

Background: Iztli peptides (IP) are a new class of antibacterial peptides that could be internalized by receptor-mediated endocytosis in yeast.
Results: IP-1 penetrates cells independently of endocytosis and makes pores in membranes with large electric potential.
Conclusion: Antibacterial peptides like IP-1 make pores or penetrate membranes depending on the membrane potential.
Significance: Antibacterial peptides with penetrating activity are robust to control bacterial infections.
Keywords: Antimicrobial Peptides, Cell-penetrating Peptides, Computational Biology, Membrane Biophysics, Receptor Endocytosis
Abstract
Cell penetrating peptides (CPP) and cationic antibacterial peptides (CAP) have similar physicochemical properties and yet it is not understood how such similar peptides display different activities. To address this question, we used Iztli peptide 1 (IP-1) because it has both CPP and CAP activities. Combining experimental and computational modeling of the internalization of IP-1, we show it is not internalized by receptor-mediated endocytosis, yet it permeates into many different cell types, including fungi and human cells. We also show that IP-1 makes pores in the presence of high electrical potential at the membrane, such as those found in bacteria and mitochondria. These results provide the basis to understand the functional redundancy of CPPs and CAPs.
Introduction
Hunter-killer peptides (HKPs)2 combine two activities: a ligand peptide (hunter sequence) and a cationic antibacterial peptide (killer sequence) (1). Such design aims to selectively induce cell death of target eukaryotic cells. For instance, HKPs have been applied successfully to treat cancer (2) and obesity (3) in animal models. The mechanism of action of these peptides proposes that the hunter sequence recognizes a receptor on the cell surface and upon binding the hunter sequence may be internalized by receptor-mediated endocytosis carrying with it the killer sequence. Inside the cell, the killer sequence targets mitochondria, which share similar structure with bacteria, to destabilize their membrane and ultimately induce apoptosis (4).
The ability to deliver macromolecules inside cells is important both in academic research as well as in biotechnological applications such as drug delivery and gene therapy (5). Chemical and physical methods have been developed to deliver cargo molecules inside cells. These methods, e.g. electroporation and cationic lipids/liposomes, have been shown to deliver hydrophobic macromolecules, yet these induce cellular side effects and are limited to in vitro applications (6). Cell penetrating peptides (CPPs) overcome some of these limitations and HIV-Tat, penetratin, transportan, and octa-arginine among others represent extensively studied CPPs (7). Many CPPs are able to deliver themselves, small cargo, and relatively large macromolecules directly across the cellular plasma membrane (8).
Understanding the penetrating mechanism of CPPs is important to control the delivery of macromolecules by CPPs. In this sense HIV-Tat and nona-arginine peptides have been shown to be internalized by endocytosis (9), yet other CPPs use an energy-independent mechanism of internalization (5). Despite such differences, common features exist among CPPs; for instance, most CPPs do not show any cell specificity, are amphipathic, and present a net positive charge (10). Relevant to this work is that cationic antibacterial peptides (CAPs), like the killer peptides used in HKPs, are also amphipatic and cationic (11). It is not clear though, how peptides with similar physicochemical features such as CPPs and CAPs can perform two distinct functions; CPPs penetrate eukaryotic cells without any apparent toxicity, and CAPs kill bacteria. Given their similar physicochemical properties, the explanation to this functional difference may reside in the target cell and not in the peptide itself. If so, CPPs may work as CAPs in the presence of bacterial cells. In agreement with this idea, several CPPs have been shown to have antibacterial activity (12, 13). Furthermore, given that CPPs and CAPs act on cellular membranes, it can be assumed that these membranes modulate their functions. In terms of the mechanism of action proposed for the interaction with cellular membranes, CAPs are able to make pores by “barrel-stave,” “carpet” or “toroidal-pore” mechanisms (14); such pores ultimately end up killing bacterial cells. On the other hand, CPPs cross eukaryotic cellular membranes without making pores. To the best of our knowledge there is no evidence that may explain how a cellular membrane may switch a CPP to become a CAP.
We have recently described a new class of HKPs (15): Iztli peptides (IPs). IPs are CAPs that harbor a hunter sequence within; such design differs from typical HKPs that fuse a hunter sequence next to a CAP sequence (see Fig. 1). To study the mechanism of action of these peptides, IPs were designed to target Saccharomyces cerevisiae using the pheromone α-factor as the hunter sequence; only mating type A (MATa) cells expressing the receptor for the pheromone (Ste2p) would be killed by the action of IPs. Indeed, four IPs (IP-1, IP-2, IP-3, and IP-4) sharing the same pheromone sequence but differing in six residues at the N terminus were shown to have antibacterial activity, to swell mitochondria, and to induce the pheromone response pathway, and only MATa cells expressing the Ste2p receptor were killed by IPs (15). Thus, Ste2p is required for the killing as expected from the HKP model of action, yet this cannot be taken as evidence for cellular uptake via Ste2p.
FIGURE 1.

Iztli peptide 1. Embedding of the α-pheromone sequence into a cationic antibacterial peptide sequence is shown. Note that the residues added to the α-pheromone (bold letters) do not present any antibacterial or antifungal activity and were added to the pheromone to match the physicochemical properties of cationic antibacterial peptides (11).
In the current work we studied the mechanism of internalization of IP-1 at concentrations required to kill cells. Our results indicate that IP-1 is internalized into many cell types in the absence of receptor-mediated endocytosis, yet binding to the receptor is relevant for the killing. We also show that IP-1 forms pores only in the presence of high electric potential values. The relevance of our findings is discussed in terms of the biological and biotechnological advantage of multifunctional peptides, such as IPs.
EXPERIMENTAL PROCEDURES
Strains and Peptides
The strains used in this study were: S. cerevisiae BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0), Δdid4::kanMX, Δypt7::kanMX, Δrvs161::kanMX, Δpep5::kanMX, Δyck1::kanMX, and Δyck2::kanMX; BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) purchased from OpenBiosystems Candida glabrata CGM99 (Ura3D::Tn903(G418R), Trp 1D), and Aspergillus nidulans CLK43; these last two were kindly provided, respectively, by Dra. Irene Castaño (Instituto Potosino de Investigación Científica y Tecnológica, San Luis Potosi, Mexico) and Dr. Jesus Aguirre Linares (Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Mexico). See Table 1 for a detailed list of the cell lines used in this study.
TABLE 1.
Cell lines
| Organism | Strain | ORF | Genotype | Source |
|---|---|---|---|---|
| S. cerevisiae | BY4741 | WT | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Open Biosystems YKO MATa Strain Collection-Glycerol Stocks |
| YKL002W | MATa Δdid4 | |||
| YML001W | MATa Δypt7 | |||
| YCR009C | MATa Δrvs161 | |||
| YMR231W | MATa Δpep5 | |||
| YHR135C | MATa Δyck1 | |||
| Endocytosis null mutants (see supplemental Table S1) | MATa Δgene::kanMX | |||
| BY4742 | YNL154C | MATa Δyck2 | Open Biosystems YKO MATα Strain Collection-Glycerol Stocks | |
| C. glabrata | CGM99 | WT | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Domergue R., et al. (52) |
| A. nidulans | CLK43 | WT | Ura3D::Tn903(G418R), Trp 1D | Kawasaki, L, et al. (53) |
| WT | pabaA1 yA2; veA1 |
For the electrophysiological experiments 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) dissolved in chloroform and powdered cholesterol were purchased from Avanti Polar Lipids (Alabaster, AL) and stored in the dark at −20 °C. Potassium chloride (KCl, ACS grade) and calcium chloride (CaCl2, ACS grade) were purchased from Merck. All organic solvents were ACS grade and were purchased from Mallinckrodt Baker. Borosilicate glass capillaries were obtained from World Precision Instruments (Sarasota, FL). Praxair (Cuernavaca, México) supplied high purity nitrogen gas.
