

Summary
Mono-methylation of lysine 4 on histone H3 (H3K4me1) is a well-established feature of enhancers and promoters, although its function is unknown. Here, we reveal novel roles for H3K4me1 in diverse cell types. Remarkably, we find that MLL3/4 provokes mono-methylation of promoter regions and the conditional repression of muscle and inflammatory response genes in myoblasts. During myogenesis, muscle genes are activated, lose MLL3 occupancy, and become H3K4-trimethylated through an alternative COMPASS complex. Mono-methylation mediated repression was not restricted to skeletal muscle. Together with H3K27me3 and H4K20me1, H3K4me1 was associated with transcriptional silencing in embryonic fibroblasts, macrophages, and human ES cells. On promoters of active genes, we find that H3K4me1 spatially demarcates the recruitment of factors that interact with H3K4me3, including ING1, which, in turn, recruits Sin3A. Our findings point to a unique role for H3K4 mono-methylation in establishing boundaries that restrict the recruitment of chromatin-modifying enzymes to defined regions within promoters.
Introduction
Genome-wide mapping of histone modifications in diverse cell types has revealed distinct chromatin signatures (e.g., active and repressed euchromatin, facultative and constitutive heterochromatin) and allowed the identification and characterization of distal and proximal transcriptional regulatory elements (Ernst et al., 2011). Mono-methylation of H3K4 (H3K4me1) is found at both transcriptionally active promoters and distal regulatory elements. Promoters of active genes are characterized by an intriguing, but poorly understood, combination of all three methylated forms of H3K4. H3K4me3 localizes closest to the TSS, whereas H3K4me1 extends furthest downstream (Barski et al., 2007). H3K4 methylation at promoters results from the combined activity of Set1a/b (COMPASS) and MLL1-4 (COMPASS-like) complexes. Set1a/b are thought to regulate genome-wide H3K4 methylation, whereas MLL1/2 specifically targets the Hox genes (Wu et al., 2008, Wang et al., 2009a). Although these complexes are distinguished by unique subunits, Wdr5, RbBP5, and Ash2 are commonly found in all COMPASS and COMPASS-like complexes (Milne et al., 2002). The activity of H3K4 methylating enzymes is counter-balanced by histone de-methylases, including LSD1, which is capable of reversing the mono- and di-methylated states (Shi et al., 2004). Furthermore, in ES cells, LSD1 is recruited to enhancers and plays a role in differentiation through enhancer decommissioning (Whyte et al., 2012).
A chromatin signature for enhancers has been studied extensively. Enhancers are distinguished by robust levels of H3K4me1 and H3K27 acetylation (H3K27ac), as well as recruitment of RNA polymerase II (Pol II) and the histone acetyl-transferase, p300 (Blum et al., 2012, Visel et al., 2008, Heintzman et al., 2007). In contrast with promoters, enhancers exhibit relatively low levels of H3K4me3. The MLL3/4 complex has recently been shown to promote H3K4 mono-methylation at enhancers (Herz et al., 2012, Hu et al., 2013). Therefore, it is likely that a single histone modification (H3K4me1) serves multiple context-dependent functions at distal and proximal regulatory elements.
Despite extensive studies related to H3K4 mono-methylation at enhancers or promoters, a clear function for this mark has not emerged. In contrast, H3K4me2/3 has been shown to function as a beacon for recruitment of chromatin “readers” or interactors, proteins with canonical motifs that facilitate binding to H3K4me2 and me3 (Yun et al., 2011). As an example, the PHD fingers of ING1 and ING2 have been shown to bind to H3K4me2/me3, with a preference for H3K4me3 (Shi et al., 2006). Although ING1 itself does not serve any enzymatic function, it is known to associate with the Sin3A/histone deacetylase (HDAC) complex via an N-terminal SAP30-interacting (SAID) domain (Peña et al., 2008). As H3K4me3 marks the promoters of active genes, this would paradoxically suggest that ING1 recruits a co-repressor (Sin3A) to transcriptionally active genes. Indeed, the presence of HDACs at promoters of active genes has been previously reported (Wang et al., 2009b, van Oevelen et al., 2010). This strongly suggests that transcriptional regulators often regarded as repressors are not strictly recruited by “repressive” histone marks but are also recruited to active genes.
Here, we have uncovered novel roles for H3K4 mono-methylation and describe an association between this modification and gene repression in diverse cell types. First, we show that MLL3/4-mediated H3K4 mono-methylation of promoters is associated with conditional repression of inducible genes. Loss of MLL3/4 leads to decreases in H3K4me1 and a concomitant increase in expression of these genes. In striking contrast, on transcriptionally active genes, H3K4me1 is deposited with H3K4me2/3 on MLL1/2 target genes. We show that H3K4me1 delimits chromatin “boundaries” on active promoters by spatially restricting readers of H3K4me3, including ING1. Aberrant spreading of H3K4me1 across TSS-proximal regions of active genes leads to gene repression and diminished recruitment of H3K4me3 interactors. Our findings identify novel roles for H3K4me1 at gene promoters and shed light on mechanisms by which cells interpret the balance between H3K4 mono- and tri-methylation at promoters.
Results
H3K4 mono-methylation at promoters is associated with gene repression
Previously, we analyzed genome-wide patterns of histone modifications, factor occupancy, and gene expression in skeletal muscle (C2C12) myoblasts and differentiated myotubes (Asp et al., 2011, Blum et al., 2012). To determine if there are distinct roles for H3K4me1 and H3K4me3 at promoters, we closely examined the occurrence of both marks. We defined promoters as regions spanning 3 kb upstream and downstream of all transcription start sites (TSS) and separated them into groups based on enrichment of H3K4me1 and H3K4me3 (Figure 1A, Table S1). Promoters could be partitioned into three unique groups. Group 1 promoters were associated with both H3K4me1 and H3K4me3 and represent almost all expressed genes. Although H3K4me1 and H3K4me3 were both abundant on this set of promoters, there was a marked difference in the pattern of deposition: H3K4me3 was found on regions immediately adjacent to the TSS, whereas H3K4me1 was depleted from these regions. Group 2 promoters lacked H3K4me1, were either marked exclusively by H3K27me3 or lacked all of the activating and repressive histone modifications that we previously investigated, and were generally repressed.
Figure 1. H3K4me1 is associated with features of gene repression and facultative heterochromatin.
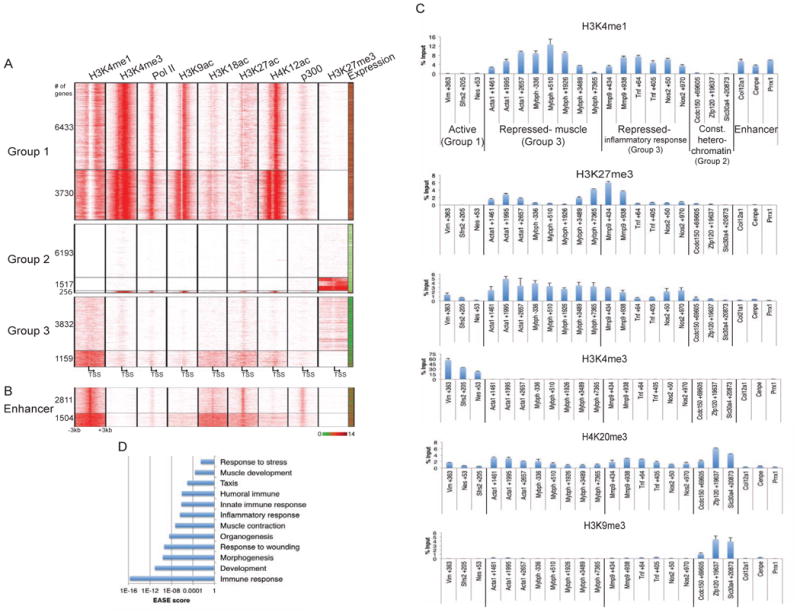
(a) Genes enriched for H3K4me1 but depleted of H3K4me3 are repressed. All genes were sorted into categories based on enrichment of H3K4me1 and H3K4me3 (Group 1: H3K4me1+/H3K4me3+; Group 2: H3K4me1-; Group 3: H3K4me1+/H3K4me3-). ChIP-seq data were plotted on regions 3 kb upstream and downstream of the TSS and (b) with respect to the midpoint of C2C12 myoblast-specific enhancers identified in Blum et al., 2012. Expression (from Liu el al., 2010) of associated genes is displayed in the far right column. Red-green key indicates relative expression. Numbers of genes in each cluster are displayed on the left. (c) A chromatin signature consisting of H3K4me1, H3K27me3, and H4K20me1 marks the promoters of muscle and inflammatory response (Group 3) genes. qChIP enrichment (presented as percent input) with the indicated antibodies in C2C12 myoblasts for Group 1, 2, and 3 genes and enhancers. Bars indicate SD. (d) H3K4me1 marks inducible genes. GO analysis of genes in Group 3 was performed, and statistically significant enrichments corresponding to EASE scores are plotted on the x-axis (logarithmic scale).
