

Abstract
We present a high-quality genome sequence of a Neandertal woman from Siberia. We show that her parents were related at the level of half siblings and that mating among close relatives was common among her recent ancestors. We also sequenced the genome of a Neandertal from the Caucasus to low coverage. An analysis of the relationships and population history of available archaic genomes and 25 present-day human genomes shows that several gene flow events occurred among Neandertals, Denisovans and early modern humans, possibly including gene flow into Denisovans from an unknown archaic group. Thus, interbreeding, albeit of low magnitude, occurred among many hominin groups in the Late Pleistocene. In addition, the high quality Neandertal genome allows us to establish a definitive list of substitutions that became fixed in modern humans after their separation from the ancestors of Neandertals and Denisovans.
In 2008, a hominin finger phalanx was discovered during excavation in the east gallery of Denisova Cave in the Altai Mountains. From this bone, a genome sequence was determined to ~30-fold coverage1. Its analysis showed that it came from a previously unknown group of archaic humans related to Neandertals which we named “Denisovans”2. Thus, at least two distinct human groups, Neandertals and the related Denisovans, inhabited Eurasia when anatomically modern humans emerged from Africa. In 2010, another hominin bone, this time a proximal toe phalanx (Fig. 1a), was recovered in the East Gallery of Denisova Cave3. Layer 11, where both the finger and the toe phalanx were found, is thought to be at least 50,000 years old. The finger was found in sublayer 11.2, which has an absolute date of 50,300 ± 2200 years (OxA-V-2359-16), while the toe derives from the lowest sublayer 11.4, and may thus be older than the finger (Supplementary Information (SI) 1, 2a). The phalanx comes from the fourth or the fifth toe of an adult individual and its morphological traits link it with both Neandertals and modern humans3.
Figure 1. Toe phalanx and location of Neandertal samples for which genome-wide data are available.
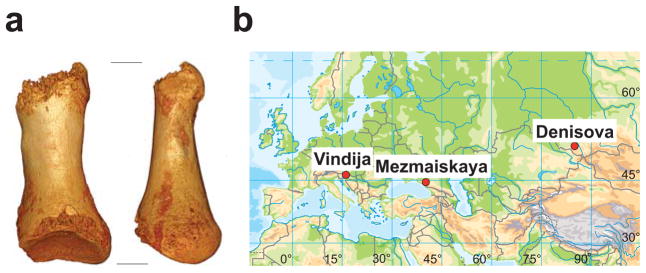
a, The toe phalanx found in the East Gallery of Denisova Cave in 2010. Left: dorsal view; Right: left view. b, Map of Eurasia showing the location of Vindija cave, Mezmaiskaya cave and Denisova cave.
Genome sequencing
In initial experiments to determine if DNA was preserved in the toe phalanx, we extracted and sequenced random DNA fragments. This revealed that about 70% of the DNA fragments present in the specimen aligned to the human genome. Initial inspection of the fragments with similarity to the mitochondrial (mt) genome suggested that its mtDNA was closely related to Neandertal mtDNAs. We therefore assembled the full mitochondrial sequence by aligning DNA fragments to a complete Neandertal mitochondrial genome4 (SI 2b). A phylogenetic tree (Fig. 2a) shows that the toe phalanx mtDNA shares a common ancestor with six previously published Neandertal mtDNAs5 to the exclusion of present-day humans and the Denisova finger phalanx. Among Neandertal mtDNAs, the toe mtDNA is most closely related to the mtDNA from infant 1 from Mezmaiskaya Cave in the Caucasus6.
Figure 2. Phylogenetic relationships of the Altai Neandertal.
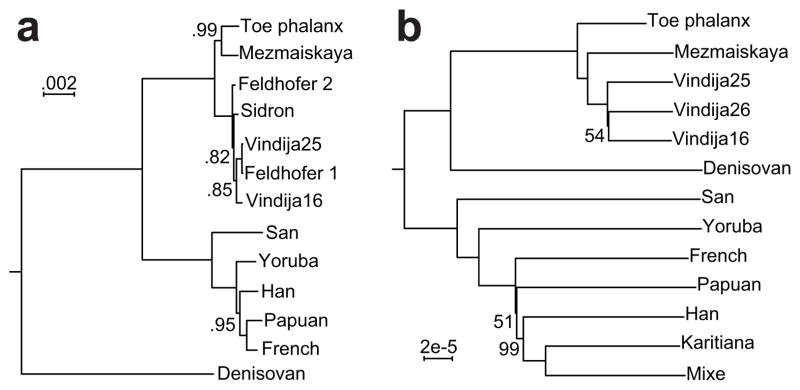
a, Bayesian tree of mitochondrial sequences of the toe phalanx, the Denisovan finger phalanx, six Neandertals, and five present-day humans. Posterior probabilities are given for branches whose support is less than one (SI 2b). b, Neighbor-joining tree based on autosomal transversion differences among the toe phalanx, four Neandertals, the Denisova genome, and seven present-day human individuals. Bootstrap values are shown for branches supported by less than 100% of 1,000 bootstrap replicates (SI 6).
We generated four DNA libraries using a recently published protocol that is particularly efficient in retrieving DNA from ancient samples1,7. These libraries, together with one library prepared using a previous protocol8, were treated with uracil-DNA-glycosylase to remove uracil residues, a common miscoding lesion in ancient DNA that results from the deamination of cytosine9–11 (SI 5a). In total, these five DNA libraries provided 52-fold sequence coverage of the genome. We estimated present-day human DNA contamination in the libraries with four complementary approaches (SI 5) using mtDNA and nuclear DNA and conclude that present-day human contamination among the DNA fragments sequenced is around 1%. After genotype calling, which is designed to be insensitive to low levels of error, we expect that the inferred genome sequence is largely free from contamination.
Relationship to other hominins
We compared the toe phalanx genome to the Denisovan genome1, the draft Neandertal genome of 1.3-fold coverage determined from three individuals from Vindija Cave, Croatia12, the genome of a Neandertal infant estimated to be 60,000 to 70,000 years old13 from Mezmaiskaya Cave in the Caucasus that we sequenced to 0.5-fold genomic coverage (SI 1; Fig. 1b) as well as 25 genomes of present-day humans: 11 previously sequenced to between 24- and 31- fold coverage1 (“Panel A”), and 14 sequenced to between 35- and 42- fold coverage for this study (“Panel B”). We used pooled fosmid sequencing to resolve the sequences of the two chromosomes carried by 13 of these individuals14 (SI 4).
