

Background: Antimicrobial properties of the anti-inflammatory neuropeptide VIP are limited by its unstable nature.
Results: The VIP derivatives protected against polymicrobial sepsis and cutaneous leishmaniasis by selectively killing pathogens through membrane-disrupting mechanisms.
Conclusion: Modification of critical residues in the native VIP sequence generates stable peptides with potent antimicrobial activities in vitro and in vivo.
Significance: This work indicates a molecular rationale for designing new agents against drug-resistant infectious diseases.
Keywords: Antimicrobial Peptides, Bacteria, Lipopolysaccharide (LPS), Neuropeptide, Sepsis, Vasoactive Intestinal Peptide, Cutaneous Leishmaniasis
Abstract
Vasoactive intestinal peptide (VIP) is an anti-inflammatory neuropeptide recently identified as a potential antimicrobial peptide. To overcome the metabolic limitations of VIP, we modified the native peptide sequence and generated two stable synthetic analogues (VIP51 and VIP51(6–30)) with better antimicrobial profiles. Herein we investigate the effects of both VIP analogues on cell viability, membrane integrity, and ultrastructure of various bacterial strains and Leishmania species. We found that the two VIP derivatives kill various non-pathogenic and pathogenic Gram-positive and Gram-negative bacteria as well as the parasite Leishmania major through a mechanism that depends on the interaction with certain components of the microbial surface, the formation of pores, and the disruption of the surface membrane. The cytotoxicity of the VIP derivatives is specific for pathogens, because they do not affect the viability of mammalian cells. Docking simulations indicate that the chemical changes made in the analogues are critical to increase their antimicrobial activities. Consequently, we found that the native VIP is less potent as an antibacterial and fails as a leishmanicidal. Noteworthy from a therapeutic point of view is that treatment with both derivatives increases the survival and reduces bacterial load and inflammation in mice with polymicrobial sepsis. Moreover, treatment with VIP51(6–30) is very effective at reducing lesion size and parasite burden in a model of cutaneous leishmaniasis. These results indicate that the VIP analogues emerge as attractive alternatives for treating drug-resistant infectious diseases and provide key insights into a rational design of novel agents against these pathogens.
Introduction
Resurgence of infections is a current worldwide problem. People from both the poorest areas and the richest countries are continuously faced with pathogens that cause different infectious diseases. For example, neglected tropical parasitic diseases affect more than 1 billion people (1). On the other hand, uncontrolled response to bacterial dissemination in sepsis is the third leading cause of death in developed societies, affecting 18 million people (2). Current conventional drugs suffer from loss of function and severe adverse effects. This highlights the urgent need for new strategies to combat microbial infections. The search for novel anti-infective therapies has lately focused on endogenous cationic peptides that regulate immunological and physiological homeostatic parameters in the context of the host response to infectious diseases. These molecules, termed host defense peptides, have emerged as potential therapeutic candidates. Recently, some neuropeptides, which are pleiotropic and integral components of the neuroendocrine system, have been recognized as host defense peptides (3).
Vasoactive intestinal peptide (VIP)2 is a neuropeptide belonging to the glucagon/secretin superfamily (4) that is widely distributed in many tissues and organs of the body, including brain, heart, lung, digestive and genitourinary tracts, immune cells, and thyroid gland (5). Through its binding to specific receptors, VIP elicits a broad spectrum of biological functions in organisms, including neuroprotection, relaxation of smooth muscle, regulation of exocrine and endocrine secretions, and modulation of the immune response (6). As a consequence, many studies have proven the therapeutic potential of VIP in several diseases, especially in inflammatory, respiratory, and neurodegenerative disorders (7–13). Moreover, it was recently reported that VIP exerts a direct antimicrobial activity against a variety of pathogens, including bacteria (14) and African trypanosomatids (15), although the involved mechanisms remain largely unclear. Similar to other neuropeptides (3), VIP shares some properties with antimicrobial peptides, such as small size, cationic charge, and amphipathic design (Table 1). In addition, VIP is abundantly present in physical barriers of the body (skin and mucosa), physiological fluids (maternal milk, tears, and saliva), and immunoprivileged sites (eyes, testis, and ovaries) (5). Furthermore, VIP is released under microbial-induced inflammation (15, 16), and it has immunomodulatory functions. These findings support a new role for VIP as a potential host defense peptide (17). However, the poor stability of VIP after systemic administration and its poor penetration to the desired site of action have limited its clinical application. To overcome these shortcomings, the development of efficacious VIP analogues and several drug delivery systems has been attempted. Thus, amino acid modifications/substitutions in the VIP sequence, its combination with endopeptidase inhibitors, design of VIP formulations based on protected nanoparticles or micelles, and even VIP-secreting viral vectors have improved the half-life of the neuropeptide and its delivery in the targeted tissue (18). Specifically, following the strategy of amino acid substitutions, Onoue et al. (19–21) recently generated various VIP derivatives in inhalable formulations that showed improved metabolic stability and higher therapeutic efficacy than the native VIP in models of inflammatory pulmonary diseases. The analysis of the sequence and structure of one of these VIP derivatives named IK312551 and referred to herein as VIP51 (Table 1 and supplemental Fig. S1) revealed that the chemical modifications made in the VIP analogue increased its cationic charge, hydrophobicity index, and percentage of α-helix (Table 1), improving its predictable profile as antimicrobial peptide in comparison with native VIP.
TABLE 1.
Sequence and structural properties of VIP and the VIP derivatives
Numbers correspond to the positions of the amino acids in the sequence of each peptide. Cationic residues are shaded, anionic residues are in italic type, and hydrophobic residues are underlined.

a Grand average of hydropathicity. Negative and positive GRAVY values correspond to hydrophilic and hydrophobic peptides, respectively. Net charges and gravy indexes were calculated by using the Bioinformatics Resource Portal and Antimicrobial Peptide Predictor database.
b Percentage of α-helix in the structure.
The aim of this study was to develop a rational design of more efficient therapeutic strategies against microbial diseases. Therefore, we investigated in vitro and in vivo the antimicrobial and immunomodulatory activities of VIP51 against different pathogens. Moreover, to discriminate in vivo between its direct effect against pathogens and its potential indirect effect in VIP receptor-expressing mammalian cells, we evaluated the effect in this system of a new chemically designed truncated version of VIP51 (named VIP51(6–30)), which lacks the first five N-terminal residues that are essential to activate the VIP receptor (22).
EXPERIMENTAL PROCEDURES
Peptides
VIP was purchased from American Peptide, and the VIP derivatives (VIP51 and VIP51(6–30)) and their FITC-labeled forms were chemically synthesized by GeneScript. All peptides were initially reconstituted in sterile water (Bio-Rad) and further diluted in phosphate-buffered saline (PBS; 1.2 mm KH2PO4, 8.1 mm Na2HPO4, 130 mm NaCl, 2.6 mm KCl, pH 7.0) or the corresponding culture medium at the indicated concentrations before their use.
Microorganisms and Growth Conditions
Streptococcus mutans (CECT 479) and Enterococcus faecalis (CECT 184) were grown under shaking in brain heart infusion broth (Difco) at 37 °C, Micrococcus luteus (CECT 243) and Enterobacter aerogenes (CECT 684) in Nutrient I at 30 °C, Escherichia coli (CECT 45) in Nutrient I at 37 °C, and Pseudomonas aeruginosa (CECT 108) and Pseudomonas pseudoalcaligenes (CECT 318) in Nutrient II at 37 and 26 °C, respectively. These strains were obtained from the Spanish Center for Type Culture Collection and prepared and grown following supplier instructions for biological group risk 1 (non-pathogenic) and 2 (pathogenic). E. coli Top 10 (Invitrogen) and E. coli K-12 mutants deficient in LPS synthesis (E. coli Genetic Stock Center, Yale University, New Haven, CT) were cultured under shaking in Luria broth medium at 37 °C. Gram-negative bacteria isolated from the peritoneum of septic mice were grown under shaking in Nutrient I at 37 °C.
For L. major, we used two different wild type (WT) and mutant strains: L. major Friedlin (MHOM/IL/80/Friedlin) clone V1 and the lipophosphoglycan (LPG)-inducible mutant glf−/+KiddGLF (23). In this inducible system, the synthesis of a key galactofuranose residue within the LPG core is controlled using a conditional “destabilization domain” fused to UDP-galactopyranosylmutase in the WT L. major Friedlin V1 background. In the presence of the inducer FK506 (or other rapamycin-like analogues) UDP-galactopyranosylmutase is stabilized, and parasites make high levels of LPG, whereas in its absence, LPG is reduced to background levels. The phosphoglycan (PG)-deficient lpg2−/− mutant (24) lacking a Golgi GDP-mannose transporter required for PG synthesis was generated in a WT L. major LV39 clone 5 (Rho/SU/59/P) background. These cell lines and L. donovani (MHOM/ET/67/HU3) promastigotes were cultured at 28 °C in M-199 medium (Invitrogen) supplemented with 40 mm HEPES, 100 μm adenosine, 0.77 μm hemin, 10 μm biopterin, and 10% heat-inactivated fetal bovine serum (FBS; Invitrogen). Promastigotes from Leishmania infantum (MHOM/ES/1993/BCN-99), L. tropica 656 cl (MHOM/IS/1998/LRC), and L. mexicana (MHOM/VE/1990/M9012) were grown in modified RPMI 1640 medium (Invitrogen) supplemented with 20% FBS at 28 °C (L. infantum and L. tropica) and at 23 °C (L. mexicana).
Antimicrobial Assays
The bactericidal activity of VIP, VIP51, and VIP51(6–30) was determined by incubation of a 5 μm concentration of every peptide (unless other concentrations are indicated) with 106 bacteria of each strain at mid-exponential growth phase in 100 μl of PBS for 3 h, under shaking at the indicated growth temperatures. We stopped the reaction by adding ice-cold PBS (dilution 1:100) for 5 min, and then we determined the number of cfu/ml after overnight incubation on agar dishes. Results were expressed as a percentage of bacterial survival, (cfu after peptide treatment/cfu in the absence of peptide) × 100. Alternatively, bacterial viability was assayed using the BacTiter-Glo microbial cell viability kit (Promega) as described below.
To determine the antimicrobial activity of the peptides on Leishmania promastigotes, we used the Alamar Blue reagent (Sigma-Aldrich). Briefly, parasites were harvested at late exponential phase and incubated in 96-well plates (100 μl/well, 2.5 × 105 cells/ml) with serial peptide dilutions at the corresponding growth temperatures. Peptides were also added 24 h after initiating the culture. After 72 h of incubation, Alamar Blue (0.11 mg/ml, 20 μl/well) was added to cultures for 4 h and then solubilized with 100 μl of 3% SDS. Cell viability was assayed using a fluorescent plate reader (550 nm excitation, 590 nm emission).
