

Abstract
The use of small molecule BRAF inhibitors has revolutionized the treatment of advanced melanoma. Despite this, resistance is commonplace and associated with a median progression-free survival of >5 months. Major resistance mechanisms include reactivation of the MAPK pathway and increased PI3K/AKT signaling. Here we review some of the combination therapeutic strategies currently undergoing evaluation for the management of acquired drug resistance in melanoma.
Keywords: melanoma, BRAF, CRAF, resistance, hairy cell
Introduction
The RAF proteins constitute a family of 3 serine/threonine kinases (ARAF, BRAF and CRAF) that form one module of the mitogen activated protein kinase (MAPK) pathway [1]. Under physiological conditions RAF signaling is initiated through receptor tyrosine kinases (RTK) and the activation of the GTPase Ras. Accumulation of Ras in its active form (GTP-bound) leads to the recruitment of RAF to the plasma membrane and the stimulation of other kinases required for RAF phosphorylation and activation [1]. Once active, RAF phosphorylates MEK1/2 at Ser217 and Ser221, which in turn phosphorylates and activates ERK (extracellular signal regulated kinase) at Thr202 and Tyr204. ERK is the major effector of the MAPK signaling pathway. Constitutive ERK activity exerts its effects through the phosphorylation of multiple cytoplasmic targets involved in cell survival, mTOR signaling and the actin cytoskeleton [1]. Activated ERK also migrates to the nucleus where it phosphorylates multiple transcription factors including ELK-1, ETS and c-Myc. The current review discusses some of the approaches used to identify mechanisms of BRAF inhibitor resistance and outlines strategies for increasing the durability of responses to these inhibitors in patients.
BRAF as a major oncogene
CRAF (or RAF-1) was the first RAF isoform to be identified and has been the subject of intensive study [1]. Despite this, mutations in CRAF (or ARAF) are extremely rare in human cancer whereas BRAF mutations are relatively common. The reason why BRAF mutations are favored in cancer compared to mutations in other RAF isoforms is not entirely clear, but may be a consequence of the different number of steps required to stimulate BRAF versus ARAF or CRAF. Activation of both CRAF and ARAF requires phosphorylation by a coordinated series of kinases at multiple residues. In contrast, BRAF is “primed” and constitutively phosphorylated at some of these same sites, allowing the kinase to become activated following the acquisition of a single point mutation [2].
High throughput sequencing of multiple cancer types identified activating mutations in BRAF in >50% of human melanoma cell lines [3]. Since this time over 50 individual BRAF mutations have been described with the majority (>80%) being a valine to glutamic acid substitution at position 600, the BRAFV600E mutation [4]. There is strong evidence that mutant BRAF is a bona fide melanoma oncogene with studies showing the introduction of oncogenic BRAF to transform immortalized melanocytes and in concert with PTEN inactivation to drive melanoma formation in transgenic mouse models [1]. Acquisition of the BRAFV600E mutation is associated with constitutive activity in the MAPK signaling pathway which leads in turn to increased growth via upregulation of cyclin D1 expression, survival through the downregulation of BIM and increases in Mcl-1 and invasion via regulation of RND3 [1](Figure 1). In addition to melanoma, BRAFV600E mutations have also been reported in 100% of hairy cell leukemias and at lower frequencies in many other cancer types including colorectal carcinoma, myeloma, ovarian cancer, thyroid carcinoma and some subtypes of lung cancer (Table 1) [5–8].
Figure 1.
