

Abstract
Fibrosis of the lung is one of the major clinical problems of cystic fibrosis and chronic obstructive pulmonary disease. However, the molecular mechanisms leading to pulmonary fibrosis are poorly characterized and require definition. Here, we demonstrate that chronic accumulation of ceramide in the lung contributes to the development of fibrosis in aged cystic fibrosis mice. Genetic or pharmacological normalization of ceramide in cystic fibrosis mice, which was achieved by heterozygosity of acid sphingomyelinase or chronic (6.5 month long) treatment of mice with pharmacological inhibitors of acid sphingomyelinase significantly decreased the development of lung fibrosis. Moreover, our studies demonstrate that long-term treatment of cystic fibrosis mice with pharmacological inhibitors of acid sphingomyelinase or genetic heterozygosity of the enzyme also minimizes pulmonary inflammatory cytokines in cystic fibrosis mice. This data identifies ceramide as a key molecule associated with pulmonary fibrosis in cystic fibrosis mice and demonstrate for the first time that prolonged inhibition of acid sphingomyelinase is able to attenuate fibrosis and inflammation in this animal model.
Keywords: Ceramide, Cystic fibrosis, Acid sphingomyelinase, Cftr
1. Introduction
Cystic fibrosis is the most common autosomal recessive genetic disorder in western countries. It is caused by mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene [1,2]. The gene frequency of mutations is approximately 1/50 and the disease affects approximately one newborn out of 2500 births [3,4]. Thus, approximately 80,000 children and young adults in the European Union and the United States are affected.
Mutations of the CFTR molecule cause a variety of clinical abnormalities, the most important of which involve the pulmonary and gastrointestinal systems. Gastrointestinal manifestations include malabsorption, intestinal obstruction, obstruction of pancreas ducts, pancreatitis, pancreatic insufficiency and cirrhosis. However, at present, the main cause of morbidity and mortality for patients with cystic fibrosis is due to complications of the pulmonary system. Almost all patients experience chronic lung infection with Pseudomonas aeruginosa, Staphylococcus aureus, Burkholderia cepacia and/or Haemophilus influenzae [3,4].
Cystic fibrosis patients display a slowly progressing fibrosis of the lung, which together with the chronic infection and inflammation, results in severe lung disease and failure [5]. Previous studies by Durie et al. demonstrated changes of the lung in Cftr-deficient mice such as areas of acinar dilation, thickening of the interstitial tissue, marked increase of interstitial connective tissue, an increase of the number of interstitial fibroblasts and focal areas of increased collagen formation and deposition – all signs of pulmonary fibrosis [6]. While lung fibrosis is a key hallmark of cystic fibrosis, the molecular events that contribute to fibrosis are largely unknown. CFTR exhibits chloride channel activity and it has been suggested that the lack of chloride secretion may result in increased water absorption from the mucus on tracheal and bronchial epithelial cells. Abnormal absorption was thought to increase mucus viscosity, reduce mucociliary clearance and possibly obstruct the flow of the airway [7,8]. However, in vivo studies involving cystic fibrosis patients and animal models of cystic fibrosis have failed to demonstrate a significant and uniform reduction of water content in the mucus [9]. Further, it is unknown whether such a change of the mucus would result in lung fibrosis. Recent studies emphasized that cystic fibrosis patients suffer from chronic inflammation in the lung with an imbalance between pro-inflammatory and anti-inflammatory cytokines in the airways [10,11]. Higher amounts of pulmonary IL-1, IL-8/keratinocyte chemoattractant (KC), TNF-α and macrophage inflammatory protein (Mip)-2 have been documented even prior to infection with P. aeruginosa [10–13]. However, it is still unknown if and how inflammation in cystic fibrosis contributes to lung fibrosis.