A neighbor-joining tree (Fig. 2b) based on transversions, i.e. purine-pyrimidine differences, among 7 present-day humans and the low-coverage Mezmaiskaya and Vindija genomes corrected for errors (SI 6a), shows that the toe phalanx nuclear genome forms a clade with the genomes of Neandertals. The average DNA sequence divergence between the toe phalanx genome and the Mezmaiskaya and Vindija Neandertal genomes is approximately a third of that between the Neandertal and Denisova genomes. We conclude that the individual from whom the toe phalanx derives is a Neandertal. In what follows we refer to it as the “Altai Neandertal”.
Branch Shortening
The length of the branches leading from the common ancestor shared with chimpanzee to the high-coverage Altai Neandertal and Denisovan genomes are 1.02% (range: 0.99–1.05%) and 0.81% (range: 0.77–0.84%) shorter, respectively, than the branches to the present-day human genomes (Table 1; SI 6b). This is expected because the archaic genomes ceased accumulating substitutions at the death of the individual tens of thousands of years ago. We previously estimated the shortening of the Denisovan lineage to be 1.16% (range: 1.13–1.27%)1. The fact that using present-day human genomes of higher quality and more stringent quality filtering reduces the estimate by about a third shows that at present, estimates of lineage lengths are unstable, probably due to differences in the error rates among the genomes used. Nevertheless, the fact that the Neandertal lineage is about 20% shorter than the Denisovan lineage suggests that the Neandertal toe phalanx is older than the Denisovan finger phalanx, consistent with the stratigraphy of the cave.
Table 1.
Dating for Branch Shortening and Population Splits
| Event | As % of human-chimp divergence | Absolute date calibration #1 in kya (μ= 1 × 10−9/bp/year) | Absolute date calibration #2 in kya (μ= 0.5 × 10−9/bp/year) | Supplement |
|---|---|---|---|---|
| Altai Neandertal Branch Shortening | 0.99% – 1.05% | 64–68 | 129–136 | SI 6b |
| Denisova Branch Shortening | 0.77% – 0.84% | 50–54 | 100–109 | SI 6b |
| San-West African split | 0.66% – 1.00% | 43–65 | 86–130 | SI 12 |
| Introgressing Neandertal – Altai split | 0.58% – 0.88% | 38–57 | 77–114 | SI 13 |
| Introgressing Denisovan – Denisovan split | 2.12% – 3.10% | 138–202 | 276–403 | SI 13 |
| Neandertal-Denisova split* | 2.93% – 3.64% | 190–236 | 381–473 | SI 12 |
| Archaic-African split* | 4.23% – 5.89% | 275–383 | 550–765 | SI 12 |
| Unknown archaic split | 7.90% – 31.12% | 450–2027 | 900–4054 | SI 16a,b |
This table gives date ranges for two calibrations. The first assumes human-chimpanzee divergence of 6.5 million years and 1.30% for human-chimp divergence, or a mutation rate of 1 x 10−9/bp/year1,2,12. The second is based on direct measurement of per generation mutation rates 15–17, corresponding to a mutation rate of 0.5 × 10−9/bp/year or 13 million years ago for human-chimpanzee divergence, and may fit better with some aspects of the fossil record45,46. Intervals give the range of values over tested human genomes for branch shortening; lowest and highest estimate for two or three methods for San-West African, Neandertal-Denisova, Neandertal-African and Denisova-African split; jackknife confidence interval over introgressed chunks for the Introgressing-Archaic - Archaic splits; and a union of the jackknife confidence interval in SI 16a and the 95% highest posterior density in SI 16b for the unknown-archaic split.
The indicated values are corrected for branch shortening where relevant as described in the supplementary notes.
Population split times
Fig. 2b reflects the average divergence between DNA sequences. The times at which the ancestral populations of archaic and modern humans separated are by necessity younger. We used two approaches to estimate these population split times (SI 12). We caution that for these and other age estimates we rely on dates for the divergence of human and chimpanzee DNA sequences that in turn depend on the human mutation rate, which is currently controversial. In the text we present estimates based on a mutation rate of 0.5 x 10−9/bp/year, estimated from comparisons of the genomes of parents and children15–19. In Table 1 we also present estimates based on a rate of 1.0 x 10−9/bp/year derived from the fossil record which was used in previous studies of archaic genomes1,2,12. We also caution that the split times are at the best approximate because the models of population history used are likely to be inaccurate.
We first estimated population split times by extending the pairwise sequentially Markovian coalescent model (PSMC) to estimate the distribution of coalescence times between single chromosomes where one comes from one and one from another population20,21 (SI 12). Using sub-Saharan African genomes that were experimentally phased (SI 14) and segments of the archaic genomes where the two chromosomes within an individual are closely related we estimate the population split time between modern humans on the one hand, and Neandertals and Denisovans on the other, to between 553,000 and 589,000 years, and the split time between Neandertals and Denisovans to 381,000 years (SI 12).
In a second approach we counted how often randomly chosen alleles in an individual from one population are derived (i.e. different from the apes) at positions where both the derived and ancestral alleles are seen in an individual from a second population1,12. Such derived alleles will be less frequent the older the population separation time is because more derived alleles in the second population will then be due to mutations that occurred after the split. Using this approach, and the demographic history inferred from the PSMC (SI 12), we estimate the population split of Neandertals and Denisovans from modern humans to 550,000–765,000 years, and the split time of Neandertals and Denisovans to 445,000–473,000 years.