The most unexpected pattern was observed on ∼5,000 promoters wherein H3K4me1 was accompanied by a lack of H3K4me3 (Group 3). Unlike Group 1 promoters, Group 3 promoters exhibited a unique pattern of H3K4 mono-methylation in which this mark spanned, and extended beyond, the entire 6 kb promoter region. This unique pattern of H3K4me1 is reminiscent of similar block-like domains of H3K27me3, which we have noted in skeletal muscle cells (Asp et al., 2011). The presence of H3K4me1 and concomitant absence of H3K4me3 has been used to define enhancers (Figure 1B; Blum et al., 2012). However, Group 3 promoters lacked other features associated with active enhancers, including recruitment of Pol II and p300, and H3K27ac deposition. Furthermore, the majority of genes in Group 3 exhibited H3K27me3, and expression analysis showed that these genes were repressed. We validated the distinct genome-wide patterns of H3K4me1 and H3K4me3 across a select group of active Group 1 and repressed Group 3 genes in myoblasts using quantitative ChIP (qChIP; Figure 1C and Figure S1A). To further characterize the Group 3 genes, we determined which gene ontology (GO) terms were most highly enriched (Figure 1D). We found that Group 3 genes, primarily in the top cluster of 3,832 H3K27-trimethylated genes, were involved in muscle contraction and development (including Acta1, Mybph, Tnnc1, and Myog), response to stress, wound healing, and the immune and inflammatory response.
Previous studies have shown that stress and inflammatory response genes can be acutely induced in response to certain stimuli in C2C12 myoblasts, whereas muscle development genes are activated during differentiation (Frost et al., 2003, Cao et al., 2010). These results suggested that Group 3 genes could be induced in a condition-dependent manner. If so, the inducibility of these genes could indicate that H3K4me1 is a feature of facultative heterochromatin or condition-specific gene repression. To test this possibility, we determined if the inflammatory response and myogenic genes were enriched for histone marks commonly associated with facultative (H3K27me3 and H4K20me1) or constitutive (H3K9me3, H4K20me3) heterochromatin (Trojer and Reinberg, 2007). H4K20me3 has also been shown to repress inflammatory response genes (Stender et al., 2012). We found that conditionally repressed (Group 3) genes exhibited a unique signature of histone modifications compared to active (Group 1) and constitutively repressed (Group 2) genes and enhancers (Figure 1C). That is, strong enrichment of H3K27me3 and H4K20me1 was more prominent on muscle development and inflammatory response genes. By contrast, enrichment of H3K9me3 was not evident on these genes, and together with H4K20me3, it was most pronounced on constitutively repressed (Group 2) genes. Interestingly, Group 3 genes exhibited higher levels of H3K4me1 than regions flanking the TSS of highly expressed genes and myoblast enhancers. Overall, these findings suggest a role for H3K4me1 in condition-dependent repression typically associated with facultative heterochromatin.
H3K4 mono-methylation depends on recruitment of MLL3/4
In order to directly investigate the role of H3K4me1 in gene repression, we sought to identify the enzyme(s) responsible for this mark. Recently, MLL3/4 complexes have been implicated in H3K4 mono-methylation at enhancers in mammalian cells (Herz et al., 2012, Hu et al., 2013). Therefore, we asked whether MLL3 and MLL4 were recruited to Group 3 repressed genes in C2C12 myoblasts. We found that MLL3 and MLL4 co-occupy promoters of both muscle development and inflammatory response genes (Figure 2A) and validated the specificity of both antibodies by performing ChIP after depletion of both proteins. Next, we determined MLL3 binding sites genome-wide using ChIP-seq. Globally, MLL3 binding mimicked the pattern of H3K4me1 across promoters of Group 1 and Group 3 genes, while MLL3 was relatively depleted from the Group 2 genes (S1B-C and S2A). Distinct, focal peaks of MLL3 and H3K4me1 can be found across Group 1 promoters, whereas widespread blocks of H3K4me1 and H3K27me3 across Group 3 promoters correlated with a similarly broad pattern of MLL3 localization (Figure S1C-D).
Figure 2. MLL3/4 mediates H3K4 mono-methylation of promoters and repression of Group 3 genes.
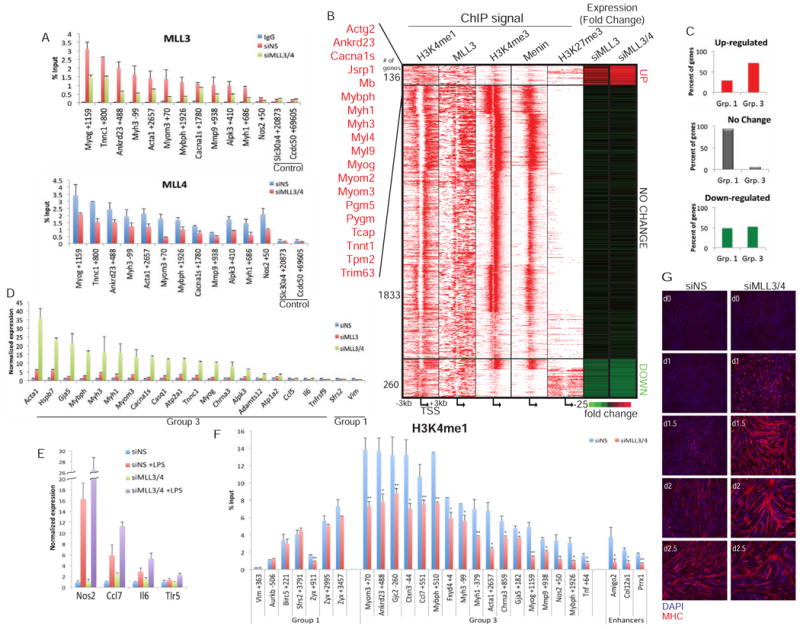
(a) Specificity of the MLL3 and MLL4 antibodies was determined through qChIP of MLL3 or MLL4 after transfection with a non-specific (NS) control or double knockdown of MLL3/4. Background ChIP signal was determined using an antibody against IgG. (b) MLL3/4 regulate the expression of muscle development Group 3 genes. Differential expression of MLL3 and H3K4me1 target genes after the knock-down of MLL3 or MLL3/4 was determined using RNA-seq (right columns). Up- and down-regulated genes exhibited a ≥2.5 fold change, expression of genes with <1.3 fold change was categorized as no change. A subset of muscle development genes was identified as significantly up-regulated after loss of MLL3/4. ChIP-seq using the indicated antibodies was plotted 3kb upstream and downstream of the TSS. (c) Genes up- or down-regulated and no change genes were identified as belonging to Group 1 or 3 from Figure 1A. Group 2 genes were unaffected by loss of MLL3 alone or MLL3/4. (d) Silencing of MLL3 and MLL3/4 increases the expression of muscle (Group 3) genes as compared to immune response genes (Ccl5, Il6, Tnfsf9) and constitutively expressed genes (Sfrs2, Vim). Expression was normalized to siNS. (e) Knock-down of MLL3/4 followed by LPS treatment leads to enhanced expression of several inflammatory response (Group 3) genes. siNS and siMLL3/4 transfected cells were treated with 100ng/ml LPS for 3 hours. Expression was normalized to siNS. (f) qChIP analysis was carried out to determine the impact of MLL3/4 depletion on deposition of H3K4me1 at promoters. (g) Loss of MLL3/4 leads to enhanced myogenic differentiation. Cells were stained with MHC (red) and DAPI (blue). Day 0 (D0) represents fully confluent cells and the onset of differentiation. Error bars indicate SD. For all qChIP, y-axis represents enrichment as percent input. Student's t test was performed to indicate significance: * indicates p-values <0.05 and ** <0.01, respectively.