When indicated, we adjusted the pH of PBS (for bacteria) or the pH of HPMI buffer (20 mm HEPES, 132 mm NaCl, 3.5 mm KCl, 0.5 mm MgCl2, 5 mm glucose, 1 mm CaCl2, pH 7.4; for parasites) to acid (pH 5.0, pH 6.0) or basic (pH 8.0, pH 9.0) conditions. To elucidate the effects of NaCl and MgCl2, the bacteria-peptide and parasite-peptide reaction solution consisted of PBS or HPMI buffer, respectively, supplemented with 20 mm (physiological dose) or 50 mm NaCl or with 1 mm (physiological dose) or 50 mm MgCl2. In these assays, bacteria and parasites were incubated with the peptides for 3 and 4 h, respectively.
We calculated EC50, which represents the concentration of the peptide that reduces to 50% the viability of each bacteria and parasite strain, by using four-parameter logistic curve-fitting software (SigmaPlot).
Assays of Hemolysis and Cytotoxicity in Mammalian Cells
The cytotoxicity assay was performed using mouse macrophages and splenocytes. Peritoneal macrophages were allowed to adhere for 4 h in plastic in complete RPMI medium (RPMI 1640 containing 10% FBS, 2 mm l-glutamine, 100 units/ml penicillin, 100 mg/ml streptomycin, and 50 μm β-mercaptoethanol) at 37 °C and then washed to remove non-adherent cells. A cell suspension from spleen was obtained after mechanical homogenization of the tissue and depletion of erythrocytes by incubating the whole suspension with the erythrocyte lysis buffer (0.15 mm NH4Cl, 10 mm KHCO3, 0.1 mm EDTA) for 10 min at room temperature. In both cases, macrophages and splenocytes were seeded at 105 cells/well in 96-well microtiter plates (100 μl/well) and then incubated in the absence or presence of 12 μm VIP51 and VIP51(6–30) for 72 h at 37 °C and 5% CO2 atmosphere. Subsequently, Alamar Blue was added to each well, and cell viability was determined.
To determine the hemolytic activity of VIP51 and VIP51(6–30), we incubated mouse red blood cells (107 cells/well, 100 μl) obtained by cardiac puncture with a 12 μm concentration of the peptides at 37 °C under shaking. After 1 h of incubation, samples were centrifuged (1,000 × g, 10 min), and the concentration of hemoglobin in the supernatants was determined by measuring the absorbance at 450 nm in a spectrophotometer (Molecular Devices). We used samples with erythrocytes without peptide treatment (control of basal hemolysis) and samples with erythrocytes incubated with 1% Triton X-100 (control of 100% of hemolysis). We calculated the percentage of hemolysis using the formula, % hemolysis = (absorbance units of the erythrocytes exposed to peptide × 100)/absorbance units of the erythrocytes exposed to Triton X-100.
Binding Assay of the VIP Derivatives to Pathogens
To determine the binding capacity of VIP51 and VIP51(6–30) to both pathogens, we co-treated 107 parasites with 8 μm VIP51 or VIP51(6–30) and 4 μm FITC-labeled VIP51 or FITC-VIP51(6–30) in HPMI buffer or 5 × 106 E. coli with 3 μm VIP51 or VIP51(6–30) and 2 μm FITC-labeled VIP51 or FITC-VIP51(6–30) in PBS. After different time periods of incubation, cells were washed with ice-cold PBS, fixed in 2% formaldehyde, and mounted in DAPI-containing ProLong medium (Molecular Probes). Images were acquired with a fluorescence microscope (Olympus IX81) and then deconvolved using the Huygens Professional program (Scientific Volume Imaging).
Determination of Changes in the Plasma Membrane Potential and Permeabilization
To monitor changes in the plasma membrane potential, we used bisoxonol (Molecular Probes), a potentially sensitive anionic dye that increases fluorescence after its insertion into the depolarized membrane. E. coli (5 × 106) were cultured in 100 μl of PBS at 37 °C for 1 h in the absence or presence of 10 μm VIP or the VIP derivatives. Log phase L. major promastigotes (2 × 106) were cultured in 100 μl of HPMI buffer in the absence or presence of 12 μm VIP or the VIP derivatives for different time periods at 28 °C. Then bacteria and parasites were incubated with 0.025 μm bisoxonol for 10 min at room temperature. Fluorescence changes were monitored using a fluorescent plate reader (544 nm excitation, 584 nm emission). We used heated bacteria (10 min at 80 °C) and parasites treated with 0.8 μm gramicidin D as controls of maximal membrane depolarization.
To evaluate changes in the permeability of the plasma membrane, we used SYTOX Green (Mr 600; Molecular Probes). E. coli and L. major were pretreated with VIP or the VIP derivatives as described above and then incubated with SYTOX Green (0.05 μm for bacteria, 2 μm for parasite) at room temperature. After 10 min, we measured the increase in fluorescence (485 nm excitation, 520 nm emission) that reflects substantial damage to the plasma membrane. We used heated bacteria (10 min at 80 °C) or 0.1% Triton X-100-treated parasites as controls of maximal membrane permeabilization.
Electron Microscopy
E. coli (106; incubated with 5 μm VIP51 or VIP51(6–30) at 37 °C) or log phase promastigotes of L. major (5 × 106; incubated with 12 μm VIP51 or VIP51(6–30) at 28 °C) were harvested by centrifugation (2,000 × g, 15 min, at 4 °C) after different incubation time periods and then subjected to the high pressure freezing method (25) and embedded in Lowicryl HM20 resin. Ultrathin cryosections (65 nm) were stained with 2% uranyl acetate (pH 7.0); collected with 2% methylcellulose, 4% uranyl acetate (pH 7.0); and observed on a JEM-1010 electron microscope (Jeol).
Determination of ATP Levels
Log phase L. major (2 × 106) were treated with 12 μm VIP and VIP derivatives in HPMI buffer, as described above, and mixed with 50 μl of CellTiter-Glo reagent (Promega). After 10 min at room temperature, luminescence signal, which is proportional to the ATP amount, was monitored by an Infinite F200 microplate reader. We used parasites incubated with 10 mm sodium azide, 5 mm 2-deoxyglucose, and 5 mm pyruvate as a control of ATP deprivation (without glucose). ATP levels in bacteria cultures were determined by luminescence using the BacTiter-Glo microbial cell viability kit (Promega) following the manufacturer's instructions.
Homology Modeling and Docking
The molecular modeling of VIP used for simulations was obtained from the Protein Data Bank (code 2RRH/2RRI). VIP51 and VIP51(6–30) structures were resolved in the I-TASSER server (26) using the NMR construction of human pituitary adenylate cyclase-activating polypeptide-38 (Protein Data Bank code 2D2P) as a template.
The confidence score for estimating the quality of predicted models was calculated based on the significance of threading template alignments and the convergence parameters of the structure assembly simulations. From all theoretical predicted models found for each peptide, we selected those with the highest C-scores that indicate higher confidence (VIP, 0.18; VIP51, 0.28; VIP51(6–30), 0.29). We also determined the structural similarity between structures using the TM-score parameter (27) for each peptide: VIP, 0.74 ± 0.11; VIP51 and VIP51(6–30), 0.75 ± 0.10 (TM > 0.5 indicates correct topology, and TM < 0.17 indicates random similarity).
We conducted docking simulations with AutoDockVina version 4.2 (Scripps Research Institute, La Jolla, CA) (28), using VIP, VIP51, and VIP51(6–30) as receptor molecule and the bacterial lipopolysaccharide (LPS; Protein Data Bank code 1FI1) as the ligand. For all simulations, the grid size was x = 80, y = 80, and z = 80, centered at x = −5.7, y = −3.4, and z = 6.6. Each simulation contained eight conformations (exhaustiveness = 8). For the analysis of interactions, we focused only on polar hydrogen bonds between oligopeptides and LPS/truncated LPS mutants. We calculated how many times each amino acid from VIP and the VIP analogues was involved in hydrogen bonds in each predicted conformation and which residues of LPS were targeted. The figures were constructed using PyMOL (version 1.5.0.4; Schrödinger, LLC, New York).
In Vivo Experimental Models
Female BALB/c mice (6–8 weeks old, Charles River) were used in all experiments. Animal experimental protocols were performed according to the National/European Union Guidelines for the Care and Use of Laboratory Animals in Research and approved by the Experimental Animal Committee of the University of Seville and by the Ethics Committee of the Spanish Council of Scientific Research (file P09-CTS-4705).
To induce endotoxemia, mice were injected intraperitoneally with LPS (E. coli 055:B5, Sigma; 400 μg/mouse). To induce sepsis, the cecum of anesthetized mice was ligated 5.0 mm from the cecal tip and punctured once with a 22-gauge needle. PBS (controls), VIP, or the VIP derivatives (1 nmol in 200 μl of PBS) were administered intraperitoneally at 1, 12, and 24 h after cecal ligation and puncture (CLP) or at 1 and 18 h after LPS injection. The effective concentrations of the peptides used in the study were chosen based on previous experiments performed in our laboratory. Animals were monitored daily for survival and clinical signs. Sera and peritoneal fluid were collected by cardiac puncture and peritoneal lavage, respectively, 6 and 24 h post-CLP. The bacterial load was determined by dilution plating onto trypticase soy agar with 5% sheep blood (BD Biosciences) and cfu counting after overnight incubation. Peritoneal exudates and sera were assayed for cytokine/chemokine contents by using a specific sandwich ELISA (BD Pharmingen and Peprotech). To evaluate histopathology, lungs were collected 24 h after CLP, fixed in 10% buffered formalin phosphate, and processed for paraffin embedding and sectioning (Leica). Cross-sections (4-μm thickness) were stained with hematoxylin/eosin using standard techniques.
Colitis was induced as described previously (29). In brief, mice were lightly anesthetized with halothane, and a 3.5F catheter was inserted intrarectally 4 cm from the anus. 2,4,6-Trinitrobenzene sulfonic acid (3 mg; Sigma) dissolved in 50% ethanol (to break the intestinal epithelial barrier) was administered into the lumen via the catheter. Animals were treated intraperitoneally with vehicle or with 1 nmol/mouse VIP, VIP51, and VIP51(6–30) at 1, 12, and 36 h after 2,4,6-trinitrobenzene sulfonic acid injection. Animals were monitored daily for the appearance of diarrhea, loss of body weight, and survival. Scores for stool consistency and rectal bleeding were assessed according to procedures published previously (30).