Inbreeding
We noticed that the Altai Neandertal genome contains several long runs of homozygosity, suggesting that her parents were closely related (Fig. 3a). To estimate the extent of their relatedness, we scanned the genome for 1Mb regions where most non-overlapping 50-kb-windows were devoid of heterozygous sites and merged adjacent regions (SI 10). The Neandertal genome has 20 such regions longer than 10cM whereas the Denisovan genome has one. We performed simulations of inbreeding scenarios that can result in regions of this number and length, and find that the inbreeding coefficient is 1/8, indicating that the parents were as closely related as half-siblings. Since the Altai individuals is a female (SI 5) and the X chromosome also has long runs of homozygosity, we can exclude parental relationships where none or only one of the two X chromosomes was inherited from closely-related common ancestor(s), i.e. scenarios that include two successive males in the pedigree. We conclude that the parents of this Neandertal individual were either half-siblings who had a mother in common, double first cousins, an uncle and a niece, an aunt and a nephew, a grandfather and a granddaughter, or a grandmother and a grandson (Fig. 3b).
Figure 3. Indications of inbreeding in the Altai Neandertal individual.

a, Time since the most recent common ancestor in log-scale for the two alleles of a French, the Denisovan and the Altai Neandertal individual (SI 12) along 40 Mb of chromosome 21. b, Pedigrees showing four possible scenarios of parental relatedness for the Altai Neandertal (i.e. the child at the bottom of each pedigree). Two additional scenarios can be derived by switching the sex of the parents for the panels marked with an asterisk. c, Fraction of the genome in runs of homozygosity between 2.5 and 10cM in length for Altai Neandertal, Denisovan and the three present-day human individuals with the largest fractions (grey bars). The fractions for the Altai Neandertal (bottom four bars) are reduced by the fraction expected from the four inbreeding scenarios in a.
To investigate whether mating between closely related individuals may have been typical of the Altai Neandertal population, we examined the distribution of runs of homozygosity between 2.5 and 10 cM in length. After removing the runs expected from recent inbreeding, the Altai Neandertal genome still contains more runs than the Denisovan genome (P < 2.2 × 10−16), and both archaic genomes contain more than the Karitiana, a present-day population known to have a small effective size22 (Fig. 3c; SI 10). The sequencing of additional Neandertal genomes to high quality will address whether breeding among close relatives was common also among Neandertals in other geographic areas.
Heterozygosity and population size
The Neandertal autosomal genome carries 1.7–1.8 heterozygous sites per 10,000 bp (SI 9). This is 84% of the number of heterozygous sites in the Denisovan genome, 22–30% of that in present-day non-African genomes, and 16–18% of that in present-day African genomes (Extended Data Fig. 1). When regions of homozygosity longer than 2.5cM stemming from recent as well as long-term inbreeding in the Neandertal are removed, 2.1–2.2 sites per 10,000 are heterozygous, similar to what is observed in the Denisovan genome. Thus, heterozygosity in Neandertals as well as Denisovans appears to have been lower than in present-day humans and is among the lowest measured for any organism23.
Extended Data Figure 1.
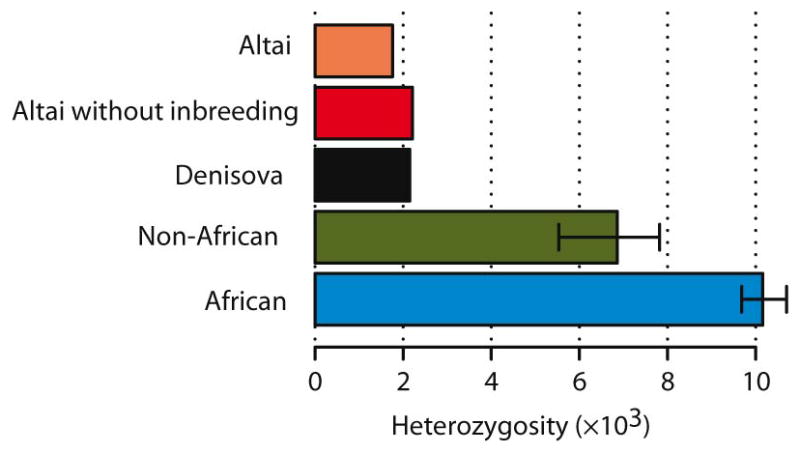
Heterozygosity estimates for the Altai Neandertal individual, the Denisovan individual, non-Africans and Africans.
The bars for the latter two give the range of heterozygosity observed among 15 non-African and 10 African individuals, respectively (SI 9).
The demographic history of the population can be reconstructed from the distribution of the times since the most recent common ancestor of the two copies of the genome that a single person carries. We use the PSMC20 to infer changes in the size of the Neandertal population over time and compare this to inferences from the Denisovan and present-day human genomes (Fig. 4) (SI 12). All genomes analyzed show evidence of a reduction in population size that occurred sometime before 1.0 million years ago. Subsequently, the population ancestral to present-day humans increased in size while the Altai and Denisovan ancestral populations decreased further in size. It is thus clear that the demographic histories of both archaic populations differ drastically from that of present-day humans.
Figure 4. Inference of population size change over time.

The y-axis specifies a number proportional to the population size Ne. The x-axis specifies time in units of divergence per base pair (along the top in years for mutation rates of 0.5 × 10−9 to 1.0 × 10−9 per site per year). The analysis assumes that the Neandertal and Denisova remains are of the same age, whereas archaeological evidence and the branch shortening suggest that the Neandertal bone is older than the Denisovan bone. However, because the exact difference in ages is not known, it is not possible to determine whether the reduction in population size experienced by both archaic groups (but not by modern humans) coincided in time.
Neandertal gene flow into modern humans
We have previously shown that Neandertals contributed parts of their genomes to present-day populations outside Africa12 and that Denisovans contributed to the genomes of present-day populations in Oceania2,24 (used here to refer Australia, Melanesia and the Philippines). Using the high-coverage Neandertal genome in conjunction with the two other Neandertal genomes, we now estimate that the proportion of Neandertal-derived DNA in people outside Africa is 1.5–2.1% (SI 14; Extended Data Table 1). Second, we find that the Neandertal-derived DNA in all non-Africans is more closely related to the Mezmaiskaya Neandertal from the Caucasus than it is to either the Neandertal from Siberia (Extended Data Table 2; SI 14) (Z-score range: 4.0–6.4) or to the Vindija Neandertals from Croatia12 (Z-score range: 1.7–3.9). These results cannot be explained by present-day human contamination in the Mezmaiskaya Neandertal data, as a contamination level on the order of 2.0–5.4% would be needed to account for the excess relatedness to the Mezmaiskaya Neandertal while the contamination in the Mezmaiskaya data is estimated to be 0–1.1% (SI 5a).