To determine if MLL3 regulates the expression of H3K4-mono-methylated target genes, especially the repressed Group 3 genes, we conducted genome-wide expression profiling (RNA-seq) after depleting MLL3 alone or MLL3 and MLL4 together (Figure 2B, Figure S2B). By examining changes in the expression of mono-methylated genes that bound MLL3, we found that loss of MLL3 alone strongly induced expression of a cluster of genes that are primarily in Group 3 (Figure 2B-C; Table S2). Interestingly, Group 3 genes were further de-repressed upon depletion of both MLL3 and MLL4 (by ≥2.5-fold), a result that we further validated with qPCR (Figure 2B-D). These results reinforce previous studies indicating that MLL3 and MLL4 are functionally redundant (Hu et al., 2013). Strikingly, the cluster of genes up-regulated upon loss of MLL3 or MLL3/4 together was enriched in muscle development genes (Figure 2B). On the other hand, Group 2 genes, which lack H3K4me1, were unaffected by MLL3 or MLL3/4 depletion. We note that a subset of Group 1 and 3 genes were down-regulated. This subset contained genes were highly H3K27 tri-methylated, suggested that these genes were expressed at low levels even before the loss of MLL3/4. Genes in these groups may also be positively regulated by MLL3/4 through an established role for these proteins at enhancers. Alternatively, expression of these genes, including a cluster involved in cell cycle control, could diminish as a result of accelerated differentiation in MLL3/4-depleted cells (see below).
H3K4-trimethylated Group 1 genes were largely unaffected by loss of MLL3 or MLL3/4, perhaps because H3K4 methylation is mediated by MLL1-Menin (Figure 2B-C). In support of this conclusion, loss of Wdr5 consistently resulted in reduced expression of active (Group 1), H3K4-trimethylated genes (Figure S2C). In stark contrast, we found that ablation of Wdr5 resulted in the increased expression of H3K4-mono-methylated, muscle development (Group 3) genes. Interestingly, although loss of MLL3/4 was not sufficient to de-repress the inflammatory response genes, combined ablation of MLL3/4 and treatment with a known pro-inflammatory inducer, lipopolysaccharide (LPS), resulted in enhanced induction of mRNA levels as compared to LPS alone (Figure 2E).
Next, we determined if MLL3 plays a role in H3K4-mono-methylation of the Group 3 repressed genes. Since MLL3 and MLL4 are functionally redundant, we silenced both proteins and observed significant reductions in H3K4me1 on promoters of muscle and inflammatory response (Group 3) genes, as well as enhancers, although Group 1 genes were largely refractory to depletion (Figure 2F). This suggests that, in addition to their role at enhancers, MLL3/4 complexes are involved in the H3K4-mono-methylation of certain repressed genes. To address the possibility that loss of MLL3/4 indirectly de-repressed Group 3 genes through modulation of histone occupancy or other modifications associated with facultative heterochromatin or active genes, we investigated enrichment of H3K27me3, H4K20me1, and H3K4me3 and found that they were not altered in the absence of MLL3/4 (Figure S2D-F). Furthermore, we examined the impact of MLL3/4 loss on myogenic differentiation and found that depletion of these proteins dramatically accelerated terminal differentiation, consistent with the up-regulation of the master regulator, Myog, as well as skeletal muscle-specific proteins (Figure 2G).
To extend our findings regarding H3K4me1 and MLL3 to another cell type, we took advantage of genome-wide studies using MLL3-/- MEFs (Herz et al., 2012). We identified a group of ∼750 genes that showed loss of TSS-proximal H3K4me1 in MLL3-/- versus wild-type MEFs, and we validated a subset of these genes as MLL3 targets by qChIP using wild-type MEFs (Figure S2G-H). Notably, MLL3-dependent methylation was preferentially enriched over the TSS. Importantly, genes that lose H3K4 mono-methylation are involved in synaptic transmission, expressed at very low levels as compared to genes that are H3K4-monomethylated independently of MLL3, and are significantly de-repressed in MLL3-/- cells, reminiscent of Group 3 genes (Figure S2G, I). Taken together, these results lead to several key conclusions: (1) MLL3/4 complexes mediate H3K4 mono-methylation of Group 3 target genes, attenuating expression of both acutely inducible genes and muscle-related genes activated during differentiation; (2) loss of MLL3/4 promotes stimulus-dependent gene expression without impacting H3K27me3, H4K20me1, or H3K4me3; and (3) on a subset of active promoters, MLL3/4 does not play an observable role in H3K4-monomethylation or maintenance of expression.
Loss of gene repression coincides with a switch from H3K4me1 to H3K4me3
Currently, the exact role of H3K4me1 in gene expression is unclear, and connections to gene repression have not been previously established. A pattern in which H3K4me1 spanned the entire promoter of repressed genes (Group 3), including the TSS, which is typically depleted of nucleosomes, suggested that H3K4me1 could mark regions with high nucleosome occupancy. To determine how H3K4me1 associates with individual nucleosomes, we performed deep sequencing of MNase-digested chromatin and used sequence tags to map nucleosome positions (MNase-seq) (see Supplemental Experimental Procedures). We also mapped nucleosomes enriched for H3K4me1, H3K4me3, H3K27ac, and H3K27me3 in myoblasts. We limited our analysis to regions 500 bp upstream and downstream of the TSS, identified ten clusters marked by distinct patterns of nucleosomes, and further sorted genes within each cluster by expression (Figure S3A). Through MNase-seq and ChIP-seq analyses, we detected two patterns of nucleosomes: (1) positioned nucleosomes across a promoter containing a nucleosome free region and (2) nucleosomes that span the TSS in an uninterrupted manner. Unlike Group 1, Group 3 (and Group 2) genes were strikingly similar to the latter pattern wherein there is uniform distribution of nucleosomes throughout the TSS region (Figure S3C). This suggests that H3K4me1 can mark regions with relatively high nucleosome occupancy and that promoters that are highly enriched for H3K4me1 may therefore be refractory to transcription.
Since many transcription factors have a tendency to localize to regions depleted of nucleosomes, we hypothesized that the loss of H3K4me1 and acquisition of H3K4me3 (a switch from the Group 3 to Group 1 pattern of histone modifications) might accompany a transition from gene repression to activation. We identified 247 genes that shift from a Group 3 to Group 1 profile during muscle differentiation (Figure S4A; Figure 3A; Table S3). Within this group, we focused on a cluster of 71 genes, which primarily are (1) known to play a critical role in muscle development and differentiation (Figure 1D), (2) were identified as repressed by MLL3/4 (Figure 2B), and (3) were up-regulated during differentiation as indicated by expression and enrichment of Pol II (Figure 3A). Upon activation of these Group 3 genes during myogenesis, we observed dramatic decreases in H3K4me1 across TSS-proximal regions, which we confirmed by qChIP (Figure 3B), coinciding with the acquisition of H3K4me3. By contrast, genes that were constitutively expressed at low levels in myoblasts and myotubes showed persistently high levels H3K4me1 (and low levels of H3K4me3) across promoter regions.
Figure 3. Loss of gene repression coincides with dynamic changes in H3K4me1.