The mating pheromone α-factor from S. cerevisiae was purchased from Sigma (catalogue number T6901). Anaspec, Inc. produced the IP-1 and IP-labeled with Hilytefluor 488, whereas we synthesized TAMRA-IP-1. See Table 2 for the sequences of the peptides used in this study.
TABLE 2.
Synthetic peptides
| Name | Sequence | Measured molecular weight |
|---|---|---|
| g/mol | ||
| α-Pheromone | NH2-WHWLQLKPGQPMY-COOH | 1684 |
| IP-1 | NH2-KFLNRFWHWLQLKPGQPMY-COOH | 2490.8 |
| TAMRA-IP-1 | TAMRA-KFLNRFWHWLQLKPGQPMY-COOH | 2903.2 |
| Hilytefluor 488-IP-1 | NH2-KFLNRFWHWLQLK(GGG-CO-(CH2)2S-maleimido Hilytefluor 488)-PGQPMY-COOH | 3335.4 |
Measuring the Cell Death Induced by Iztli Peptide 1
An early stationary culture for every assay was obtained from a single colony of BY4741, and every endocytosis null mutant grew for 12 h in YPD medium (yeast extract 1%, peptone 2%, glucose 2%). These cultures were used to inoculate a fresh YPD medium to reach a 0.03 optical density in a total volume of 200 μl. BY4741 strain was tested against TAMRA-IP-1 (62.9 μm) or Hilytefluor 488-IP-1 (26.9 μm), and every endocytosis null mutant was tested against the pheromone α-factor (10 μm) and PI-1 (10 μm) in a 100-well honeycomb plate, each containing 200 μl, incubating at 30 °C with shaking using a Bioscreen C (Oy Growth Curves Ab Ltd). The activity of these peptides on each strain was determined by measuring the A600 nm every hour for 24 h.
Modeling the Internalization of Iztli Peptides
The models were implemented based on the processes depicted in Fig. 3, A and B. The biochemical reactions were assumed to follow mass-action kinetics and resulted in the ordinary differential equations provided below. Model simulations were performed using the LSODA solver and an evolutionary optimization algorithm performed fitting to the measured time course of dead cells. Here, we used the implementations within the modeling software Copasi. For each model only two parameters had to be fitted (denoted as kE/kD and kIn/kD in Fig. 3, A and B). The remaining parameters were obtained from previously published measurements.
FIGURE 3.

Models for Iztli peptides internalization on yeast cells. A, the model assuming an endocytic import of Iztli peptide via Ste2p (× denotes degradation). B, the model assuming permeation of Iztli peptide where the peptide may cross the membrane independent of Ste2p-mediated endocytosis. C, comparison of the two models fitted to the measured colony-forming unit time course of dead cells (n = 8, bars denote S.D.). The model assuming direct import generally performed better, noticeable for early time points. Mio, millions of cells.
Models were considered significant when they could reproduce the measured time course significantly better than a zero-intercept linear reference model. Thus, we obtained p values for the entire time course, or parts of it, from F tests comparing the linear model to the fit provided by the optimization of the models. Those analyses were performed in R (The R Project for Statistical Computing).
Endocytosis Model
The endocytosis model comprised the reactions from Fig. 3A. This resulted in the following set of ordinary differential equations (square brackets denote concentrations, and the indices e, m, v, and c denote locations in the extracellular medium, the membrane, (pre)vacuole, and cytosol, respectively).
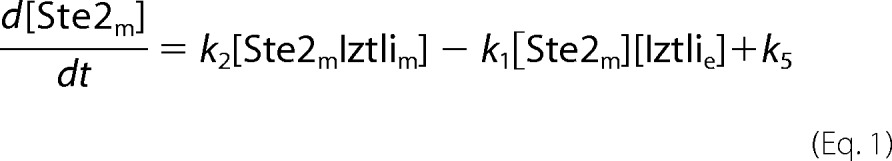 |
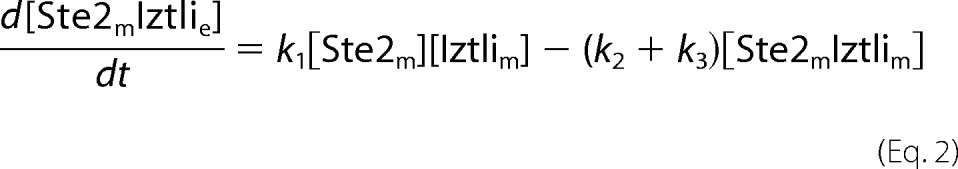 |
 |
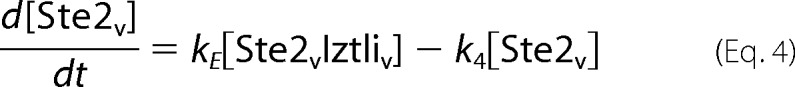 |
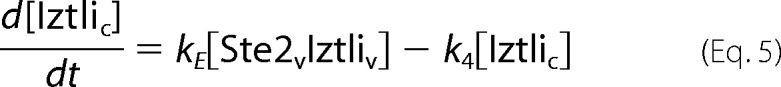 |
The cell volume was chosen as 4.9·10−15 liters. Due to the large volume of the extracellular medium, the extracellular concentration of IP-1 was treated as a constant referring to the amount used in the experiments. The number of dead cells, nD, in the population was expressed by the minimal concentration of IP-1 within the cells to kill a cell, kD. nD was evaluated at each time-step of the ordinary differential equations, yielding an algebraic system where nD was given by the formula,
 |
Here, ntot denotes the total number of cells in the population and was obtained by fitting a fourth-order polynomial in time to the colony-forming unit measurements of untreated cells. Iztlic denoted the concentration of IP-1 inside the cells that had escaped the (pre)vacuole. All simulations and fits were performed using Copasi.
The parameters k1 through k5 were obtained from literature. k2 was obtained as 10−3 s−1 from a mean of 3 direct measurements (16–18). k1 was then calculated from k2 and the mean kD for Ste2p binding to α-factor from 9 experiments, resulting in k1 = 0.185 μm−1s−1 (18–26). We assumed that dissociation of α-factor from Ste2p has the same kinetics in the (pre)vacuole before degradation. The rate of endocytosis of Ste2p in presence of α-factor, k3, was derived from the published half-time of 5 min as 2.3·10−3 s−1 (27). The degradation rate of internalized Ste2p, k4, was obtained from the half-life of total Ste2p in the presence of α-factor in cycloheximide-treated cultures as 5.8·10−4 s−1 (24). Because α-factor is normally degraded along with Ste2p, we assumed that the degradation rate of IP-1 within the cells happens with the same rate as for Ste2p. The synthesis rate of Ste2p, k5, was chosen as 7·10−5 μm·s−1 to counteract the degraded Ste2p after about 90 min as reported earlier (20).