To induce cutaneous leishmaniasis, mice were injected subcutaneously in the left hind footpads with 104 L. major purified metacyclic promastigotes isolated from stationary cultures by negative selection with peanut agglutinin as described below. Vehicle or peptides (1.5 nmol/mouse) were administered subcutaneously in the infected footpad for 7 weeks (3 times/week) starting 2 weeks postinfection. Disease progression was monitored by measuring the inflammatory edema, and the area of the cutaneous lesion of the infected footpad was measured by using a digital caliper. Values obtained in the uninfected contralateral footpad were used as a reference.
Parasite burden was determined 7 weeks postinfection by the presence of viable parasites in homogenates of the footpad, spleen, and lymph nodes by using a limiting dilution assay (31). Briefly, 12–24 serial 10-fold dilutions of the non-sedimented fraction of the homogenates were seeded on flat-bottom 96-well plates and incubated in modified RPMI 1640 medium supplemented with 20% FBS at 28 °C for 7–10 days to allow transformation of amastigotes into promastigotes. We then determined the number of positive wells (presence of motile parasites) and negative wells using an inverted microscope. Results were expressed as parasite burden (i.e. the highest dilution yielding growth of viable parasites). For histological analysis, paws were fixed in 10% buffered formalin, decalcified in Decalcifier I (Surgipath Europe Ltd.), embedded in paraffin, sectioned, and stained with hematoxylin-eosin.
Purification of Metacyclic Parasites
Stationary phase L. major promastigotes (108) were collected after 4 days in culture, washed with fresh supplemented M199 medium, and incubated with 100 μg of peanut agglutinin (Vector Labs) for 15 min at room temperature. Cells were separated by centrifugation (500 × g, 10 min, 4 °C), and non-agglutinated metacyclic promastigotes were collected from the supernatant and diluted at 2.5 × 105 cells/ml in PBS supplemented with 0.5 mm MgCl2 and 1 mm CaCl2.
Assessment of T Cell Response
Single-cell suspensions (106 cells/ml) from draining lymph nodes isolated after 5 weeks postinfection were incubated in complete RPMI medium and restimulated with 2.5 μg/ml concanavalin A for 48 h (for cytokine determination) or for 72 h (for proliferative response). Cytokine and chemokine contents in culture supernatants were determined by specific sandwich ELISA. Cell proliferation was evaluated by the addition of 2.5 μCi/ml [3H]thymidine during the last 8 h of culture and determination of cpm in a Microbeta counter 1450 (Wallac).
Statistical Analysis
All data are expressed as the mean ± S.E. Statistical analysis was carried out using Student's t test or one-way analysis of variance followed by Bonferroni's post-test to evaluate differences between groups. We assumed significance at p < 0.05.
RESULTS
The VIP Derivatives Showed Higher Bactericidal Activity than Native VIP
We first investigated the bactericidal activity of the two synthesized derivatives of VIP, VIP51 and the fragment VIP51(6–30). The chemical modifications achieved to increase the stability of native VIP resulted in peptides with higher positive charge and hydrophobic nature than VIP (Table 1 and supplemental Fig. S1). VIP51 and VIP51(6–30) showed potent antimicrobial activities against a wide panel of Gram-negative and Gram-positive bacteria, including pathogenic strains such as P. aeruginosa, E. aerogenes, E. faecalis, and E. coli expressing complete, O-antigen-containing LPS (Fig. 1). In general, both VIP derivatives showed EC50 values below 1 μm (Table 2). As expected, the VIP derivatives were more potent as antibacterial peptides than VIP (Fig. 1). Whereas the VIP derivatives showed complete bactericidal effect (100% mortality) against P. pseudoalcaligenes, P. aeruginosa, and E. coli O-antigen, VIP treatment never reached more than 70% bacterial death (Fig. 1). Moreover, the VIP derivatives showed lower EC50 values than VIP for all of the bacteria strains assayed (Table 2). Interestingly, the VIP derivatives, but not VIP, were able to efficiently kill Gram-positive bacteria (Fig. 1B and Table 2). The lack of recovery of bacterial viability after removal of the VIP derivatives indicated that these peptides were bactericidal as opposed to bacteriostatic.
FIGURE 1.

Bactericidal activity of VIP and the VIP derivatives. Cell viability of non-pathogenic (E. coli Top 10, P. pseudoalcaligenes) and pathogenic (E. coli expressing complete O-antigen-containing LPS, P. aeruginosa, E. aerogenes) Gram-negative bacteria (A) and non-pathogenic (M. luteus) and pathogenic (E. faecalis) Gram-positive bacteria (B) incubated with various concentrations of native VIP, VIP51, or VIP51(6–30) for 3 h. Data are expressed as the percentage of survival (by counting cfu) relative to untreated bacteria. The mean ± S.E. (error bars) (n = 4, each performed in duplicate) is shown. *, p < 0.05; **, p < 0.001 versus untreated bacteria; #, p < 0.05 versus VIP-treated bacteria (dose curve).
TABLE 2.
Bactericidal activity of VIP, VIP51, and VIP51(6–30)
Mean ± S.E. of four independent experiments is shown.
| Bacteria tested | EC50 |
||
|---|---|---|---|
| VIP | VIP51 | VIP51(6–30) | |
| μm | |||
| Gram-negative | |||
| E. coli (Top10) | 1.50 ± 0.53 | 0.66 ± 0.09 | 0.32 ± 0.03 |
| E. coli | 0.42 ± 0.20 | 0.16 ± 0.01 | 0.10 ± 0.03 |
| P. aeruginosa | 0.70 ± 0.34 | 0.21 ± 0.10 | 0.20 ± 0.02 |
| E. aerogenes | >20 | 2.00 ± 0.23 | 0.59 ± 0.52 |
| P. pseudoalcaligenes | 2.66 ± 0.95 | 0.01 ± 0.00 | 0.01 ± 0.00 |
| Gram-positive | |||
| S. mutans | 19.06 ± 0.76 | 15.08 ± 0.24 | 2.92 ± 1.72 |
| M. luteus | >20 | 0.03 ± 0.01 | 0.15 ± 0.04 |
| E. faecalis | >20 | 1.17 ± 0.02 | 1.01 ± 0.01 |
To confirm the selectivity of these antimicrobial peptides for microbes and to confirm that they are not toxic for mammalian cells, we performed toxicity and hemolytic assays. Neither VIP51 nor VIP51(6–30) affected the viability of mouse primary macrophages and splenocytes (97 ± 3 and 98 ± 1% cell survival, respectively) or lysed erythrocytes (2.5 ± 0.5 and 3.1 ± 0.1% hemolysis for VIP51 and VIP51(6–30), respectively) at the highest concentration assayed as antimicrobials in this study.
The VIP Derivatives Altered the Integrity of the Bacterial Plasma Membrane
Most of the cationic antimicrobial peptides target bacteria through electrostatic interactions with the microbial membrane. This generally results in formation of transmembrane pores and loss of membrane integrity that causes cell death. In this study, we used E. coli to investigate the potential mechanisms involved in the bactericidal effect of VIP51 and VIP51(6–30) because of the higher susceptibility of Gram-negative bacteria to both peptides and the accessibility to mutants of this strain and because Gram-negative bacteria are the major cause of sepsis (an experimental model later used in our study). We used two complementary approaches to investigate bactericidal effect of the two VIP derivatives. First, we monitored membrane depolarization by using the bisoxonol dye, a lipophilic anion that enters into membranes only if their potential has collapsed. Second, we tested the access of the vital dye SYTOX Green, which requires large size lesions in the membrane to enter into the cell, being precluded in intact cells. We found that bacteria cultured with the VIP derivatives showed significant increases in the uptake of bisoxonol (Fig. 2A, left) and SYTOX (Fig. 2A, right). Interestingly, native VIP increased the depolarization, but not the permeabilization, of bacterial membrane (Fig. 2A). As a consequence of this effect, we observed a significant depletion on ATP levels in bacteria treated with the VIP derivatives (82.5% for VIP51, 78.6% for VIP51(6–30); Fig. 2B). However, VIP caused a marginal ATP depletion (28.2%; Fig. 2B). These results suggest that only the VIP derivatives affect simultaneously the potential and integrity of the bacterial membrane. This could explain the higher antimicrobial activity of the VIP derivatives compared with VIP.
FIGURE 2.

The VIP derivatives bind to the bacterial surface and disrupt the membrane integrity. A, the potential (left) and integrity (right) of plasma membrane of E. coli Top 10 cultured with PBS (control), VIP, or the VIP derivatives (each at 10 μm for 1 h) were determined by changes in bisoxonol and SYTOX Green fluorescence, respectively. Results are expressed as -fold change in depolarization or permeabilization, (F/Fm)/(Fo/Fm), where F is the fluorescence of peptide-treated bacteria, Fo is the fluorescence of untreated bacteria, and Fm is the maximal fluorescence achieved by heated E. coli. B, levels of bacterial ATP (expressed as luminescence units) in E. coli Top 10 treated with PBS (control), VIP, or the VIP derivatives (each at 10 μm for 1 h). C, binding of FITC-labeled VIP derivatives to E. coli Top 10. Bacteria were DAPI-counterstained (blue corresponds to nucleic acids). Insets, Normarski images. Scale bar, 2 μm. D, ultrastructural analysis of E. coli incubated with PBS (controls) or with VIP51 (5 μm) for the indicated times. Arrows point to intracytoplasmic vacuoles and sites of membrane damage. Scale bar, 0.5 μm. Images are representative of three independent experiments in which more than 95% of the cells displayed this pattern. In A and B, the mean ± S.E. (error bars) (n = 4, each performed in duplicate) is shown. *, p < 0.05; **p < 0.001 versus untreated bacteria (control).
To further characterize the bactericidal activity of the VIP derivatives, we incubated bacteria with FITC-labeled VIP51/VIP51(6–30). We observed that both peptides rapidly targeted the whole bacterial surface and exerted an agglutinating effect that resulted in multiple bacteria aggregates in the culture (Fig. 2C). Moreover, ultrastructural images of bacteria treated with VIP51 showed a complete disorganization of the plasma membrane and the presence of visible pores throughout the surface, multiple intracellular vacuoles, and cytoplasmic content surrounding bacteria (Fig. 2D). These cellular changes were even more drastic in VIP51(6–30)-treated bacteria, which appeared completely disintegrated in the samples (data not shown).