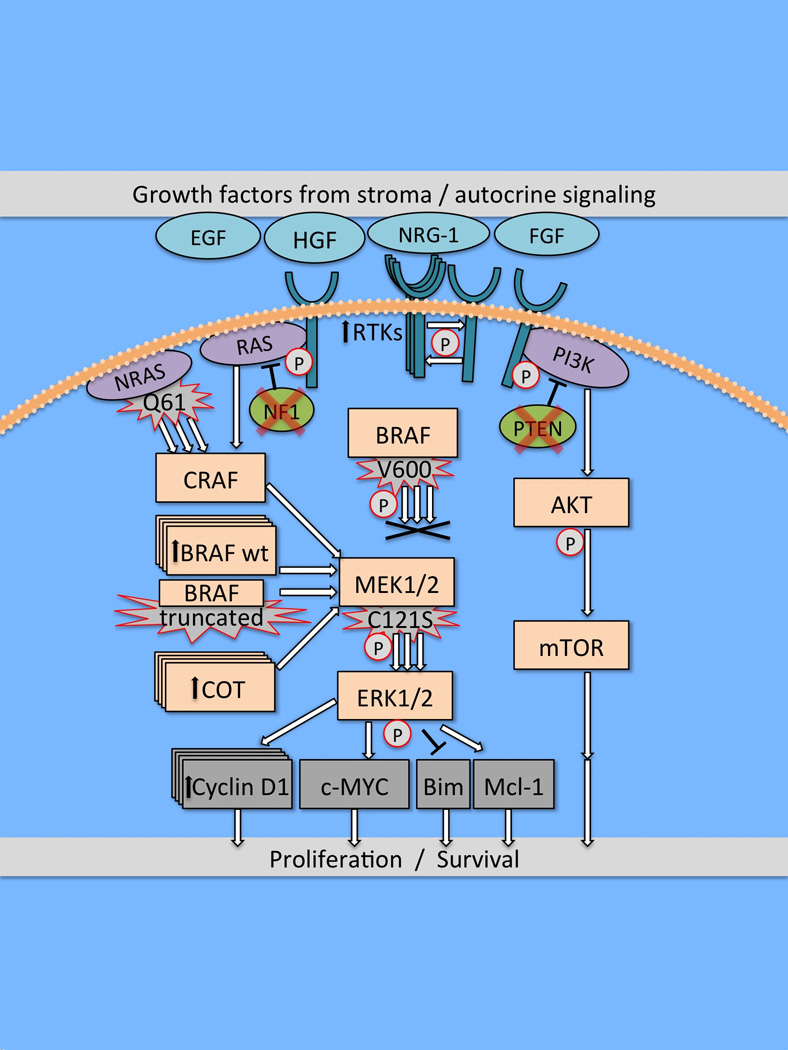
Outline of the major signaling pathways in melanoma and potential mechanisms of acquired/intrinsic BRAF inhibitor resistance. Reactivation of MAPK signaling is a common feature of acquired RAF inhibitor resistance and can be mediated through increased RTK signaling, CRAF/BRAF amplification, increased Cot expression and the dimerization of BRAF splice-mutants as well as via activating mutations in NRAS and MEK1/2. Increased PI3K/AKT signaling, resulting from RTK signaling and genetic lesions in PTEN and NF1 can also contribute to BRAF inhibitor resistance.
Table 1. Frequency of BRAFV600 mutations in patients.
| Malignancy | % BRAF* | n, BRAF | n, Total |
|---|---|---|---|
| Hairy Cell Leukemia | 100% | 48 | 48 |
| Pleomorphic xanthoastrocytoma | 66% | 42 | 64 |
| Pleomorphic xanthoastro. w/ anaplasia | 65% | 15 | 23 |
| Langerhans Cell Histiocytosis | 57% | 35 | 61 |
| Melanoma | 47% | 320 | 677 |
| Papillary Thyroid Carcinomas | 46% | 845 | 1849 |
| Ganglioglioma | 18% | 14 | 77 |
| Pilocytic astrocytoma | 9% | 9 | 97 |
| Colorectal cancer | 8% | 56 | 711 |
| Lung Adenocarcinoma | 5% | 36 | 739 |
Table outlines the % of particular tumor given histologies with activating BRAF (BRAF*) mutations. Data also shows the number of patients analyzed and the number of those with oncogenic BRAF
The discovery of mutant BRAF as an important oncogenic driver in multiple cancer types has led to the development of small molecule inhibitors of the BRAF kinase. Of these, dabrafenib and vemurafenib (IC50 values against BRAFV600E, 0.6 nM and 31 nM, respectively) have been the most intensively studied [9,10]. In preclinical experiments both inhibitors potently inhibit MAPK signaling in cell lines harboring oncogenic BRAF - an effect associated with the inhibition of growth and survival. Similarly impressive results were also seen in xenograft models of BRAFV600E mutant melanoma and both drugs rapidly advanced into clinical trials [9,10]. In large randomized phase III clinical trials, both vemurafenib and dabrafenib outperformed the previous standard-of-care – the alkylating agent dacarbazine - and were associated with an increased progression-free-survival (PFS) of 5.3 and 5.1 months, respectively [11,12]. Pharmacodynamic analyses showed anti-tumor responses to be restricted to patients whose melanomas harbored BRAF mutations with >90% MAPK signaling inhibition being required for any tumor shrinkage to be seen [13]. Off-target effects were generally mild compared to those seen to chemotherapy with pyrexia, fatigue, headache and gastrointestinal effects being the most common [12]. More unexpectedly, BRAF inhibition was also associated in some cases with the development of squamous cell carcinomas (SCC), new nevi and secondary (BRAF wild-type) melanomas [14]. The emergence of these secondary lesions on therapy was the result of the paradoxical MAPK signaling that is known to occur in cells with upstream RTK signaling or RAS mutations [14]. Similarly impressive results to vemurafenib have also been reported in a limited number of hairy cell leukemia patients, with complete responses being demonstrated [5].