The rate of IP-1 escaping from the endosome, kE, as well as the lethal IP-1 concentration, kD, were obtained by fitting the model to the measured time course of dead cells by an evolutionary optimization algorithm. We required the parameter values to stay in biologically reasonable intervals. kE was required to stay in the range of 0 to 1 s−1 and kD within 0 to 10 μm. The best fit estimated kE and kD as 1 s−1 and 0.13 μm, respectively. One should note that the fit forces the model to realize an almost immediate escape of IP-1 from the vacuole. See Table 3 for a list of parameters and values used in these models.
TABLE 3.
Data for modeling MATa killing by IP-1 dependent on cellular internalization
| Model | Parameter | Value |
|---|---|---|
| Endocytosis/direct | Cell volume | 4.9 × 10−15 l |
| Endocytosis/direct | k1 | 0.185 μm−1s−1 |
| Endocytosis/direct | k2 | 10−3s−1 |
| Endocytosis/direct | k3 | 2.3 × 10−3s−1 |
| Endocytosis/direct | k4 | 5.8 × 10−4s−1 |
| Endocytosis/direct | k5 | 7 × 10−5μMs−1 |
| Endocytosis | kD | 0.13 μm |
| Endocytosis | kE | 1s−1 |
| Direct | kIn | 2.5 × 10−4s−1 |
| Direct | kD | 8.9 μm |
Direct Model
The direct model comprised only an internalization of IP-1, which depends on its affinity for Ste2p as indicated in Fig. 3B. The resulting ordinary differential equation is,
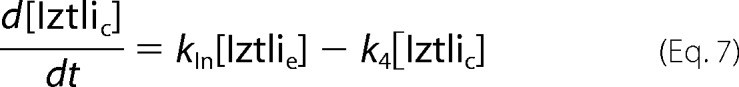 |
The same assumptions as in the endocytosis model were made, and we fitted kIn and kD as before. Here kD was again bound between 0 and 10 μm, and kIn was kept between 0 and infinity. The fitted parameters were kIn ≈ 2.5·10−4s−1 and kD ≈ 8.9 μm.
Confocal Microscopy of Labeled Iztli Peptide
For the internalization images presented in this study, S. cerevisiae MATa (BY4741), MATα (BY4742), or C. glabrata cells were grown at 30 °C for 24 h in synthetic complete (SC) media (yeast nitrogen base w/o amino acids 0.67%, potassium phosphate monobasic 0.1%, glucose 2%, amino acids 0.079%); afterward they were diluted to a final absorbance of 0.4 at 600 nm and treated with 68.9 μm TAMRA-IP-1 or 89.9 μm Hilytefluor 488-IP-1 for 1 h at 30 °C. In the case of A. nidulans these were grown as previously reported (28); briefly, cells were grown in supplemented glucose minimal nitrate medium, and 2 × 105 spores were suspended in 2 ml of water and incubated 60 min at 37 °C at 300 rpm with TAMRA-IP-1 (13.77 μm). In every case 80 μl from these samples were centrifuged at 300 rpm for 5 min, the supernatant was removed, and the samples were observed under the confocal microscope (Olympus FluoView FV1000) at 60× amplification. HEK239 and NL20 human cell lines were grown to 80% confluence in DMEM medium supplemented with 2 mm Glutamax, 50 units/ml penicillin, 50 μg/ml streptomycin, and 15% fetal bovine serum at 30 °C and 5% CO2. Cells were treated with trypsin and washed twice with PBS and recovered by centrifugation. The pellet was resuspended in Opti-MEM® to reduce serum fluorescence, and 2 × 104 cells were cultured in a Chamber Slide® system (NUNC® Lab-Tek® II-C2TM) and incubated for 1 h to allow the adhesion of cells to the bottom of the slide to form a monolayer. Afterward HEK239 cells where treated with 13 μm, and NL20 cells where treated with 24 μm TAMRA-IP-1 and visualized using confocal microscopy (Olympus FLUOVIEW FV-1000) at an emission wavelength of 570 nm for different times. For such purpose a CO2 chamber was used during the analysis. All images were reconstructed using the software FV viewer provided by the manufacturer of the microscope.
Small Unilamellar Vesicle Preparation
POPC and cholesterol were stored at - 20 °C and used without further purification. Stock chloroform solutions for every lipid were prepared once a week and stored at −20 °C. Cholesterol and POPC solutions were mixed to obtain the desired mole fraction of sterol (30%). Evaporation of the solvent was done in a rotative evaporator (B-177; Büchi Labortechnik, Flawil, Switzerland). The suspension was prepared by adding the working solution (5 mm KCl, 1 mm CaCl2, 10 mm HEPES, pH 8.0) to the film deposited in the flask in an N2-enriched atmosphere and then treated in an ultrasonic bath to produce dispersion and obtain unilamellar vesicles (29). The suspension was stored for 2 h under refrigeration (4–6 °C) before the bilayer formation and peptide incorporation.
Micropipette Fabrication
Glass capillaries with filament (1.00-mm external diameter and 0.58-mm internal diameter) from World Precision Instruments were pulled using the P2000 instrument from Sutter Instruments (Novato, CA). Micropipettes were then silanized and filled with the same working solution used for the preparation of small unilamellar vesicles and used within the following 60–120 min. The average resistance of micropipettes in the working solution was 3.46 ± 2.80 gigaohms.
Solvent-free Tip-dip Lipid Bilayer Formation
The controlled-temperature chamber was electrically insulated within a Faraday cage and suspended in elastic bands to reduce the mechanical vibration. A sample of the small unilamellar vesicle was put in the chamber. The sample was settled for 10 min at the desired temperature, and then a bilayer was formed at the tip of the micropipette by consecutive immersion in small unilamellar vesicle suspension until a capacitive response to the square potential applied was observed (30, 31). The standard seals obtained in this manner were of ∼400 gigaohms with a 2-kHz low pass filter and a current root mean square (Ir.m.s.) ∼0.12 pA. When the bilayer was formed, the proper amount of Iztli peptide stock solution was added to obtain the desired concentration, and after the equilibrium conditions the 5-min experiments were done.
Pore Formation of Iztli Peptide 1 on Artificial Membranes
Small unilamellar vesicles, micropipettes, and solvent-free tip-dip lipid bilayers were prepared as described above. To observe the possible formation of pores in these membranes, the electrical measurements of the current signal were amplified with an Axopatch 200B set to voltage-clamp mode, 2-kHz low pass filter, and maximal scaling output, digitally converted with a Digidata 1200A, acquired with the aid of pClamp 8.2 software at a frequency of 10 kHz (all from Molecular Devices Analytical Technologies), and stored in a personal computer. The bilayer seal was measured by applying a square 5-mV step potential. The sampling rate was set to 100 μs. Once the seal was formed, the Ir.m.s. obtained was consistently 0.12 ± 0.03pA. The temperature of the experiments was 25 ± 2 °C. The Iztli peptide was added in solution to have a concentration of 5, 10, 15, 20, and 25 μm. The potentials applied in all of the reported experiments were 150 and 200 mV. Care must be taken to be sure that experiments are performed under equilibrium conditions. We determined the average conductance as a function of time and discovered that this property needs 15–20 min to attain equilibrium. All records were a posteriori base-line-corrected using a Fortran program. The occupancies obtained were defined as the ratio of the time expended in one state to the total time of experiment. The average currents reported correspond to the average value of all points registered in a given time. The average currents reported correspond to an average current in a 5-min experiment obtained from an all points histogram. All graphics were done with Grace software, a two-dimensional graph-plotting tool, for Unix-like operating systems.
