

Background: Selenoproteins are important enzymes involved in antioxidant defense, redox homeostasis, and signaling.
Results: We report that, in HEK293 cells, several selenoproteins are selectively up-regulated in response to H2O2 treatment.
Conclusion: We identify a novel translational control mechanism for selenoproteome regulation.
Significance: Antioxidant selenoproteins are regulated by oxidative stress when selenium is limiting.
Keywords: Glutathione Peroxidase, Oxidative Stress, Selenium, Thioredoxin Reductase, Translation Regulation, SECIS, UGA Recoding, Selenoproteome
Abstract
Selenocysteine is inserted into selenoproteins via the translational recoding of a UGA codon, normally used as a stop signal. This process depends on the nature of the selenocysteine insertion sequence element located in the 3′ UTR of selenoprotein mRNAs, selenium bioavailability, and, possibly, exogenous stimuli. To further understand the function and regulation of selenoproteins in antioxidant defense and redox homeostasis, we investigated how oxidative stress influences selenoprotein expression as a function of different selenium concentrations. We found that selenium supplementation of the culture media, which resulted in a hierarchical up-regulation of selenoproteins, protected HEK293 cells from reactive oxygen species formation. Furthermore, in response to oxidative stress, we identified a selective up-regulation of several selenoproteins involved in antioxidant defense (Gpx1, Gpx4, TR1, SelS, SelK, and Sps2). Interestingly, the response was more efficient when selenium was limiting. Although a modest change in mRNA levels was noted, we identified a novel translational control mechanism stimulated by oxidative stress that is characterized by up-regulation of UGA-selenocysteine recoding efficiency and relocalization of SBP2, selenocysteine-specific elongation factor, and L30 recoding factors from the cytoplasm to the nucleus.
Introduction
Selenium is an essential trace element implicated in muscular, thyroid, immune, and brain functions (1, 2). Numerous epidemiological and intervention studies have recognized the link between low levels of selenium in human body fluids and an increased risk of cancers, including prostate, lung, and colon cancer. Selenium is incorporated as selenocysteine, a rare amino acid, into at least 25 human selenoproteins, an essential family of redox enzymes (3). Many selenoproteins are involved in antioxidant defense, redox homeostasis, and redox signaling through the action of glutathione peroxidase (Gpx1-Gpx6), methionine sulfoxide reductase (MsrB1 or SelX), thioredoxin reductases (TR1-TR3), and endoplasmic reticulum (ER)3 selenoproteins (Sel15, SelS, SelK, and SelM). Nevertheless, about half of the selenoproteins are without a known function, and the regulation of the selenoproteome in response to oxidative stress remains uncharacterized.
Selenocysteine is cotranslationally inserted by the ribosome using an UGA codon, normally read as a stop signal. The 3′ UTRs of selenoprotein mRNAs contain a secondary structure called the selenocysteine insertion sequence (SECIS) element. The SECIS controls the faithful recoding of the UGA codon as selenocysteine, thereby preventing the formation of truncated proteins (for reviews, see Refs. 2, 4–7). Two categories of recoding factors have been characterized as components of the selenocysteine insertion machinery, including tRNASec binding proteins (selenocysteine-specific elongation factor (EFsec), SECp43, O-phosphoseryl-tRNA(Sec) kinase (PSTK), and selenocysteine synthase (8–13)) and SECIS binding proteins (SECIS binding protein 2 (SBP2), ribosomal protein L30 (rpL30), translation initiation factor 4A3 (eIF4A3), and nucleolin (14–17)). However, despite much effort, the precise mechanism of selenoprotein synthesis and regulation remains elusive. Many studies have shown that selenium levels differentially control the expression of the selenoproteome in mammals and cultured cells. This regulation occurs moderately at the level of mRNA transcription/stability, but mostly during translation, to maintain essential selenoproteins at the expense of the others (18–21). This has been particularly described for Gpx1 and Gpx4, two members of the Gpx family that are ubiquitously expressed. Gpx1 expression was more affected than Gpx4 by selenium availability (22), essentially via a translational control mechanism (23). We established HEK293 cell lines stably expressing luciferase-SECIS constructs to quantitatively evaluate the translational regulation of selenoprotein expression (23, 24). Indeed, we demonstrated that, in response to selenium level variations, selenocysteine insertion was driven by the nature of the SECIS element. To date, no stimulus other than selenium has been shown to stimulate selenoprotein expression.
Reactive oxygen species (ROS) are generated either endogenously during cellular metabolism, mostly by mitochondria, or exogenously from environmental stress. ROS are also emerging as essential signaling molecules at low concentrations that transduce signals from the mitochondrial compartment to other compartments of the cell. To maintain ROS homeostasis, cells possess enzymatic antioxidant systems such as Gpxs, TRs, catalase, superoxide dismutase, and non-enzymatic antioxidant systems, including glutathione (GSH), vitamin E, and ascorbate. Among the selenoproteins, the Gpxs are implicated in the elimination of peroxides, whereas the TRs are critical for many cellular functions involving thiol-dependent redox mechanisms. Gpxs reduce hydrogen and lipid peroxides using GSH as a cofactor, which is subsequently recycled by glutathione reductases (25). TRs catalyze the NADPH-dependent reduction of oxidized thioredoxin and, therefore, participate in the defense against oxidative stress during DNA synthesis and in redox signaling (26). In addition to thioredoxin, a vast range of small molecules can also be reduced by TRs and participate in its antioxidant function. In parallel, the ER-resident selenoproteins, which include SelS, SelK, Sel15, and SelM, have important functions in protein folding and the ER stress response (27). ROS can severely damage a variety of macromolecules, including proteins, lipids, and nucleic acids. Depending on the nature of oxidative damage, these oxidized molecules can be repaired by various cellular machineries, several of which involve selenoproteins such as Gpxs, SelX, or TRs. When ROS production is overwhelming, oxidized macromolecules accumulate, hamper biological functions, and lead to pathological conditions. Down-regulation of selenoprotein expression is expected to increase ROS production and ER stress, which would lead to accumulation of oxidized molecules and DNA damage, and to impair cellular homeostasis and cell cycle progression. Accumulation of ROS-induced cellular damage is associated with cancer, neurological disorders, atherosclerosis, inflammation, and aging. ROS have been proposed to link the selenium activity in mammals with these pathologies (28).
The transcriptional regulation of the antioxidant defense has been well described in response to oxidative stress. ROS induce a rapid activation of the NRF2 and NF-κB transcription factors (29). Their targets include a multitude of genes involved in the stress response, antioxidant activity, the anti-inflammatory response, DNA repair, molecular chaperones, proteasome systems, and two selenoproteins, Gpx2 (mostly expressed in the intestine) and TR1 (an essential cytoplasmic enzyme). The degree of expression of these genes involves other transcription factors (30, 31). A link between selenium, selenoprotein expression, and the antioxidant response has been established via NRF2, which is induced in certain cell lines during severe selenium deficiency or complete loss of selenoproteins obtained by tRNASec gene inactivation (32). The remarkable feature of selenoprotein expression lies in the many levels of control. Indeed, the complex network of regulation during transcription, translation, and mRNA stabilization allows a hierarchical expression of these antioxidant components (6). Whether and how the selenoproteins that are involved in antioxidant defense are regulated by oxidative stress remains to be studied.