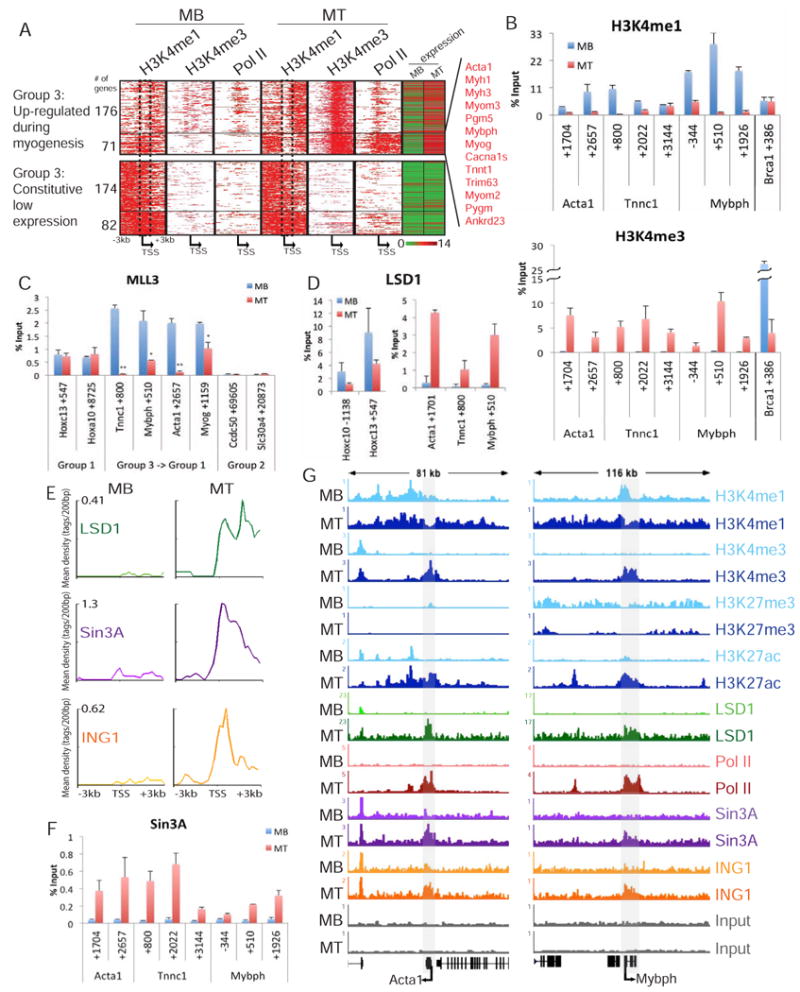
(a) Transcriptional activation of a subset of Group 3 genes during myogenic differentiation. H3K4me1, H3K4me3, and Pol II ChIP-seq data was plotted for all Group 3 genes up-regulated during myogenesis (top) or a representative subset of genes constitutively expressed at low levels (bottom), as indicated by expression (right column). The bottom cluster of (71) up-regulated genes is enriched with muscle development genes. The dashed box highlights a region that loses H3K4me1 in the 71 genes up-regulated genes. (b, f) Dynamic regulation of H3K4me1, H3K4me3, and Sin3A on muscle development genes in myoblasts (blue) and myotubes (red). (c) Enrichment of MLL3 on constitutively active Group 1, muscle genes that switch from a Group 3 to Group 1 signature, and Group 2 genes during differentiation. (d) Enrichment of LSD1 on Group 3 muscle genes during differentiation compared to constitutively expressed Hoxc cluster genes. (e) Distribution of LSD1, Sin3A, and ING1 ChIP-seq data plotted 3kb upstream and downstream of the TSS of the 71 up-regulated genes identified in Figure 3A (g) Pattern of histone modifications switches from a Group 3 to Group 1 signature across Acta1 and Mybph during differentiation. Read density profile for H3K4me1, H3K4me3, H3K27me3, H3K27ac, LSD1, Pol II, Sin3A, and ING1 across Acta1 and Mybph in myoblasts and myotubes indicated by gray shading. The y-axis corresponds to ChIP-seq signal density. All ChIP data, expressed as percentage of input, are plotted on the y-axis. Error bars indicate SD. Student's t test was performed to indicate significance: * indicates p-values <0.05 and ** <0.01, respectively.
Importantly, the decrease in H3K4me1 in myotubes coincided with reduced recruitment of MLL3 to Group 3 muscle development genes (Figure 3C). Interestingly, the loss of MLL3 during myogenesis was not accompanied by changes in Wdr5 recruitment to Acta1, Mybph, or Myog (Figure S4B). Previously, we had shown that Set1a is recruited to Acta1 and Myog, trimethylating these genes specifically in myotubes (Vethantham et al., 2012). Therefore, we conclude that muscle development genes recruit MLL3/4 in myoblasts, leading to H3K4 mono-methylation and repression. The loss of this mark in myotubes coincides with decreased MLL3/4 enrichment, gene activation, and the recruitment of a distinct COMPASS complex for deposition of H3K4me3. Loss of H3K4me1 during myogenesis may be further mediated by the activity of LSD1, an H3K4-mono-methyl demethylase (Shi et al., 2004). Remarkably, by ChIP-seq and qChIP, we find that LSD1 specifically localizes to the muscle development genes in myotubes, suggesting that H3K4me1 may be regulated through the concerted loss of the mono-methyltransferase, MLL3, and recruitment of the mono-methyl-demethylase (Figure 3D-E, 3G).
Next, we sought to examine the mechanistic consequences of this transition from a Group 3 to Group 1 profile. In agreement with previous studies, we showed that Sin3A is recruited to muscle development genes upon differentiation, concomitant with deposition of H3K4me3 (Figure 3B, 3F; van Oevelen et al. 2008). Previous studies demonstrated that recruitment of Sin3A was necessary for the proper expression of these genes (van Oevelen et al., 2010). Indeed, although Sin3A was originally identified as a transcriptional repressor, we found that it was strikingly enriched on ∼70% of the 71 muscle development genes (Figure 3A) exclusively in myotubes, when they are H3K4-tri-methylated but essentially devoid of H3K4me1 over the TSS (Figure 3E). In contrast to inflammatory response genes, such as Nos2, which remain H3K4-monomethylated during differentiation, transcriptional activation of Acta1 and Mybph coincided with loss of H3K4me1, acquisition of H3K4me3, and recruitment of LSD1 and Sin3A (Figure 3G, Figure S4C).
A change in methylation state is necessary to recruit ING1 and Sin3A to promoters
Although factors specifically recognizing the H3K4me1 state have not been reported, an array of proteins that recognize H3K4me3 have been identified. Since Sin3A is not thought to bind chromatin directly, we investigated which proteins could recognize H3K4me3 in concert with Sin3A. We subjected solubilized myoblast chromatin to immuno-affinity purification using anti-Sin3A antibodies and performed mass spectrometry to identify proteins known to interact with chromatin and specific DNA sequences. These interactors included the paralogous ING1 and ING2 proteins (which recognize H3K4me2/3), Wdr5, and Mxi1 and Max (sequence-specific factors known to recruit Sin3A) (Figure S5A; C.B., D. Ayer, and B.D.D., in prep.; Alland et al., 2007). We validated interactions between endogenous Sin3A and both ING1/2 and Wdr5 and by enforced expression of flag-tagged ING1 and ING2 in myoblasts (Figure S5B-C). These data indicated that Sin3A could be recruited to chromatin in skeletal muscle cells via distinct mechanisms that include recognition of specific DNA binding sites and methylated H3K4.
Next, we performed ChIP-seq on Sin3A, ING1, LSD1, and Wdr5, and merged these data with our genome-wide maps of histone modifications and gene expression profiles. We also identified regions bound by MLL1 and Menin, a component of MLL1/2 complexes, because (1) Sin3A has been shown to interact with MLL1 (Nakamura et al., 2002), and (2) we sought to determine if MLL1/2 is responsible for methylating H3K4 at the promoters of active genes. We plotted enrichment of all factors across a 6 kb region spanning the TSS and sorted all genes based on expression profiles (Figure 4A, Table S4). We found that Sin3A co-localized with ING1, LSD1, Wdr5, MLL1, and Menin on the majority of active genes (Cluster 1). In striking contrast, these factors were largely depleted from genes that were not expressed and exhibited high levels of H3K4me1 (cluster 2A) or H3K27me3 (cluster 2B). Although the MLL1/2 complex is thought to tri-methylate H3K4 on a small fraction (<5%) of all promoters in MEFs (Wang et al., 2009a), our data indicate that MLL1 and Menin co-localize extensively on the majority of active promoters with H3K4me3 (Figure 4A, Figure S5D). Furthermore, superimposition of Sin3A and ING1 ChIP-seq tags upon MNase-seq data indicated that Sin3A and ING1 localize to nucleosomes with reduced H3K4 mono-methylation but significant H3K4-trimethylation (Figure S3B; asterisks denote nucleosomes with low H3K4me1/high H3K4me3).
Figure 4. Sin3A is recruited to regions that lack H3K4me1.