Extended Table 1.
Neandertal ancestry estimate
| Other Neandertal = Mezmaiskaya | Other Neandertal = Vindija | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Panel A | Panel B | Panel A | Panel B | |||||
| â | Std. Err. | â | Std. Err. | â | Std. Err. | â | Std. Err. | |
| French | 0.020 | 0.003 | 0.019 | 0.003 | 0.016 | 0.002 | 0.017 | 0.002 |
| Sardinian | 0.019 | 0.002 | 0.017 | 0.003 | 0.018 | 0.002 | 0.018 | 0.002 |
| Han | 0.022 | 0.003 | 0.018 | 0.003 | 0.023 | 0.002 | 0.019 | 0.002 |
| Dai | 0.019 | 0.003 | 0.016 | 0.003 | 0.019 | 0.002 | 0.016 | 0.002 |
| Karitiana | 0.020 | 0.003 | 0.019 | 0.003 | 0.018 | 0.002 | 0.019 | 0.002 |
| Mixe | - | - | 0.018 | 0.003 | - | - | 0.017 | 0.002 |
Extended Table 2.
Selected “D-statistics” supporting inferences about gene flows
| Statistic | D | Z | Interpretation |
|---|---|---|---|
| D(French, Dinka; Altai, Chimp) | 5.4% | 9.2 | SI 14: Neandertals share more derived alleles with non-Africans than with Africans. |
| D(Han, Dinka; Altai, Chimp) | 7.3% | 11.4 | |
| D(Han, Papuan; Denisova, Chimp) | −7.0% | −9.5 | SI 14: Denisovans share more derived alleles with Oceanian populations than with other non Africans. |
| D(Han, Australian; Denisova, Chimp) | −7.7% | −10.7 | |
| D(Altai, Mezmaiskaya; French, Dinka) | −16.4% | −5.8 | SI 14: The archaic material in non-Africans falls within late Neandertal variation: Non-Africans share more alleles with some Neandertals (Mezmaiskaya/Vindija) than others (Altai). |
| D(Altai, Vindija; French, Dinka) | −7.0% | −4.3 | |
| D(Altai, Mezmaiskaya; Denisova, Chimp) | 13.2% | 5.9 | SI 15: Gene flow between Altai related Neandertals and Denisovans (Denisovans share more derived alleles with Altai than with Mezmaiskaya) |
| D(Altai, Vindija; Denisova, Chimp) | 7.9% | 5.6 | |
| D(Altai, Denisova; 12 Africans, Chimp) | 7.0% | 11.6 | SI 16: Unknown archaic gene flow intoDenisova: Africans share more derived alleles with Altai than with Denisova, a signal that strengthens for fixed derived alleles |
| D(Altai, Denisova; 12 Africans Fixed, Chimp) | 13.4% | 10.0 |
Denisovan gene flow in mainland Asia
We used the two high-coverage archaic genomes and a hidden Markov model (HMM) to identify regions of specifically Neandertal and specifically Denisovan ancestry in 13 experimentally phased present-day human genomes1,14 (SI 4; SI 13). In the Sardinian and French genomes from Europe we find genomic regions of Neandertal origin and few or no regions of Denisovan origin. In contrast, in the Han Chinese, the Dai in southern China, and the Karitiana and Mixe in the Americas, we find, in addition to regions of Neandertal origin, regions that are consistent with being of Denisovan origin (Z-score = 4.3 excess relative to the Europeans) (SI 13), in agreement with previous analysis based on low-coverage archaic genomes25. These regions are also more closely related to the Denisova genome than the few regions identified in Europeans (SI 13). We estimate that the Denisovan contribution to mainland Asian and Native American populations is ~0.2% and thus about 25 times smaller than the Denisovan contribution to populations in Papua New Guinea and Australia. The failure to detect any larger Denisovan contribution in the genome of a 40,000-year-old modern human from the Beijing area26 suggests that any Denisovan contribution to modern humans in mainland Asia was always quantitatively small. In fact, we cannot, at the moment, exclude that the Denisovan contribution to people across mainland Asia is due to gene flow from ancestors of present-day people in Oceania after they mixed with Denisovans. We also note that in addition to this Denisovan contribution, the genomes of the populations in Asia and America appear to contain more regions of Neandertal origin than populations in Europe1,27 (SI 13, SI 14).
Archaic population differentiation
To estimate how closely related the archaic populations that contributed DNA to present-day humans were to the archaic individuals from which high-coverage genomes have been determined, we compared the regions of Neandertal and Denisovan ancestry in present-day human genomes identified by an HMM to the sequenced archaic genomes (SI 13). We find that the DNA sequence divergence in the regions that are most similar between the Altai Neandertal genome and the Neandertals that contributed DNA to present-day Eurasians is ~1.35% of the human-chimpanzee divergence, while the regions with the smallest sequence divergence between the Denisovan genome and the Denisovans that contributed DNA to present-day Papuans and Australians is ~3.18%. Regions of similarly low divergence are also identified by a window-based comparison (Fig. 5).
Figure 5. Relatedness of introgressing archaic and sequenced archaic samples.

Divergence of phased present-day human genomes to archaic genomes in windows of size 0.01cM with a minimum of 25,000 analysed bases. Windows are sorted by sequence divergence measured on the archaic side of the tree (SI 13) and the y-axis reports the divergence relative to human-chimpanzee divergence for cumulative fractions of the sorted windows over the entire genomes. Regions of low divergence between non-Africans and Neandertals (a) and between Oceanians and Denisovans (b) indicate gene flow between these groups and the relative divergences between the introgressing archaic and sequenced archaic samples.
We estimate the population split time between the introgressing Neandertal and the Altai Neandertal genome to 77,000–114,000 years ago, and the split time between the introgressing Denisovan and the Denisovan genome to 276,000–403,000 years ago (SI 13) (Table 1). This is consistent with the Denisovan population being larger, more diverse and/or more subdivided than Neandertal populations, and with the idea that Denisovans may have populated a wide geographical area. It is also in agreement with the low diversity among Neandertal nuclear2 and mitochondrial5 genomes.