Several recent studies investigated the role of sphingolipids, in particular ceramide, in the pathogenesis of cystic fibrosis. Ceramide is generated by the hydrolysis of sphingomyelin by the activity of acid, neutral or alkaline sphingomyelinase or by de novo synthesis. We and others have shown that ceramide accumulates in the bronchial and tracheal epithelial cells of cystic fibrosis patients and mice [11,14–17]. We also detected increased levels of ceramide in intestinal epithelial cells of cystic fibrosis mice [14]. We demonstrated that ceramide increases death of epithelial cells, triggers a deposition of DNA in cystic fibrosis-bronchi, causes aseptic inflammation and mediates susceptibility to P. aeruginosa [10]. All of these changes are corrected by a normalization of ceramide in the airways of cystic fibrosis mice [10,18]. In these studies the reduction of pulmonary ceramide concentrations was achieved by acute inhibition of acid sphingomyelinase. Although these studies demonstrate an important role of ceramide in inflammation and infection of cystic fibrosis mice, its role in the development of lung fibrosis in cystic fibrosis was not examined.
In the present study we determined the role of pulmonary ceramide for the pathogenesis of lung fibrosis in cystic fibrosis mice using genetic and pharmacological approaches [18–20]. The data demonstrate that long-term inhibition of acid sphingomyelinase (by approximately 50%) is sufficient to normalize ceramide in the lung of cystic fibrosis mice to levels observed in wild-type mice. Most importantly, correction of ceramide levels by long-term inhibition of acid sphingomyelinase minimized fibrosis, reduced inflammation and abrogated the increased susceptibility to infection in 6–8 month old cystic fibrosis mice.
2. Methods
2.1. Mice
We used B6.129P2(CF/3)-CftrTgH(neoim)Hgu (abbreviated Cftr−/−) congenic mice on a C57BL/6 background and syngenic C57BL/6 wildtype mice as controls. Cftr−/− mice were crossed with acid sphingomyelinase-deficient mice, also on a C57BL/6 background, to obtain animals lacking Cftr and being heterozygous for acid sphingomyelinase (called Cftr−/−/Smpd1+/−; Smpd1 is the gene symbol for acid sphingomyelinase). Amitriptyline or fluoxetine were applied to the mice via the drinking water at 180 mg amitriptyline/L or 120 mg fluoxetine/L. All mice were housed in the Central Laboratory Animal Facility of the University Hospital Essen, University of Duisburg-Essen, Germany, in isolator cages that provided a pathogen-free environment. The hygienic status of the mice was repeatedly tested by a panel of common murine pathogens according to the 2002 recommendations of the Federation for Laboratory Animal Science Associations. Bacterial and parasite culturing and serology were always negative. All procedures performed on mice were approved by the Animal Care and Use Committee of the Bezirksregierung Duesseldorf, Duesseldorf, Germany.
2.2. Accustain trichrom-stains
Stainings to evaluate for collagen deposition were performed with the Accustain Trichrom-Stains (Masson) kit from Sigma Aldrich. Stainings were performed directly per kit directions: Slides were deparaffinized in a series of xylol and ethanol gradiants, warmed in Bouin's solution at 56 °C for 15 min, and cooled via tap water wash. Slides were incubated in working Weigert's Iron Hematoxylin solution for five minutes and again washed under running tap water. Samples were washed in Millipore water, stained with Biebrich Scarlet-Acid Fuchsin for five minutes, rinsed in Millipore water, and then subjected to a five minute stain with working phosphotungstic/phosphomolybdic acid solution followed by Aniline blue solution for five minutes and 1% acetic acid solution for two minutes. Slides were dehydrated via ethanol and xylol gradients and embedded in Eukitt mounting medium for microscopy.