Specific Factors Influencing Antibacterial Activity of VIP51 and VIP51(6–30)
The bactericidal activities of the antimicrobial peptides are strongly influenced by the ionic strength and pH of the medium. Whereas a previous study reported that VIP almost completely loses its antibacterial effect against E. coli at physiological concentrations of 150 mm NaCl (32), we found that both VIP derivatives fully kept their bactericidal effects (E. coli survival rate, 6.0 ± 1.2% for VIP51 and 0.9 ± 0.5% for VIP51(6–30)) at this concentration of NaCl. Furthermore, the presence of high salt concentrations (180 mm NaCl) attenuates the activity of VIP51 slightly, but not that of VIP51(6–30), against both the Gram-negative E. coli and the Gram-positive E. faecalis (Fig. 3A), supporting the functional relevance of these peptides in physiological fluids. However, an increase of the ionic strength of the medium (with 50 mm MgCl2) significantly abolished the bactericidal activity of the VIP derivatives in both strains (Fig. 3A). These results indicate that electrostatic interactions between the VIP derivatives and bacterial targets might play a critical role in their antibacterial activity.
FIGURE 3.

Determinants influencing the antibacterial effect of the VIP derivatives. A, effect of the ionic strength of the medium on the activity of native VIP and the VIP derivatives against E. coli and E. faecalis. Bacteria were incubated with VIP, VIP51, and VIP51(6–30) (each at 5 μm) in the absence or presence of 180 mm NaCl or 50 mm MgCl2 for 3 h. *, p < 0.05; **, p < 0.001 versus peptide-treated bacteria; #, p < 0.05 versus untreated bacteria at each condition. B, effect of pH of the medium on the antimicrobial activity of VIP and the VIP derivatives (5 μm, 3 h of culture) against E. coli and E. faecalis.**, p < 0.001 versus controls at each pH; #, p < 0.05 versus peptide-treated bacteria at pH 7.0. In both panels, survival was determined as described in the legend to Fig. 1. The mean ± S.E. (error bars) (n = 3, each performed in duplicate) is shown.
On the other hand, we demonstrated that the antimicrobial activities of VIP and its derivatives in Gram-negative but not in Gram-positive bacteria depend on the pH of the medium (Fig. 3B). Interestingly, the three peptides gained and lost effectiveness killing E. coli at basic and acidic pH conditions, respectively (Fig. 3B). Whereas the three peptides are positively charged at pH values between 5.0 and 9.0, the net charge of bacterial membrane is more negative when the pH of the medium raises. Therefore, a basic environment could favor stronger interactions of the bacteria with the more cationic VIP51 and VIP51(6–30).
Another important modulator of the antimicrobial activity is LPS, the main outer membrane component of Gram-negative bacteria (33, 34). In order to study the role of LPS in the antimicrobial effect of the VIP derivatives, we tested their action in five mutant strains derived from E. coli K-12 D21, which are differentially defective in various components of the LPS molecule (Fig. 4A). Our data showed that the antibacterial activity of VIP and its derivatives against the mutants expressing the truncated forms of LPS did not significantly change in comparison with that shown by the wild type D21“Ra” strain, with the exception of the mutant D21e7“Rc” that lacks the outer network of residues of α-d-glucose (GLC), which significantly reversed the activity of VIP51(6–30) (p < 0.01 by analysis of variance; Fig. 4B). This suggests that, although it could participate, the oligosaccharide core of LPS is not essential for the bactericidal activity of VIP and the VIP derivatives against E. coli. In fact, the three peptides showed the best EC50 values against the D21f2“Rd” strain, which only retains the portion of the molecule containing Lipid A and 3-deoxy-d-manno-octulosonic acid (KDO) groups (Fig. 4B).
FIGURE 4.

Antimicrobial activity against E. coli mutants deficient in LPS synthesis. A, scheme of LPS from E. coli K-12 D21 chemotype. GLC, α-d-glucose; GLA, alpha-d-galactose; GMH, l-glycero-d-mannoheptopyranose; NAG, N-acetylgalactosamine; DPO, diphosphate; PA1, 2-amino-2-deoxy-α-d-glucopyranose; GCN, d-glucosamine; OM, outer membrane. B, antimicrobial activity of VIP, VIP51, and VIP51(6–30) (added at 5 μm for 3 h) against mid-log phase growth E. coli chemotypes (D21Ra and defective strains D21e19, D21e7, D21f1, and D21f2). Cell viability was determined with the BacTiterGlo kit and expressed as the percentage of survival versus untreated control bacteria. Numbers in parenthesis show EC50 for each peptide. The mean ± S.E. (error bars) (n = 4, each performed in duplicate) is shown. *, p < 0.05; **, p < 0.001 versus untreated bacteria; #, p < 0.05 versus VIP-treated bacteria; &, p < 0.05 versus VIP51(6–30)-treated bacteria at the indicated dose.
To investigate the potential molecular interactions of VIP analogues with LPS, we performed a series of docking simulations by using the LPS derived from E. coli K-12 D21 strain as a model. We observed that less than 25% of the hydrogen bonds established between VIP or the VIP derivatives and LPS were made with the outer network of residues of α-d-glucose and α-d-galactose, whereas most of the interactions (67–72%) occurred with the polar groups of Lipid A and KDO residues of LPS (supplemental Tables S1–S3). These findings were confirmed by docking simulations of the peptides with the truncated LPS containing only Lipid A and KDO (Tables S4–S6). Interestingly, almost all the amino acids of VIP and its derivatives that established hydrogen bonds with LPS are located in the middle portion of the peptide, a domain that concentrates the majority of the cationic residues (Tables S1–S3). Moreover, the substitution K15R made in VIP51(6–30) increased its interaction with α-d-galactose residues of LPS (supplemental Table S3). This could be relevant to explain the attenuation in the killing activity of VIP(6–30) against the D21e7“Rc” mutant (Fig. 4B).
The VIP Derivatives Protected against Severe Sepsis by Inducing Bacterial Clearance
Herein, we aimed to investigate the potential therapeutic effect of VIP51 and VIP51(6–30) in a clinically relevant experimental model of polymicrobial sepsis induced in mice by CLP. We found that after cecal perforation, all untreated animals died (Fig. 5A) as a consequence of a focal infection (peritoneum) that subsequently became systemic (blood) (Fig. 5B), which induced the secretion of cytotoxic and inflammatory factors by immune cells (Fig. 5C). These responses caused inflammatory infiltration; tissue damage and disseminated coagulation in target organs, such as lung (Fig. 5D); and the subsequent multiorgan failure characteristic for sepsis. The therapeutic efficacy of VIP on lethal sepsis by down-regulating inflammatory mediators was reported previously (35). As expected, VIP treatment reduced mortality, bacterial loads, and inflammatory mediators in septic animals (Fig. 5, A–C). Similarly, the administration of the VIP derivatives into the peritoneum of septic animals significantly increased the survival (Fig. 5A) and drastically reduced the local and systemic bacterial loads (Fig. 5B). Subsequently, treatment with the VIP derivatives reduced inflammatory mediators and the histopathologic alterations associated with sepsis (Fig. 5, C and D). These results suggested that the protective effect exerted by the VIP analogues might be primarily mediated through a local bactericidal effect. Indeed, it is widely known that persistence of the local bacterial infection plays a critical role in the severity and progression of sepsis. To support this hypothesis, we performed additional experiments to separate the direct antimicrobial activity of the VIP derivatives from their potential anti-inflammatory action. First, we analyzed the direct antibacterial activity of VIP and the VIP analogues against four different Gram-negative strains isolated from the peritoneal fluid of mice with CLP-induced sepsis. Only the VIP derivatives were effective against these pathogenic bacterial strains (Table 3). Second, we compared the effect of VIP51 and VIP51(6–30) in a severe form of septic shock induced by LPS, which causes exacerbated systemic inflammatory responses and multiorgan failure in the absence of bacterial infection. Also, we investigated the effect of the VIP derivatives in a model of chemically induced colitis characterized by uncontrolled colonic inflammation. As demonstrated previously (36, 37), treatment with native VIP was protective in both inflammatory disorders (Fig. 6). However, only VIP51, and not VIP51(6–30), reduced mortality caused by the bacterial endotoxin (Fig. 6A) and improved clinical evolution of colitis caused by bowel inflammation in the absence of infection (Fig. 6B). Together, these results indicate a dual role for VIP51 that combines the equipotent immunomodulatory role of VIP with an improved stability and higher antimicrobial activity. However, VIP51(6–30) does not share with VIP its capacity to regulate host immune responses in inflammatory conditions, although it protected efficiently from sepsis by neutralizing living bacteria.
FIGURE 5.

The VIP derivatives improve survival and bacterial clearance in sepsis. A, survival of mice subjected to sepsis by CLP and treated with vehicle (control) or with VIP, VIP51, and VIP51(6–30) (n = 20 mice/group). B and C, bacterial load (B, n = 10 mice/group) and cytokine contents (C, n = 5 mice/group) were determined in sera and peritoneal fluids isolated 24 h after sepsis induction. The mean ± S.E. (error bars) (n = 4, each performed in duplicate) is shown. n.d, not detectable. *, p < 0.05; **, p < 0.001 versus untreated control mice. D, histological sections of lungs isolated 24 h after CLP induction. Images are representative of 10 mice/group. Scale bar, 100 μm.
TABLE 3.
Bactericidal activity of VIP, VIP51, and VIP51(6–30) against peritoneal bacterial isolated from septic mice
Mean ± S.E. of four independent experiments is shown.
| Bacteria tested (Gram-negative) | EC50 |
||
|---|---|---|---|
| VIP | VIP51 | VIP51(6–30) | |
| μm | |||
| B1 | >20 | 0.41 ± 0.07 | 1.25 ± 0.36 |
| B2 | >20 | 0.52 ± 0.14 | 0.28 ± 0.05 |
| B3 | >20 | 0.59 ± 0.06 | 0.16 ± 0.07 |
| B4 | >20 | 0.04 ± 0.01 | 0.15 ± 0.08 |
FIGURE 6.