Neandertal gene flow into Denisovans
If gene flow occurred between Neandertals and Denisovans, we would expect that regions of the genome where the divergence between Denisovan and Neandertal haplotypes is low would carry many differences between the two haplotypes of the individual who harbors the introgressed genetic material. This is because this individual carries two haplotypes that have accumulated differences independently in the two populations. In contrast, in the absence of gene flow, regions of low divergence between a Neandertal and a Denisovan haplotype are not expected to have particularly elevated diversity (SI 15).
We plotted the number of differences between the Neandertal genome and the closest inferred DNA sequences in the Denisovan genome against Denisovan heterozygosity (Fig. 6). We find that Denisovan heterozygosity is increased in regions where the Neandertal and one Denisovan allele are close, indicating that gene flow from Neandertals into Denisovans occurred, and estimate that a minimum of 0.5% of the Denisovan genome was contributed by Neandertals. The Denisovan genome shares more derived alleles with the Altai Neandertal genome than with the Croatian or Caucasus Neandertal genomes (Z-score range: 5.6–10.2) (Extended Data Table 2; SI 15), suggesting that the gene flow into Denisovans came from a Neandertal population more related to the Altai Neandertal than to the other two Neandertals. In the reciprocal analysis, we find no corresponding increase in Neandertal heterozygosity.
Figure 6. Neandertal gene flow into Siberian Denisovans.

Divergence in 0.01cM sized windows with at least 50kb analyzed bases between a “test”-archaic genome and effectively haploid regions of the other archaic genome archaic plotted against the most recent-common ancestor of the two alleles of the “test”-archaic. The plot shows 50 equally sized bins of windows for the “test”-archaic Denisovan against the effectively haploid Neandertal (red) and for the “test”-archaic Altai Neandertal against the effectively haploid Denisovan (blue). Divergence is given as percentage of human-chimpanzee divergence. Windows that show a close relationship between the effective haploid Altai Neandertal and the closest inferred Denisovan haplotype show a deep divergence to the second Denisovan haplotype, indicating gene flow from Neandertal into Denisovan.
Particularly strong signals of Neandertal gene flow into Denisovans are found in the human leucocyte antigen (HLA) region and the CRISP gene cluster on chromosome 6 (Extended Data Fig. 2), where we find many segments for which one of the Denisova haplotypes and the Altai Neandertal share a common ancestor within a few tens of thousands of years before the death of the Altai individual (SI 15). This suggests the possibility that introgressed Neandertal alleles may have contributed to the Denisovan functional variation at the HLA and the CRISP cluster, which are involved in immunity and sperm function, respectively. This is interesting since it has been suggested that HLA alleles from Neandertals and Denisovans have been of functional relevance in modern humans28.
Extended Data Figure 2.
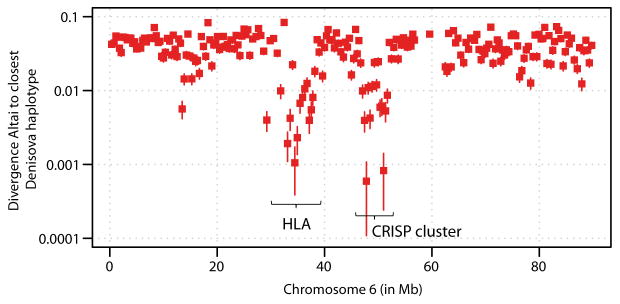
Neandertal-introgressed loci in Denisova
Divergence of the Altai Neandertal to the most closely related Denisovan haplotype in windows of at least 200kb on chromosome 6. Divergence is given as percentage of human-chimpanzee divergence and bars represent ± 1 standard error.
Unknown archaic gene flow into Denisovan
Since the ancestors of both Neandertals and Denisovans left Africa before the emergence of modern humans, one might expect present-day Africans to share equal proportions of derived alleles with these two archaic groups. However, we find that African genomes share about 7% more derived alleles with the Neandertal genome than with the Denisova genome (Z = 11.6 to 13.0; Extended Data Table 2; SI 16a) and that this is particularly the case for derived alleles that are fixed in Africans, of which 13–16% more are shared with the Neandertal than with the Denisovan genome (Fig. 7).
Figure 7. Altai and Denisovan allele sharing with Africans stratified by African allele frequency.
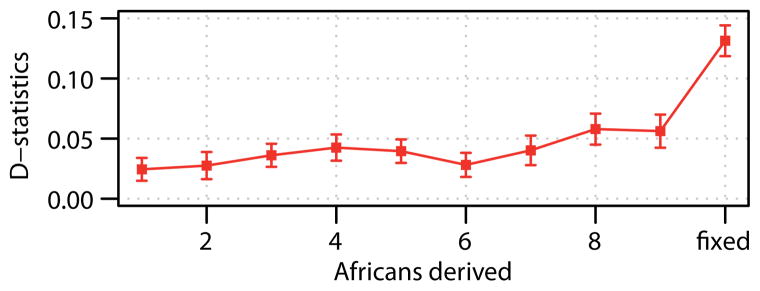
The plot shows the D-statistic of the form D(Neandertal, Denisova; Africa, Chimpanzee) binned by derived allele count in 10 deeply sequenced African genomes. Error-bars represent ± 1 standard error. High-frequency and fixed derived alleles in Africa are more often shared with the Neandertal than with Denisovan genome.
We tested three non-mutually exclusive scenarios that could explain these observations. First, gene flow from the ancestor of Neandertals after the split from Denisovans into the ancestors of all present-day humans would result in more sharing of derived alleles between present-day Africans and Neandertals. However, because gene flow contributes alleles at low frequency the sharing of derived alleles with Neandertals would grow weaker with higher African derived allele frequency (SI 16a), whereas we observe the opposite (Fig. 7). Second, gene flow from the ancestors of present-day humans to Neandertals after their split from Denisovans would also result in more sharing of derived alleles. However, the amount of allele frequency change (genetic drift) that has occurred in present-day Africans since the split from Neandertals is too small to explain the extent of sharing of derived alleles fixed in Africans (SI 16a). Third, we considered a scenario where Denisovans received gene flow from a hominin whose ancestors diverged deeply from the lineage leading to Neandertals, Denisovans and present-day humans. We find that this scenario is consistent with the data, as also suggested by others29, and estimate that 2.7–5.8% (jackknife 95% confidence interval) of the Denisova genome comes from this putative archaic hominin which diverged from the other hominins 0.9–1.4 million years ago (SI 16a). An approximate Bayesian computation30 again supports the third scenario (SI 16b) and estimates that 0.5–8% of the Denisovan genome comes from an unknown hominin which split from other hominins 1.1 and 4 million years ago.