2.3. Collagen assay
Complete right and lower left lobes of murine lungs were harvested and snap frozen for in vivo immunoblot analysis. Following protein quantification with Bradford assay and dilution with Millipore water for protein standardization, the lysates were analyzed employing the Sircol Collagen assay kit. The protocol provided with the kit was followed exactly. Sixty microgram of protein per sample was incubated with 1 ml of Sircol Dye reagent for 30 min at room temperature. During this step, the collagen absorbs the dye. While shaking, the tubes were also regularly inverted by hand to effectively mix the complex. The samples were then centrifuged at 12,000 rpm for 10 min. The supernatant was drained and the pellet washed in 750 μL of ice-cold acid-salt wash reagent (acetic acid, sodium chloride, surfactants and deionized water) to remove any unbound dye. The samples were again centrifuged at 12,000 rpm for 10 min. and further drained. Pellets were resuspended in 250 μL alkali reagent (0.5 M sodium hydroxide) and again vortexed to mix the pellet of dye bound collagen. After five min of dissolution, 200 μL of each sample was loaded into a 96 well plate. Each sample was measured with a 555 nm spectrum filter on a BMG Labtech microplate reader. It is important to note that because of the extreme sensitivity of the assay, it was necessary to harvest and lyse identical parts of the lung to ensure an accurate comparison.
2.4. Asm activity
The lungs were removed, shock frozen, and lysed in 250 mM sodium acetate (pH 5.0), 1% NP40, and 1.3 mM EDTA for 15 min. The tissues were then homogenized with a tip sonicator. Aliquots of the lysates were diluted to 250 mM sodium acetate (pH 5.0), 0.1% NP40, and 1.3 mM EDTA and incubated with 50 nCi per sample [14−C]sphingomyelin for 30 min at 37 °C. The substrate was dried prior to the assay, resuspended in 250 mM sodium acetate (pH 5.0), 0.1% NP40, and 1.3 mM EDTA and bath-sonicated for 10 min to obtain micelles. The enzyme reaction was terminated by the addition of 800 μL chloroform/methanol (2:1, v/v), phases were separated by centrifugation and radioactivity of the aqueous phase was measured by using liquid scintillation counting to determine the release of [14C]phosphorylcholine from [14C]sphingomyelin as a measure of Asm activity.
2.5. Ceramide measurements
Ceramide concentrations in the lung were determined by the diacylglycerol (DAG) kinase method. The lungs were removed, shock frozen, and homogenized under liquid nitrogen. The homogenates were transferred to 300 μL CH3OH and an aliquot was removed to normalize for protein by a Bradford assay. Homogenates were then brought to 600 μL CHCl3:CH3OH:1 N HCl (100:100:1, v/v/v), 200 μL H2O was added, the samples were centrifuged for 5 min at 14,000 rpm, and the lower phase was collected and dried. The samples were resuspended in 20 μL of a detergent solution (7.5% [w/v] n-octyl glucopyranoside, 5 mM cardiolipin in 1 mM diethylenetriaminepentaacetic acid) and sonicated for 10 min, and the kinase reaction was initiated by the addition of 70 μL of a reaction mixture consisting of 10 μL DAG kinase (GE Healthcare Europe, Munich, Germany), 0.1 M imidazole/HCl (pH 6.6), 0.2 mM diethylenetriaminepentaacetic acid (pH 6.6), 70 mM NaCl, 17 mM MgCl2 and 1.4 mM EGTA, 2 mM DTT, 1 μM ATP, and 10 μCi [32P]γATP. The kinase reaction was performed for 30 min at room temperature and terminated by the addition of 1 mL CHCl3:CH3OH:1 N HCl (100:100:1, v/v/v), 170 μL buffered saline solution (135 mM NaCl, 1.5 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, 10 mM HEPES, pH 7.2), and 30 μL of a 100 mM EDTA solution. The samples were vortexed and centrifuged, the phases were separated, and the lower phase was collected, dried, dissolved in 20 μL CHCl3:CH3OH (1:1, v/v), and separated on Silica G60 Thin Layer Chromatography (TLC) plates by using chloroform, acetone, methanol, acetic acid, and H2O (50:20:15:10:5, v/v/v/v/v). The TLC plates were analyzed by autoradiography. Ceramide spots were identified by co-migration with a ceramide standard and were removed from the plates. The incorporation of [32P] into ceramide was quantified by liquid scintillation counting. Ceramide amounts were determined by comparison with a standard curve using C16-ceramide as substrate.