In this work, we investigated the regulation of selenoprotein expression in the context of H2O2-induced oxidative stress as a function of selenium concentration in the culture media, defined as control (Ctrl) and supplemented (Sup) conditions. We used HEK293 cells in our study because selenoproteins are expressed at significant levels in this cell line (23, 24) and also because we developed cellular tools to quantitatively study the recoding of UGA as selenocysteine in this particular cell line (23). We found that down-regulation of the antioxidant defense in the Ctrl condition was linked to a selective reduction of several selenoproteins. Thus, HEK293 cells grown in selenium-limiting conditions produced more ROS, accumulated more oxidized proteins, and were more sensitive to oxidative challenge. Remarkably, in this condition (i.e. Ctrl), several selenoproteins, including Gpx1, Gpx4, TR1, SelS, SelK, and Sps2, were specifically up-regulated by H2O2 treatment. However, only limited changes were observed in the Sup condition. We determined that H2O2-induced up-regulation of selenoproteins is due to a novel translational control mechanism leading to the stimulation of UGA recoding efficiency in selenoprotein mRNAs.
EXPERIMENTAL PROCEDURES
Material and Chemicals
Luciferase reporter plasmids and cell lines were generated as in Refs. 23, 24). Cell culture media, FCS, and supplements were purchased from Invitrogen. H2O2, t-BHP, Cu-OOH, NADPH, thioredoxin, L-GSH, glutathione reductase, and 5,5′-dithio-bis(2-dinitrobenzoic acid) were purchased from Sigma-Aldrich. Antibodies were purchased from Euromedex (Gpx1, catalog no. 2971; Gpx4, catalog no. 3649-1; TR1, catalog no. LF-MA0015; Sel15, catalog no. 3364; and SelP, catalog no. 3718), Sigma-Aldrich (SelS, catalog no. HPA010025; TR2, catalog no. HPA003323; actin, catalog no. A1978; tubulin, catalog no. T9026), Covalab (EFsec (23)), and ProteinTech Group (SBP2, catalog no. 1289-1-AP).
Cell Lines and Cell Culture Conditions
HEK293 cells (Invitrogen) were grown and maintained as described in Ref. 23, 24. Ctrl and Sup culture media were made according to Refs. 23, 24) and the references therein. The same lot number of FCS was used in all experiments because selenium is provided by this source. The selenium concentration in the Ctrl medium was determined by inductively coupled plasma mass spectrometry (ICP-MS) to be 15 nm. Sup medium was made by adding 30 nm sodium selenite to the Ctrl medium, and, therefore, it contained 45 nm selenium. One day prior to H2O2 exposure, cells were plated in 100-mm dishes in either Ctrl or Sup medium. When the cells reached 70% confluency, the medium was changed to a freshly prepared solution of FCS-free DMEM that contained different concentrations of H2O2, Cu-OOH, or t-BHP as indicated. After 30 min of exposure, the media were removed and replaced by the appropriate ones. Cell extracts were harvested 24 h post-treatment in 300 μl of lysis buffer (23, 24). Cellular fractionations were performed with the ProteoJET cytoplasmic and nuclear protein extraction kit (Fermentas) following the instructions of the manufacturer. Protein concentrations were measured using the DC kit (Bio-Rad) in microplate assays.
Measurement of Mitochondrial Superoxide Production
HEK293 cells that were grown in Ctrl or Sup medium were incubated with 5 μm MitoSOXTM Red (Invitrogen) for 15 min at 37 °C in 5% CO2. Antimycine A (2.5 μg/ml), an inhibitor of respiratory complex III, was added as indicated. Oxidation of MitoSOXTM Red reagent by superoxide produced red fluorescence that was measured as a function of time on a Partech flow cytometer according to the instructions of the manufacturer. Fluorescence kinetics were determined from three independent experiments.
Cell Cycle Analysis
HEK293 cells were seeded in 6-well plates and grown in either Ctrl or Sup medium for 1 day. Cells were treated with 0 or 300 μm H2O2 for 30 min in serum-free medium and then harvested 24 h later. Cells were fixed in 70% ethanol/PBS for 2 h at −20 °C, rehydrated in PBS, and stained for 2 h at 37 °C with propidium iodide (50 μg/ml) and RNase A (250 μg/ml). Cell fluorescence was then analyzed by flow cytometry. Cell cycle analyses were done using Flowmax and Multicycle software.
Immunoblotting
Protein extracts (50 μg) were separated in BisTris NuPAGE Novex Midi gels (Invitrogen). Proteins were transferred onto nitrocellulose membranes that were probed with primary antibodies (as indicated) and HRP-conjugated anti-rabbit or anti-mouse secondary antibodies. Detection of total carbonyl contents in cell extracts was performed with the OxyblotTM protein oxidation detection kit (Millipore). Protein carbonyls from cell extracts (5 μg) were derived using 2,4-dinitrophenylhydrazine, separated on a 10% SDS-PAGE, transferred to a nitrocellulose membrane, and detected with an anti-2,4-dinitrophenol antibody according to the instructions of the manufacturer. The chemiluminescence signal was detected using an ECL Advance Western blotting detection kit (GE Healthcare) and an LAS3000 charge-coupled device (CCD) camera (GE Healthcare).
Enzymatic Activities
Gpx activity was measured in an enzymatic coupled assay according to methods described previously (19). The reaction mixture was composed of 50 μg of protein extract, 0.25 mm NADPH, 2 mm reduced l-glutathione, and 1.5 IU of glutathione reductase adjusted to a total volume of 250 μl with 50 mm potassium phosphate buffer (pH 7.5). The reaction was started by the addition of 300 nm t-BHP, and consumption of NAPDH was followed at 340 nm with a Fluostar reader. Enzymatic activities were expressed as nanomoles of glutathione per minute per milligram (milliunits per milligram of protein). TR activity was measured as described in Ref. 33. Briefly, the catalytic reduction of oxidized thioredoxin by TR is coupled with the oxidation of one molecule of NADPH. In this work, the recycling of NADP in NADPH is associated with the conversion of 5,5′-dithio-bis(2-dinitrobenzoic acid) to 5-thio-2-nitrobenzoic acid, which is visible at 410 nm. The reaction mixture was composed of 50 μg of protein extract, 0.2 mm NADPH, 10 mm EDTA, and 0.2 mg/ml BSA, adjusted to a total volume of 250 μl with 50 mm potassium phosphate buffer (pH 7.5). The reaction was started by the addition of 25 mm 5,5′-dithio-bis(2-dinitrobenzoic acid), and the production of 5-thio-2-nitrobenzoic acid was followed at 340 nm with a Fluostar reader. Enzymatic activities were expressed as nanomoles of NADPH per minute per milligram (milliunits per milligram of protein). In every enzymatic analysis, the assays were performed in duplicate in three independent experiments.
RNA Extraction and RT Quantitative PCR
Total RNAs were extracted using the Nucleospin RNA II kit (Macherey Nagel). Synthesis of cDNA was carried out using a Transcriptor high-fidelity cDNA synthesis kit (Roche Applied Science) according to the instructions of the manufacturer. Real-time PCR was performed in triplicate using LightCycler® 480 SYBR Green I Master (Roche Applied Science). The PCR program was 95 °C for 5 min, 45 cycles of 95 °C for 10 s, 60 °C for 20 s, and 72 °C for 20 s. Data were analyzed using LightCycler® 480 software and normalized relative to the mRNA levels of Hrpt. The primers used are described in Ref. 34.
Evaluation of UGA Recoding Efficiency Using Luciferase Activity
HEK293 cells that stably express the Luc UGA/Gpx4 or Luc UGA/Gpx1 constructs (23) were plated in the different culture media 1 day prior to H2O2, Cu-OOH, or t-BHP exposure. Cells were harvested 24 h later in lysis buffer and analyzed for luciferase activity as described in Refs. 23, 24 with a Lumistar reader (BMGLabtech). Luciferase activity in arbitrary units was expressed relative to micrograms of protein.