GO Term Enrichment Estimation
To determine the participation of endocytosis on the killing mediated by IP-1, we analyzed the enrichment of GO terms in the 291 genes known to affect endocytosis (see supplemental Table S1). For this analysis we used as a control set the set of genes in yeast. We used the service provided by GO.
RESULTS
Iztli Peptide 1 Is Not Internalized through Receptor-mediated Endocytosis
Based on the proposed mechanism of action for HKPs (4), it would be expected that the rate of killing yeast cells by IPs would be proportional to the rate of internalization. If so, reduction or elimination of Ste2p endocytosis may affect the rate of killing by IP-1. To test this we compared the estimated overall number of growing cells in the absence and presence of IP-1 for wild-type and six strains reported to fail endocytic internalization of Ste2p in MATa cells (see Fig. 2). The figure shows the effect on cell growth expressed as the area under the growth curve recorded at 600 nm. Our results show that there is no difference in the cell growth in the presence of IP-1 for the wild-type and the mutants; consequently we conclude that Ste2p-mediated endocytosis does not play a role in the killing of MATa cells. Note that even though Ste2p-mediated endocytosis is discarded, Ste2p signaling is required for the killing of MATa by IP-1 (15); null mutants for STE2 or other critical genes involved in the pheromone signaling cascade in a MATa genotype are resistant to killing by IP-1, and thus binding to Ste2p may play a role in the killing mechanism of action of IP-1.
FIGURE 2.
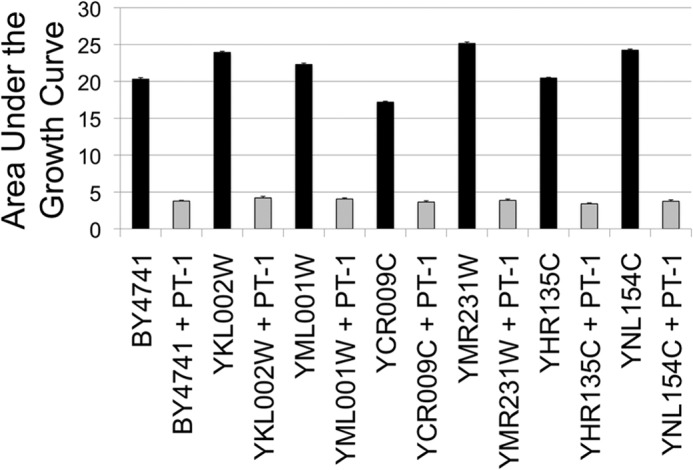
Cell death effect of six null mutants preventing internalization of Ste2p. Six strains carrying gene null mutants (YKL002W (www.ncbi.nlm.nih.gov), YML001W, YCR009C, YMR231W, YHR135C, and YNL154C) reported to reduce the internalization of Ste2p were compared in their sensitivity to be killed by IP-1 with the wild-type strain (BY4741). To estimate the number of cells remaining in the presence of IP-1, the figure presents the area under the growth curves (summation of A600 values recorded every hour is presented as the percentage) of these six mutants plus the wild-type strain in the presence (ORF name+IP-1; gray bars) and in the absence (ORF name; black bars) of IP-1. The figures present the average data obtained from four independent experiments; the lines on top of each bar represent the S.D. calculated for each data set. The optical density was followed in the Synergy MX plate-reader (BioTek) incubated at 30 °C.
To test whether endocytosis might explain the observed kinetics of cell death induced by IPs, we built a kinetic model for internalization of the IPs using the reported rates related to the internalization of the pheromone (32) (see Fig. 3A). In this model the number of IPs inside cells depends on the affinity of the pheromone to its receptor (Ste2p or any other with similar affinity), the degradation rate of the pheromone, and the endocytosis rate of the pheromone; all those rates have been reported and were used for modeling (see “Experimental Procedures”). On the other hand, the rate of escape from endosomes and the number of IP molecules required to kill cells are not known; these two quantities were estimated by fitting the model to the observed number of dead cells (quantified from the number of colony forming units; see “Experimental Procedures”). The adjustment between the observed and calculated values for the number of dead cells is presented in Fig. 3C. We observed that the model assuming receptor-mediated endocytosis could not reproduce the observed time course of dead cells, as it predicts a plateau phase due to depletion of free receptors at the membrane by endocytosis; such a plateau was not observed in the experimental data (p > 0.14, see Fig. 3C). In contrast, when we did not consider endocytosis of the peptide but still binding of IP-1 to Ste2p (see Fig. 3B), the model fit reproduces the number of dead cells over time (p < 4.2e−3; see Fig. 3C). These results are in agreement with our observation about a mechanism of internalization different from Ste2p-mediated endocytosis yet requiring Ste2p binding for killing activity. Note that the use of all-D enantiomers to discard receptor-mediated endocytosis does not apply to the study of IP-1 considering the requirement of Ste2p binding for killing MATa cells.
To further analyze the involvement of endocytosis in the killing by IP-1 (beyond receptor-mediated endocytosis), we carried out a genetic screening on 291 out of 441 null mutants annotated on SGD (Saccharomyces Genome Database) to affect endocytosis in yeast (see supplemental Table S1). Four out of these 291 ORFs (YHR161C, YKL081W, YKL119C, YOR141C) fully protected MATa cells from the killing by IP-1 (see supplemental Table S1). To evaluate if the effect of these four ORFs could implicate the functioning of the core endocytosis machinery for the killing activity of IP-1, we conducted two complementary analyses. First, we identified those mutants with the largest annotated effect on endocytosis (YDR017C, YDR173C, YJL204C, YOR089C, YKL126W; see supplemental Table S1) and observed that none protected against the killing by IP-1; these results suggest that the core machinery of endocytosis is not participating in the killing by IP-1. Second, we were able to group 30 out of the 291 ORFs by the endocytosis GO term and noticed that only one of these fully protected against IP-1 killing activity (YHR161C, involved in clathrin cage assembly; see supplemental Table S1), further supporting that the core machinery of endocytosis does not play a role on the killing of MATa by IP-1. Thus, even though four genes related to endocytosis protect against the killing of IP-1, overall endocytosis does not seem to play a role on the killing.