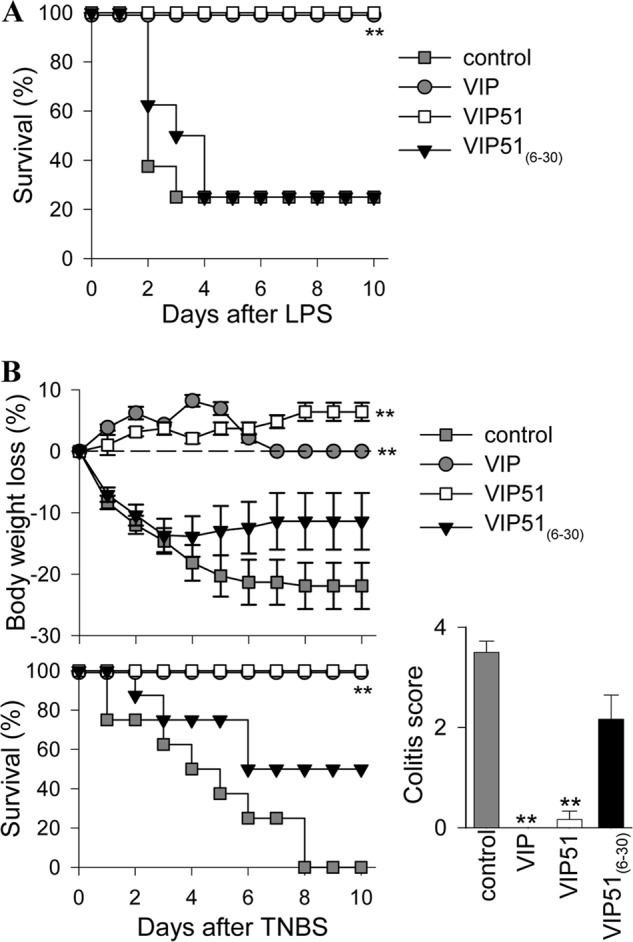
Modulation of the host immune response in endotoxemia and inflammatory bowel disease. A, survival of mice subjected to LPS-induced endotoxemia and treated with PBS (control) or with VIP, VIP51, and VIP51(6–30). n = 12–16 mice/group, from three independent experiments. B, body weight loss, colitis scores, and survival of mice subjected to 2,4,6-trinitrobenzene sulfonic acid-induced colitis and treated with PBS (control) or with VIP, VIP51, and VIP51(6–30). n = 12–16 mice/group, from three independent experiments. **, p < 0.001 versus untreated control mice. Error bars, S.E.
The VIP Derivatives Killed Promastigotes of Leishmania by Inducing the Formation of Pores in the Plasma Membrane
We next investigated whether the antimicrobial effect of VIP51 and VIP51(6–30) could be extended to other pathogens, such as tropical trypanosomatid parasites. Among them, Leishmania represents a global health problem with ineffective drug treatments. This is an obligate intracellular organism with a digenetic life cycle alternating between the flagellated, extracellular promastigote form in the digestive tract of the sandfly vectors and the aflagellated amastigote form that lives inside mononuclear phagocytic cells and causes the disease symptoms in mammals. This protozoan parasite is one of the most used models to assay antiparasitic strategies. Importantly, Leishmania species have a highly negative glycocalix that makes them attractive targets for antimicrobial peptides (38).
Therefore, we assayed the antiparasitic activity of the three peptides against different species of Leishmania. The two VIP derivatives, but not VIP, significantly reduced the viability of promastigote forms of L. major, which causes cutaneous leishmaniasis (Fig. 7A and Table 4). With the exception of the effect of VIP51(6–30) in L. tropica, the other species of the parasite responsible of visceral manifestations of leishmaniasis were resistant to the VIP derivatives (Table 4). Therefore, we selected L. major to further characterize the mechanisms involved in the leishmanicidal actions of the VIP derivatives. Similar to our observation in bacteria, VIP51 and VIP51(6–30) bound in less than 5 min to the whole surface of the parasite, induced a complete disorganization of the parasite structure (reflected by the acquisition of a rounded morphology), and caused leakage of the cellular content (Fig. 7B). Moreover, incubation of L. major promastigotes with VIP51 or VIP51(6–30) resulted in a rapid dissipation of the membrane potential and an increase in the influx of SYTOX Green (Fig. 7C). These findings suggest that the VIP derivatives kill L. major by disrupting the plasma membrane of the promastigote through the formation of large size lesions. The membrane disruption caused by VIP51 and VIP51(6–30) led to a collapse of the bioenergetic metabolism of the parasite monitored by a sustained decrease in intracellular ATP levels (Fig. 7D). To study how these effects translated into morphological changes, we analyzed the peptide-treated parasites by transmission electron microscopy. Treatment with VIP51 induced the formation of pores in the plasma membrane, a complete disruption of cell integrity, and loss of cytoplasmic material that was found in the extracellular medium (Fig. 7E).
FIGURE 7.

The VIP derivatives induce the formation of disrupting membrane pores in L. major promastigotes. A, cell viability of L. major Friedlin clone V1 incubated with medium (control) or with various concentrations of VIP and the VIP derivatives determined by reduction of Alamar Blue and expressed as a percentage of untreated control samples. B, binding of the FITC-labeled VIP derivatives to L. major promastigotes (bottom panels). Top panels, Normarski images. Arrows point to sites of plasma membrane damage. Bars, 4 μm. C, variation in plasma membrane potential (top) and integrity (bottom) of parasites cultured with medium (control), VIP51, or VIP51(6–30). Data are expressed as a percentage of fluorescence intensity relative to that achieved by maximal depolarization or permeabilization. D, changes in ATP levels (expressed as luminescence units) in L. major promastigotes cultured with medium (control), VIP51, or VIP51(6–30). Parasites incubated in the absence of glucose were used as negative controls. E, ultrastructural analysis of the cell damage caused by VIP51 in L. major. The arrows point to membrane breakages (at 1 h) and depletion of electron-dense cytoplasmic material, severe cellular damage, and apparent leakage of the cytoplasmic content (at 4 h). Bars, 1 μm. The mean ± S.E. (error bars) (n = 3, each performed in duplicate) is shown. *, p < 0.05; **, p < 0.001 versus untreated parasites; ##, p < 0.001 versus VIP-treated parasites.
TABLE 4.
Antiparasitic activity of VIP, VIP51, and VIP51(6–30) against Leishmania sp.
Mean ± S.E. of four independent experiments is shown.
| Parasite | EC50 |
||
|---|---|---|---|
| VIP | VIP51 | VIP51(6–30) | |
| μm | |||
| L. major | >24 | 3.39 ± 0.24 | 1.74 ± 0.45 |
| L. mexicana | >24 | >24 | >24 |
| L. donovani | >24 | >24 | >24 |
| L. tropica | >24 | >24 | 16.31 ± 0.18 |
| L. infantum | >24 | >24 | >24 |
Next, we evaluated determinants influencing the antiparasitic activity of the VIP analogues. We observed that the leishmanicidal effect of the VIP derivatives strongly depended on the ionic strength of the medium because it was completely abolished in the presence of high salt concentrations (Fig. 8A). Moreover, the pH of the medium influenced the killing effect of the VIP derivatives. Whereas the leishmanicidal activities of both peptides increased at basic conditions, VIP51 completely lost its killing effect at the acidic pH (which is similar to the pH found inside of the parasitophorous vacuole) (Fig. 8B). Interestingly, VIP51(6–30) still maintained (over 50% of) the leishmanicidal activity at this pH 5.0 (Fig. 8B).
FIGURE 8.

Determinants influencing leishmanicidal activity of the VIP derivatives. A, effect of the ionic strength of the medium on the activity of VIP analogues against L. major. Survival of parasites incubated with VIP51 and VIP51(6–30) (12 μm) in the absence or presence of 180 mm NaCl or 50 mm MgCl2 for 4 h. **, p < 0.001. B, effect of pH of the medium on the antiparasitic effect of the VIP derivatives (12 μm, 4 h of culture) against L. major. **, p < 0.001 versus controls at each pH; #, p < 0.05 versus peptide-treated parasites at pH 7.0. C, VIP51 requires the interaction with LPG and PG of the parasite surface to kill L. major promastigotes. The effect of VIP51 on the cell viability of the mutant strain of L. major glf−/Kidd GLF-1F lacking LPG (glf−/Kidd GLF-1F L. major Friedlin V1 parasites treated with FK506 that induces LPG expression were used as the WT strain for comparison) or the L. major lpg2−/− and lpg2+/− mutants lacking PG (LV39wt was used as the WT strain for comparison) is shown. Survival was expressed as a percentage of cell viability of their corresponding WT strains. Data are the mean ± S.E. (error bars) of three independent experiments, each performed in duplicate. *, p < 0.05; **, p < 0.001 versus untreated parasites; #, p < 0.01; ##, p < 0.001 versus WT treated parasites.
To investigate potential targets for the VIP derivatives in the parasite, we considered the LPG, a major anionic component of the glycocalix of Leishmania promastigotes, as a potential target. LPG structurally contains repeated PG units (Gal-Man-P) anchored by a glycan core to a phosphatidylinositol anchor (39). In addition, both promastigotes and infecting amastigotes contain moderate levels of proteophosphoglycan, a mucin-like protein also modified by the PG repeats. To determine the involvement of LPG and proteophosphoglycan in the leishmanicidal activity of the VIP derivatives, we used an inducible system of LPG and an lpg2−/− mutant (24) that lacks a Golgi GDP-mannose transporter required for PG synthesis (40). We observed that the absence of LPG and proteophosphoglycan significantly impaired the leishmanicidal activity of VIP51 (Fig. 8C), suggesting that both anionic molecules are essential for its action in L. major.
Therapeutic Activity of the VIP Derivatives in Experimental Cutaneous Leishmaniasis
Finally, we evaluated the capacity of the VIP derivatives to protect BALB/c mice against L. major infection. Treatment with native VIP was included as a negative control because it did not show leishmanicidal activity. Mice infected in the hind footpad with purified metacyclic promastigotes of L. major developed marked and progressive inflammatory edema and cutaneous lesions around the site of injection (Fig. 9A). Analysis of histopathology of the cutaneous lesions showed extended areas of necrosis and ulceration in epidermis as well as erosion and destruction of cartilage and bone caused by a severe granulomatous inflammation as a consequence of the accumulation of parasite-infected macrophages in the dermis (Fig. 9B). Although the injection of the three peptides diminished paw swelling and lesion size, only the treatment with VIP51(6–30) significantly controlled the clinical manifestations and progression of this disease (Fig. 9A) and abolished the histopathologic signs in the infected footpads (Fig. 9B). The protective effect of VIP51(6–30) in cutaneous leishmaniasis correlated with a striking decrease in the number of living parasites in cutaneous lesions (Fig. 9C). It is noteworthy that treatment with VIP51(6–30) also prevented the systemic dissemination of this pathogen to immune and visceral organs (Fig. 9C). Moreover, treatment with VIP51(6–30) normalized the exacerbated immune response observed in the infected animals (Fig. 10), probably as a consequence of a reduced parasite load. Although treatment with VIP51 limited the presence of parasites in target organs, such as liver and draining lymph nodes (Fig. 9C), this was not enough to significantly ameliorate the clinical course of the disease (Fig. 9A).
FIGURE 9.