Fluorescence Microscopy
Cells were seeded on coverslips in 6-well plates, treated as described above, washed with PBS, fixed in 4% paraformaldehyde/PBS for 15 min at room temperature, permeabilized with 0.5% Triton/PBS for 8 min, and blocked in 1% BSA/PBS for 30 min. Coverslips were incubated with anti-SBP2 antibody at 4 °C overnight, washed with PBS, and incubated with an Alexa Fluor 488 dye-conjugated anti-rabbit secondary antibody. Nuclei were stained with DAPI. Epifluorescence images were captured in 10 z-sections on an AxioOberver Z1 microscope (Zeiss) equipped with an Evolve electron multiplying charge-coupled device (EMCCD) camera (Roper Scientific). Image analysis was performed with National Institutes of Health ImageJ software.
RESULTS
The Proliferation of HEK293 Cells Is Induced in Response to Oxidative Stress
We grew HEK293 cells in Ctrl and Sup culture media that contained 15 and 45 nm of selenium, respectively. These conditions have been shown previously to regulate selenoprotein expression in various cell lines after 24 h (18–21, 23). In most cases, Ctrl medium is considered to be a selenium-limiting condition (16, 35). To evaluate the impact of H2O2 treatment on cell proliferation at two selenium levels, we analyzed cell morphology by optical microscopy (Fig. 1A) and the cell cycle profile by flow cytometry (Fig. 1, B and C). First, we noted that H2O2-treated cells reached a higher confluence 24 h post-treatment (Fig. 1A), in comparison with untreated cells, under both Ctrl and Sup conditions. This result suggested that H2O2 treatment stimulated cell growth and did not induce cell cycle arrest. To confirm this observation, we analyzed the influence of both selenium and oxidative stress on the cell cycle profile by quantifying the DNA amount per cell using propidium iodide staining (Fig. 1, B and C). We noted that selenium concentration had virtually no effect on the cell cycle profile. However, a profound alteration of the cell cycle profile was found in cells treated with H2O2. Cell proliferation was stimulated strongly, as illustrated by the increased ratio of cells in G2 versus G1 phase (Fig. 1C). Our results show that cell proliferation is highly sensitive to H2O2 treatment, independent of the selenium concentration.
FIGURE 1.
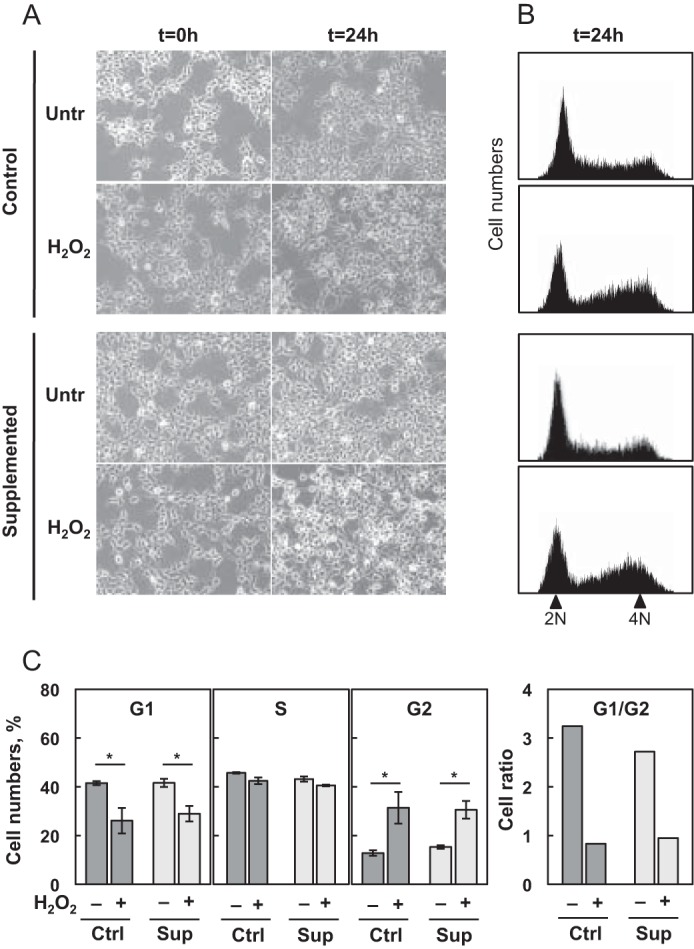
Proliferation of HEK293 cells as a function of selenium levels and H2O2 treatment. HEK293 cells were grown under Ctrl or Sup conditions for 24 h and then submitted to 0 or 300 μm H2O2 treatment (30 min). A, cells were observed in phase-contrast microscopy at time 0 and 24 h post-treatment to compare respective confluency and estimate cell proliferation and phenotype. Untr, untreated. B, cell cycle profiles were determined by propidium iodide staining 24 h post-treatment. 2N represents relative diploid, not replicated equivalent of DNA (G1), 4N represents relative tetraploid, replicated equivalent of DNA (G2/M). C, the relative proportions of cells in G1, S, or G2 phases were obtained by Flowmax and Multicycle software analyses. The results represent the mean ± S.D. of three independent experiments. *, p < 0.05.
Selenium Concentration Modified the Oxidative Status and Antioxidant Capacity of HEK293 Cells
The levels of selenium greatly regulate the expression of several selenoproteins involved in antioxidant defense and, therefore, could alter the steady-state levels of ROS produced in living cells. ROS are essential signaling molecules that transduce signals from the mitochondria to other compartments of the cell and alter cell cycle progression. To evaluate the steady-state levels of ROS produced in mitochondria of living cells as a function of selenium levels, we used a fluorescent probe (Fig. 2A). We observed that the rate production of the MitoSOX red signal was similar under Ctrl and Sup conditions. To evaluate how mitochondrial function in antioxidant defense is modulated by selenium levels, we challenged the HEK293 cells grown initially in the different media with a complex III inhibitor. Under Ctrl conditions, the addition of antimycine led to a 2-fold increase in the rate production of ROS, whereas no effect was detected under Sup conditions. Taken together, our results suggest that selenium levels, which modulate selenoprotein expression, greatly impact ROS production and the antioxidant capacity of the cell. Therefore, the increased expression levels of several selenoproteins under Sup conditions may participate in the efficient scavenging of ROS when cells are challenged with oxidative stress.
FIGURE 2.
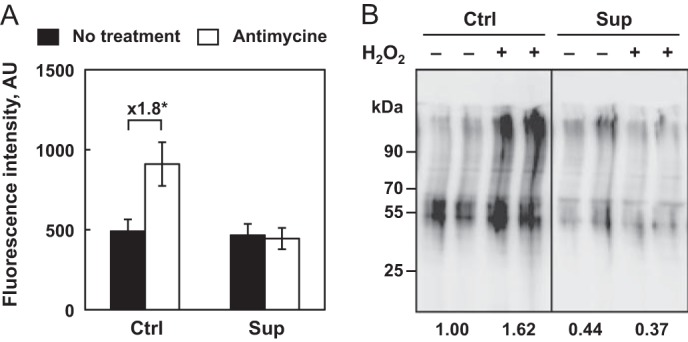
Rate of ROS production and protein damage as a function of selenium levels and oxidative stress. A, rate of mitochondrial ROS production. HEK293 cells were grown under Ctrl or Sup conditions for 24 h and analyzed for MitoSOX Red fluorescence intensity in the absence or presence of antimycine, an inhibitor of mitochondrial complex III. This assay allows rapid quantification of ROS production in the mitochondria of live cells using a flow cytometer. The data represent the mean ± S.D. of three independent experiments. The differences between fluorescence intensities expressed in arbitrary units (AU), that are significant (*, p < 0.05) are indicated above the respective brackets in fold change. B, HEK293 cells were grown and treated as described in Fig. 1. Cell extracts were harvested 24 h post-treatment and evaluated for the presence of protein carbonyl using an Oxiblot commercial kit. The numbers below the gel represent the mean intensity of the duplicate samples relative to the untreated cells under the Ctrl condition, which is set as 1.