Despite the presence of a BRAF mutation being a pre-requisite for a BRAF inhibitor response, only ~50% of patients whose melanomas harbored oncogenic BRAF met the RECIST (response evaluation criteria in solid tumors) criteria for responses to vemurafenib or dabrafenib. Melanomas have complex mutational profiles, with lesions in other genes responsible for Ras and PI3K signaling such as NF1 and PTEN, as well as genomic amplification of MAPK pathway mediators such as BRAF, CRAF and cyclin D1 being implicated in intrinsic BRAF inhibitor resistance [15,16] (Figure 1). Studies are ongoing to address how the co-operation between multiple genetic hits can predict for intrinsic sensitivity or resistance to RAF kinase inhibitors.
Acquired RAF inhibitor resistance
Although the responses to BRAF inhibitors in patients with BRAF mutant melanomas were highly impressive and out-performed every previous therapy tried in this disease, resistance was common for the majority of patients [11,12]. Despite this, limited numbers of individuals have been identified who show durable responses to BRAF inhibitors (median duration 35.9 months) and studies are ongoing to identify the unique genetic characteristics of this patient sub-group [17]. Acquired resistance to other small molecule kinase inhibitors, such as imatinib in chronic myeloid leukemia and EGFR inhibitors in non-small cell lung cancer is usually associated with the acquisition of mutations - so-called “gatekeeper” mutations - in the kinase domain of the RTK that prevents drug binding. Despite preclinical work identifying Thr-529 as the potential gatekeeper site in BRAF, no studies to date have identified this mutation in any melanoma specimens from patients failing BRAF inhibitor therapy [18]. Instead, a complex picture of resistance has emerged implicating multiple potential mechanisms, with some being co-existent within the same tumor [1]. Common to all the resistance mechanisms reported thus far, and a finding that has now been extensively validated clinically, is reactivation of the MAPK signaling pathway [1].
One of the first studies to address the issue of acquired BRAF inhibitor resistance was an unbiased screen in which >600 open reading frames (ORFs) encoding for kinases and kinase-like proteins from the Broad Institute library were overexpressed in BRAFV600E mutant melanoma cells [19]. It was found that 9 candidate ORFs including AXL, CRKL, ERBB2, FGR, MAP3K8, PAK3, CRAF and PKC (epsilon and eta subunits) conveyed resistance to BRAF inhibition. Some limited clinical evidence was provided implicating MAP3K8 or Cot in acquired vemurafenib resistance and it was noted that overexpression of MAP3K8 in preclinical studies strongly upregulated MAPK signaling and limited the responses to BRAF inhibition [19].
Most initial investigations into acquired RAF inhibitor resistance were in vitro studies in which resistant BRAFV600E mutant cell lines were generated through chronic treatment with either vemurafenib and its analogues or dabrafenib. The resistant cell lines were then characterized through commercially available RTK arrays. Together these studies identified increased growth factor signaling, through RTKs including PDGFRβ, IGF1R, EGFR and ERBB3 as being potential modulators of response to BRAF inhibition [20,21] (Figure 1). Another study, by Settleman and colleagues also identified hepatocyte growth factor (HGF), fibroblast growth factor (FGF) in addition to neuregulin (NRG1) and epithelial growth factor (EGF) as having the capacity to induce resistance to targeted therapies, while IGF and PDGF were mostly ineffective [22]. Activation of the target RTK and one downstream effector was not sufficient to reduce drug sensitivity, and successful rescue was instead associated with activation of both the MAPK and PI3K pathways [22]. In all cases, the rescue observed with a particular growth factor could be successfully overcome by co-treatment with an inhibitor of its target RTK [22]. Although targeting RTKs could offer new opportunities for the development of combination therapy strategies it was noted that several cell lines responded to three or four of the growth factors tested.