Iztli Peptides Are Cell-penetrating Peptides
To determine the presence of IP-1 inside cells, the peptide was modified with two different fluorophores: TAMRA and Hilytefluor 488. The Hilytefluor derivative was based on previous results on the α-pheromone (33); note that the use of these two fluorophores was to discard that any possible observed internalization was dependent on the nature of the fluorophores. Both peptides maintained antifungal activity (see Fig. 4A) and were internalized by MATa cells (see Fig. 4B). Although IP-1 fully inhibited the growth of MATa cells at 10 μm (15), the TAMRA derivate of IP-1 (TAMRA-IP-1) had almost half the activity (minimal inhibitory concentration = 40 μm) of the Hilytefluor 488 derivative (minimal inhibitory concentration = 27 μm). We used the TAMRA derivative for the rest of our studies. MATa cells exposed to TAMRA-IP-1 for 15 min showed strong staining of the cell membrane, and after 30 min the peptide was also localized inside the cells (see Fig. 5, A–D). To evaluate whether the internalization was dependent on the Ste2p receptor, the MATα cells were also exposed to this labeled peptide (see Fig. 6A). Finally, other fungi (see Fig. 6B) and human cells (see Fig. 6C) were exposed to the TAMRA-IP-1, and in all cases the peptide was internalized without any observable toxicity (data not shown). These results further support the notion that IP-1 internalization is independent of Ste2p-mediated endocytosis.
FIGURE 4.

Analysis of two fluorogenic derivates of Iztli peptide 1. The antifungal activity (A) and the ability to get internalized by S. cerevisiae MATa cells (B) are shown for the TAMRA-IP-1 (left side, red) and Hilytefluor 488-IP-1 (right side, green). The change in cell density over time is presented in the presence (black symbols) or in the absence of these peptides (white symbols): squares, TAMRA-IP-1; triangles, Hilytefluor 488-IP-1. Scale bars in B are 5 μm.
FIGURE 5.
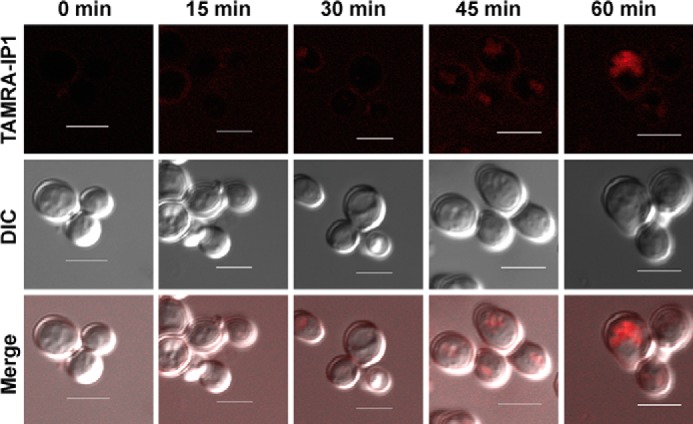
Time-dependent internalization of Iztli peptide 1 in yeast cells. TAMRA-IP1 was used to follow the internalization of the peptide in MATa cells. The sites where the peptide is found near or within cells appear as red dots. Scale bars are 5 μm. DIC, differential interference contrast.
FIGURE 6.
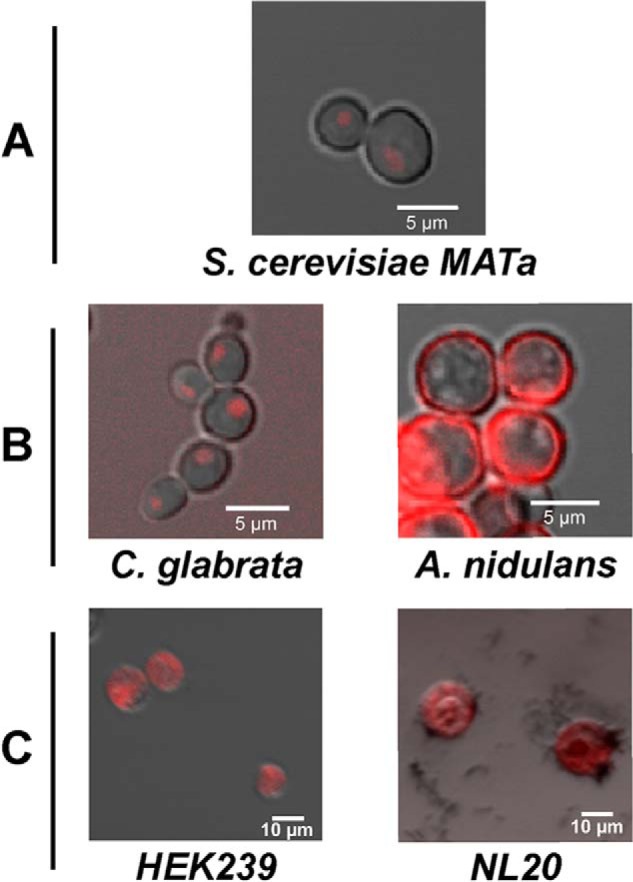
Iztli peptide 1 gets internalized by different cell types. S. cerevisiae MATα cells (A), C. glabrata (left in panel B), A. nidulans (right in panel B), HEK239 (left in panel C), and NL20 (right in panel C) human cells were exposed to TAMRA-IP-1 (red) to test for its internalization.
Iztli Peptides Make Channels at High Membrane Potentials
We determined the ability of IP-1 to induce ion transport across lipidic bilayers. For this we considered its action on POPC plus 30% cholesterol artificial membranes. We did not observe any electrical conductance induced by the peptide at 50 or 100 mV (data not shown). Yet at 150 and 200 mV there was observable activity. The dependence of the ionophoretic activity on peptide concentration is presented in Fig. 7, A and B. At low concentration (5 μm) there are mainly small channels that can barely be distinguished from the noise of the base line (see Fig. 7B). At medium concentration (10 μm) there is clear activity (see Fig. 7A) mainly of a channel of 4.5 picosiemens even if there is a multitude of larger channels with small occurrence (see Fig. 7B). At large concentrations (15 μm) the activity is increased, and a multitude of channels in the range from 3 to 20 picosiemens was observed; large channels were also observed but less frequently than at 10 μm. At 20 μm frequent seal ruptures and closings appeared, and at 25 μm there was an immediate rupture of the membrane patch (data not shown). This latter behavior could be explained by the peptide entering the seal region to disturb it, or else it affected the membrane integrity. We can quantify the activity at each concentration with the following expression,
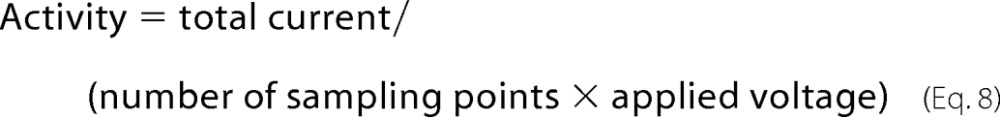 |
where total current is the sum of the current value at each sampling point. This yields an average conductance, i.e. a virtual channel that would be open all the time. The corresponding activities are then 0.64, 1.63, and 3.31 picosiemens for 5, 10, and 15 μm, respectively.
FIGURE 7.