When ROS are generated, they react with different components in various cellular compartments to generate oxidized macromolecules that accumulate thereafter. Among the various oxidative lesions, protein carbonylation has been used extensively to monitor oxidative damage. Therefore, to evaluate the impact of selenium levels on ROS-induced cellular damage, we analyzed the presence of carbonylated proteins in extracts from cells grown under Ctrl and Sup conditions with or without H2O2 treatment. As illustrated in Fig. 2B, the levels of oxidized proteins detected 24 h post-treatment were inversely correlated to the levels of selenium in the culture media. An approximate 2-fold increase in carbonylated proteins was observed between the Ctrl and Sup extracts, demonstrating the important role of selenium in protecting cells against oxidative damage. Then, when we challenged HEK293 cells with H2O2, we confirmed that selenium supplementation conferred almost complete resistance to oxidative stress, in contrast to the Ctrl conditions (Fig. 2B).
Selenium Levels Influenced the Response of Selenoprotein Expression to Oxidative Stress
The response to H2O2-induced oxidative stress is highly dependent on selenium levels in the growth media. We wanted to identify which selenoproteins were altered by selenium concentration and how H2O2 treatment subsequently modified their expression. Therefore, we performed extensive analyses of the well characterized members of the selenoproteome at the levels of mRNA, protein, and enzymatic activities. Among the Gpxs, only Gpx1 and Gpx4, which are expressed ubiquitously in the cytoplasm and mitochondria, respectively, are abundant in HEK293 cells. TR1 and TR2 are expressed ubiquitously in the cytoplasm and mitochondria, respectively, whereas TR3 is only detected in the testis in the nucleus or ER. Selenoproteins that are present in the ER have been shown recently to participate in protein folding with the assistance of ER chaperones and/or ER-associated degradation of misfolded proteins (27). Sps2 (selenophosphate synthetase 2) participates in selenocysteine tRNA biosynthesis and, therefore, controls selenoprotein expression.
Given the availability of enzymatic assays, Gpx and TR activities were evaluated 24 h post-treatment under the different growth conditions (Fig. 3, A and B, respectively). First, we confirmed that these selenoprotein activities were stimulated by selenium bioavailability of the culture media. Interestingly, Gpx was approximately two times more sensitive to selenium variation than TR. In the context of H2O2 treatment, a significant stimulation of Gpx and TR activities was detected under Ctrl but not Sup conditions. This result indicates that Gpx and TR were induced via the antioxidant defense mechanism in response to oxidative stress. However, as observed in Fig. 2, this activation was not sufficient to control the rate of ROS production or prevent the protein oxidation under Ctrl conditions. On the contrary, selenium supplementation, which induced the levels of Gpx and TR activities, seemed sufficient to cope with oxidative stress.
FIGURE 3.

Analysis of selenoprotein levels and enzymatic activities as a function of selenium concentration and H2O2-induced oxidative stress. A and B, HEK293 cells were grown and treated as described in Fig. 1. Cell extracts were evaluated for Gpx (A) and TR (B) activities. The data represent the mean ± S.D. of three independent experiments. The fold changes in activities are indicated above the respective brackets. *, p < 0.05. C and D, selenoprotein expression was investigated in protein extracts by immunoblotting. The results were normalized to the signal for actin (E). The relative expression is indicated with the untreated Ctrl condition set as 1. Selenoproteins are sorted as up-regulated (C) and poorly affected (D) in response to oxidative stress.
To understand the hierarchy of the selenoprotein expression in response to selenium variation and oxidative stress, we performed Western blot analyses for TR1, TR2, Gpx1, Gpx4, SelS, SelM, SelK, SelP, and Sps2 under different conditions (Fig. 3C). Our first observation was that, even though Western immunoblotting is a semiquantitative methodology, selenoproteins are differentially regulated by selenium levels, with the following hierarchy (from highly to poorly regulated): SelK > Gpx1 > Gpx4, TR1 > SelS, SelP, SelM, TR2 > Sps2 (Fig. 3, C and D). Then, in agreement with enzymatic activities for Gpx and TR, we observed that selenoprotein expression was differentially regulated by H2O2 treatment in Ctrl medium. Indeed, although Gpx1, Sps2 and SelK were highly stimulated by oxidative stress treatment, Gpx4, SelS, and TR1 were increased only moderately, but significantly (see Fig. 3C). Strikingly, the levels of TR2, which is the only mitochondrial TR, was decreased moderately in response to H2O2 treatment under both Ctrl and Sup conditions (Fig. 3D). In contrast, the expression of SelP remained unchanged (Fig. 3D). Interestingly, for all selenoproteins except for Sps2 and TR2, no variation was noted under the Sup condition in response to H2O2, again demonstrating a strong influence of selenium bioavailability in the response to oxidative stress.
Selenoprotein mRNA Levels Were Regulated Poorly by Selenium Availability and Oxidative Stress
Regulation of gene expression at the level of transcription or mRNA stability is a well characterized response to oxidative stress. First, the activation of NRF2 and NF-κB transcription factors by H2O2 treatment could potentially target several selenoprotein genes (36). In addition, nonsense-mediated mRNA decay, which is a quality control mechanism that selectively degrades mRNAs harboring a premature stop (also referred as nonsense) codon, could also alter selenoprotein expression. In fact, depending on the animal tissue or mammalian cell line, only a limited subset of selenoprotein mRNAs were sensitive to selenium variation, including Gpx1, SelH, SelW (37), and Gpx4 (38). In HEK293 cells, 20 selenoprotein mRNA levels were measured over several orders of magnitude among the 25 encoded in the genome, with Gpx2, Gpx6, and Dio1/2 and 3 being undetectable. Surprisingly, we observed very modest changes in selenoprotein mRNA levels in response to selenium and/or H2O2 treatment (Fig. 4). First, we found that only the levels of SelT mRNA were significantly up-regulated between Ctrl and Sup conditions, whereas the other 19 selenoprotein mRNAs remained insensitive to selenium variation (Fig. 4A). In addition, we found that only a few mRNAs were moderately affected by H2O2 exposure (Fig. 4, B and C). SelN, SelX, Sel15, and SelT mRNAs were down-regulated after oxidative stress. Interestingly, this alteration was only observed in Sup medium for the TR1 and Sel15 mRNAs. SelX and SelN mRNAs were reduced by 50–70% in response to H2O2 treatment under both Ctrl and Sup conditions. Our data indicate a selective but limited redox control of selenoprotein mRNA abundance and suggest a more pronounced translational control of selenoprotein expression.
FIGURE 4.

Evolution of selenoprotein mRNA levels as a function of selenium concentration and H2O2-induced oxidative stress. HEK293 cells were grown and treated as described in Fig. 1. The steady-state levels of mRNAs were measured by RT quantitative PCR and normalized to Hrpt mRNA. The data represent the mean ± S.D. of three independent experiments in a logarithmic scale. Differences greater than 50% between levels that are significant (*, p < 0.05) are indicated within the histograms. The influence of selenium is shown in A. The responses to oxidative stress under Ctrl and Sup conditions are represented in B and C, respectively.