The tumor microenvironment plays an important role in the resistance to many anticancer agents. A screen of 35 anticancer drugs against 45 cancer cell lines cultured either alone or with each of 23 different stromal cell lines demonstrated the ability of stromal cells to render otherwise sensitive cancer cell lines resistant to both chemotherapy and targeted therapies [23]. Microenvironment-mediated resistance was observed against 15/23 (65%) of targeted therapies, including the Her2 inhibitor lapatinib in ERBB2 overexpressing breast cancer and vemurafenib for BRAFV600E mutated melanoma and colorectal cancer cell lines. Six of 18 stromal cell lines induced resistance against vemurafenib in BRAFV600E mutant melanoma by direct contact co-culture, which could be recapitulated by treatment with conditioned medium from the same stromal cells [23]. Quantification of 567 secreted factors by an antibody array showed that the level of hepatocyte growth factor (HGF) displayed the highest correlation with the resistance to vemurafenib. Addition of HGF to BRAFV600E mutant melanoma cells was sufficient to induce resistance to both vemurafenib and the MEK inhibitor PD184352, while the resistance conferred by conditioned medium, was abolished by HGF neutralizing antibodies or the MET inhibitor crizotinib [23]. HGF induced persistent activation of both the MAPK and PI3K pathways in the presence of either vemurafenib or PD184352. Despite some correlation being seen between HGF levels in the tumor and response to a BRAF inhibitor in a small patient cohort, a follow-up analysis of a larger patient group (n=209) did not show any association between MET expression and objective response, PFS or overall survival on BRAF inhibitor therapy [23,24].
Although many studies have now implicated RTK signaling as a potential modulator of RAF inhibitor resistance, melanoma cells are not typically responsive to exogenous growth factors. Under baseline conditions, BRAF mutant melanoma cells have high levels of feedback inhibition in the MAPK signaling pathway that suppresses their sensitivity to growth factor mediated Ras signaling through increased Spry2 expression [25]. Following RAF inhibition, feedback inhibition is relieved and responsiveness to growth factors is restored. It thus seems that addition of RAF or MEK kinase inhibitors may subsequently allow melanoma cells to respond to both tumor intrinsic and host-derived growth factors allowing for drug tolerance. The adaptive RTK signaling seen following BRAF inhibition may also be critical in understanding the low vemurafenib response rates (~5%) seen in BRAFV600E mutant colorectal carcinoma [26]. A number of studies have now shown that inhibition of BRAF in colon cancer cell lines triggers a strong survival signal that drastically limits drug efficacy. This intrinsic resistance could be overcome through co-targeting of EGFR and BRAF or BRAF and PI3K and combination studies are ongoing to validate this hypothesis clinically [26]. There is also very recent evidence that melanocyte lineage specific cyclic AMP (cAMP) signaling can also mediate acquired BRAF inhibitor resistance [27].
Genetic approaches have also been used to characterize mechanisms of acquired and intrinsic BRAF inhibitor resistance. One of the major methods used thus far has been whole exome sequencing (WES). Although WES is much less comprehensive than whole genome sequencing, it offers the advantages of being both cheaper and quicker while still capturing all of the protein coding regions implicated in cancer. In one of the first genetic studies, acquired NRAS (Q61K) mutations were shown to be a potential driver of BRAF inhibitor resistance [20]. Other sequencing studies have since identified amplification of BRAF and activating C121S MEK1 mutations as mediators of therapeutic escape in both cell lines and melanoma patient specimens [28] (Figure 1). There is also evidence that mutations in MEK2 (Q60P) in concert with BRAF amplification can also blunt the responses to both BRAF and MEK inhibitors [29]. Another mechanism that melanoma cells can use to reactivate their MAPK signaling is through the alternate splicing of BRAF. It was reported that a resistance-associated BRAF mutant splice-form (p61BRAFV600E) lacking exons 4–8 (the Ras-binding domain) formed dimers in melanoma cells with low Ras activity [30]. When dimerized the splice-form mediated downstream MAPK signaling and conveyed resistance to BRAF inhibitors [30]. A number of patients failing BRAF inhibitor therapy were found to harbor the BRAF splice-form mutant. The mechanisms of acquired BRAF inhibitor resistance are now being addressed in large cohorts of melanoma patients failing BRAF inhibitor therapy. An analysis of pre and post-treatment biopsies from 91 patients on the BRIM-2 phase II clinical trial of vemurafenib showed the reactivation of MAPK signaling to be a common feature of acquired drug resistance [31]. Mutational analysis of the specimens showed the acquisition of NRAS (Q61) and MEK1 (Q56P and E203K) to be a common occurrence. A modest association was noted between decreased expression of PTEN and reduced response [31,32]. Pre-existing mutations in MEK at P124 were not predictive of reduced anti-tumor response. A second analysis of 41 patients receiving dabrafenib therapy was performed to determine the association between anti-tumor response and the status (copy number/mutational status) of PTEN, cyclin D1 and CDKN2A [32]. Baseline PTEN loss/mutation was found to be significantly associated with shorter PFS, as was increased copy number of cyclin D1 and lower copy number of CDKN2A [32]. Two patients who showed progressive disease as their best response were found to have a higher copy number in BRAF.