2.6. IL-1 and KC-derived chemokine measurements
Murine lungs were homogenized in liquid nitrogen and lysed in 25 mM Tris/HCl, pH 7.4, 2% Nonidet P40, 125 mM NaCl, 10 mM EDTA and 10 mM sodiumpyrophosphate. Cytokine concentrations were determined by commercial ELISA assays according to the manufacturer's instructions (R&D, Wiesbaden-Nordenstedt, Germany).
2.7. Statistics
Data were analyzed by ANOVA followed by post hoc T test and are expressed as arithmetic means ± SD. Statistical significance was set at the level of p ≤ 0.05.
3. Results
To determine the role of ceramide in the development of pulmonary fibrosis in cystic fibrosis, we employed Cftr-deficient mice crossed with mice lacking acid sphingomyelinase to obtain mice lacking Cftr and heterozygous for acid sphingomyelinase. These mice show an approximately 50% reduction of the activity of acid sphingomyelinase in the lung compared to wt mice or Cftr-deficient mice (Fig. 1). In addition, we treated Cftr-deficient mice with functional inhibitors of acid sphingomyelinase, i.e. amitriptyline or fluoxetine, starting at 1.5 months of age. The mice were continuously treated via the drinking water until the age of 8 months. The two drugs reduced the activity of acid sphingomyelinase by 40–50% throughout the duration of treatment (Fig. 1).
Fig. 1.
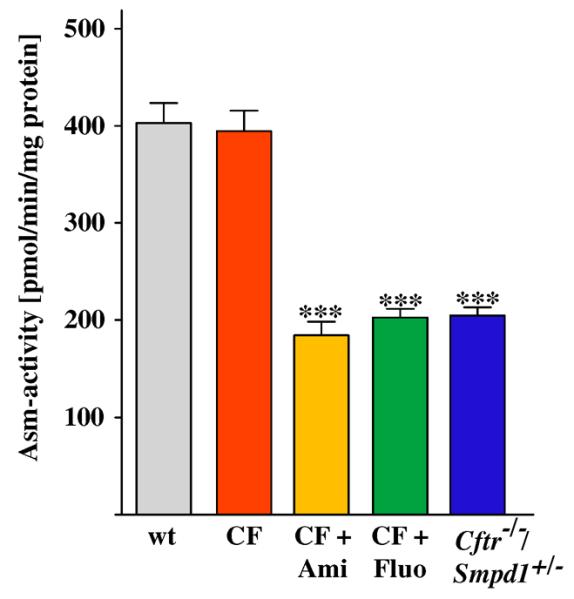
Heterozygosity of or long-term treatment with functional acid sphingomelinase inhibitors reduces the enzyme activity in the lung. Cftr-deficient mice (CF) were crossed with mice lacking acid sphingomyelinase (gene symbol Smpd1) to obtain mice lacking Cftr and heterozygous for acid sphingomyelinase (Cftr−/−/ Smpd1+/−) or treated for 6.5 months with amitriptyline (Ami) or fluoxetine (Fluo) applied via the drinking water. Untreated Cftr or wild-type (wt) mice served as controls. The activity of acid sphingomyelinase was determined in lung extracts by an in vitro enzyme assay. Both, genetic heterozygosity or treatment with functional acid sphingomyelinase inhibitors reduced the activity of acid sphingomyelinase. Data are the mean ± SD with n = 5 per group, ***p < 0.001 compared to wt.
Next, we analyzed whether genetic heterozygosity of acid sphingomyelinase or functional inhibition of acid sphingomyelinase with amitriptyline or fluoxetine reduced ceramide levels in the lungs of Cftr-deficient mice. The results demonstrate that genetic or long-term pharmacological inhibition of acid sphingomyelinase (by approximately 50%) reduced pulmonary ceramide levels in 8 month-old Cftr-deficient mice to a similar level observed in age matched wild-type mice (Fig. 2). This remarkable finding indicates that long-term treatment of Cftr-deficient mice with functional inhibitors of acid sphingomyelinase is able to reduce the activity of the enzyme and normalize ceramide levels in ageing Cftr-deficient mice.