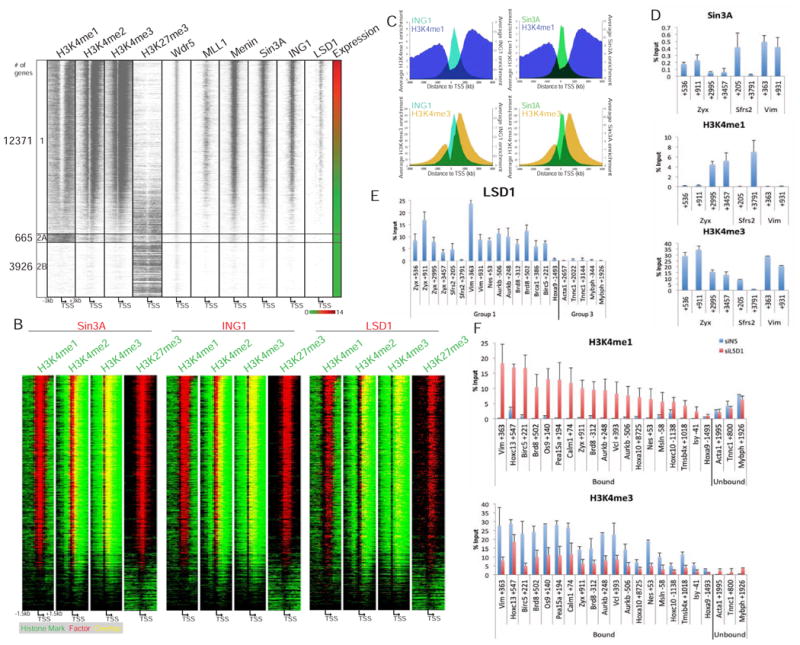
(a) Genome-wide, Sin3A is recruited to H3K4-trimethylated regions of active promoters. Genes in heat map were sorted based on expression in myoblasts (right-most column). Red-green key indicates relative expression. ChIP-seq data for the indicated antibodies was plotted for regions 3 kb upstream and downstream of the TSS. Cluster 1 includes all expressed genes. Cluster 2A/B includes all non-expressed genes sorted based on pattern of H3K4me1. (b) Heat map indicates overlap of Sin3A, ING1, and LSD1 (red) with H3K4me1, H3K4me2, H3K4me3, and H3K27me3 peaks (green) in a 1.5 kb window upstream and downstream of the TSS of genes in Cluster 1 in a. Regions containing an overlap of factors with histone marks are shown in yellow. (c) ING1 and Sin3A localize to a valley of H3K4me1. ING1 (teal; left panels), Sin3A (green; right panels), H3K4me1 (blue; top panels), and H3K4me3 (yellow; bottom panels) enrichment by ChIP-seq was plotted for regions 3 kb upstream and downstream of the TSS of all myoblast Sin3A target genes. The x- and y-axes represent the distance from the TSS of Sin3A target genes, and average enrichments, respectively. (d) H3K4me1 localization is anti-correlated with H3K4me3 and Sin3A across Zyx, Sfrs2, and Vim. (e) LSD1 localizes to regions on active (Group 1) genes with low H3K4me1 in myoblasts. (f) LSD1 depletion leads to increased enrichment of H3K4me1 (top) and decreased enrichment of H3K4me3 (bottom). qChIP signals expressed as percentage of input are plotted on the y-axis. Error bars indicate SD.
We overlapped our enrichment profiles for histone marks with Sin3A, ING1, and LSD1 (from Figure 4A). We found that Sin3A and ING1 peaks (red) were completely enveloped by H3K4me3 (green, H3K4me3; yellow, overlap) (Figure 4B). However, Sin3A and ING1 binding did not extend throughout the entire H3K4me3 domain, suggesting that H3K4me3 cannot solely dictate ING1 recruitment. More importantly, Sin3A, ING1, and LSD1 occupancy anti-correlated with the presence of H3K4me1, strongly indicating that recruitment of these factors and deposition of this histone mark are mutually exclusive. We also plotted the average enrichment of Sin3A, ING1, H3K4me1, and H3K4me3 across all Sin3A target genes, which reinforced these observations (Figure 4C). Moreover, Sin3A and ING1 localized to regions depleted of H3K4me1 (as indicated by gray shading) across the Hoxa and Hoxc clusters and selected Group 1 genes (Figure 4D, Figure S6). Notably, as we had observed previously (Figure 1A), H3K4me3 was highly enriched within regions depleted of H3K4me1 and strongly correlated with binding of LSD1, MLL1, Wdr5, and Menin. These analyses indicated (1) that ING1 (and to a lesser extent, Sin3A) pervasively overlapped with H3K4me3 and (2) that both factors, together with LSD1, were situated within regions depleted of H3K4me1. We conclude that on active genes, H3K4 mono-methylation could limit the binding of Sin3A and ING1 to distinct TSS-proximal regions marked by tri-methylation.
Impact of LSD1 and ING1/2 depletion on Sin3A recruitment
Our observations suggested that the balance between H3K4me1 and H3K4me3 might limit the recruitment of ING1/Sin3A complexes. Therefore, we speculated that enzymes able to influence the me1/me3 ratio, such as LSD1, could impact Sin3A binding. Indeed, we consistently detected very robust LSD1 binding to regions with low levels of H3K4me1 and high levels of H3K4me3 by ChIP-seq and qPCR, reminiscent of Sin3A binding (Figure 4A, 4E: compare with Figures 1D and Figure 4D).
We next sought to determine the impact of depleting LSD1 on the levels of H3K4me1 and H3K4me3, and we asked whether factor recruitment and gene expression were also perturbed. Loss of LSD1 in myoblasts provoked a dramatic increase in H3K4me1 on promoters that recruit this enzyme, whereas regions that did not recruit LSD1 (Acta1, Tnnc1, and Mybph) did not exhibit any change after LSD1 ablation (Figure 4F; Figure S5E). Unlike H3K4me1, LSD1 target genes exhibited consistent reductions in H3K4me3 after LSD1 depletion. Most importantly, the loss of LSD1 coincided with reduced expression of many of these LSD1 target genes (Figure 5A). Next, we investigated the impact of LSD1 knock-down on ING1 and Sin3A recruitment to active genes. We found that ING1 and Sin3A occupancy decreased after LSD1 knock-down, concomitant with loss of H3K4me3 and increased H3K4me1 (Figure 5B-C). However, the reductions in Sin3A recruitment were not universal. Interestingly, we found that promoters could be segregated into two groups based on their response to LSD1 knock-down: genes bound by E2F4, a factor known to recruit Sin3A to DNA, were relatively insensitive to changes in H3K4me1 and H3K4me3, despite robust alterations brought about by LSD1 depletion (Ren et al., 2002, van Oevelen et al., 2008). On the other hand, Sin3A recruitment was significantly reduced on target genes unable to recruit E2F4 (“Sin3-only” group). We propose that E2F4, and other sequence-specific factors, may be sufficient to recruit and stabilize Sin3A on chromatin, and Sin3A recruited through this mechanism may be less sensitive to changes in the H3K4me1/me3 ratio.
Figure 5. Sin3A recruitment depends on H3K4 methylation state and ING1.
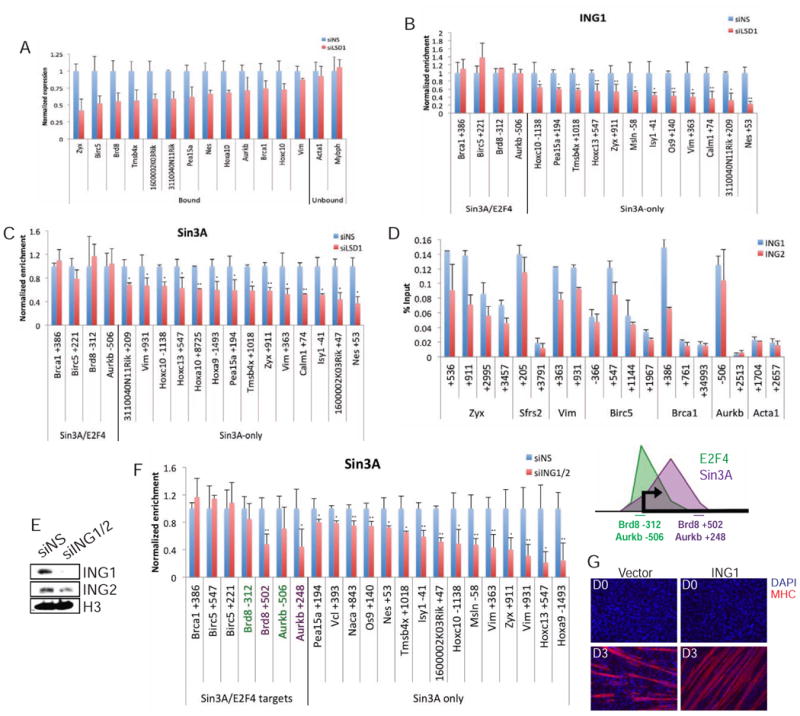
(a) qRT-PCR analysis after LSD1 knock-down on genes that differentially recruit this protein. Expression was normalized to the siNS control. (b) LSD1 depletion leads to decreased enrichment of ING1 and Sin3A (c). (d) ING1 and ING2 co-localize on chromatin. (e) After ablation of ING1/2 with siRNA, myoblast extracts were immuno-blotted for ING1, ING2, and H3 (loading control). (f) (Left) ING1/2 depletion leads to strong reductions in Sin3A recruitment to regions bound by Sin3A-only. (Right) Schematic indicating locations of Sin3A/E2F4 target gene primers. (g) The effect of ING1 over-expression on myogenic differentiation. Cells were stained with MHC (red) and DAPI (blue). Day 0 (D0) represents fully confluent cells and the onset of differentiation; D3 indicates differentiation for three days. Student's t test was performed to indicate significance: * indicates p-values <0.05 and ** <0.01, respectively. Error bars indicate SD.