Combination therapy strategies
The evidence to date suggests reactivation of the MAPK signaling pathway to be the major mechanism of resistance to BRAF inhibitors in melanoma. Adaptations to BRAF inhibition in tissue culture occur rapidly following drug treatment, with early preclinical data suggesting that the combination of a BRAF and MEK inhibitor may be one strategy to limit the onset of resistance. This idea has been explored in a phase II clinical trial in which the BRAF inhibitor dabrafenib (150 mg twice daily) was combined with the MEK inhibitor trametinib (2 mg daily) [33]. The combination outperformed BRAF inhibitor alone with respect to response rates (complete or partial responses in 76% versus 54% of patients) and median PFS (9.4 months versus 5.8 months, respectively) [33]. Importantly, the combination limited the frequency of SCCs emerging on therapy, most likely through the MEK inhibitor-mediated suppression of paradoxical MAPK pathway activation [14]. In light of this data both dabrafenib and trametinib were recently FDA-approved in the single agent setting, with approval of the combination expected in 2014. Despite the improved responses seen to the double combination, resistance still emerged with preliminary analyses again suggesting MAPK pathway reactivation to be the major resistance mechanism. Interestingly, the mechanisms of therapeutic escape reported thus far to the combination are similar to those seen with single agent BRAF inhibitors, with mutations in MEK1, MEK2 and BRAF splice forms being reported [34].
The continued reliance upon MAPK signaling has suggested a future need for triple therapy combinations. One option being explored is the addition of an inhibitor of ERK, and there is already some preclinical evidence that the ERK inhibitor SCH772984 can overcome resistance to the combination of vemurafenib and trametinib [35]. The ability of SCH772984 to prevent or delay the onset of resistance to the BRAF/MEK inhibitor combination has not been evaluated. One other strategy to suppress the recovery of MAPK signaling is through combination with an inhibitor of HSP90, and there is already good preclinical evidence that this doublet may be effective at abrogating resistance to vemurafenib [36,37]. One further strategy under investigation is to target mitogenic signaling further downstream at the level of the cyclin dependent kinases and trials have been initiated where BRAF inhibitors are combined with CDK inhibitors (Table 2).
Table 2. Inhibitor combinations in clinical trials.
Summary of ongoing BRAF inhibitor based combination therapy trials. Data derived from clinicaltrials.gov on 18/9/13. Table shows primary BRAF inhibitor, combination partners, phase of trial, target accrual and start/end dates.