Fig. 2.
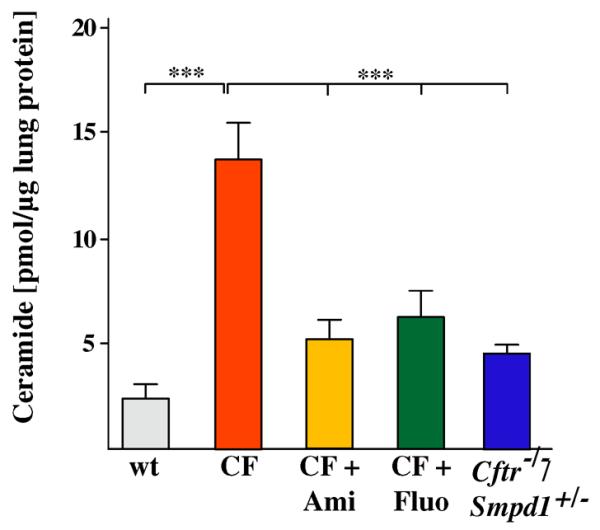
Genetic heterozygosity or functional inhibition of acid sphingomyelinase normalizes ceramide in lungs of cystic fibrosis mice. Ceramide was measured in lung extracts of wildtype, Cftr-deficient mice, Cftr-deficient mice heterozygous for acid sphingomyelinase or Cftr-deficient mice treated for 6.5 months with amitriptyline or fluoxetine. Ceramide was analyzed by a ceramide kinase assay. Data are the mean ± SD with n = 5 per group, ***p < 0.001 as indicated.
To investigate whether long-term correction of ceramide levels in Cftr-deficient mice also prevents important pathophysiological hallmarks of cystic fibrosis, we examined the development of fibrosis in 8 month-old Cftr-deficient mice by quantification of collagen in the lung and by qualitative analysis with masson trichrome staining. The results show a marked increase of collagen in the lungs of 8 month-old Cftr-deficient mice (Fig. 3A) and patchy increases of masson trichrome staining, in particular in peribronchial areas of the lung (Fig. 3B). These findings are consistent with development of lung fibrosis in Cftr-deficient mice. Analysis of younger and older mice (data not shown) showed a progression of the fibrosis with greatest contrast visualized at 8 months of age. Eight month-old Cftr-deficient mice that were heterozygous for acid sphingomyelinase or treated with amitriptyline for 6.5 months were almost completely protected from the development of lung fibrosis (Fig. 3A and B), suggesting that ceramide is a critical factor in the pathogenesis of pulmonary fibrosis in cystic fibrosis mice.
Fig. 3.
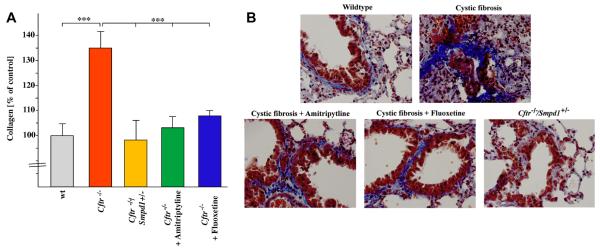
Ceramide mediates lung fibrosis in cystic fibrosis mice. Fibrosis in the lung of 8 month old Cftr-deficient mice, Cftr-deficient mice heterozygous for acid sphingomyelinase, Cftr-deficient mice treated with amitriptyline or fluoxetine and wildtype mice was determined by measuring collagen using the Sircol Dye technique (A) and masson-trichrome staining (B). Data are the mean ± SD with n = 6 per group. *p < 0.001 compared as indicated. (B). Genetic or pharmacological inhibition of acid sphingomyelinase minimized lung fibrosis in Cftr-deficient mice. Representative lung sections from 6 mice per group are shown. Magnification is 400×.