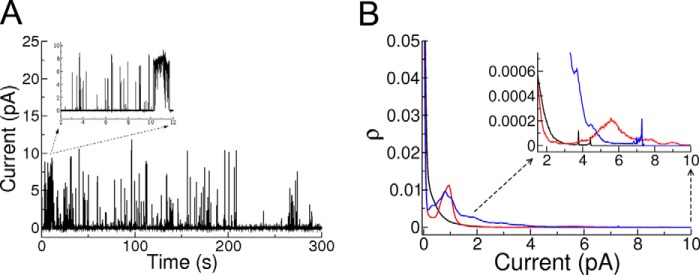
Iztli peptide 1 makes pores. A, example of a 5-min record of electrical activity for IP-1 (10 μm) in POPC membrane with 30% mol cholesterol in a transmembrane electric potential of 200 mV. The inset corresponds to a 10-s section showing an example of the variability of the opening times of the pores formed. B, average all point histograms for five experiments at the concentrations of 5 μm (black), 10 μm (red), and 15 μm (blue) on the same bilayer and at 200 mV of applied potential; the duration of each experiment was 5 min. The all points histogram is constructed by filling the histogram buckets with the number of points, i.e. the number of times that a particular current was registered during the experiment with a 100-μs sampling. This histogram is normalized by the time duration of the experiment, and therefore, the ρ value corresponds to the number of counts in each bucket divided by the total number of counts. The records were base-line-corrected and filtered for peaks with duration of <30 μs (to suppress electric noise of the bath) with a homemade suite of programs. The inset corresponds to large conductance channels that present small occurrence, which depends on peptide concentration.
These results raised the question that even for eukaryotic-like membranes, IP-1 could make pores provided a large electric potential. Plants, like bacteria and mitochondria, maintain a large transmembrane potential due to the action of plasma membrane H+ pumps fuelled by ATP (34). IP-1 did not alter the resting membrane potential of soybean roots with low potentials (<100 mV); however, we could not find cells with high potentials (>100 mV) (data not shown).
DISCUSSION
HKPs have been shown to have the potential to treat cancer and obesity among others (1). The basis for their use as therapeutics is their selectivity; only the cells expressing the receptor for the hunter peptide should internalize the killer peptide, and consequently only those cells should be eliminated. Even when the results in animal models have shown the effectiveness of these peptides, some toxicity has been reported for other tissues besides those targeted in animals (3, 4). This toxicity could be explained considering the presence of the receptor for the hunter peptide in other tissues than the target one. Our results suggest an alternative explanation for the observed toxicity of HKPs. That is, we showed that when a killer peptide presents CPP activity (e.g. IP-1, see Fig. 1) it may loose the specificity provided by the hunter peptide (see Figs. 4, A–C, 5, A–D, and 6, A–C). Accordingly, we showed that IP-1 is not internalized via receptor-mediated endocytosis (see Figs. 2 and 3) or endocytosis at all (see supplemental Table S1). However, IP-1 is still able to kill MATa cells as long as the pheromone receptor is activated by the peptide (15). Thus, the observed specificity displayed by IP-1 (or other HKPs) may be explained by the requirement to activate the hunter receptor (e.g. Ste2p) and/or by increasing the avidity for IP-1 in MATa cells. In this last case our simulation results show that the attraction of IP-1 by Ste2p accounts for the kinetics of cell death induced by IP-1 in MATa cells. Additional experiments are required to validate these observations, which may provide the basis to explain the observed killing specificity of HKPs.
The design of HKPs combines CAP and CPP in separate domains, yet such design may sometimes be redundant considering that some CPPs (35) by themselves may have CAP activity. In such a case, these CPPs when internalized carry along a killer peptide (CAPs) into cells. If so, those CPPs may affect mitochondria function. In agreement with this idea, a recent metabolomic analysis comparing the toxic potential of four CPPs (transportan, penetratin, HIV Tat derived peptide, and nona-arginine) showed the toxicity of transportan (36), suggesting the potential CAP activity of transportan. The toxicity was estimated by the effect on cellular redox potential and energy depletion, both related to mitochondria function. In a similar fashion, some CAPs may as well display CPP activity. Indeed, magainin 2 has been shown to translocate across phospholipid bilayers (37). Thus, IP design may avoid redundancy of activities, rendering shorter HKPs.
Beyond the design of HKPs, it is interesting to study the basis of this redundant function of CPPs and CAPs. We hypothesized that the membrane potential may play a role on switching between these activities; particularly, it is noticeable that mitochondria and bacteria maintain larger membrane potentials than mammalian cells, and such a difference may explain the toxicity of CAPs to bacteria and not to mammalian cells. In the case of IPs, we have previously observed that these alter the membrane stability of mitochondria and kill bacteria but do not affect the viability of mammalian cells (15). Here we showed that IP-1 is capable of making pores in artificial membranes only when a large electric potential (>100 mV) is present (see Fig. 7, A and B). These results are in agreement with recent reports showing that some CPPs may switch from penetrating mammalian cells with no apparent toxicity to peptides capable of killing/swelling bacterial cells (12, 13). Understanding the mechanism involved in this functional switch may assist in designing CPPs.
In that sense it has been observed that natural antibiotics, peptides, and polyenes can form different membrane structures that allow for transmembrane transport, detergent-like carpet disruption, pores, and toroidal pores independently of other intracellular actions (see for example Refs. 38 and 39). It is also clear that their activities are strongly dependent on the environmental conditions (40). For example, the dependence on the transmembrane electric potential to form pores has been observed for polyenes (41) and for Alamethicin (42). Hence, it is not surprising that IP-1 presents this behavior. IP-1 forms transmembrane channels of different conductivity, similar to Amphotericin B (43) and Alamethicin (44), and also detergent-like activity, similar to Nystatin (41) and Alamethicin (42). In either case, the environmental conditions can affect helix formation for peptides (39) and/or the aggregation of monomers for polyenes (45). Such a situation may prevail for IP-1 and may explain the dual role observed in this study.
Considering the complexity of biological systems (46), our observations about the overlapping roles of CPPs and CAPs represent a special case of functional redundancy. Functional redundancy in genes occurs when one gene has the ability to perform the function of another gene. A common example of such genetic redundancy is found in gene duplications (47). A consequence for a system with these redundant capabilities is the emergence of robustness (48). In the case of antibacterial peptides, the robustness this redundancy confers to CAPs can be appreciated; by acquiring the ability to get internalized by eukaryotic cells, CAPs can become more effective antibiotics by killing bacteria cells residing both at the extracellular and intracellular spaces of eukaryotic cells. Additionally, under certain circumstances some bacteria may reduce their membrane potential (e.g. sporulation), thus preventing CAPs to disrupt their membrane (49). In such cases, the possibility that CAPs may permeate cellular membranes with low electric potential and act upon an intracellular target may provide an alternative mechanism to maintain their antibiotic activity.
The multifunctionality of CAPs has been recognized as well in the context of neurodegenerative diseases; for instance, the β-amyloid peptide that is commonly associated with Alzheimer disease exerts antimicrobial activity against common infectious bacteria (50); another neuropeptide, substance P, displays features of CAP and has antibacterial activity (11). Yet neither every peptide involved in neurodegeneration can be expected to have an antibacterial activity (51) nor every CPP may have a CAP activity. Identifying the different functionalities of CAPs may have an impact on hunter-killer peptide design as discussed before or in the etiology of different diseases where peptides (e.g. amyloid, penetrating) may be present, such as in Alzheimer disease.