To further explore this hypothesis, we asked whether recruitment of the Sin3A complex relied directly on recognition of H3K4me3. Our proteomics data and immunoprecipitation experiments showed that Sin3A interacts with ING1 and ING2, factors that specifically recognize H3K4me2/3. We observed that both proteins co-localized with Sin3A on chromatin (Figure 5D and Figure 3F). Next, we knocked down both ING1 and ING2 and determined whether there was an effect on Sin3A recruitment to Sin3A/ING1 target genes. Remarkably, Sin3A recruitment was perturbed on all Sin3A-only genes after ING1/ING2 depletion, but regions bound by Sin3A/E2F4 were unaffected, similar to loss of LSD1 (Figure 5E-F). As a more direct test of this phenomenon, we showed that Sin3A recruitment to regions downstream of E2F4 binding sites was significantly diminished by loss of ING1/2, whereas regions bound by E2F4 and Sin3 <1 kb upstream were considerably less sensitive to loss of ING1/2 (compare Aurkb and Brd8; Figure 5F, right panel). Interestingly, similar to Sin3A, which has been shown to play a role in the activation of muscle development genes, the over-expression of ING1 led to enhanced differentiation (van Oevelen et al., 2010; Figure 5G). These observations suggest that the Sin3A complex can be recruited by sequence-specific transcription factors, but its recruitment also strongly depends on recognition of H3K4me3 and attenuation by H3K4me1.
H3K4 mono-methylation is associated with gene repression in diverse cell types
Overall, our data support a model in which H3K4me1 at promoters is not only linked to gene repression but also plays a role in limiting the recruitment of H3K4me3-interacting proteins. To test whether this is a general phenomenon, we examined H3K4me1 and H3K4me3 deposition at promoters in other cell types, including bone marrow-derived macrophages (BMDM). Whereas LPS treatment of skeletal muscle myoblasts serves as an in vitro model for studying muscle damage, the recruitment of macrophages and expression of Il6 by these cells in muscle tissue is necessary for proper muscle regeneration (Heredia et al., 2013, Zhang et al., 2013). By taking advantage of genome-wide studies using LPS-treated murine BMDM (Figure S7A-B; Ostuni et al., 2013), we sought to determine if LPS-inducible genes were H3K4-mono-methylated in untreated BMDMs, as seen in C2C12s. Genes were assigned to the LPS-inducible category based on Pol II recruitment after 4 hours of LPS exposure, but not before treatment. Genes exhibiting similar enrichment of Pol II before and after LPS treatment were considered constitutively active and served as a control. We identified 89 genes as LPS-inducible. Surprisingly, although genes in the control group showed no differences in H3K4me1 enrichment, LPS-inducible genes showed a decrease in TSS-proximal mono-methylation (Figure S7A-B, indicated by gray). Conversely, H3K4me3 increased after LPS treatment across the same locations on LPS-inducible genes. Overall, we conclude that in skeletal muscle, embryonic fibroblasts, and macrophages, H3K4me1 is associated with context-depending gene repression, and gene activation correlates with a loss of H3K4me1.
Lastly, we investigated whether H3K4me1-associated repression plays a cell-type specific role using genome-wide histone modification and factor (Sin3A, Pol II) binding data from human ES cells (Figure 6A, Table S5). Like skeletal muscle cells, promoters in ES cells could be segregated into three groups based on patterns of H3K4 mono- and trimethylation. Furthermore, there was again a strong anti-correlation between Sin3A binding and H3K4me1 on one hand, and a highly robust positive correlation between Sin3A and H3K4me3 on the other. Interestingly, we identified 367 genes (Cluster 7) that were densely marked with H3K4me1 and indicators of facultative heterochromatin (H3K27me3 and H4K20me1) but lacked Pol II and Sin3A. To determine any shared biological function for this group of genes, we searched for enriched Gene Ontology (GO) terms. Interestingly, we found that these genes regulate transcription, pattern specification, cell fate commitment, and development and differentiation of various cell lineages and signify the pluripotency of ES cells (Figure 6B). However, in contrast with skeletal muscle, inflammatory response genes were not observed in this cluster. Instead, genes involved in defense and inflammatory response and taxis lacked all of these marks, consistent with the notion that they are not inducible in ES cells (Földes et al., 2010; Figure 6C). These findings indicate that the combination of H3K4me1, H3K27me3, and H4K20me1 is a general signature of repression and facultative heterochromatin and that Sin3 recruitment to H3K4me3-rich/H3K4me1-depleted regions surrounding the TSS is conserved across cell types and between species. Importantly, genes harboring this signature are expressed in an inducible and highly dynamic, cell-type and condition-specific manner.
Figure 6. Profiles of H3K4me1, H3K4me3 and Sin3A in H1-hESC.
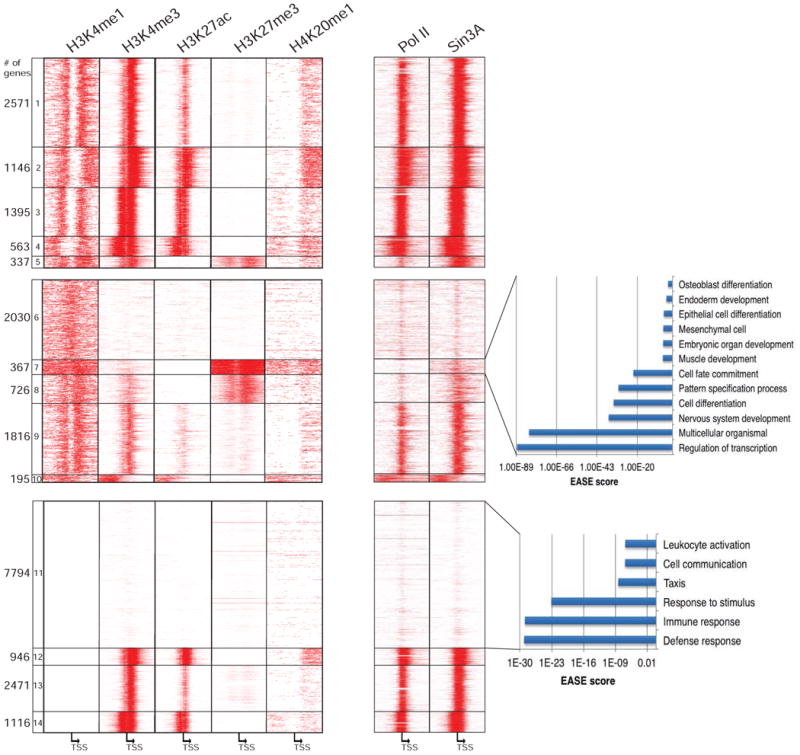
(a) ChIP-seq data for H3K4me1, H3K4me3, H3K27ac, H3K27me3, H4K20me1, Pol II, and Sin3A were obtained from the ENCODE Project Consortium (see Supplemental Experimental Procedures). Genes were sorted as in Figure 1A. (b, c) GO analysis of genes with (b) H3K4me1, H3K27me3, and H4K20me1 and (c) H3K4me1-/H3K4me3- signatures using EASE. The analysis was conducted as in Figure 1C.
Discussion
A role for H3K4me1 in gene repression
Although all three methylation states of histone H3 lysine 4 are thought to act in concert to mark the promoters of actively transcribed genes, no functional distinctions have been drawn for each methylation state until now. Our detailed analyses suggest a model in which (1) MLL3/4-mediated H3K4me1 marks the promoters of a subset of repressed genes and (2) H3K4me1 demarcates the occupancy of H3K4me3 interactors on active genes (Figure 7).
Figure 7. Model in which H3K4 methylation state dictates recruitment of ING1 and Sin3A to chromatin.