We caution that these analyses make several simplifying assumptions. Despite these limitations we show that the Denisova genome harbors a component that derives from a population that lived prior to the separation of Neandertals, Denisovans and modern humans. This component may be present due to gene flow, or to a more complex population history such as ancient population structure maintaining a larger proportion of ancestral alleles in the ancestors of Denisovans over hundreds of thousands of years.
The putative admixture into Denisovans from an unknown archaic group raises the possibility that the apparent Denisovan contribution to the genomes of Papuans and Australians could originate from admixture with the same unknown archaic population instead of with Denisovans. However, we tested this hypothesis and found that the archaic component in the genomes of people in Papua New Guinea and Australia comes from a group related to the Denisovans and not from an unknown archaic hominin (SI 17).
Copy number differences
The high-quality archaic genomes allow us to identify genetic changes that may have been relevant for putative biological traits that set modern humans apart from archaic humans. To identify genomic regions that have changed in copy number during hominin evolution, we used the variation of coverage along the two archaic genomes and 25 present-day human genomes (SI 8). We find three regions that have been duplicated only on the modern human lineage (Extended Data Table 3). One region overlaps BOLA2, which occurs as a single copy per haploid genome in the archaic genomes but has two to five copies in all but one of 675 present-day humans analyzed, and which is near a microdeletion associated with developmental delay, intellectual disability and autism31.
Extended Table 3.
Lineage specific segmental duplications along each of the terminal branches and genes encompassed
| Locus | Length | Lineage | Genotypes
|
||||
|---|---|---|---|---|---|---|---|
| Genes | Modern Humans (median) | Denisova | Altai | Mezmaiskaya | |||
| chr12:122079832–122087495 | 7663 | Altai-Neandertal | ORAI1 | 2 | 2 | 4 | 3 |
| chr12:132295389–132391442 | 96053 | Altai-Neandertal | MMP17, ULK1 | 2 | 2 | 4 | 2 |
| chr19:9284044–9291195 | 7151 | Altai-Neandertal | 2 | 2 | 4 | 4 | |
| chr20:281880–290717 | 8837 | Altai-Neandertal | 2 | 2 | 10 | 9 | |
| chr3:12639069–12641393 | 2324 | Altai-Neandertal | RAF1 | 2 | 2 | 7 | 3 |
| chr6:95473793–95532866 | 59073 | Altai-Neandertal | 2 | 2 | 3 | 2 | |
| chr11:39901956–39909545 | 7589 | Denisova | 2 | 4 | 2 | 2 | |
| chr1:161272681–161274838 | 2157 | Denisova | MPZ | 2 | 4 | 2 | 2 |
| chr12:49894191–49897733 | 3542 | Denisova | SPATS2 | 2 | 4 | 2 | 2 |
| chr19:55302094–55315197 | 13103 | Denisova | KIR3DP1, KIR2DL4 | 2 | 4 | 2 | 2 |
| chr2:48781187–48787915 | 6728 | Denisova | 2 | 3 | 2 | 2 | |
| chr4:68542692–68577288 | 34596 | Denisova | UBA6, LOC550112 | 2 | 3 | 2 | 2 |
| chr4:68579206–68581585 | 2379 | Denisova | LOC550112 | 2 | 3 | 2 | 2 |
| chr7:140872574–140879065 | 6491 | Denisova | LOC100131199 | 2 | 6 | 2 | 2 |
| chr1:108924526–108990191 | 65665 | Modern Human | 4 | 2 | 2 | 2 | |
| chr16:30200098–30206185 | 6087 | Modern Human | CORO1A, LOC606724, BOLA2 | 6 | 2 | 2 | 2 |
| chr2:87417089–87420544 | 3455 | Modern Human | 4 | 2 | 2 | 2 | |
Catalog of modern human changes
We compiled a genome-wide catalog of sites where all or nearly all of 1,094 present-day humans32 carry the same nucleotide but differ from the Neandertal, Denisovan and great ape genomes (SI 18). In the regions of the genome to which short fragments can be mapped, there are 31,389 such single nucleotide substitutions and 4,113 short insertions and deletions (indels) shared by all present-day humans analyzed, and a further 105,757 substitutions and 3,900 indels shared by 90% of present-day humans. This list of simple DNA sequence changes that distinguish modern humans from our nearest extinct relatives is thus comparatively small. For example, it contains only 96 fixed amino acid substitutions in a total of 87 proteins and in the order of three thousand fixed changes that potentially influence gene expression in present-day humans (SI 18).
Because the manner in which modern and archaic humans may have differed in aspects of their cognition is particularly interesting, we focused on the expression in the developing human brain of transcripts encoding the 87 proteins with fixed amino acid changes (SI 20). In comparison to a control set of transcripts that carry 108 silent substitutions fixed in present-day humans, there is a tendency for genes carrying fixed amino acid changes to be expressed in the ventricular zone of the developing neocortex (P=0.06, corrected for multiple testing). Out of the five genes which are expressed in the proliferative layers (ventricular and subventricular zones combined) during mid-fetal development (CASC5, KIF18A, TKTL1, SPAG5, VCAM1), three (CASC5, KIF18A, SPAG5) are associated with the kinetochore of the mitotic spindle. This may be interesting since the orientation of the mitotic cleavage plane in neural precursor cells during cortex development is thought to influence the fate of the daughter cells and the number of neurons generated (see e.g. ref.33). Another of these five genes, VCAM1, is essential for maintenance of neural stem cells in the adult subventricular zone34.