Finally, the present work provides an experimental system (IP targeted against S. cerevisiae) and mathematical model (see Fig. 3 and “Experimental Procedures”) that may be useful to further characterize the mechanism of internalization of CPPs and to explore alternatives to control the selectivity of CPPs and HKPs.
In summary, our results show that IP-1, a HKP targeted against S. cerevisiae, penetrates cells in an endocytosis-independent fashion. Additionally, our results show that CAP activity may be tailored to act as CPP by large membrane potentials. These results provide the basis to explain the observed toxicity of HKPs and the biological relevance of the observed functional redundancy of some cationic antibacterial peptides and cell-penetrating peptides as well as to emphasize the relevance to improve on the classification of antimicrobial peptides.
Supplementary Material
Acknowledgments
We thank the technical support from the IT core facility and the microscopy core facility at the Institute of Cellular Physiology/Universidad Nacional Autónoma de México. We also thank Biol. Olivia Sánchez and Dr. Jesus Aguirre at the Institute of Cellular Physiology/Universidad Nacional Autónoma de México for providing the materials and expertise to perform the experiments with A. nidulans.
This work was supported in part by the Alexander von Humboldt Foundation, Consejo Nacional de Ciencia y Tecnologia (82308 and 128575), Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica Grants IN205911 and IG100513 (to G. D. R. and I. O. B.), and German Research Council Grant CRC740 (to E. K. and A. H.).

This article contains supplemental Table S1.
- HKP
- Hunter-killer peptide
- IP-1
- Iztli peptide 1
- CPP
- cell penetrating peptide
- CAP
- cationic antibacterial peptide
- MATa
- mating type A
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- TAMRA
- 6-carboxytetramethylrhodamine.
REFERENCES
- 1. Ellerby H. M., Bredesen D. E., Fujimura S., John V. (2008) Hunter-killer peptide (HKP) for targeted therapy. J. Med. Chem. 51, 5887–5892 [DOI] [PubMed] [Google Scholar]
- 2. Arap W., Haedicke W., Bernasconi M., Kain R., Rajotte D., Krajewski S., Ellerby H. M., Bredesen D. E., Pasqualini R., Ruoslahti E. (2002) Targeting the prostate for destruction through a vascular address. Proc. Natl. Acad. Sci. U.S.A. 99, 1527–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kolonin M. G., Saha P. K., Chan L., Pasqualini R., Arap W. (2004) Reversal of obesity by targeted ablation of adipose tissue. Nat. Med. 10, 625–632 [DOI] [PubMed] [Google Scholar]
- 4. Ellerby H. M., Arap W., Ellerby L. M., Kain R., Andrusiak R., Rio G. D., Krajewski S., Lombardo C. R., Rao R., Ruoslahti E., Bredesen D. E., Pasqualini R. (1999) Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 5, 1032–1038 [DOI] [PubMed] [Google Scholar]
- 5. Vives E. (2005) Present and future of cell-penetrating peptide mediated delivery systems: “is the Trojan horse too wild to go only to Troy?” J. Control Release 109, 77–85 [DOI] [PubMed] [Google Scholar]
- 6. Deshayes S., Morris M. C., Divita G., Heitz F. (2005) Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell. Mol. Life Sci. 62, 1839–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones A. T., Sayers E. J. (2012) Cell entry of cell penetrating peptides: tales of tails wagging dogs. J. Control Release 161, 582–591 [DOI] [PubMed] [Google Scholar]
- 8. Pooga M., Langel U. (2005) Synthesis of cell-penetrating peptides for cargo delivery. Methods Mol. Biol. 298, 77–89 [DOI] [PubMed] [Google Scholar]
- 9. Duchardt F., Fotin-Mleczek M., Schwarz H., Fischer R., Brock R. (2007) A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 8, 848–866 [DOI] [PubMed] [Google Scholar]
- 10. Zorko M., Langel U. (2005) Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv. Drug Deliv. Rev. 57, 529–545 [DOI] [PubMed] [Google Scholar]
- 11. del Rio G., Castro-Obregon S., Rao R., Ellerby H. M., Bredesen D. E. (2001) APAP, a sequence-pattern recognition approach identifies substance P as a potential apoptotic peptide. FEBS Lett. 494, 213–219 [DOI] [PubMed] [Google Scholar]
- 12. Li L., Shi Y., Su G., Le G. (2012) Selectivity for and destruction of Salmonella typhimurium via a membrane damage mechanism of a cell-penetrating peptide ppTG20 analogue. Int. J. Antimicrob. Agents 40, 337–343 [DOI] [PubMed] [Google Scholar]
- 13. Nan Y. H., Park I. S., Hahm K. S., Shin S. Y. (2011) Antimicrobial activity, bactericidal mechanism, and LPS-neutralizing activity of the cell-penetrating peptide pVEC and its analogs. J. Pept. Sci. 17, 812–817 [DOI] [PubMed] [Google Scholar]
- 14. Brogden K. A. (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3, 238–250 [DOI] [PubMed] [Google Scholar]
- 15. Rodríguez Plaza J. G., Villalon Rojas A., Herrera S., Garza-Ramos G., Torres Larios A., Amero C., Zarraga Grandos G., Gutierrez Aguilar M., Lara Ortiz M. T., Polanco Gonzalez C., Uribe Carvajal S., Coria R., Peña Díaz A., Bredesen D. E., Castro-Obregon S., del Rio G. (2012) Moonlighting peptides with emerging function. PloS One 7, e40125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raths S. K., Naider F., Becker J. M. (1988) Peptide analogues compete with the binding of α-factor to its receptor in Saccharomyces cerevisiae. J. Biol. Chem. 263, 17333–17341 [PubMed] [Google Scholar]
- 17. Jenness D. D., Burkholder A. C., Hartwell L. H. (1983) Binding of α-factor pheromone to yeast a cells: chemical and genetic evidence for an α-factor receptor. Cell 35, 521–529 [DOI] [PubMed] [Google Scholar]
- 18. Bajaj A., Celić A., Ding F. X., Naider F., Becker J. M., Dumont M. E. (2004) A fluorescent α-factor analogue exhibits multiple steps on binding to its G protein-coupled receptor in yeast. Biochemistry 43, 13564–13578 [DOI] [PubMed] [Google Scholar]
- 19. Blumer K. J., Reneke J. E., Thorner J. (1988) The STE2 gene product is the ligand-binding component of the α-factor receptor of Saccharomyces cerevisiae. J. Biol. Chem. 263, 10836–10842 [PubMed] [Google Scholar]
- 20. Jenness D. D., Spatrick P. (1986) Down regulation of the α-factor pheromone receptor in S. cerevisiae. Cell 46, 345–353 [DOI] [PubMed] [Google Scholar]
- 21. Dube P., Konopka J. B. (1998) Identification of a polar region in transmembrane domain 6 that regulates the function of the G protein-coupled α-factor receptor. Mol. Cell. Biol. 18, 7205–7215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. David N. E., Gee M., Andersen B., Naider F., Thorner J., Stevens R. C. (1997) Expression and purification of the Saccharomyces cerevisiae α-factor receptor (Ste2p), a 7-transmembrane-segment G protein-coupled receptor. J. Biol. Chem. 272, 15553–15561 [DOI] [PubMed] [Google Scholar]