| Trial ID | BRAFi | 2nd/3rd targets |
2nd/3rd drug | Phase | Enroll | Start | End |
|---|---|---|---|---|---|---|---|
| NCT01909453 | LGX818 | MEK | MEK162 | 3 | 900 | 9/13 | 6/17 |
| NCT01689519 | vemurafenib | GDC-0973 | 3 | 500 | 1/13 | 8/16 | |
| NCT01726738 | dabrafenib | trametinib | 2 | 20 | 10/12 | 10/14 | |
| NCT01616199 | vemurafenib | PI3K | PX-866 | 1/2 | 146 | 8/12 | 12/14 |
| NCT01512251 | vemurafenib | BMK120 | 1/2 | 46 | 6/12 | 6/14 | |
| NCT01673737 | vemurafenib | SAR260301 | 1 | 75 | 8/12 | 1/16 | |
| NCT01841463 | vemurafenib | CDK4/6 | P1446A-05 | 1/2 | 100 | 7/13 | 3/15 |
| NCT01777776 | LGX818 | LEE011 | 1/2 | 150 | 7/13 | 12/15 | |
| NCT01835184 | vemurafenib | c-MET | XL184 | 1 | 34 | 5/13 | 5/14 |
| NCT01820364 | LGX818* | MEK | MEK162 | 2 | 100 | 8/13 | 7/16 |
| PI3K | BMK120 | ||||||
| CDK4/6 | LEE011 | ||||||
| c-MET | INC280 | ||||||
| FGFR | BGJ398 | ||||||
| NCT01902173 | dabrafenib | AKT | GSK2141795 | 1/2 | 66 | 7/13 | 10/15 |
| NCT01495988 | vemurafenib | VEGFR | bevacizumab | 2 | 180 | 8/13 | 8/13 |
| NCT01657591 | vemurafenib | HSP90 | XL888 | 1 | 36 | 7/12 | 1/15 |
| NCT01543698 | LGX818 | MEK CDK4/6 | MEK162 LEE011 | 1b/2 | 179 | 5/12 | 3/15 |
| NCT01750918 | dabrafenib | MEK EGFR | trametinib panitumumab | 2 | 200 | 12/12 | 12/12 |
| NCT01719379 | LGX817 | PI3K EGFR | BYL719 Cetuximab | 1/2 | 124 | 1/13 | 11/15 |
All patient start treatment with LGX818 only, and the 2nd agent is added if patients progress.
Although most attention has focused upon the role of MAPK pathway in melanoma, signaling through other pathways is also required. Of these, the PI3K/AKT signal transduction cascade is known to be critical for melanoma initiation and maintenance as well being an important mediator of intrinsic BRAF inhibitor resistance [38]. Preclinical studies have already demonstrated the utility of dual MAPK/PI3K targeting in BRAFV600E mutant melanoma and a number of clinical trials are ongoing to evaluate this, as well as the BRAF/mTOR inhibitor combination (Table 2) [10,39]. Increased RTK signaling has been identified as a potential host-derived and tumor-intrinsic mechanism of adaptation to BRAF inhibition. This hypothesis is currently being explored through clinical trials in which RTK signaling (either VEGFR or multiple RTKs) is targeted along with BRAF (Table 2). In non-melanoma cancers, combination studies are also currently accruing. In BRAF mutant colorectal carcinoma, the combination of a BRAF inhibitor with a PI3K inhibitor or a BRAF inhibitor with a methyltransferase inhibitor was found to overcome innate resistance in preclinical models [40]. Clinical trials are also ongoing to evaluate the efficacy of BRAF inhibitors in combination with EGFR targeting agents.
Conclusion
Raf inhibition is rapidly becoming the paradigm for targeted therapy in cancer. Despite this, significant challenges remain. We do not yet fully grasp the complexity of adaptive signaling that occurs when important oncogenes such as BRAF are therapeutically targeted. Resistance is also likely to be complex and multi-factorial even within individual patients and this will require novel assays to monitor response and relapse in real time. Improved strategies to delineate how genotype predicts for adaptive signaling changes will allow personalized RAF-inhibitor based combination therapy strategies to be designed that will deliver long-term responses to cancer patients.
Acknowledgments
Grant support: Work in the Smalley lab is supported by R01 CA161107-01 from the National Institutes of Health
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The author(s) have no conflict of interest to declare.
References
- 1.Fedorenko IV, et al. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem Pharmacol. 2011;82(3):201–209. doi: 10.1016/j.bcp.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wellbrock C, et al. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5(11):875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 3.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 4.Wan PT, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 5.Tiacci E, et al. BRAF Mutations in Hairy-Cell Leukemia. New England Journal of Medicine. 2011;364(24):2305–2315. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singer G, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95(6):484–486. doi: 10.1093/jnci/95.6.484. [DOI] [PubMed] [Google Scholar]
- 7.Andrulis M, et al. Targeting the BRAF V600E Mutation in Multiple Myeloma. Cancer Discovery. 2013;3(8):862–869. doi: 10.1158/2159-8290.CD-13-0014. [DOI] [PubMed] [Google Scholar]
- 8.Namba H, et al. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J Clin Endocrinol Metab. 2003;88(9):4393–4397. doi: 10.1210/jc.2003-030305. [DOI] [PubMed] [Google Scholar]
- 9.Bollag G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467(7315):596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greger JG, et al. Combinations of BRAF, MEK, and PI3K/mTOR Inhibitors Overcome Acquired Resistance to the BRAF Inhibitor GSK2118436 Dabrafenib, Mediated by NRAS or MEK Mutations. Molecular Cancer Therapeutics. 2012;11(4):909–920. doi: 10.1158/1535-7163.MCT-11-0989. [DOI] [PubMed] [Google Scholar]
- 11.Hauschild A, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 12. Chapman PB, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England journal of medicine. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. **Landmark clinical study showing the efficacy of vemurafenib in a randomized phase III clinical trial.