Efficiency of Selenocysteine Insertion in Selenoproteins Was Highly Regulated in Response to Oxidative Stress
The unusual translational mechanism involved in selenoprotein synthesis is a checkpoint that can be modulated by environmental stimuli. It is well documented that selenium stimulates selenoprotein expression by increasing the UGA-selenocysteine recoding efficiency. To determine whether the change in selenoprotein expression observed in response to oxidative stress is linked to a change in selenocysteine insertion efficiency, we used our HEK293 cell lines that stably express luciferase reporter constructs (23). These cells were specifically designed to quantify UGA recoding activity. Briefly, the luciferase coding sequence is interrupted by a UGA codon that can be recoded as selenocysteine when a SECIS element is cloned into the 3′ UTR (Fig. 5A), allowing the production of a fully active enzyme. However, when the UGA codon is read as a stop signal, a truncated and, therefore, inactive protein is produced. The nature of the SECIS determines the response to selenium variation (23, 39). Our cell lines transfected with the reporter containing the SECIS from either Gpx4 or Gpx1 were named Luc UGA/gpx4 and Luc UGA/gpx1, respectively. As observed in previous work (23) and in Fig. 5, luciferase activity was highly dependent on selenium availability. A dose-response experiment with 50, 150, and 300 μm H2O2 was performed with the Luc UGA/gpx4 cell line in comparison to untreated cells. Interestingly, as illustrated in Fig. 5B for Ctrl medium (dark gray bars), oxidative stress induced a 5-fold increase in UGA recoding efficiency. In contrast, in cells grown in Sup medium (Fig. 5B, light gray bars), the luciferase activity was only slightly stimulated by H2O2 treatment. To take into account a possible variation in luciferase transcript levels, we performed RT quantitative PCR analyses (Fig. 5C). We observed a modest but significant increase in luciferase mRNA levels in response to H2O2 treatment. These changes minimize the effect of oxidative stress on UGA recoding in Sup medium (Fig. 5D). Therefore, the differential response of UGA recoding to oxidative stress as a function of selenium levels is in agreement with our observation from enzymatic activities and Western blot analyses. It is clear that the enhancement of the selenocysteine insertion efficiency in response to H2O2 treatment is the main regulator of selenoprotein expression under Ctrl conditions.
FIGURE 5.

The efficiency of recoding UGA as selenocysteine is influenced by selenium concentration and H2O2-induced oxidative stress. A, representation of the luciferase reporter assay used for the analysis. The open reading frame of the firefly luciferase with a UGA/Sec codon at position 258 is linked to either the gpx1 or gpx4 SECIS element. HEK293 cells that stably express the Luc UGA/gpx4 reporter construct were grown and treated for 30 min with increasing concentrations of H2O2 (B), cumene hydroperoxide (E), and ter-butyl hydroperoxide (F). Luciferase activities were normalized to the protein concentration of the extract. UGA recoding efficiencies are calculated as the percentage of the normalized luciferase activity obtained in the untreated Ctrl extract. C, relative luciferase mRNA levels as a function of selenium concentration and H2O2 treatment in Luc UGA/Gpx4-expressing cells normalized to the mRNA levels of Hrpt. D, ratio of UGA recoding efficiency obtained in B over the mRNA levels obtained in C. The results were expressed relative to untreated Ctrl conditions. The cell lines expressing the Luc UGA/gpx1 (G) and Luc UGA/gpx4 (H) reporter constructs were treated with 300 μm of H2O2, harvested at time 0 and 8, 24, 48, and 72 h post-treatment, and analyzed as described above. Cells were either grown in Ctrl (gray squares) or Sup (white squares) conditions. UGA recoding efficiencies are calculated as the percentage of the normalized luciferase activity obtained in the untreated Ctrl extract at time 0. Data represent the mean ± S.D. of three independent experiments.
To extend our analysis to different sources of oxidative stress, we used cumene hydroperoxide (Cu-OOH) or t-BHP instead of H2O2. As illustrated in Fig. 5, E and F, we observed very similar effects for the different molecules with a strong stimulation of luciferase activity. Similar to H2O2 treatment, the enhancement is greater under Ctrl conditions than under Sup conditions. Interestingly, when we performed time course analyses, as shown in Fig. 5, G and H, the H2O2-induced up-regulation of luciferase activities was not detected 8 h post-treatment but, instead, only after 24 h, lasting for at least 48 h. In addition, we observed that the kinetics of UGA recoding stimulation in response to H2O2 treatment were very similar between Luc UGA/gpx4 and Luc UGA/gpx1, suggesting an up-regulation of selenocysteine insertion independent of the nature of the SECIS element. The kinetic response was also very similar regardless of which selenium condition was used. Our results demonstrate that exogenous oxidative stress stimulates UGA recoding efficiency within 24–48 h post-treatment.
Cellular Relocalization of Recoding Factors Was Induced by Oxidative Stress
Previous work performed in different cell lines (HEK293, MSTO-211, HeLa, and COS7) identified a functional nuclear localization signal and nuclear export signals in SBP2 and EFsec (40, 41). The nucleocytoplasmic shuttling of selenocysteine insertion machinery members was dependent on the CRM1 transporter and ensured the proper assembly of selenoprotein mRNP complexes in the nucleus for efficient cytoplasmic translation (40, 41). Interestingly, in response to oxidative stress, a nuclear translocation of SBP2 was transiently observed in HEK293 and HeLa cells in culture medium containing high levels of selenium, similar to our Sup condition (41, 42). In HEK293 cells, we found that the expression levels of SBP2, EFsec, and L30 recoding factors are insensitive to selenium variation and to oxidative stress (Fig. 6A and Ref. 23). However, using immunohistochemistry and fluorescence microscopy, we observed changes in the cellular localization of endogenous SBP2 (Fig. 6B). The signal was essentially cytoplasmic under Ctrl condition, as observed previously (40). Interestingly, under our conditions, H2O2 treatment induced a partial nuclear translocation of SBP2 (Fig. 6B), confirming the nucleocytoplasmic shuttling of this recoding factor, as reported initially in Ref. 41. To confirm this relocalization and extend our analyses to EFsec and L30, we performed a Western blot analysis on nuclear and cytoplasmic fractions (Fig. 6C). Interestingly, although the overall levels of recoding factors remained constant under all conditions (Fig. 6A), we noted a difference in the nuclear abundance of SBP2 and L30 in response to H2O2 treatment. A modest nuclear relocalization of EFsec upon oxidative stress was only observed in Ctrl medium. Our results suggested that the nucleocytoplasmic shuttling of several components involved in the selenocysteine insertion machinery modified the efficiency of UGA recoding and, therefore, selenoprotein levels.
FIGURE 6.

Expression levels and subcellular localization of the recoding factors SBP2, EFSec, and L30. A, HEK293 cells that were grown and treated as described in Fig. 1 were analyzed by immunoblotting for SBP2, EFsec, and L30. B, cells grown under the Ctrl condition, treated with 0 or 300 μm H2O2, were fixed and observed under fluorescence microscopy after immunostaining with anti-SBP2 antibodies. C, immunoblotting was performed on fractionated extracts to investigate the relative localization of SBP2, EFSec, and L30 in the nucleus and cytoplasm. Cell fractionation was verified with histone H1 and Cox2 biomarkers. Protein expression levels were normalized to H1 (nucleus) or Cox2 (cytoplasm) and expressed relative to the lane with the highest levels.
DISCUSSION
In this work, we demonstrate the preponderant role of the selenoproteome in the antioxidant capacity and cellular response to oxidative stress in HEK293 cells. Among the nine selenoproteins studied here, six (Gpx1, SelK, Sps2, Gpx4, TR1, SelS, and Sps2) were differentially up-regulated by H2O2 treatment. Strikingly, a more effective response was obtained when selenium was limiting. This novel regulation of selenoprotein expression occurred at the level of translation by a stimulation of UGA recoding efficiency. As hypothesized before, but never proven, our studies establish oxidative stress as a new stimulus of selenoprotein expression.