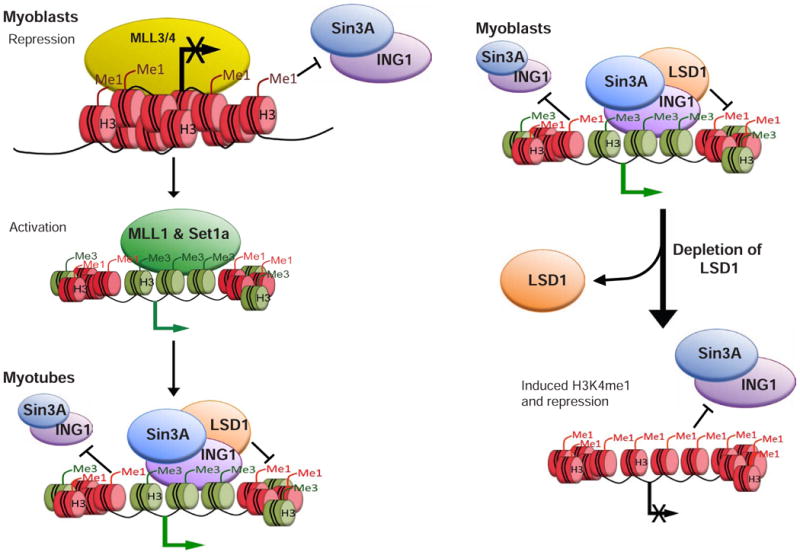
H3K4me1 covers the promoters of a subset of repressed, but inducible, genes. MLL3/4-mediated H3K4me1 is necessary for gene repression. Gene activation coincides with a change in H3K4me1 levels and loss of MLL3/4. On some genes, MLL3/4 are replaced with a distinct COMPASS complex (Set1a or MLL1) able to tri-methylate H3K4 on chromatin linked to the TSS. Although H3K4me3 is necessary for ING1 and Sin3A recruitment, H3K4me1 at the same promoters might restrict the localization of ING1 and Sin3A. Alterations in the ratio of H3K4me1 and H3K4me3 on active genes lead to loss of Sin3A recruitment to chromatin and repression. Multi-valent interaction with sequence-specific factors also contributes to Sin3 recruitment (omitted for simplicity). See text for details.
A surprising finding of our work is that a chromatin signature consisting of H3K4me1, H3K27me3, and H4K20me1 marks a unique subset of promoters. The H3K4-mono-methylated genes (Group 3, Figure 1A) identified in our study display a similar context-dependent expression pattern, wherein muscle development genes are silenced in skeletal muscle myoblasts but expressed in myotubes. The ability of these genes to adopt a chromatin conformation conducive to expression during differentiation suggests that TSS-proximal H3K4me1 is related to conditional gene repression. We further propose that H3K4me1 marks a subset of promoters with elevated nucleosome occupancy at the TSS and suggest that this mark may be linked to nucleosome compaction commonly associated with gene repression (Figure S3), although the mechanism by which compaction could be effected remains to be determined.
A role for MLL3 in promoter mono-methylation and repression
We further show that MLL3/4 deposits H3K4me1 on the promoters of repressed muscle genes in myoblasts, whereas MLL1/2 or Set1a/b deposit H3K4me3 on the same genes in myotubes. The MLL3/4 complex, but not other COMPASS complexes, contains the H3K27me3 demethylase, UTX (Agger et al., 2007, Lee et al., 2007, and Herz et al, 2012). Interestingly, our study indicates that muscle development and inflammatory response genes are simultaneously marked with H3K4me1 and H3K27me3 in myoblasts, and loss of MLL3/4 has little effect on H3K27me3 (Figure 1, Figure S2D). While the co-occurrence of both marks requires further study, we note that other proteins, besides MLL3/4, are able to recruit UTX to chromatin. For example, during myogenic differentiation, the trans-activator Six4 has been shown to recruit UTX to Myog (Seenundun et al., 2007). Further, MLL3/4 belong to large multi-subunit complexes that interact with other transcriptional regulators (Lee et al., 2008). Therefore, it remains formally possible that the repressive activity of MLL3/4 could arise from a combination of catalytic and non-catalytic activities.
We also found that the pattern of H3K4me1 distinguishes active and repressed promoters. This pattern of H3K4me1 may be dictated by nucleosome positioning, where active promoters have both nucleosome and H3K4me1 free regions, resulting in a bi-modal peak of H3K4me1, while repressed Group 3 promoters instead exhibit broad, block-like peaks (Figure 1A, Figure S3C). Although we have yet to determine how H3K4me1 is linked to nucleosome density, enhanced nucleosome density may prevent the recruitment of transcriptional activators. Uni- and bi-modal peaks of H3K4me1 within enhancers have been previously reported. At enhancers, bi-modal H3K4me1 peaks correlate with the binding of pioneering factors, including Pax7 and Foxa2 (Hoffman et al., 2010, Budry et al., 2012). Hoffman et al. also showed that a uni-modal peak of H3K4me1 correlated with lower gene expression, although distinctions between enhancers and promoters were not drawn. Our data suggest that at promoters, bi-modal H3K4me1 peaks result from recruitment of Set1a or MLL1/2 complexes, which tri-methylate nucleosomes very close to the TSS, whereas uni-modal peaks are generated by MLL3/4. As enhancers are defined as regions with relatively low H3K4me3 levels, bi-modal peaks of H3K4me1 are likely to be generated through distinct mechanisms at enhancers and promoters. Furthermore, in our genome-wide studies, we found that, irrespective of whether the peak of H3K4me1 at enhancers is bi- or uni-modal, Sin3A is not recruited.
A multi-valent binding model for Sin3A in mammalian cells
Previously, we found that Sin3A occupancy extended into the coding region of active genes, and such binding could not be accounted for by the presence of E2F4, prompting the prediction that this spreading was due to recognition of a histone mark (van Oevelen et al., 2008). Indeed, studies in yeast, have strongly suggested a role for H3K36me3 in the recruitment of Sin3 (Carrozza et al., 2005). In striking contrast, our genome-wide mapping of H3K36me3 in C2C12 cells suggests that H3K36me3 deposition is located significantly downstream of regions bound by Sin3A or ING1 (Asp et al., 2011, and data not shown). These findings underscore an important difference between Sin3 recruitment in yeast and mammalian cells. Instead, we propose that Sin3A complexes could be recruited to certain promoters through “multi-valent” binding by sequence-specific transcription factors on one hand, and by the presence of H3K4me3 and absence of H3K4me1 on the other (Figure 7). Nevertheless, this recruitment strategy may be analogous to mechanisms in yeast, wherein Sin3 is recruited via PHD and chromo-domains of Rco1 and Eaf3, respectively, which recognize H3K36me3 (Li et al., 2007).
How H3K4 mono-methylation is “interpreted” at promoters
ING1 was shown to recognize H3K4me2/3, and it has been suggested that this mechanism could thereby recruit Sin3A (Kuzmichev et al., 2002). Nevertheless, our data clearly demonstrate that for a given promoter marked by H3K4me2/me3, ING1 and Sin3A binding is spatially constrained. Although the localization of ING1 and Sin3A to valleys of H3K4me1 does not unequivocally demonstrate a functionally antagonistic relationship, we have used three scenarios to definitively show that H3K4me1 limits the recruitment of Sin3A and ING1: (1) muscle development and inflammatory response genes are repressed and heavily enriched with H3K4me1 in myoblasts and do not recruit Sin3A or ING1; (2) Sin3A and ING1 localize to TSS-proximal regions on active promoters characterized by high H3K4me3 and low H3K4me1; and (3) loss of LSD1 induced H3K4me1 at TSS-proximal regions and disrupted Sin3A and ING1 recruitment to chromatin (Figure 7). Furthermore, although H3K4me1 is incompatible with Sin3A and ING1 binding, loss of H3K4me1 alone through ablation of MLL3/4 is not sufficient to recruit Sin3A and ING1 to muscle development genes in myoblasts (data not shown). Instead, only the combination of low H3K4me1 and robust H3K4me3 is sufficient to recruit Sin3A to these genes. Such a model--whereby the recruitment of Sin3A and ING1 to chromatin is defined by the dynamic enzymatic activity of various histone methyltransferases and demethylases—may be a useful paradigm for studying other chromatin-interacting factors that could adopt similar modes of recruitment—and restrictions--to diverse histone modifications.
Experimental Procedures
Quantitative RT-PCR and transfections
RNA isolation and RT-qPCR were performed as described (Blum et al. 2012). Briefly, transfections of synthetic oligonucleotides obtained from Dharmacon were performed in myoblasts using Lipofectamine RNAiMAX (Invitrogen). siRNA transfected cells were harvested 24 hours after transfection. In each case, expression was normalized to a control gene. Nuclear extracts were prepared as described (Blum et al. 2012).