- 13.Bollag G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibney GT, et al. Paradoxical oncogenesis--the long-term effects of BRAF inhibition in melanoma. Nature reviews. Clinical oncology. 2013;10(7):390–399. doi: 10.1038/nrclinonc.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maertens O, et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discovery. 2012 doi: 10.1158/2159-8290.CD-12-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smalley KS, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther. 2008;7(9):2876–2883. doi: 10.1158/1535-7163.MCT-08-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim K, et al. Significant long-term survival benefit demonstrated with vemurafenib in ongoing phase I study. Pigment Cell & Melanoma Research. 2012;25(6):866. [Google Scholar]
- 18.Whittaker S, et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci Transl Med. 2010;2(35):35ra41. doi: 10.1126/scitranslmed.3000758. [DOI] [PubMed] [Google Scholar]
- 19.Johannessen CM, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010 doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nazarian R, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villanueva J, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson TR, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Straussman R, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408) doi: 10.1038/nature11183. 500-U118. ** Interesting paper demonstrating the role of host cells in mediating drug resistance.
- 24.Jubb AM, et al. Impact of MET expression on outcome in BRAF advanced melanoma. Histopathology. 2013 doi: 10.1111/his.12169. [DOI] [PubMed] [Google Scholar]
- 25. Lito P, et al. Relief of Profound Feedback Inhibition of Mitogenic Signaling by RAF Inhibitors Attenuates Their Activity in BRAFV600E Melanomas. Cancer Cell. 2012;22(5):668–682. doi: 10.1016/j.ccr.2012.10.009. ** Intriguing study indicating that melanoma cells are only sensitive to exogenous growth factors following the relief of feedback inhibition in the BRAF/MEK/ERK signaling pathway.
- 26.Prahallad A, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 27.Johannessen CM, et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504(7478):138–142. doi: 10.1038/nature12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi HB, et al. Preexisting MEK1 Exon 3 Mutations in (V600E/K)BRAF Melanomas Do Not Confer Resistance to BRAF Inhibitors. Cancer Discovery. 2012;2(5):414–424. doi: 10.1158/2159-8290.CD-12-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villanueva J, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Reports. 2013 doi: 10.1016/j.celrep.2013.08.023. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poulikakos PI, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480(7377) doi: 10.1038/nature10662. 387-U144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trunzer K, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(14):1767–1774. doi: 10.1200/JCO.2012.44.7888. [DOI] [PubMed] [Google Scholar]
- 32. Nathanson KL, et al. Tumor Genetic Analyses of Patients with Metastatic Melanoma Treated with the BRAF Inhibitor Dabrafenib (GSK2118436) Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(17):4868–4878. doi: 10.1158/1078-0432.CCR-13-0827. * clinical study demonstrating that PTEN impairment may be a negative prognostic factor for response to BRAF inhibition.
- 33. Flaherty KT, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. The New England journal of medicine. 2012;367(18):1694–1703. doi: 10.1056/NEJMoa1210093. ** Phase II clinical trial demonstrating that vertical pathway inhibition is more effective than single agent BRAF inhibition.
- 34.Wagle N, et al. Whole exome and whole transcriptome sequencing in melanoma patients to identify mechanisms to combined RAF/MEK inhibition. Journal of Clinical Oncology. 2013;31:9015. [Google Scholar]
- 35.Morris EJ, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discovery. 2013;3(7):742–750. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 36.Paraiso KH, et al. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(9):2502–2514. doi: 10.1158/1078-0432.CCR-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu X, et al. Activity of the heat shock protein 90 inhibitor ganetespib in melanoma. PLoS ONE. 2013;8(2):e56134. doi: 10.1371/journal.pone.0056134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi H, et al. A Novel AKT1 Mutant Amplifies an Adaptive Melanoma Response to BRAF Inhibition. Cancer Discovery. 2013 doi: 10.1158/2159-8290.CD-13-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smalley KS, et al. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5(5):1136–1144. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- 40.Mao M, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(3):657–667. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]