Another way to prioritize changes in the catalog for functional studies is to identify those that show signs of having risen to high frequency rapidly since they may have been affected by positive selection. We implemented an HMM to scan the genome for regions where the Neandertal and Denisovan genomes fall outside of the variation of present-day humans (SI 19a). We ranked these regions, which cover less than 100Mb of the genome, according to genetic length, because regions that rose rapidly to fixation are expected to be longer as they have been less affected by recombination events. A set of 63 regions likely to have been affected by positive selection were identified (S19.3). They contain 2,123 substitutions and 61 indels that are fixed or of high-frequency (>90%) in modern humans (SI 19b). They include, for example, the gene RB1CC1 (also called FIP200) which encodes a transcription factor which, like VCAM1, is essential for maintenance of neuronal stem cells in the adult subventricular zone35. In present-day humans, but not Neandertals and Denisovans, RB1CC1 carries a substitution inferred to change an amino acid in the encoded protein as well as a substitution that affects a conserved site in a motif that occur across the genome36. Functional investigations will be necessary to clarify whether these and other such changes affect any phenotypes in present-day humans.
Discussion
We present evidence for three to five cases of interbreeding among four distinct hominin populations (Fig. 8). Clearly the real population history is likely to have been even more complex. For example, most cases of gene flow are likely to have occurred intermittently, often in both directions and across a geographic range. Thus, combinations of gene flow among different groups and substructured populations may have yielded the patterns detected rather than the discrete events considered here. Nevertheless, our analyses show that hominin groups met and had offspring on many occasions in the Late Pleistocene, but that the extent of gene flow between the groups was generally low.
Figure 8. A possible model of gene flow events in the late Pleistocene.

The direction and estimated magnitude of inferred gene flow events are shown. Branch lengths and ages gene flows are not drawn to scale. The dashed line indicates that it is uncertain if Denisovan gene flow into modern humans occurred once or more times. D.I. denotes the introgressing Denisovan, N.I. the introgressing Neandertal. Note that the age of the archaic genomes precludes detection of gene-flow from modern humans into the archaic hominins.
We note that the observation that the Neandertal DNA sequences in non-Africans share more derived alleles with the Neandertal from the Caucasus than with Neandertals from either Croatia or the Altai indicates that the archaic gene flow into non-Africans occurred at a time when Neandertal populations had separated from each other. We also note that the introgressed Neandertal DNA sequences suggest a population split from the Altai Neandertal between 77,000 and 114,000 years ago (SI 13), well after ~230,000 years ago when Neandertal features appear in the fossil record37. These and other results38,39 show that the allele sharing between Neanderthals and non-African populations is due to recent admixture rather than ancient population subdivision, an alternative which we and others previously considered possible12,40.
The evidence suggestive of gene flow into Denisovans from an unknown hominin is interesting. The estimated age of 0.9 to 4 million years for the population split of this unknown hominin from the modern human lineage is compatible with that it contributed its mtDNA to Denisovans since the Denisovan mtDNA diverged from the mtDNA of the other hominins about 0.7–1.3 million years ago41. The estimated population split time is also compatible with the possibility that this unknown hominin was what is known from the fossil record as Homo erectus. This group started to spread out of Africa around 1.8 million years ago42, but Asian and African H. erectus populations may have become finally separated only about one million years ago43. However, further work is necessary to establish if and how this gene flow event occurred.
Methods
Sequences were generated on the Illumina HiSeq 2500 and base-calling was carried out using Ibis44. Reads were merged and adapter trimmed as described1 and mapped to the human reference genome using BWA (version: 0.5.10). Genotyping was carried out using GATK (version 1.3). We restrict analyses to regions of the genome that are non-repetitive (excluding tandem repeats), unique (requiring at least 50%, or all, overlapping 35-mers covering a position to map uniquely, allowing for one mismatch), and fall within the central 95% of the coverage distribution corrected for GC bias (SI 5b). The supplementary information describes the details of data processing and other analyses.
Supplementary Material
Acknowledgments
We thank Michael Hammer, Cheryl Winkler and William Klitz for sharing DNA samples, Wieland Huttner and his group, Benjamin Peter, Joshua G. Schraiber and Melinda A. Yang for helpful discussions, and Alexandra Lewis and Ruolan Qiu for technical assistance. NP and DR are grateful for the chance to discuss these results with Peter Waddell who independently found evidence of a deeply diverged hominin admixing into the Denisova genome. DR and NP were supported by NSF grant #1032255 and NIH grant GM100233; JS by grant HG006283 from the National Genome Research Institute (NHGRI); SS by a post-doctoral fellowship from the Harvard University Science of the Human Past Program; FJ and MS in part by a grant from the NIH (R01-GM40282); PHS by an HHMI International Student Fellowship. We thank the team at the NIH Intramural Sequencing Center, and Alice Young in particular, for generating some of the sequence reported here. This research was supported in part by the Paul G. Allen Family Foundation. Major funding support came from the Presidential Innovation Fund of the Max Planck Society.
Footnotes
Contributions
S.Saw., A.H. and Q.F. performed the experiments; K.P., F.R., N.P., F.J., S.San., S.Saw., A.H., G.R., P.H.S., C.dF., M.D., Q.F., M.Ki., M.Ku., M.L., M.M., M.O., M.Si., C.T., H.L., S.M., A.T., P.M., J.P., J.C.M., S.H.V., R.E.G., I.H., P.L.F.J., J.O.K., J.S., E.E.E., E.S.L., T.E.B., M.Sl., D.R., J.K., and S.P. analysed genetic data; L.V.G., V.B.D., M.V.S., A.P.D. and B.V. analyzed archaeological and anthropological data; H.B. and H.C. provided samples and reagents; K.P., J.K. and S.P. wrote and edited the manuscript with input from all authors.
Competing financial interests
The authors declare no competing financial interests.
Accession codes
All sequence data have been submitted to the European Nucleotide Archive (ENA) and are available under the following accessions: Altai Neandertal: ERP002097, Mezmaiskaya Neandertal: ERP002447. The data from the 25 present-day human genomes and 13 experimentally phased present-day genomes are available as a public dataset from http://aws.amazon.com/datasets/ and from http://cdna.eva.mpg.de/neandertal/altai/.