The expression of the selenoproteome is tightly controlled by selenium levels in the blood for mammals or culture medium for cell models. Here we provide evidence for a link between selenium concentration in the growth medium and the antioxidant potential of HEK293 cells. Our results strongly suggest that cells are much more responsive to damage from exogenous stimuli in Ctrl than in Sup media. In response to oxidative stress, we observed changes in the cell cycle profile, the mitochondrial rate of ROS production, the relative amount of ROS-induced damage to proteins, and selenoprotein expression because of modulation of translational efficiency and relocalization of recoding factors. We report that cells grown in Ctrl medium failed to actively cope with oxidative stress despite the stimulation of many selenoproteins involved in peroxide elimination (Gpx), thiol metabolism (TR), ER stress response (SelS and SelK), and selenoprotein biosynthesis. The most likely explanation is that this up-regulation often did not reach the levels observed under Sup conditions. When supplemented with selenium, HEK293 cells seem to express optimal levels of selenoproteins to reduce oxidative stress and were, therefore, responding poorly to this stimulus. The narrow range of control of antioxidant capacity by selenium must be seriously considered because its concentration in commercial calf serum is highly variable. It follows that our Sup medium could correspond to control conditions in other studies. As reported in Ref. 35, the concentration of selenium in seven independent calf sera varied from 164.6–582.6 nm. This variability in selenium content of cell culture serum could greatly influence redox-regulated gene expression and may be responsible for generating contradictory results. For example, the response to oxidative stress treatment induced by 150 μm t-BHP on reporter constructs was higher in less concentrated calf sera than in the naturally abundant counterpart in which the effect was barely detectable (35). This is in agreement with our differential up-regulation of the selenoproteome by H2O2 treatment as a function of selenium levels in Sup and Ctrl media. Therefore, this variability could explain why, unexpectedly, H2O2 treatment did not up-regulate any selenoproteins (41) because the serum used (Supreme serum, Cambriex) was reported in Ref. 35 to have the highest selenium content (582.6 nm). This concentration is higher than our Sup medium where the cells were virtually insensitive to moderate oxidative stress (Fig. 2). In light of the information in Ref. 35 and this study, we respectfully advisee the research community working with oxidative stress to consider the concentration of selenium in the culture serum for the proper interpretation of their data.
The role of selenoproteins in antioxidant defense and redox homeostasis is mediated by the presence of a selenocysteine residue at the active site of enzymes that are usually involved in oxidoreduction catalysis. The physiological relevance of the whole selenoproteome or its individual members has been partially elucidated by knockout/transgenic animal models, human genetic inherited diseases, and cellular models. With animal models, it appeared that, although many different phenotypes were characterized, selenoproteins operate at many levels to protect the cells against oxidative stress and maintain redox homeostasis (43, 44). Concerning human inherited diseases, only mutations in SBP2 and SelN have been characterized to date (45–48). Mutations in the recoding factor SBP2 gene that caused a defect in almost all selenoprotein expression led to a severe and complex phenotype, including increased cellular ROS and susceptibility to ultraviolet radiation-induced oxidative damage in skin (46). Mutations in the SelN gene caused a novel early-onset muscle disorder and led to a severe myopathy. Fibroblast and myoblast primary cultures from these patients displayed abnormal susceptibility to H2O2 stress and increased oxidative damage (49). In parallel, in cellular models, SBP2 knockdown also increased the sensitivity to oxidative stress in terms of cell viability, ROS levels, DNA damage, stress granules, and lipid peroxidation (50). In this study, we demonstrated that the selenoproteome is implicated in the antioxidant response in several crucial cell compartments: mitochondria (Gpx4), the cytoplasm (Sps2, Gpx1 and TR1), the ER (SelS, SelK), and the nucleus (Gpx4), with many cellular consequences. The use of cellular or animal models that are deficient in one or several selenoproteins would be important to understand the precise role of this antioxidant response in cell physiology. In addition, our luciferase reporter construct is particularly useful for rapidly and precisely monitoring selenocysteine insertion capacity in response to exogenous stimuli. It would be of particular interest to analyze whether this antioxidant response is similar in other models, such as cancerous cells or redox-related pathologies in which selenoproteins are particularly involved. Our results could pave the way for a better understanding of the role of selenium in antioxidant defense through the comprehensive analysis of the selenoproteome.
The antioxidant response has been largely described as a drastic change in gene expression through the activation of transcription factors that include NF-κB and NRF2. However to date, the transcriptional regulation of mammalian selenoproteins by oxidative stress has been only studied in silico or in restricted contexts (as reviewed in Ref. 36). A link between NRF2 and selenium metabolism has been characterized, but this depended on the tissue analyzed (51, 52). In HEK293 cells, we observed very little regulation of selenoprotein mRNA levels as a function of selenium bioavailability or oxidative stress, even though selenoprotein expression was modulated dramatically, indicating a specific translational control of this family of proteins in the cell line analyzed.
The regulation of selenoprotein expression has been mostly studied in response to selenium status. Selenium selectively controls the relative expression of different selenoproteins, primarily at the translational level, presumably to maintain vital enzymes at the expense of others (18–21). Very little work has been done on selenoprotein regulation by oxidative stress or in selenium-rich conditions (41, 42). The SECIS element, present in the 3′ UTRs of selenoprotein mRNAs, is considered the main player in this translational regulation, together with its interacting partners. Indeed, the SECIS serves as a platform for mRNP assembly that subsequently dictates the efficiency of selenoprotein mRNA translation by the ribosome (23, 24, 39). The stoichiometry and the nature of mRNP complexes present in the nucleus and cytoplasm remain to be determined. Several studies suggest that the nuclear assembly of selenocysteine incorporation complexes on selenoprotein mRNAs prior to nucleocytoplasmic export could contribute significantly to the efficiency of selenoprotein synthesis, as illustrated in Fig. 7 (12, 41). SBP2, EFsec, and L30 have nuclear and cytoplasmic localization (40, 41), but the change in nucleocytoplasmic shuttling in response to exogenous stimuli has only been characterized for SBP2. The mechanism of relocalization has not yet been elucidated, but the reversible oxidation of redox-sensitive cysteine residues has been proposed (40, 41). Here, we found that H2O2 treatment induced an increase in the nuclear localization of recoding factors SBP2, L30, and, to a lesser extent, EFsec. This finding supports the hypothesis that redox control of recoding factors subsequently influences the efficiency of selenoprotein expression. One could envision that, depending on the complexes formed upon the nuclear assembly of selenoprotein mRNPs, a selective regulation could be driven.
FIGURE 7.

Schematic of the nuclear selenoprotein mRNPs assembly. Recoding factors are imported into the nucleus to assemble with selenoprotein mRNAs as described in Refs. 40, 50, 53. mRNPs are built and exported to the cytoplasm to promote efficient translation by the ribosome. An increase in nucleocytoplasmic shuttling in response to H2O2 induced oxidative stress is proposed to improve selenocysteine insertion complex nuclear assembly and, consequently, selenoprotein expression.
Acknowledgments
We thank Dr. Marion Segalen for technical assistance with microscopy imaging, Michael Bourge from the Imagif platform for help with the flow cytometry analyses, and Dr. Donna Driscoll for critical reading of the manuscript.
This work was supported by the CNRS (ATIP program) (to L. C.), by the Fondation pour la Recherche Médicale (to L. C.), by the Ligue Contre le Cancer (Comité de l'Essonne) (to L. C.), by Association pour la Recherche sur le Cancer grant 4849 (to L. C.), by the Programme Interdisciplinaire de Recherche du CNRS Longévité et Vieillissement (to L. C.), and by Agence Nationale de la Recherche grant ANR-09-BLAN-0048 (to L. C.).
