

Background: The role of GOLPH3 in mammalian glycosylation is not well studied.
Results: GOLPH3 binds to and controls the Golgi localization of POMGnT1.
Conclusion: GOLPH3 regulates the glycosylation of α-dystroglycan by anchoring POMGnT1 at the Golgi.
Significance: The result provides a further understanding of the role of GOLPH3 in mediating the Golgi localization of glycosyltransferases in mammalian cells.
Keywords: Glycosylation, Glycosyltransferases, Protein Targeting, Protein-protein Interactions, Skeletal Muscle, GOLPH3, POMGnT, Muscle-eye Brain Disease
Abstract
GOLPH3 is a highly conserved protein found across the eukaryotic lineage. The yeast homolog, Vps74p, interacts with and maintains the Golgi localization of several mannosyltransferases, which is subsequently critical for N- and O-glycosylation in yeast. Through the use of a T7 phage display, we discovered a novel interaction between GOLPH3 and a mammalian glycosyltransferase, POMGnT1, which is involved in the O-mannosylation of α-dystroglycan. The cytoplasmic tail of POMGnT1 was found to be critical for mediating its interaction with GOLPH3. Loss of this interaction resulted in the inability of POMGnT1 to localize to the Golgi and reduced the functional glycosylation of α-dystroglycan. In addition, we showed that three clinically relevant mutations present in the stem domain of POMGnT1 mislocalized to the endoplasmic reticulum, highlighting the importance of identifying the molecular mechanisms responsible for Golgi localization of glycosyltransferases. Our findings reveal a novel role for GOLPH3 in mediating the Golgi localization of POMGnT1.
Introduction
GOLPH3 was discovered in proteomic studies designed to identify novel resident proteins of the Golgi (1, 2). GOLPH3 does not display homology to any known mammalian functional domains or proteins, yet it is highly conserved across the entire eukaryotic lineage (1). GOLPH3 is recognized as an oncoprotein and is amplified in various forms of cancers, including melanoma, breast, non-small cell lung cancer, gliomas, and connective tissue tumors (3–5). Its protumorigenic activity was linked to enhanced signaling through mTOR (mammalian target of rapamycin) (3), which is known to act as a primary regulator of protein synthesis and cell growth (6). GOLPH3 has also been implicated in other biological roles, including the efficient trafficking of proteins from the Golgi to the plasma membrane (7) as well as in the recycling of transmembrane receptors from the endosome to the trans-Golgi network (3).
Importantly, the yeast homolog of GOLPH3, Vps74p, was shown to mediate interactions with Golgi glycosyltransferases, and these interactions are subsequently critical in determining the outcome of N- and O-glycosylation in yeast (8, 9). GOLPH3 corrected for phenotypic defects in vps74-deleted yeast cells (8); thus, it was speculated that GOLPH3 and Vps74p perform a similar function at the Golgi. This hypothesis is supported by a recent finding demonstrating that GOLPH3 is indeed involved in glycosylation as it interacts with the core 2 N-acetylglucosaminyltransferase 1 (C2GnT1), a key enzyme involved in the synthesis of core 2-associated Lewis X (C2-O-sLex) (10). Subsequently, reduction in GOLPH3 led to alterations in glycosylation of P selectin-associated ligand (10).
α-Dystroglycan is a component of the dystrophin glycoprotein complex found in the sarcolemma membrane surrounding muscle fibers. The dystrophin glycoprotein complex acts to physically couple the extracellular matrix that surrounds each myofiber to the intracellular actin cytoskeleton (11–13). α-Dystroglycan represents a vital component of the dystrophin glycoprotein complex as it mediates interactions with several ligands present in the extracellular matrix, the most important of which is laminin (12). These interactions are critically dependent on the glycosylation status of the α-dystroglycan glycoprotein (14–16). Consequently, perturbations in the glycosylation of α-dystroglycan disrupts the vital interaction between α-dystroglycan and its ligands and impairs its attachment to the extracellular matrix subsequently leading to various forms of congenital muscular dystrophies (14, 17, 18). To date, mutations in several glycoysltransferases or glycosyltransferase-like genes have been reported to alter the glycosylation of α-dystroglycan resulting in the development of congenital muscular dystrophy (19). One of these genes is POMGnT1, which catalyzes the second step in the O-mannosylation of α-dystroglycan by mediating the transfer of N-acetylglucosamine (GlcNAc)2 from UDP-GlcNAc onto the mannose in the 2-OH position. POMGnT1 encodes a 660-amino acid type II transmembrane protein located in the Golgi and is predicted to have four domains, an N-terminal cytoplasmic tail, a transmembrane domain, a stem domain, and a catalytic domain facing the Golgi lumen (20). Loss of function mutations in POMGnT1 leads to the development of a specific form of CMD termed muscle-eye-brain disease (20).
The T7 phage display is based on the surface expression of peptide sequences fused to phage capsid protein. Although the main application for the T7 phage display has been the identification of natural binders to antibodies (21–23), it is increasingly being used for the identification of protein-protein binding partners (24–26). Using such a platform, we have identified a novel interaction between POMGnT1 and GOLPH3. We showed that GOLPH3 is responsible for mediating the Golgi localization of POMGnT1 and consequently regulates the glycosylation of α-dystroglycan.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
HeLa cells and HEK 293 cells were obtained from American Type Culture Collection and cultured at 37 °C with 5% CO2 in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen).