References
- 1.Meyer M, et al. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338:222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reich D, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mednikova MB. A proximal pedal phalanx of a paleolithic hominin from Denisova cave, Altai. Archaeology Ethnology & Anthropology of Eurasia. 2011;39:129–138. http://dx.doi.org/10.1016/j.aeae.2011.06.017. [Google Scholar]
- 4.Green RE, et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134:416–426. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Briggs AW, et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325:318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 6.Golovanova LV, Hoffecker JF, Kharitonov VM, Romanova GP. Mezmaiskaya cave: A Neanderthal occupation in the Northern Caucasus. Curr Anthropol. 1999;40:77–86. doi: 10.1086/515805. [DOI] [Google Scholar]
- 7.Gansauge MT, Meyer M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nature protocols. 2013;8:737–748. doi: 10.1038/nprot.2013.038. [DOI] [PubMed] [Google Scholar]
- 8.Kircher M. Analysis of high-throughput ancient DNA sequencing data. Methods Mol Biol. 2012;840:197–228. doi: 10.1007/978-1-61779-516-9_23. [DOI] [PubMed] [Google Scholar]
- 9.Briggs AW, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Briggs AW, et al. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic acids research. 2010;38:e87. doi: 10.1093/nar/gkp1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofreiter M, Jaenicke V, Serre D, von Haeseler A, Paabo S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic acids research. 2001;29:4793–4799. doi: 10.1093/nar/29.23.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skinner AR, et al. ESR dating at Mezmaiskaya Cave, Russia. Appl Radiat Isotopes. 2005;62:219–224. doi: 10.1016/j.apradiso.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 14.Kitzman JO, et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nature biotechnology. 2011;29:59–63. doi: 10.1038/nbt.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abecasis GR, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Awadalla P, et al. Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. American journal of human genetics. 2010;87:316–324. doi: 10.1016/j.ajhg.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roach JC, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong A, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campbell CD, et al. Estimating the human mutation rate using autozygosity in a founder population. Nature genetics. 2012;44:1277–1281. doi: 10.1038/ng.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prado-Martinez J, et al. Great ape genetic diversity and population history. Nature. 2013 doi: 10.1038/nature12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirin M, et al. Genomic runs of homozygosity record population history and consanguinity. PloS one. 2010;5:e13996. doi: 10.1371/journal.pone.0013996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leffler EM, et al. Revisiting an old riddle: what determines genetic diversity levels within species? PLoS biology. 2012;10:e1001388. doi: 10.1371/journal.pbio.1001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reich D, et al. Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. American journal of human genetics. 2011;89:516–528. doi: 10.1016/j.ajhg.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skoglund P, Jakobsson M. Archaic human ancestry in East Asia. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18301–18306. doi: 10.1073/pnas.1108181108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu Q, et al. DNA analysis of an early modern human from Tianyuan Cave, China. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2223–2227. doi: 10.1073/pnas.1221359110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wall JD, et al. Higher levels of neanderthal ancestry in East aSians than in Europeans. Genetics. 2013;194:199–209. doi: 10.1534/genetics.112.148213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abi-Rached L, et al. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science. 2011;334:89–94. doi: 10.1126/science.1209202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waddell PJ, Tan X. New g%AIC, g%AICc, g%BIC, and Power Divergence Fit Statistics Expose Mating between Modern Humans, Neanderthals and other Archaics. 2012. arXiv:1212.6820. [Google Scholar]
- 30.Wegmann D, Leuenberger C, Neuenschwander S, Excoffier L. ABCtoolbox: a versatile toolkit for approximate Bayesian computations. BMC bioinformatics. 2010;11:116. doi: 10.1186/1471-2105-11-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar RA, et al. Recurrent 16p11.2 microdeletions in autism. Human molecular genetics. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 32.Abecasis GR, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fietz SA, Huttner WB. Cortical progenitor expansion, self-renewal and neurogenesis-a polarized perspective. Current opinion in neurobiology. 2011;21:23–35. doi: 10.1016/j.conb.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 34.Kokovay E, et al. VCAM1 is essential to maintain the structure of the SVZ niche and acts as an environmental sensor to regulate SVZ lineage progression. Cell stem cell. 2012;11:220–230. doi: 10.1016/j.stem.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 35.Wang C, Liang CC, Bian ZC, Zhu Y, Guan JL. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nature neuroscience. 2013;16:532–542. doi: 10.1038/nn.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rios D, et al. A database and API for variation, dense genotyping and resequencing data. BMC bioinformatics. 2010;11:238. doi: 10.1186/1471-2105-11-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hublin JJ. Out of Africa: modern human origins special feature: the origin of Neandertals. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16022–16027. doi: 10.1073/pnas.0904119106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sankararaman S, Patterson N, Li H, Paabo S, Reich D. The date of interbreeding between Neandertals and modern humans. PLoS genetics. 2012;8:e1002947. doi: 10.1371/journal.pgen.1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang MA, Malaspinas AS, Durand EY, Slatkin M. Ancient structure in Africa unlikely to explain Neanderthal and non-African genetic similarity. Molecular biology and evolution. 2012;29:2987–2995. doi: 10.1093/molbev/mss117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eriksson A, Manica A. Effect of ancient population structure on the degree of polymorphism shared between modern human populations and ancient hominins. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:13956–13960. doi: 10.1073/pnas.1200567109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krause J, et al. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature. 2010;464:894–897. doi: 10.1038/nature08976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gabunia L, et al. Dmanisi and dispersal. Evol Anthropol. 2001;10:158–170. doi: 10.1002/Evan.1030. [DOI] [Google Scholar]
- 43.Asfaw B, et al. Remains of Homo erectus from bouri, Middle Awash, Ethiopia. Nature. 2002;416:317–320. doi: 10.1038/416317a. [DOI] [PubMed] [Google Scholar]
- 44.Kircher M, Stenzel U, Kelso J. Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome biology. 2009;10:R83. doi: 10.1186/gb-2009-10-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langergraber KE, et al. Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:15716–15721. doi: 10.1073/pnas.1211740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scally A, Durbin R. Revising the human mutation rate: implications for understanding human evolution. Nature reviews. Genetics. 2012;13:745–753. doi: 10.1038/nrg3295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.