Next, we tested whether long-term treatment with functional acid sphingomyelinase inhibitors or genetic heterozygosity also protects old Cftr-deficient mice from chronic (aseptic) lung inflammation. The results of these studies show increased levels of IL-1 and KC in the lungs of untreated, eight month old Cftr-deficient mice. These levels were corrected by long-term treatment with a pharmacological inhibitor of acid sphingomyelinase or heterozygosity of acid sphingomyelinase (Fig. 4).
Fig. 4.
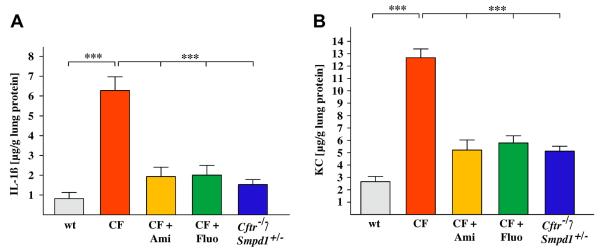
Long-term treatment with functional acid sphingomyelinase inhibitors or genetic heterozygosity protects cystic fibrosis mice from lung inflammation. Concentrations of IL-1 and KC are elevated in lungs of untreated, 8 month-old Cftr-deficient mice. Levels of these inflammatory markers are corrected by long-term treatment with amitriptyline or fluoxetine or heterozygosity of acid sphingomyelinase. IL-1 and KC levels were measured in lung homogenates by ELISA. Displayed is the mean ± SD from each 5 mice, ***p < 0.001 compared as indicated.
4. Discussion
In the present study we provide a novel mechanism for the pathogenesis of pulmonary fibrosis in cystic fibrosis. We demonstrate that genetic or long-term pharmacological normalization of ceramide levels in lungs of Cftr-deficient mice is sufficient to prevent the development of fibrosis. Further, long-term treatment with acid sphingomyelinase inhibitors or genetic heterozygosity of acid sphingomyelinase also decreases molecular markers for chronic inflammation. These studies establish that ceramide plays a critical role in the development of lung fibrosis in cystic fibrosis. The studies also suggest that pharmacological inhibitors of acid sphingomyelinase might be safely used for long-term maintenance therapy and that the effect of the drugs on acid sphingomyelinase, normalization of ceramide and protection of Cftr-deficient mice from progression of the disease is not blunted by time or a counteracting mechanism.
Our data are consistent with previous studies by Petrache et al., demonstrating the role of ceramide in the development of fibrosis and lung emphysema in chronic obstructive pulmonary disease (COPD). Those studies demonstrated that application of ceramide into the lung induced emphysema [21]. Further, it was shown that cigarette smoke triggered the accumulation of ceramide in humans and mice, while inhibition of ceramide formation attenuated the development of emphysema and fibrosis [21]. The molecular mechanisms associated with COPD are uniquely complex, as inhibition of acid sphingomyelinase was not sufficient to completely prevent the development of emphysema.
While these studies establish an important function of ceramide in the pathophysiology of lung fibrosis in cystic fibrosis and COPD, the mechanisms by which ceramide influences fibrotic development in the lung are presently unknown. Ceramide has been shown to trigger clustering of NADPH-oxidase subunits and a release of reactive oxygen species [22]. However, we were unable to show a significant increase of ROS in cystic fibrosis epithelia prior to infection with P. aeruginosa [Henry et al., unpublished data]. Ceramide may also activate protein kinase C [23,24], Jun-N terminal kinase [25] and NF-κB [26], which may lead to altered gene transcription and, for instance, in an increased expression of IL-1 and KC. However, it is unknown whether the continuous release of these pro-inflammatory mediators induces lung fibrosis in cystic fibrosis mice. We have previously shown that the increase of ceramide in cystic fibrosis epithelia triggers the constitutive clustering and activation of CD95, which results in increased death of bronchial epithelial cells [27]. Epithelial cell death resulted in the release of DNA into the bronchial lumen and facilitated infection with P. aeruginosa. Although the situation in humans with chronic P. aeruginosa infections is certainly more complex than in mice, the failure of Pulmozyme, an inhaled DNase administered to CF patients, to prevent lung fibrosis, suggests that cell death may only be a minor factor in the development of lung fibrosis. Further, it may be possible that ceramide induces activation of signaling pathways in epithelial cells that eventually contribute to epithelial-mesenchymal transition and the associated development of a fibrosis. Future studies are required to answer questions regarding the molecular mechanisms involved in the role of ceramide in lung fibrosis.