In vivo effects in experimental cutaneous leishmaniasis. A, inflammatory edema (top) and lesion size (bottom) in footpads of mice infected with metacyclic L. major and treated locally with saline (control), VIP, VIP51, or VIP51(6–30). Contralateral uninfected footpads were used as basal controls. Pictures show representative images of footpad inflammation and necrotic cutaneous lesions at 7 weeks postinfection. Data are the mean ± S.E. (error bars) of one representative experiment of five (n = 10 mice/group). B, histopathologic analysis of the footpad lesions of uninfected and infected mice treated with saline (control) or peptides. The arrows point to parasites inside and outside of infected macrophages. Left scale bars, 100 μm; right scale bars, 5 μm. Magnified images show amastigote forms of Leishmania (stars) lining the walls of the vacuoles following a typical arrangement. They can be distinguished as the darkly stained nucleus and kinetoplast closely associated. Images are representative of 5 mice/group, three independent experiments. C, parasite loads in footpads, liver, spleen, and draining lymph nodes in infected mice treated with saline (control), VIP, or the VIP derivatives (at 7 weeks postinfection). Data are the mean ± S.E. of one representative experiment of three (n = 5 mice/group). *, p < 0.05; **, p < 0.001 versus untreated infected mice.
FIGURE 10.
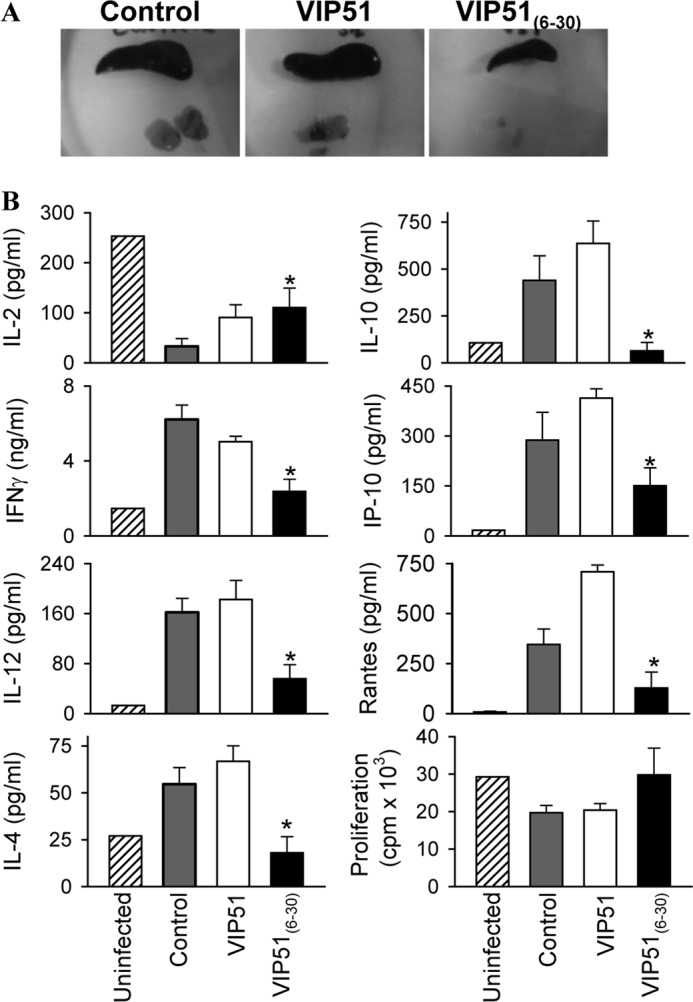
Immunological responses of animals suffering cutaneous leishmaniasis. A, mice infected with L. major and treated with saline (control), VIP, or VIP51 showed enlarged lymph nodes and spleen compared with the normal size of these lymphoid organs isolated from VIP51(6–30)-treated mice. B, proliferation and cytokine production by draining lymph node cells isolated (at 5 weeks postinfection) from mice infected with L. major and treated with saline (control) or the VIP derivatives. The mean ± S.E. (error bars) of three independent experiments (10 mice/group) is shown. *, p < 0.05 versus untreated infected mice.
DISCUSSION
With microbial drug resistance becoming a global public health problem, it is imperative to develop new anti-infective strategies. In this sense, nature has chosen a different concept for the evolution of the innate host defense, favoring the appearance of compounds able to modulate many biological functions that simultaneously affect the host and the pathogens. Thus, neuropeptides with antimicrobial properties could represent one of the most promising future strategies for combating infections. However, the potential therapeutic possibilities of these peptides are usually limited by some drawbacks concerning metabolic stability, bioavailability, immunogenicity, routes of administration, and production cost that must be overcome (41). Therefore, research has been focused on avoiding these disadvantages and improving the therapeutic efficacy of these molecules. Our study supports the view considering the design principles of neuropeptides as primitive and effective antimicrobial molecules to develop novel drugs. Here, we demonstrate that VIP51, a metabolic stable analogue of VIP generated by substitutions/additions of critical residues, and an N-terminal truncated derivative of VIP51 (VIP51(6–30)) were more potent than native VIP as antimicrobial peptides against various non-pathogenic and pathogenic Gram-positive and Gram-negative bacteria. Moreover, both VIP derivatives, but not VIP, were able to kill tropical protozoa parasites from the genus Leishmania.
Besides the increase in their stability, the chemical changes made in VIP51 and VIP51(6–30) increased the cationic charge and hydrophobicity of the molecule and improved their features as antimicrobial peptides. Moreover, these modifications produced relevant changes at the structural level. Thus, both VIP derivatives adopted a clear amphipathic structure consisting of a highly cationic N-terminal middle domain and a C-terminal domain enriched in hydrophobic residues. This structure could explain the potent antimicrobial activity observed for VIP51 and VIP51(6–30) in comparison with the native VIP and their selectivity for pathogens versus mammalian cells. Both VIP derivatives are able to recognize and interact with anionic residues exposed in the outer surface of bacteria and parasites but not with the neutral membrane of mammalian cells. After their binding to the surface of the pathogen, VIP51 and VIP51(6–30) induced the depolarization of the membrane through the formation of disrupting pores, which led to a collapse of the intracellular ATP levels and the subsequent death of the pathogens. In general, the mechanism(s) through which antimicrobial peptides may permeabilize and traverse microbial membranes are not entirely clear and probably vary for different peptides. Our data suggest that a translocation mechanism termed “self-promoted uptake” described for other antimicrobial peptides against Gram-negative bacteria (54) could fit with the way of action of the VIP derivatives. According to this theory, the VIP derivatives would initially bind to the LPS of the outer membrane, causing displacement of divalent cations that bridge and neutralize LPS. This would generate destabilized outer membrane areas through which peptides could translocate. After the peptide adsorption to the outer leaflet of membrane and after acquiring a certain concentration, the peptides could insert into the membrane surface through their hydrophobic domain. This could cause a chemical potential imbalance across the bilayer and result in the translocation of the peptides to the inner membrane. Then peptides could aggregate and insert deeper into the hydrophobic membrane core, facilitating membrane rupture and pore formation. Besides the disruption of the membrane integrity through the formation of pores or other aggregated structures, changes in the influx/release of ions through ion channels may cause depolarization of cell membranes. Interestingly, the native VIP only causes depolarization and not disruption of the bacterial membrane, leading to a bacteriostatic action more than to a bactericidal effect. Indeed, depolarization of the cytoplasmic membrane is not, per se, a lethal event in bacteria (55). In this sense, we cannot discard the involvement of modifications of ion channel activity by native VIP. Regarding this, large scale genome analyses showed that prokaryotes express ion channels belonging to molecular families expressed in neurons (42), and VIP modulates different ion channels in neurons, in the range of high concentrations used in our study (43).
Because LPS is a highly anionic molecule and represents up to 50% of the outer membrane of Gram-negative bacteria, it emerges as a potential target for the VIP derivatives. Our data indicate that, although it could facilitate the antibacterial activity of the VIP derivatives, the oligosaccharide core of LPS is not essential. In fact, docking simulations supported the capacity of VIP, VIP51, and VIP51(6–30) to bind to LPS. However, the three peptides interacted minimally with the outer network of carbohydrates of LPS and established most of the hydrogen bonds with other inner anionic moieties of LPS, such as KDO and various components of Lipid A, in close proximity with the bacterial membrane surface. Most of the amino acid modifications made in the derivatives seem to have roles in establishing new hydrogen bonds with an LPS molecule. Moreover, the fact that the substitutions N24A and S25A at the C-terminal region of the native peptide generated a strong hydrophobic domain in the two VIP derivatives could be even more critical in the formation of disrupting pores in the bacterial membrane by VIP51 and VIP51(6–30) but not by VIP, which only caused membrane depolarization. Furthermore, the addition of the motif GRR at the C terminus in the VIP derivatives increased the proportion of α-helix in the molecule and changed the distribution of the amino acids in the molecule, favoring the orientation of the cationic residues on the same side (supplemental Fig. S1), which could improve their binding capacity to LPS.
Interestingly, VIP and its derivatives showed the strongest antimicrobial activities against pathogenic E. coli strains that express complete O-antigen-containing LPS. This suggests that, far from being a permeability barrier and a cause of resistance, as occurs with other antimicrobial peptides (33), the anionic nature of the O-antigen and oligosaccharide core could facilitate the recruitment of the VIP derivatives to the surface of bacteria and allow a critical peptide concentration to form pores. This finding could be also related to the capacity of the two derivatives to induce bacterial aggregation, which represents an ancient mechanism of defense to neutralize pathogens (56). Some peptides, like the eosinophil cationic protein, decrease their antimicrobial activity with a progressively truncated LPS core (57), a fact that is related to the involvement of a complete LPS in their agglutinating activity.
On the other hand, whereas native VIP failed to kill any of the Gram-positive bacteria strains tested, both derivatives showed potent antimicrobial activity against non-pathogenic S. mutans and M. luteus and pathogenic E. faecalis, a strain that is normally resistant to many cationic peptides. The differential bactericidal effects found between the VIP derivatives and native VIP could be due to the higher net positive charge and hydrophobicity of the former because both characteristics seem to be critical in the susceptibility of some Gram-positive bacteria, such as S. mutans, to many antimicrobial peptides (44). Because VIP51(6–30) has a higher positive charge and α-helix percentage than VIP51, VIP51(6–30) could interact better with S. mutans and cause its death at lower peptide concentrations. The killing effect against Gram-positive bacteria is especially relevant from a therapeutic point of view because these bacteria have a thick cell wall and develop sophisticated resistance mechanisms such as the absence of uncharged lipids (45). However, the mechanisms involved in this effect by the VIP derivatives are still unknown.