- ER
- endoplasmic reticulum
- SECIS
- selenocysteine insertion sequence
- ROS
- reactive oxygen species
- Gpx
- glutathione peroxidase
- TR
- thioredoxin reductase
- Ctrl
- control
- Sup
- supplemented
- t-BHP
- ter-butyl hydroperoxide
- Luc
- luciferase
- Cu-OOH
- cumene hydroperoxide.
REFERENCES
- 1. Rayman M. P. (2012) Selenium and human health. Lancet 379, 1256–1268 [DOI] [PubMed] [Google Scholar]
- 2. Papp L. V., Holmgren A., Khanna K. K. (2010) Selenium and selenoproteins in health and disease. Antioxid. Redox Signal. 12, 793–795 [DOI] [PubMed] [Google Scholar]
- 3. Kryukov G. V., Castellano S., Novoselov S. V., Lobanov A. V., Zehtab O., Guigó R., Gladyshev V. N. (2003) Characterization of mammalian selenoproteomes. Science 300, 1439–1443 [DOI] [PubMed] [Google Scholar]
- 4. Allmang C., Wurth L., Krol A. (2009) The selenium to selenoprotein pathway in eukaryotes: more molecular partners than anticipated. Biochim. Biophys. Acta 1790, 1415–1423 [DOI] [PubMed] [Google Scholar]
- 5. Berry M. J., Tujebajeva R. M., Copeland P. R., Xu X. M., Carlson B. A., Martin G. W., 3rd, Low S. C., Mansell J. B., Grundner-Culemann E., Harney J. W., Driscoll D. M., Hatfield D. L. (2001) Selenocysteine incorporation directed from the 3′UTR: characterization of eukaryotic EFsec and mechanistic implications. Biofactors 14, 17–24 [DOI] [PubMed] [Google Scholar]
- 6. Driscoll D. M., Copeland P. R. (2003) Mechanism and regulation of selenoprotein synthesis. Annu. Rev. Nutr. 23, 17–40 [DOI] [PubMed] [Google Scholar]
- 7. Hatfield D. L., Carlson B. A., Xu X. M., Mix H., Gladyshev V. N. (2006) Selenocysteine incorporation machinery and the role of selenoproteins in development and health. Prog. Nucleic Acid Res. Mol. Biol. 81, 97–142 [DOI] [PubMed] [Google Scholar]
- 8. Fagegaltier D., Hubert N., Yamada K., Mizutani T., Carbon P., Krol A. (2000) Characterization of mSelB, a novel mammalian elongation factor for selenoprotein translation. EMBO J. 19, 4796–4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tujebajeva R. M., Copeland P. R., Xu X. M., Carlson B. A., Harney J. W., Driscoll D. M., Hatfield D. L., Berry M. J. (2000) Decoding apparatus for eukaryotic selenocysteine insertion. EMBO Rep. 1, 158–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ding F., Grabowski P. J. (1999) Identification of a protein component of a mammalian tRNA(Sec) complex implicated in the decoding of UGA as selenocysteine. RNA 5, 1561–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu X. M., Mix H., Carlson B. A., Grabowski P. J., Gladyshev V. N., Berry M. J., Hatfield D. L. (2005) Evidence for direct roles of two additional factors, SECp43 and soluble liver antigen, in the selenoprotein synthesis machinery. J. Biol. Chem. 280, 41568–41575 [DOI] [PubMed] [Google Scholar]
- 12. Small-Howard A., Morozova N., Stoytcheva Z., Forry E. P., Mansell J. B., Harney J. W., Carlson B. A., Xu X. M., Hatfield D. L., Berry M. J. (2006) Supramolecular complexes mediate selenocysteine incorporation in vivo. Mol. Cell Biol. 26, 2337–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carlson B. A., Xu X. M., Kryukov G. V., Rao M., Berry M. J., Gladyshev V. N., Hatfield D. L. (2004) Identification and characterization of phosphoseryl-tRNA[Ser]Sec kinase. Proc. Natl. Acad. Sci. U.S.A. 101, 12848–12853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Copeland P. R., Fletcher J. E., Carlson B. A., Hatfield D. L., Driscoll D. M. (2000) A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J. 19, 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chavatte L., Brown B. A., Driscoll D. M. (2005) Ribosomal protein L30 is a component of the UGA-selenocysteine recoding machinery in eukaryotes. Nat. Struct. Mol. Biol. 12, 408–416 [DOI] [PubMed] [Google Scholar]
- 16. Budiman M. E., Bubenik J. L., Miniard A. C., Middleton L. M., Gerber C. A., Cash A., Driscoll D. M. (2009) Eukaryotic initiation factor 4a3 is a selenium-regulated RNA-binding protein that selectively inhibits selenocysteine incorporation. Mol. Cell 35, 479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miniard A. C., Middleton L. M., Budiman M. E., Gerber C. A., Driscoll D. M. (2010) Nucleolin binds to a subset of selenoprotein mRNAs and regulates their expression. Nucleic Acids Res. 38, 4807–4820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bermano G., Arthur J. R., Hesketh J. E. (1996) Selective control of cytosolic glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase mRNA stability by selenium supply. FEBS Lett. 387, 157–160 [DOI] [PubMed] [Google Scholar]
- 19. Bermano G., Nicol F., Dyer J. A., Sunde R. A., Beckett G. J., Arthur J. R., Hesketh J. E. (1995) Tissue-specific regulation of selenoenzyme gene expression during selenium deficiency in rats. Biochem. J. 311, 425–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weiss Sachdev S., Sunde R. A. (2001) Selenium regulation of transcript abundance and translational efficiency of glutathione peroxidase-1 and -4 in rat liver. Biochem. J. 357, 851–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wingler K., Böcher M., Flohé L., Kollmus H., Brigelius-Flohé R. (1999) mRNA stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. Eur. J. Biochem. 259, 149–157 [DOI] [PubMed] [Google Scholar]
- 22. Lei X. G., Evenson J. K., Thompson K. M., Sunde R. A. (1995) Glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase are differentially regulated in rats by dietary selenium. J. Nutr. 125, 1438–1446 [DOI] [PubMed] [Google Scholar]
- 23. Latrèche L., Duhieu S., Touat-Hamici Z., Jean-Jean O., Chavatte L. (2012) The differential expression of glutathione peroxidase 1 and 4 depends on the nature of the SECIS element. RNA Biol. 9, 681–690 [DOI] [PubMed] [Google Scholar]
- 24. Latrèche L., Jean-Jean O., Driscoll D. M., Chavatte L. (2009) Novel structural determinants in human SECIS elements modulate the translational recoding of UGA as selenocysteine. Nucleic Acids Res. 37, 5868–5880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brigelius-Flohé R., Maiorino M. (2013) Glutathione peroxidases. Biochim. Biophys. Acta 1830, 3289–3303 [DOI] [PubMed] [Google Scholar]
- 26. Arnér E. S. (2009) Focus on mammalian thioredoxin reductases: important selenoproteins with versatile functions. Biochim. Biophys. Acta 1790, 495–526 [DOI] [PubMed] [Google Scholar]
- 27. Shchedrina V. A., Zhang Y., Labunskyy V. M., Hatfield D. L., Gladyshev V. N. (2010) Structure-function relations, physiological roles, and evolution of mammalian ER-resident selenoproteins. Antioxid. Redox Signal. 12, 839–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steinbrenner H., Sies H. (2009) Protection against reactive oxygen species by selenoproteins. Biochim. Biophys. Acta 1790, 1478–1485 [DOI] [PubMed] [Google Scholar]