Our studies are consistent with previous data published by P. Durie et al., which also described the development of fibrosis in Cftr-deficient mice (6). However, our studies are novel in that they identify ceramide as a critical factor in the development of lung fibrosis in cystic fibrosis. The current studies not only identify a novel molecular mechanism of fibrosis development in cystic fibrosis, but also provide a novel treatment option to prevent lung fibrosis. We demonstrate that Cftr-deficient mice can be treated long-term with functional inhibitors of acid sphingomyelinase without overt side effects and that this treatment prevents the development of lung fibrosis. Since both amitriptyline and fluoxetine are approved for the treatment of major depression and have been used successfully in the clinic for decades, our data is easily transferable to a clinical study.
Acknowledgments
The study was supported by DFG-grant GU 16–2 within the DFG-SPP 1267 and the B is BF.
References
- [1].Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- [2].Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Ianuzzi MC, Collins FS, Tsui L-C. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- [3].Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361:681–689. doi: 10.1016/S0140-6736(03)12567-6. [DOI] [PubMed] [Google Scholar]
- [4].CF Foundation . Patient Registry Annual Report. CF Foundation; Bethesda: 2011. [Google Scholar]
- [5].Sheppard DN, Welsh MJ. Structure and function of the CFTR channel. Physiol. Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- [6].Durie PR, Kent G, Phillips MJ, Ackerley CA. Characteristic multiorgan pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am. J. Pathol. 2004;164:1481–1493. doi: 10.1016/S0002-9440(10)63234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis lung disease in mice. Nat. Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- [8].Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu. Rev. Med. 2007;58:157–170. doi: 10.1146/annurev.med.58.071905.105316. [DOI] [PubMed] [Google Scholar]
- [9].Chen JH, Stoltz DA, Karp PH, Ernst SE, Pezzulo AA, Moninger TO, Rector MV, Reznikov LR, Launspach JL, Chaloner K, Zabner J, Welsh MJ. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell. 2010;143:911–923. doi: 10.1016/j.cell.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Teichgräber V, Ulrich M, Endlich N, Riethmüller J, Wilker B, De Oliveira-Munding CC, van Heeckeren AM, Barr ML, von Kürthy G, Schmid KW, Weller M, Tümmler B, Lang F, Grassmé H, Döring G, Gulbins E. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 2008;14:382–391. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- [11].Ulrich M, Worlitzsch D, Viglio S, Siegmann N, Iadarola P, Shute JK, Geiser M, Pier GB, Friedel G, Barr ML, Schuster A, Meyer KC, Ratjen F, Bjarnsholt T, Gulbins E, Döring G. Alveolar inflammation in cystic fibrosis. J. Cyst. Fibros. 2010;9:217–227. doi: 10.1016/j.jcf.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Inoue H, Massion PP, Ueki IF, Grattan KM, Hara M, Dohrman AF, Chan B, Lausier JA, Golden JA, Nadel JA. Pseudomonas stimulates interleukin-8 mRNA expression selectively in airway epithelium, in gland ducts, and in recruited neutrophils. Am. J. Respir. Cell Mol. Biol. 1994;11:651–663. doi: 10.1165/ajrcmb.11.6.7946394. [DOI] [PubMed] [Google Scholar]
- [13].Tirouvanziam R, de Bentzmann S, Hubeau C, Hinnrasky J, Jacquot J, Peault B, Puchelle E. Inflammation and infection in naive human cystic fibrosis airway grafts. Am. J. Respir. Cell Mol. Biol. 2000;23:121–127. doi: 10.1165/ajrcmb.23.2.4214. [DOI] [PubMed] [Google Scholar]