It is noteworthy that the treatment with either of the two VIP derivatives significantly increased the survival in an experimental model of polymicrobial sepsis, a major cause of mortality and morbidity that remains difficult to treat. Our hypothesis is that VIP51 protects against sepsis through two non-redundant actions. VIP51 may act as an agonist of VIP receptors expressed in immune cells, and like VIP (46), it would regulate the inflammatory immune response of the host by reducing a plethora of inflammatory mediators that cause the multiorgan failure characteristic of sepsis. At the same time, VIP51 may exert a potent antimicrobial activity by directly killing pathogenic bacteria. However, VIP51(6–30) seems to prevent mortality caused by sepsis mainly by reducing the bacterial load. This effect could be enough to reduce the later storm of inflammatory and cytotoxic factors. Because VIP51(6–30) lacks the N-terminal residues that are critical to activate VIP receptors, we assume that VIP51(6–30) is unable to directly regulate the inflammatory response. In fact, we present evidence that VIP51(6–30) failed to protect against pathogen-free inflammatory disorders, in which VIP and VIP51 were effective. Relevant for a therapeutic application was the fact that VIP51 and VIP51(6–30) resisted high salt concentrations (up to 180 mm NaCl), contrary to VIP, which completely lost its antimicrobial capacity in a physiological saline environment (32).
In the case of the parasite Leishmania, the cytolytic activity of the two VIP derivatives consisted of their initial interaction with specific components of the parasite surface, followed by the damage of the plasma membrane through the induction of large-sized lesions. This is a death mechanism shared with other leishmanicidal peptides (47). Importantly, we identified specific targets for the VIP derivatives on the parasite surface. The plasma membrane of the promastigote stage possesses a thick anionic glycocalix made up mostly of LPG (48) and the external GPI-anchored metalloproteinase gp63 (49). Both components are involved in the interaction with and partial resistance to antimicrobial peptides (50). Using different mutants of L. major (24), we demonstrated the involvement of LPG and PGs in the leishmanicidal action of VIP51. Interestingly, both mutants continued to express LPG fragments and an abundant class of glycophosphatidylinositol lipids (GIPLs), which is expressed in both the promastigote and amastigote stages (48). The lpg2−/− mutant expresses a Gal-Gal-Galf-Man-Man-GlcN-Inos-P-lyso-alkyl glycerol (24) and normal levels of GIPLs, whereas the GLF mutant should continue to express the LPG fragment Man-Man-GlcN-Inos-P-lyso-alkyl glycerol as well as truncated GIPLs, which differ from the LPG fragment in bearing an acyl-alkyl glycerol anchor (51). However, none of these molecules (GIPL, truncated LPG, or truncated GIPLs) seemed to be the target of the VIP analog. Further research will define the selectivity of the peptides for certain Leishmania strains based on the different complexity that exists in the surface coat composition of these parasites. In contrast to the VIP derivatives, VIP failed to kill any of the Leishmania strains assayed, although it was able to bind to their cell surface (not shown). Therefore, the capacity of the VIP derivatives to induce the formation of pores in the membrane (probably through their hydrophobic domains) must be a determinant in their leishmanicidal activity. In fact, VIP was reported to kill other trypanosomatids through a mechanism that does not involve disruption of the cell membrane but rather the endocytosis of VIP by the parasite and later disruption of intracellular targets (15).
Our study has important therapeutic implications because we demonstrated the efficacy of VIP51(6–30) in a model of severe cutaneous leishmaniasis, in which the local injection of VIP51(6–30) drastically reduced the cutaneous lesions, parasite loads, and dissemination of the disease to visceral organs. Surprisingly, despite its potent leishmanicidal action observed in vitro, VIP51 did not efficiently protect from cutaneous leishmaniasis. A possible explanation for this finding is that the parasite could use the anti-inflammatory and Th2 responses induced by VIP51 to counteract the immune response mounted by the host against this intracellular parasite (52). However, because VIP51(6–30) does not regulate immune responses in the host, the therapeutic effect of VIP51(6–30) in vivo is probably exerted by killing the parasites released from infected cells during lysis and reducing the density of the new infective population.
In summary, due their effectiveness, salt resistance, and speed of action against a variety of pathogens, VIP51 and VIP51(6–30) show good potential for further studies and rational design for development as antimicrobial agents. Moreover, contrary to some current treatments, these novel target-specific peptides have shown minimal toxicity toward host cells, as demonstrated in our study (hemolytic and cytotoxicity assays) and in models of airway inflammatory diseases (20, 21). Moreover, we did not observe side effects, according to the criteria established in the Guide for Laboratory Animal Care, with special emphasis in the evaluation of symptoms related to diarrhea and hypotension, which are two of the potential severe complications attributable to VIP overexposure. However, taking into account the relevance of VIP51 to the modulation of immune responses, precautions should be taken in considering how, when, and which infections could be preferentially treated with each of these VIP derivatives. Targeting of the innate immunity is an attractive approach because disease is a manifestation of the ability of pathogens to overcome or subvert host immune responses. However, although therapeutic agents with an immunomodulatory role are very attractive for treatment of patients with severe sepsis and high risk of death (53), such agents may interrupt protective host defense mechanisms or produce other deleterious effects that result in a worse outcome. On the other hand, the infection with Leishmania can be frequently accompanied by immunosuppression. This indicates that therapies directed to clear the primary infection and to avoid the development of secondary infections may be more attractive. Finally, by targeting membrane components that are critical for the pathogen, these peptides may overcome the resistance observed with existing drugs and could be used in combination with other therapeutics.
Supplementary Material
Acknowledgments
We thank Dr. F. Gamarro, Dr. S. Castanys, Dr. M. C. Lopez-Lopez, and M. Thomas (Institute of Parasitology and Biomedicine, Granada, Spain) for providing Leishmania strains, Dr. C. López-Iglesias (Barcelona University) for assistance in electron microscopy studies, Dr. M. Torrent (Pompeu Fabra University, Barcelona, Spain) for providing bacterial mutant strains, and members of the laboratory of S. M. B. for assistance and discussions. We also thank Drs. P. Anderson and V. Neubrand for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 AI031078 (to S. M. B.). This work was also supported by Excellence Grants from Junta de Andalucia (P09/CTS-4705) (to E. G.-R.) and European Cost Action (BM0802) (to E. G.-R.).

This article contains supplemental Fig. S1 and Tables S1–S6.
- VIP
- vasoactive intestinal peptide
- CLP
- cecal ligation and puncture
- LPG
- lipophosphoglycan
- PG
- phosphoglycan
- GIPL
- glycophosphatidylinositol lipid
- Man
- mannose
- KDO
- 3-deoxy-d-manno-octulosonic acid.
REFERENCES
- 1. Hotez P., Raff S., Fenwick A., Richards F., Jr., Molyneux D. H. (2007) Recent progress in integrated neglected tropical disease control. Trends Parasitol. 23, 511–514 [DOI] [PubMed] [Google Scholar]
- 2. Hotchkiss R. S., Karl I. E. (2003) The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348, 138–150 [DOI] [PubMed] [Google Scholar]
- 3. Augustyniak D., Nowak J., Lundy F. T. (2012) Direct and indirect antimicrobial activities of neuropeptides and their therapeutic potential. Curr. Protein Pept. Sci. 13, 723–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Said S. I., Mutt V. (1970) Polypeptide with broad biological activity: isolation from small intestine. Science 169, 1217–1218 [DOI] [PubMed] [Google Scholar]
- 5. Vaudry D., Gonzalez B. J., Basille M., Yon L., Fournier A., Vaudry H. (2000) Pituitary adenylate cyclase-activating polypeptide and its receptors: from structure to functions. Pharmacol. Rev. 52, 269–324 [PubMed] [Google Scholar]
- 6. Harmar A. J., Fahrenkrug J., Gozes I., Laburthe M., May V., Pisegna J. R., Vaudry D., Vaudry H., Waschek J. A., Said S. I. (2012) Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br. J. Pharmacol. 166, 4–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abad C., Martinez C., Juarranz M. G., Arranz A., Leceta J., Delgado M., Gomariz R. P. (2003) Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of Crohn's disease. Gastroenterology 124, 961–971 [DOI] [PubMed] [Google Scholar]
- 8. Delgado M., Abad C., Martinez C., Leceta J., Gomariz R. P. (2001) Vasoactive intestinal peptide prevents experimental arthritis by downregulating both autoimmune and inflammatory components of the disease. Nat. Med. 7, 563–568 [DOI] [PubMed] [Google Scholar]
- 9. Gonzalez-Rey E., Fernandez-Martin A., Chorny A., Martin J., Pozo D., Ganea D., Delgado M. (2006) Therapeutic effect of vasoactive intestinal peptide on experimental autoimmune encephalomyelitis: down-regulation of inflammatory and autoimmune responses. Am. J. Pathol. 168, 1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Said S. I. (1991) Vasoactive intestinal polypeptide: biologic role in health and disease. Trends Endocrinol. Metab. 2, 107–112 [DOI] [PubMed] [Google Scholar]
- 11. Onoue S., Ohmori Y., Endo K., Yamada S., Kimura R., Yajima T. (2004) Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide attenuate the cigarette smoke extract-induced apoptotic death of rat alveolar L2 cells. Eur. J. Biochem. 271, 1757–1767 [DOI] [PubMed] [Google Scholar]
- 12. Delgado M., Ganea D. (2003) Vasoactive intestinal peptide prevents activated microglia-induced neurodegeneration under inflammatory conditions: potential therapeutic role in brain trauma. FASEB J. 17, 1922–1924 [DOI] [PubMed] [Google Scholar]