ChIP, ChIP-seq and MNase-seq
ChIP was performed as described (Blais et al. 2007) using antibodies listed in the Supplemental Experimental Procedures. The optimal antibody/chromatin ratio was determined through serial titrations. All ChIP experiments were carried out using excess antibody. Real-time quantitative PCR after ChIP was performed as previously described (Asp et al., 2011). In all experiments presented, background IgG enrichment was generally ≤0.02% of input levels. All primer sequences are listed in Supplemental Table S6 and the 5′ end of each amplicon relative to the TSS is denoted. ChIP-seq and MNase-seq experiments were performed and analyzed as described previously (Asp et al. 2011) and in Supplemental Experimental Procedures.
Statistical Analysis
All experiments were repeated at least twice independently, and the data are presented as mean ± SD. Statistical significance was determined using the Student's t test. A p value of < 0.05 was considered to be statistically significant and is presented as *p < 0.05 or **p < 0.01.
Supplementary Material
Highlights.
H3K4me1 marks inducible genes in ESC, macrophages, and muscle
MLL3/4 recruitment to promoters mediates H3K4me1, which leads to gene silencing
H3K4me3 and sequence-specific factors “multi-valently” recruit Sin3 to chromatin
H3K4me1 at promoters establishes a “boundary” for Sin3 recruitment
Acknowledgments
We are grateful to D. Reinberg for useful discussions and for generously providing several antibodies used in this study. We thank D. Ayer for providing Sin3A antibodies, K. Riabowol for providing ING1/2 antibodies, W. Lane for assistance with mass spectrometric analysis of Sin3A complexes, and S.E. Millman for technical assistance. We thank A. Heguy and the NYU Genome Techology Center for assistance with Illumina sequencing. This work was supported by NIH grant 2R01CA077245 to B.D.D.
Footnotes
Accession Numbers: All ChIP-seq and RNA-seq data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE50590).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alland L, Muhle R, Hou H, Jr, Potes J, Chin L, Schreiber-Agus N, DePinho RA. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- Asp P, Blum R, Vethantham V, Parisi F, Micsinai M, Cheng J, Bowman C, Kluger Y, Dynlacht BD. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc Natl Acad Sci USA. 2011;108:149–158. doi: 10.1073/pnas.1102223108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Blais A, van Oevelen CJ, Margueron R, Acosta-Alvear D, Dynlacht BD. Retinoblastoma tumor suppressor protein-dependent methylation of histone H3 lysine 27 is associated with irreversible cell cycle exit. J Cell Biol. 2007;179:1399–1412. doi: 10.1083/jcb.200705051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum R, Vethantham V, Bowman C, Rudnicki M, Dynlacht BD. Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1. Genes Dev. 2012;26:2763–2779. doi: 10.1101/gad.200113.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budry L, Balsalobre A, Gauthier Y, Khetchoumian K, L'honoré A, Vallette S, Brue T, Figarella-Branger D, Meij B, Drouin J. The selector gene Pax7 dictates alternate pituitary cell fates through its pioneer action on chromatin remodeling. Genes Dev. 2012;26:2299–2310. doi: 10.1101/gad.200436.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, Parker MH, MacQuarrie KL, Davison J, Morgan MT, Ruzzo WL, et al. Genome-wide MyoD binding in skeletal muscle cells a potential for broad cellular reprogramming. Dev Cell. 2010;18:662–674. doi: 10.1016/j.devcel.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földes G, Liu A, Badiger R, Paul-Clark M, Moreno L, Lendvai Z, Wright JS, Ali NN, Harding SE, Mitchell JA. Innate immunity in human embryonic stem cells comparison with adult human endothelial cells. PLoS One. 2010;5:e10501. doi: 10.1371/journal.pone.0010501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost RA, Nystrom GJ, Lang CH. Lipopolysaccharide and proinflammatory cytokines stimulate interleukin-6 expression in C2C12 myoblasts: role of the Jun NH2-terminal kinase. Am J Physiol Regul Integr Comp Physiol. 2003;285:1153–1164. doi: 10.1152/ajpregu.00164.2003. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, Chawla A. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. 2013;153:376–88. doi: 10.1016/j.cell.2013.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, Voets O, Verrijzer CP, Shilatifard A. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 2012;26:2604–2620. doi: 10.1101/gad.201327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman BG, Robertson G, Zavaglia B, Beach M, Cullum R, Lee S, Soukhatcheva G, Li L, Wederell ED, Thiessen N, et al. Locus co-occupancy, nucleosome positioning, and H3K4me1 regulate the functionality of FOXA2-, HNF4A-, and PDX1-bound loci in islets and liver. Genome Res. 2010;20:1037–51. doi: 10.1101/gr.104356.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. The MLL3/MLL4 Branches of the COMPASS Family Function as Major Histone H3K4 Monomethylases at Enhancers. Mol Cell Biol. 2013;33:4745–54. doi: 10.1128/MCB.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A, Zhang Y, Erdjument-Bromage H, Tempst P, Reinberg D. Role of the Sin3-histone deacetylase complex in growth regulation by the candidate tumor suppressor p33(ING1) Mol Cell Biol. 2002;22:835–848. doi: 10.1128/MCB.22.3.835-848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- Lee J, Saha PK, Yang QH, Lee S, Park JY, Suh Y, Lee SK, Chan L, Roeder RG, Lee JW. Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc Natl Acad Sci U S A. 2008;105:19229–19234. doi: 10.1073/pnas.0810100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Gogol M, Carey M, Pattenden SG, Seidel C, Workman JL. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 2007;21:1422–1430. doi: 10.1101/gad.1539307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chu A, Chakroun I, Islam U, Blais A. Cooperation between myogenic regulatory factors and SIX family transcription factors is important for myoblast differentiation. Nucleic Acids Res. 2010;38:6857–6871. doi: 10.1093/nar/gkq585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Peña PV, Hom RA, Hung T, Lin H, Kuo AJ, Wong RP, Subach OM, Champagne KS, Zhao R, Verkhusha VV, et al. Histone H3K4me3 binding is required for the DNA repair and apoptotic activities of ING1 tumor suppressor. J Mol Biol. 2008;380:303–312. doi: 10.1016/j.jmb.2008.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seenundun S, Rampalli S, Liu QC, Aziz A, Palii C, Hong S, Blais A, Brand M, Ge K, Dilworth FJ. UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 2010;29:1401–1411. doi: 10.1038/emboj.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Peña P, Lan F, Kaadige MR, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, Boutros M, Perrimon N, Rosenfeld MG, Glass CK. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol Cell. 2012;48:28–38. doi: 10.1016/j.molcel.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Reinberg D. Facultative heterochromatin: is there a distinctive molecular signature? Mol Cell. 2007;28:1–13. doi: 10.1016/j.molcel.2007.09.011. [DOI] [PubMed] [Google Scholar]
- van Oevelen C, Wang J, Asp P, Yan Q, Kaelin WG, Jr, Kluger Y, Dynlacht BD. A role for mammalian Sin3 in permanent gene silencing. Mol Cell. 2008;32:359–370. doi: 10.1016/j.molcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oevelen C, Bowman C, Pellegrino J, Asp P, Cheng J, Parisi F, Micsinai M, Kluger Y, Chu A, Blais A, David G, Dynlacht BD. The mammalian Sin3 proteins are required for muscle development and sarcomere specification. Mol Cell Biol. 2010;30:5686–5697. doi: 10.1128/MCB.00975-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Prabhakar S, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Afzal V, Rubin EM, Pennacchio LA. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet. 2008;40:158–160. doi: 10.1038/ng.2007.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vethantham V, Yang Y, Bowman C, Asp P, Lee JH, Skalnik DG, Dynlacht BD. Dynamic loss of H2B ubiquitylation without corresponding changes in H3K4 trimethylation during myogenic differentiation. Mol Cell Biol. 2012;32:1044–1055. doi: 10.1128/MCB.06026-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Lin C, Smith ER, Guo H, Sanderson BW, Wu M, Gogol M, Alexander T, Seidel C, Wiedemann LM, et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol Cell Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Wang PF, Lee JS, Martin-Brown S, Florens L, Washburn M, Shilatifard A. Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol Cell Biol. 2008;28:7337–7344. doi: 10.1128/MCB.00976-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Res. 2011;21:564–578. doi: 10.1038/cr.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Li Y, Wu Y, Wang L, Wang X, Du J. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J Biol Chem. 2013;288:1489–1499. doi: 10.1074/jbc.M112.419788. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.