- [14].Becker KA, Tümmler B, Gulbins E, Grassmé H. Accumulation of ceramide in the trachea and intestine of cystic fibrosis mice causes inflammation and cell death. Biochem. Biophys. Res. Commun. 2010;403:368–374. doi: 10.1016/j.bbrc.2010.11.038. [DOI] [PubMed] [Google Scholar]
- [15].Bodas M, Min T, Mazur S, Vij N. Critical modifier role of membrane-cystic fibrosis transmembrane conductance regulator-dependent ceramide signaling in lung injury and emphysema. J. Immunol. 2011;186:602–613. doi: 10.4049/jimmunol.1002850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brodlie M, McKean MC, Johnson GE, Gray J, Fisher AJ, Corris PA, Lordan JL, Ward C. Ceramide is increased in the lower airway epithelium of people with advanced cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2010;182:369–375. doi: 10.1164/rccm.200905-0799OC. [DOI] [PubMed] [Google Scholar]
- [17].Bodas M, Min T, Vij N. Critical role of CFTR-dependent lipid rafts in cigarette smoke-induced lung epithelial injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011;300:L811–L820. doi: 10.1152/ajplung.00408.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Becker KA, Riethmüller J, Lüth A, Döring G, Kleuser B, Gulbins E. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2010;42:716–724. doi: 10.1165/rcmb.2009-0174OC. [DOI] [PubMed] [Google Scholar]
- [19].Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, Desnick RJ, Stewart CL, Schuchman EH. Acid sphingomyelinase deficient mice. a model of types A and B Niemann-Pick disease. Nat. Genet. 1995;10:288–293. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- [20].Kornhuber J, Tripal P, Reichel M, Terfloth L, Bleich S, Wiltfang J, Gulbins E. Identification of new functional inhibitors of acid sphingomyelinase using a structure-property-activity relation model. J. Med. Chem. 2008;51:219–237. doi: 10.1021/jm070524a. [DOI] [PubMed] [Google Scholar]
- [21].Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM. Ceramide upregulation causes pulmonary cell apoptosis and emphysema like disease in mice. Nat. Med. 2005;11:491–498. doi: 10.1038/nm1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Y, Li X, Carpinteiro A, Gulbins E. Acid sphingomyelinase amplifies redox signaling in Pseudomonas aeruginosa-induced macrophage apoptosis. J. Immunol. 2008;181:4247–4254. doi: 10.4049/jimmunol.181.6.4247. [DOI] [PubMed] [Google Scholar]
- [23].Lozano J, Berra E, Municio MM, Diaz-Meco MT, Dominguez I, Sanz L, Moscat J. Protein kinase C zeta isoform is critical for kappa B-dependent promoter activation by sphingomyelinase. J. Biol. Chem. 1994;269:19200–19202. [PubMed] [Google Scholar]
- [24].Bourbon NA, Yun J, Kester M. Ceramide directly activates protein kinase C zeta to regulate a stress-activated protein kinase signaling complex. J. Biol. Chem. 2000;275:35617–35623. doi: 10.1074/jbc.M007346200. [DOI] [PubMed] [Google Scholar]
- [25].Brenner B, Koppenhoefer U, Weinstock C, Linderkamp O, Lang F, Gulbins E. Fas- or ceramide-induced apoptosis is mediated by a Rac1-regulated activation of Jun N-terminal kinase/p38 kinases and GADD153. J. Biol. Chem. 1997;272:22173–22181. doi: 10.1074/jbc.272.35.22173. [DOI] [PubMed] [Google Scholar]
- [26].Schütze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, Krönke M. TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell. 1992;71:765–776. doi: 10.1016/0092-8674(92)90553-o. [DOI] [PubMed] [Google Scholar]
- [27].Becker KA, Henry B, Ziobro R, Tümmler B, Gulbins E, Grassmé H. Role of CD95 in pulmonary inflammation and infection in cystic fibrosis. J. Mol. Med. 2012;90:1011–1023. doi: 10.1007/s00109-012-0867-2. [DOI] [PubMed] [Google Scholar]