- 23. Dosil M., Schandel K. A., Gupta E., Jenness D. D., Konopka J. B. (2000) The C terminus of the Saccharomyces cerevisiae α-factor receptor contributes to the formation of preactivation complexes with its cognate G protein. Mol. Cell. Biol. 20, 5321–5329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Q., Konopka J. B. (1996) Regulation of the G-protein-coupled α-factor pheromone receptor by phosphorylation. Mol. Cell. Biol. 16, 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee B. K., Khare S., Naider F., Becker J. M. (2001) Identification of residues of the Saccharomyces cerevisiae G protein-coupled receptor contributing to α-factor pheromone binding. J. Biol. Chem. 276, 37950–37961 [DOI] [PubMed] [Google Scholar]
- 26. Weiner J. L., Guttierez-Steil C., Blumer K. J. (1993) Disruption of receptor-G protein coupling in yeast promotes the function of an SST2-dependent adaptation pathway. J. Biol. Chem. 268, 8070–8077 [PubMed] [Google Scholar]
- 27. Hicke L., Zanolari B., Riezman H. (1998) Cytoplasmic tail phosphorylation of the α-factor receptor is required for its ubiquitination and internalization. J. Cell Biol. 141, 349–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soid-Raggi G., Sánchez O., Aguirre J. (2006) TmpA, a member of a novel family of putative membrane flavoproteins, regulates asexual development in Aspergillus nidulans. Mol. Microbiol. 59, 854–869 [DOI] [PubMed] [Google Scholar]
- 29. Paternostre M., Ollivon M., Bolard J. (1996) in Manual on Membrane Lipids (Prasat R., ed) pp. 202–247, Springer, Berlin [Google Scholar]
- 30. Coronado R., Latorre R. (1983) Phospholipid bilayers made from monolayers on patch-clamp pipettes. Biophys. J. 43, 231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suarez-Isla B. A., Wan K., Lindstrom J., Montal M. (1983) Single-channel recordings from purified acetylcholine receptors reconstituted in bilayers formed at the tip of patch pipets. Biochemistry 22, 2319–2323 [DOI] [PubMed] [Google Scholar]
- 32. Thomson K. B., Faeder J., Sytchev I., Endy D. (2012) Yeast Pheromone Model, Stanford University, Palo Alto, CA [Google Scholar]
- 33. Toshima J. Y., Toshima J., Kaksonen M., Martin A. C., King D. S., Drubin D. G. (2006) Spatial dynamics of receptor-mediated endocytic trafficking in budding yeast revealed by using fluorescent α-factor derivatives. Proc. Natl. Acad. Sci. U.S.A. 103, 5793–5798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Palmgren M. G. (2001) Plant Plasma Membrane H+-ATPases: powerhouses for nutrient uptake. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 817–845 [DOI] [PubMed] [Google Scholar]
- 35. Henriques S. T., Melo M. N., Castanho M. A. (2006) Cell-penetrating peptides and antimicrobial peptides: how different are they? Biochem. J. 399, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kilk K., Mahlapuu R., Soomets U., Langel U. (2009) Analysis of in vitro toxicity of five cell-penetrating peptides by metabolic profiling. Toxicology 265, 87–95 [DOI] [PubMed] [Google Scholar]
- 37. Matsuzaki K., Murase O., Fujii N., Miyajima K. (1995) Translocation of a channel-forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry 34, 6521–6526 [DOI] [PubMed] [Google Scholar]
- 38. Cohen B. E. (2010) Amphotericin B membrane action: role for two types of ion channels in eliciting cell survival and lethal effects. J. Membr. Biol. 238, 1–20 [DOI] [PubMed] [Google Scholar]
- 39. Morgera F., Vaccari L., Antcheva N., Scaini D., Pacor S., Tossi A. (2009) Primate cathelicidin orthologues display different structures and membrane interactions. Biochem. J. 417, 727–735 [DOI] [PubMed] [Google Scholar]
- 40. González-Damián J., Ortega-Blake I. (2010) Effect of membrane structure on the action of polyenes II: nystatin activity along the phase diagram of ergosterol- and cholesterol-containing POPC membranes. J. Membr. Biol. 237, 41–49 [DOI] [PubMed] [Google Scholar]
- 41. Récamier K. S., Hernández-Gómez A., González-Damián J., Ortega-Blake I. (2010) Effect of membrane structure on the action of polyenes: I. nystatin action in cholesterol- and ergosterol-containing membranes. J. Membr. Biol. 237, 31–40 [DOI] [PubMed] [Google Scholar]
- 42. Mak D. O., Webb W. W. (1995) Two classes of alamethicin transmembrane channels: molecular models from single-channel properties. Biophys. J. 69, 2323–2336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Venegas B., González-Damián J., Celis H., Ortega-Blake I. (2003) Amphotericin B channels in the bacterial membrane: role of sterol and temperature. Biophys. J. 85, 2323–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harriss L. M., Cronin B., Thompson J. R., Wallace M. I. (2011) Imaging multiple conductance states in an alamethicin pore. J. Am. Chem. Soc. 133, 14507–14509 [DOI] [PubMed] [Google Scholar]
- 45. Huang W., Zhang Z., Han X., Tang J., Wang J., Dong S., Wang E. (2002) Ion channel behavior of amphotericin B in sterol-free and cholesterol- or ergosterol-containing supported phosphatidylcholine bilayer model membranes investigated by electrochemistry and spectroscopy. Biophys. J. 83, 3245–3255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oltvai Z. N., Barabási A. L. (2002) Systems biology. Life's complexity pyramid. Science 298, 763–764 [DOI] [PubMed] [Google Scholar]
- 47. Birchler J. A., Veitia R. A. (2012) Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc. Natl. Acad. Sci. U.S.A. 109, 14746–14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kafri R., Springer M., Pilpel Y. (2009) Genetic redundancy: new tricks for old genes. Cell 136, 389–392 [DOI] [PubMed] [Google Scholar]
- 49. McBride S. M., Sonenshein A. L. (2011) The dlt operon confers resistance to cationic antimicrobial peptides in Clostridium difficile. Microbiology 157, 1457–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Soscia S. J., Kirby J. E., Washicosky K. J., Tucker S. M., Ingelsson M., Hyman B., Burton M. A., Goldstein L. E., Duong S., Tanzi R. E., Moir R. D. (2010) The Alzheimer's disease-associated amyloid β-protein is an antimicrobial peptide. PloS ONE 5, e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kagan B. L., Jang H., Capone R., Teran Arce F., Ramachandran S., Lal R., Nussinov R. (2012) Antimicrobial properties of amyloid peptides. Mol. Pharm. 9, 708–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Domergue R., Castaño I., De Las Peñas A., Zupancic M., Lockatell V., Hebel J. R., Johnson D., Cormack B. P. (2005) Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Science 308, 866–870 [DOI] [PubMed] [Google Scholar]
- 53. Kawasaki L, (2002) SakA MAP kinase is involved in stress signal transduction, sexual development, and spore viability in Aspergillus nidulans. Mol Microbiol. 45, 1153–1163 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.