, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81(5):873–883. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Couch FJ, Wang X, McGuffog L, Lee A, Olswold C, Kuchenbaecker KB, Soucy P, Fredericksen Z, et al. Genome-wide association study in BRCA1 mutation carriers identifies novel loci associated with breast and ovarian cancer risk. PLoS Genet. 2013;9(3):e1003212. doi: 10.1371/journal.pgen.1003212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, Schmidt MK, Chang-Claude J et al. (2013) Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nature genetics 45(4):353–361, 361e351–361e352. doi:10.1038/ng.2563 [DOI] [PMC free article] [PubMed]
- 9.Do CB, Hinds DA, Francke U, Eriksson N. Comparison of family history and SNPs for predicting risk of complex disease. PLoS Genet. 2012;8(10):e1002973. doi: 10.1371/journal.pgen.1002973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lian Y, Garner HR. Evidence for the regulation of alternative splicing via complementary DNA sequence repeats. Bioinformatics. 2005;21(8):1358–1364. doi: 10.1093/bioinformatics/bti180. [DOI] [PubMed] [Google Scholar]
- 12.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6(10):729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 13.Rockman MV, Wray GA. Abundant raw material for cis-regulatory evolution in humans. Mol Biol Evol. 2002;19(11):1991–2004. doi: 10.1093/oxfordjournals.molbev.a004023. [DOI] [PubMed] [Google Scholar]
- 14.Fondon JW, 3rd, Garner HR. Molecular origins of rapid and continuous morphological evolution. Proc Natl Acad Sci USA. 2004;101(52):18058–18063. doi: 10.1073/pnas.0408118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loukola A, Eklin K, Laiho P, Salovaara R, Kristo P, Jarvinen H, Mecklin JP, Launonen V, et al. Microsatellite marker analysis in screening for hereditary nonpolyposis colorectal cancer (HNPCC) Cancer Res. 2001;61(11):4545–4549. [PubMed] [Google Scholar]
- 16.Weber J. Informativeness of human (dC-dA)n·(dG-dT)n polymorphism. Genomics. 1990;2:524–530. doi: 10.1016/0888-7543(90)90195-Z. [DOI] [PubMed] [Google Scholar]
- 17.Ledford H. Big science: the cancer genome challenge. Nature. 2010;464(7291):972–974. doi: 10.1038/464972a. [DOI] [PubMed] [Google Scholar]
- 18.Genomes Project C. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McIver LJ, Fondon JW, 3rd, Skinner MA, Garner HR. Evaluation of microsatellite variation in the 1000 Genomes Project pilot studies is indicative of the quality and utility of the raw data and alignments. Genomics. 2011;97(4):193–199. doi: 10.1016/j.ygeno.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McIver LJ, McCormick JF, Martin A, Fondon JW, 3rd, Garner HR. Population-scale analysis of human microsatellites reveals novel sources of exonic variation. Gene. 2013;516(2):328–334. doi: 10.1016/j.gene.2012.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bojesen SE, Pooley KA, Johnatty SE, Beesley J, Michailidou K, Tyrer JP, Edwards SL, Pickett HA et al. (2013) Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nature genetics 45(4):371–384, 384e371–384e372. doi:10.1038/ng.2566 [DOI] [PMC free article] [PubMed]
- 23.Evans SN, Hower V, Pachter L. Coverage statistics for sequence census methods. BMC Bioinform. 2010;11:430. doi: 10.1186/1471-2105-11-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 26.Safran M, Dalah I, Alexander J, Rosen N, InyStein T, Shmoish M, Nativ N, Bahir I, et al. GeneCards Version 3: the human gene integrator. Database J Biol Databases Curation. 2010;2010:baq020. doi: 10.1093/database/baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40(12):1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 28.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmed J, Meinel T, Dunkel M, Murgueitio, Adams R, Blasse C, Eckert A, Preissner S, et al. CancerResource: a comprehensive database of cancer-relevant proteins and compound interactions supported by experimental knowledge. Nucl Acids Res. 2011;39(Database issue):D960–D967. doi: 10.1093/nar/gkq910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.