- 29. Buelna-Chontal M., Zazueta C. (2013) Redox activation of Nrf2 and NF-κB: a double end sword? Cell. Signal. 25, 2548–2557 [DOI] [PubMed] [Google Scholar]
- 30. Brigelius-Flohé R., Müller M., Lippmann D., Kipp A. P. (2012) The yin and yang of nrf2-regulated selenoproteins in carcinogenesis. Int. J. Cell Biol. 2012, 486147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Müller M., Banning A., Brigelius-Flohé R., Kipp A. (2010) Nrf2 target genes are induced under marginal selenium-deficiency. Genes Nutr. 5, 297–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suzuki T., Kelly V. P., Motohashi H., Nakajima O., Takahashi S., Nishimura S., Yamamoto M. (2008) Deletion of the selenocysteine tRNA gene in macrophages and liver results in compensatory gene induction of cytoprotective enzymes by Nrf2. J. Biol. Chem. 283, 2021–2030 [DOI] [PubMed] [Google Scholar]
- 33. Arnér E. S. (2002) Recombinant expression of mammalian selenocysteine-containing thioredoxin reductase and other selenoproteins in Escherichia coli. Methods Enzymol. 347, 226–235 [DOI] [PubMed] [Google Scholar]
- 34. Legrain Y., Touat-Hamici Z., Chavatte L. (2014) Interplay between selenium levels, selenoprotein expression and replicative senescence in WI-38 human fibroblasts. J. Biol. Chem. 289, 6299–6310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karlenius T. C., Shah F., Yu W. C., Hawkes H. J., Tinggi U., Clarke F. M., Tonissen K. F. (2011) The selenium content of cell culture serum influences redox-regulated gene expression. BioTechniques 50, 295–301 [DOI] [PubMed] [Google Scholar]
- 36. Stoytcheva Z. R., Berry M. J. (2009) Transcriptional regulation of mammalian selenoprotein expression. Biochim. Biophys. Acta 1790, 1429–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sunde R. A., Raines A. M., Barnes K. M., Evenson J. K. (2009) Selenium status highly regulates selenoprotein mRNA levels for only a subset of the selenoproteins in the selenoproteome. Biosci. Rep. 29, 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sun X., Li X., Moriarty P. M., Henics T., LaDuca J. P., Maquat L. E. (2001) Nonsense-mediated decay of mRNA for the selenoprotein phospholipid hydroperoxide glutathione peroxidase is detectable in cultured cells but masked or inhibited in rat tissues. Mol. Biol. Cell 12, 1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Low S. C., Grundner-Culemann E., Harney J. W., Berry M. J. (2000) SECIS-SBP2 interactions dictate selenocysteine incorporation efficiency and selenoprotein hierarchy. EMBO J. 19, 6882–6890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. de Jesus L. A., Hoffmann P. R., Michaud T., Forry E. P., Small-Howard A., Stillwell R. J., Morozova N., Harney J. W., Berry M. J. (2006) Nuclear assembly of UGA decoding complexes on selenoprotein mRNAs: a mechanism for eluding nonsense-mediated decay? Mol. Cell Biol. 26, 1795–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Papp L. V., Lu J., Striebel F., Kennedy D., Holmgren A., Khanna K. K. (2006) The redox state of SECIS binding protein 2 controls its localization and selenocysteine incorporation function. Mol. Cell Biol. 26, 4895–4910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Papp L. V., Lu J., Holmgren A., Khanna K. K. (2007) From selenium to selenoproteins: synthesis, identity, and their role in human health. Antioxid. Redox Signal. 9, 775–806 [DOI] [PubMed] [Google Scholar]
- 43. Kasaikina M. V., Hatfield D. L., Gladyshev V. N. (2012) Understanding selenoprotein function and regulation through the use of rodent models. Biochim. Biophys. Acta 1823, 1633–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Conrad M., Schweizer U. (2010) Unveiling the molecular mechanisms behind selenium-related diseases through knockout mouse studies. Antioxid. Redox Signal. 12, 851–865 [DOI] [PubMed] [Google Scholar]
- 45. Dumitrescu A. M., Liao X. H., Abdullah M. S., Lado-Abeal J., Majed F. A., Moeller L. C., Boran G., Schomburg L., Weiss R. E., Refetoff S. (2005) Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat. Genet. 37, 1247–1252 [DOI] [PubMed] [Google Scholar]
- 46. Schoenmakers E., Agostini M., Mitchell C., Schoenmakers N., Papp L., Rajanayagam O., Padidela R., Ceron-Gutierrez L., Doffinger R., Prevosto C., Luan J., Montano S., Lu J., Castanet M., Clemons N., Groeneveld M., Castets P., Karbaschi M., Aitken S., Dixon A., Williams J., Campi I., Blount M., Burton H., Muntoni F., O'Donovan D., Dean A., Warren A., Brierley C., Baguley D., Guicheney P., Fitzgerald R., Coles A., Gaston H., Todd P., Holmgren A., Khanna K. K., Cooke M., Semple R., Halsall D., Wareham N., Schwabe J., Grasso L., Beck-Peccoz P., Ogunko A., Dattani M., Gurnell M., Chatterjee K. (2010) Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J. Clin. Invest. 120, 4220–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Azevedo M. F., Barra G. B., Naves L. A., Ribeiro Velasco L. F., Godoy Garcia Castro P., de Castro L. C., Amato A. A., Miniard A., Driscoll D., Schomburg L., de Assis Rocha Neves F. (2010) Selenoprotein-related disease in a young girl caused by nonsense mutations in the SBP2 gene. J. Clin. Endocrinol. Metab. 95, 4066–4071 [DOI] [PubMed] [Google Scholar]
- 48. Ferreiro A., Quijano-Roy S., Pichereau C., Moghadaszadeh B., Goemans N., Bönnemann C., Jungbluth H., Straub V., Villanova M., Leroy J. P., Romero N. B., Martin J. J., Muntoni F., Voit T., Estournet B., Richard P., Fardeau M., Guicheney P. (2002) Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am. J. Hum. Genet. 71, 739–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Arbogast S., Beuvin M., Fraysse B., Zhou H., Muntoni F., Ferreiro A. (2009) Oxidative stress in SEPN1-related myopathy: from pathophysiology to treatment. Ann. Neurol. 65, 677–686 [DOI] [PubMed] [Google Scholar]
- 50. Papp L. V., Lu J., Bolderson E., Boucher D., Singh R., Holmgren A., Khanna K. K. (2010) SECIS-binding protein 2 promotes cell survival by protecting against oxidative stress. Antioxid. Redox Signal. 12, 797–808 [DOI] [PubMed] [Google Scholar]
- 51. Burk R. F., Hill K. E., Nakayama A., Mostert V., Levander X. A., Motley A. K., Johnson D. A., Johnson J. A., Freeman M. L., Austin L. M. (2008) Selenium deficiency activates mouse liver Nrf2-ARE but vitamin E deficiency does not. Free Radic. Biol. Med. 44, 1617–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Banning A., Deubel S., Kluth D., Zhou Z., Brigelius-Flohé R. (2005) The GI-GPx gene is a target for Nrf2. Mol. Cell Biol. 25, 4914–4923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Boulon S., Marmier-Gourrier N., Pradet-Balade B., Wurth L., Verheggen C., Jády B. E., Rothé B., Pescia C., Robert M. C., Kiss T., Bardoni B., Krol A., Branlant C., Allmang C., Bertrand E., Charpentier B. (2008) The Hsp90 chaperone controls the biogenesis of L7Ae RNPs through conserved machinery. J. Cell Biol. 180, 579–595 [DOI] [PMC free article] [PubMed] [Google Scholar]