- 13. Delgado M., Ganea D. (2003) Neuroprotective effect of vasoactive intestinal peptide (VIP) in a mouse model of Parkinson's disease by blocking microglial activation. FASEB J. 17, 944–946 [DOI] [PubMed] [Google Scholar]
- 14. El Karim I. A., Linden G. J., Orr D. F., Lundy F. T. (2008) Antimicrobial activity of neuropeptides against a range of micro-organisms from skin, oral, respiratory and gastrointestinal tract sites. J. Neuroimmunol. 200, 11–16 [DOI] [PubMed] [Google Scholar]
- 15. Delgado M., Anderson P., Garcia-Salcedo J. A., Caro M., Gonzalez-Rey E. (2009) Neuropeptides kill African trypanosomes by targeting intracellular compartments and inducing autophagic-like cell death. Cell Death Differ. 16, 406–416 [DOI] [PubMed] [Google Scholar]
- 16. Brogden K. A., Guthmiller J. M., Salzet M., Zasloff M. (2005) The nervous system and innate immunity: the neuropeptide connection. Nat. Immunol. 6, 558–564 [DOI] [PubMed] [Google Scholar]
- 17. Mayer M. L., Easton D. M., Hancock R. E. W. (2011) in Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies, Advances in Molecular and Cellular Microbiology, 18th Ed (Wang G., ed), pp. 195–220, CABI Publishing, Cambridge, MA [Google Scholar]
- 18. Onoue S., Misaka S., Yamada S. (2008) Structure-activity relationship of vasoactive intestinal peptide (VIP): potent agonists and potential clinical applications. Naunyn Schmiedebergs Arch Pharmacol. 377, 579–590 [DOI] [PubMed] [Google Scholar]
- 19. Onoue S., Misaka S., Ohmori Y., Sato H., Mizumoto T., Hirose M., Iwasa S., Yajima T., Yamada S. (2009) Physicochemical and pharmacological characterization of novel vasoactive intestinal peptide derivatives with improved stability. Eur. J. Pharm. Biopharm. 73, 95–101 [DOI] [PubMed] [Google Scholar]
- 20. Onoue S., Aoki Y., Matsui T., Kojo Y., Misaka S., Mizumoto T., Yamada S. (2011) Formulation design and in vivo evaluation of dry powder inhalation system of new vasoactive intestinal peptide derivative ([R(15, 20, 21), L(17), A(24,25), des-N(28)]-VIP-GRR) in experimental asthma/COPD model rats. Int. J. Pharm. 410, 54–60 [DOI] [PubMed] [Google Scholar]
- 21. Onoue S., Matsui T., Kato M., Mizumoto T., Liu B., Liu L., Karaki S., Kuwahara A., Yamada S. (2013) Chemical synthesis and formulation design of a PEGylated vasoactive intestinal peptide derivative with improved metabolic stability. Eur. J. Pharm. Sci. 49, 382–389 [DOI] [PubMed] [Google Scholar]
- 22. Ceraudo E., Murail S., Tan Y. V., Lacapère J. J., Neumann J. M., Couvineau A., Laburthe M. (2008) The vasoactive intestinal peptide (VIP) α-Helix up to C terminus interacts with the N-terminal ectodomain of the human VIP/pituitary adenylate cyclase-activating peptide 1 receptor: photoaffinity, molecular modeling, and dynamics. Mol. Endocrinol. 22, 147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Madeira da Silva L., Owens K. L., Murta S. M., Beverley S. M. (2009) Regulated expression of the Leishmania major surface virulence factor lipophosphoglycan using conditionally destabilized fusion proteins. Proc. Natl. Acad. Sci. U.S.A. 106, 7583–7588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Späth G. F., Lye L. F., Segawa H., Sacks D. L., Turco S. J., Beverley S. M. (2003) Persistence without pathology in phosphoglycan-deficient Leishmania major. Science 301, 1241–1243 [DOI] [PubMed] [Google Scholar]
- 25. Nevot M., Deroncele V., López-Iglesias C., Bozal N., Guinea J., Mercade E. (2006) Ultrastructural analysis of the extracellular matter secreted by the psychrotolerant bacterium Pseudoalteromonas antarctica NF3. Microb. Ecol. 51, 501–507 [DOI] [PubMed] [Google Scholar]
- 26. Zhang Y. (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y., Skolnick J. (2004) Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710 [DOI] [PubMed] [Google Scholar]
- 28. Trott O., Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neurath M. F., Fuss I., Kelsall B. L., Stüber E., Strober W. (1995) Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 182, 1281–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kihara N., de la Fuente S. G., Fujino K., Takahashi T., Pappas T. N., Mantyh C. R. (2003) Vanilloid receptor-1 containing primary sensory neurones mediate dextran sulphate sodium induced colitis in rats. Gut 52, 713–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Afonso L. C., Scott P. (1993) Immune responses associated with susceptibility of C57BL/10 mice to Leishmania amazonensis. Infect. Immun. 61, 2952–2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ohta K., Kajiya M., Zhu T., Nishi H., Mawardi H., Shin J., Elbadawi L., Kamata N., Komatsuzawa H., Kawai T. (2011) Additive effects of orexin B and vasoactive intestinal polypeptide on LL-37-mediated antimicrobial activities. J. Neuroimmunol. 233, 37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nikaido H. (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Snyder D. S., McIntosh T. J. (2000) The lipopolysaccharide barrier: correlation of antibiotic susceptibility with antibiotic permeability and fluorescent probe binding kinetics. Biochemistry 39, 11777–11787 [DOI] [PubMed] [Google Scholar]
- 35. Chorny A., Delgado M. (2008) Neuropeptides rescue mice from lethal sepsis by down-regulating secretion of the late-acting inflammatory mediator high mobility group box 1. Am. J. Pathol. 172, 1297–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abad C., Martinez C., Juarranz M. G., Arranz A., Leceta J., Delgado M., Gomariz R. P. (2003) Therapeutic effects of vasoactive intestinal peptide in the trinitrobenzene sulfonic acid mice model of Crohn's disease. Gastroenterology 124, 961–971 [DOI] [PubMed] [Google Scholar]
- 37. Delgado M., Martinez C., Pozo D., Calvo J. R., Leceta J., Ganea D., Gomariz R. P. (1999) Vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activation polypeptide (PACAP) protect mice from lethal endotoxemia through the inhibition of TNF-α and IL-6. J. Immunol. 162, 1200–1205 [PubMed] [Google Scholar]
- 38. McGwire B. S., Kulkarni M. M. (2010) Interactions of antimicrobial peptides with Leishmania and trypanosomes and their functional role in host parasitism. Exp. Parasitol. 126, 397–405 [DOI] [PubMed] [Google Scholar]
- 39. McConville M. J., Homans S. W., Thomas-Oates J. E., Dell A., Bacic A. (1990) Structures of the glycoinositolphospholipids from Leishmania major: a family of novel galactofuranose-containing glycolipids. J. Biol. Chem. 265, 7385–7394 [PubMed] [Google Scholar]
- 40. Ma D., Russell D. G., Beverley S. M., Turco S. J. (1997) Golgi GDP-mannose uptake requires Leishmania LPG2. A member of a eukaryotic family of putative nucleotide-sugar transporters. J. Biol. Chem. 272, 3799–3805 [PubMed] [Google Scholar]
- 41. Baltzer S. A., Brown M. H. (2011) Antimicrobial peptides: promising alternatives to conventional antibiotics. J. Mol. Microbiol. Biotechnol. 20, 228–235 [DOI] [PubMed] [Google Scholar]
- 42. Iyer R., Iverson T., Accardi A., Miller C. (2002) A biological role for prokaryotic ClC chloride channels. Nature 419, 715–718 [DOI] [PubMed] [Google Scholar]
- 43. Sun Q.-Q., Prince D. A., Huguenard J. R. (2003) Vasoactive intestinal polypeptide and pituitary adenylate cyclase-activating polypeptide activate hyperpolarization-activated cationic current and depolarize thalamocortical neurons in vitro. J. Neurosci. 23, 2751–2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. He J., Eckert R., Pharm T., Simanian M. D., Hu C., Yarbrough D. K., Qi F., Anderson M. H., Shi W. (2007) Novel synthetic antimicrobial peptides against Streptococcus mutans. Antimicrob. Agents Chemother. 51, 1351–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Epand R. F., Schmitt M. A., Gellman S. H., Sen A., Auger M., Hughes D. W., Epand R. M. (2005) Bacterial species selective toxicity of two isomeric alpha/beta-peptides: role of membrane lipids. Mol. Membr. Biol. 22, 457–469 [DOI] [PubMed] [Google Scholar]
- 46. Delgado M., Pozo D., Ganea D. (2004) The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol. Rev. 56, 249–290 [DOI] [PubMed] [Google Scholar]
- 47. Mor A. (2009) Multifunctional host defense peptides: antiparasitic activities. FEBS J. 276, 6474–6482 [DOI] [PubMed] [Google Scholar]
- 48. Naderer T., Vince J. E., McConville M. J. (2004) Surface determinants of Leishmania parasites and their role in infectivity in the mammalian host. Curr. Mol. Med. 4, 649–665 [DOI] [PubMed] [Google Scholar]
- 49. Yao C., Donelson J. E., Wilson M. E. (2003) The major surface protease (MSP or GP63) of Leishmania sp.: biosynthesis, regulation of expression, and function. Mol. Biochem. Parasitol. 132, 1–16 [DOI] [PubMed] [Google Scholar]
- 50. Kulkarni M. M., McMaster W. R., Kamysz E., Kamysz W., Engman D. M., McGwire B. S. (2006) The major surface metalloprotease of the parasitic protozoan, Leishmania, protects against antimicrobial peptide-induced apoptotic killing. Mol. Microbiol. 62, 1484–1497 [DOI] [PubMed] [Google Scholar]
- 51. Kleczka B., Lamerz A. C., van Zandbergen G., Wenzel A., Gerardy-Schahn R., Wiese M., Routier F. H. (2007) Targeted gene deletion of Leishmania major UDP-galactopyranose mutase leads to attenuated virulence. J. Biol. Chem. 282, 10498–10505 [DOI] [PubMed] [Google Scholar]
- 52. Reiner S. L., Locksley R. M. (1995) The regulation of immunity to Leishmania major. Annu. Rev. Immunol. 13, 151–177 [DOI] [PubMed] [Google Scholar]
- 53. Eichacker P. Q., Parent C., Kalil A., Esposito C., Cui X., Banks S. M., Gerstenberger E. P., Fitz Y., Danner R. L., Natanson C. (2002) Risk and the efficacy of antiinflammatory agents: retrospective and confirmatory studies of sepsis. Am. J. Respir. Crit. Care Med. 166, 1197–1205 [DOI] [PubMed] [Google Scholar]
- 54. Hancock R. E. (1997) Peptide antibiotics. Lancet 349, 418–422 [DOI] [PubMed] [Google Scholar]
- 55. Friedrich C. L., Moyles D., Beveridge T. J., Hancock R. E. (2000) Antibacterial action of structurally diverse cationic peptides on Gram-positive bacteria. Antimicrob. Agents Chemother. 44, 2086–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Medzhitov R., Janeway C., Jr. (2000) Innate immune recognition: mechanisms and pathways. Immunol. Rev. 173, 89–97 [DOI] [PubMed] [Google Scholar]
- 57. Pulido D., Moussaoui M., Andreu D., Nogués M. V., Torrent M., Boix E. (2012) Antimicrobial action and cell agglutination by the eosinophil cationic protein are modulated by the cell wall lipopolysaccharide structure. Antimicrob. Agents Chemother. 56, 2378–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.