Plasmid Constructs
The coding region of human GOLPH3 (NCBI sequence NP_071413.1) was amplified from HeLa cDNA by PCR and cloned into pcDNA3.1+ vector. The coding region of GOLPH3 was also cloned into pGEX4T-1 (Pharmacia Biotech) for expression in bacterial cells. The coding region of human POMGnT1 (NCBI sequence NP_060209.3) was amplified from HeLa cDNA using PCR. An HA tag was fused to the C terminus of POMGnT1 and cloned into pcDNA3.1+ vectors. Site-directed mutagenesis of POMGnT1-HA constructs was generated using the QuikChange II XL site-directed mutagenesis kit (Stratagene) following the manufacturer's instructions. All constructs were fully sequenced before being used for transfection.
Expression of Recombinant Proteins in Bacteria
For bacterial expression of recombinant proteins, the pGEX4T-1 vectors were transformed into Escherichia coli BL21(DE3) cells (Invitrogen). The cells were then cultured at 37 °C until the A600 value reached 0.6. Subsequently, isopropyl 1-thio-β-d-galactopyranoside (Invitrogen) was added into the culture medium to a final concentration of 0.5 mm to induce protein expression for 2 h at 27 °C. Cells were harvested by centrifugation and lysed by sonication in PBS. GST and GST-GOLPH3 proteins were then purified by affinity chromatography with glutathione-Sepharose beads (Amersham Biosciences) under native conditions.
T7 Phage Library Screening
An aliquot of a T7 phage display library of human liver cDNA (Novagen) was pre-cleared with 100 μg of GST protein immobilized on glutathione-Sepharose beads by incubating them together for 2 h at room temperature followed by centrifugation. The supernatant was then incubated overnight at 4 °C with 100 μg of GST-fused GOLPH3 (full-length) immobilized on glutathione-Sepharose beads. All binding experiments were performed in the presence of 1% bovine serum albumin. Unbound phages were washed four times with TBST (0.1% Tween 20 in 10 mm Tris (pH 7.4) and 150 mm NaCl). The bound phages were amplified in E. coli BL5615 (Novagen), which was preinduced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 20 min at 37 °C. After cell lysis, the phage lysate was clarified by centrifugation at 8000 × g for 10 min and stored at 4 °C for use in the next selection round. A total of five rounds of selection were performed. PCR analysis was performed on amplified phages using a pair of T7 primers to ascertain the enrichment. The phage pool from the fourth round of amplification was panned to harvest single clones. Individual phage clones were picked and subjected to sequencing analysis using a T7 primer.
Co-immunoprecipitation and Western Blotting
HeLa cells were transiently co-transfected with indicated expression plasmids, and the cell lysates were prepared 2 days post-transfection. Cells were lysed in lysis buffer (1% IGEPAL CA-630, 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 5 mm EDTA, and protease inhibitors from Roche Applied Science) for 10 min at 4 °C, and insoluble materials were removed by centrifugation at 15,000 × g and 4 °C for 10 min. Co-immunoprecipitation was performed using the anti-HA immunoprecipitation kit (Sigma) according to the manufacturer's instructions. For reverse co-immunoprecipitation, 5 μg of anti-GOLPH3 antibody was added to cleared lysate for 2 h at 4 °C. Next, 50 μl of protein A/G agarose was added and incubated overnight at 4 °C. After four washes, the immunoprecipitated complexes were analyzed by Western blotting using anti-HA (Sigma) and anti-GOLPH3 (Abcam) antibodies.
Immunofluorescence Staining
HeLa cells were plated on glass coverslips and grown overnight before transfection with indicated plasmids. 2 days after transfection, cells were fixed in 4% paraformaldehyde (Sigma-Aldrich), permeabilized in 0.1% Triton X-100 (Sigma-Aldrich), and blocked in 10% normal goat serum (Invitrogen) for 30 min. Cells were then incubated with a 1:500 dilution of mouse anti-HA monoclonal antibody (Sigma-Aldrich) and a 1:300 dilution of rabbit anti-giantin (Abcam) or a 1:160 dilution of rabbit anti-GOLPH3 (Abcam) in PBS with 1% BSA for 1 h. Cells were washed three times with PBS and subsequently incubated with goat anti-mouse conjugated with Alexa Fluor 488 and goat anti-rabbit conjugated with Alexa Fluor 594 (Invitrogen) in PBS with 1% BSA for 1 h. Cells were washed three times with PBS before mounting on glass slides with a Prolong Gold Antifade mounting medium (Invitrogen). Fluorescence images were obtained using a Carl Zeiss META confocal microscope.
siRNA Knockdown
To analyze knockdown phenotypes, GOLPH3 siRNA was obtained from Integrated DNA Technologies, whereas POMGnT1 and All Stars negative control siRNA (catalog no. 1027281) were obtained from Qiagen. siRNAs were diluted to 20 μm using RNase-free distilled water and stored at −20 °C until further use. Cells were cultured in six-well plates and transfected with siRNAs at a final concentration of 10 nm using RNAiMAX (Invitrogen) according to the manufacturer's instructions. The siRNA oligonucleotide sequences were as follows. For GOLPH3, the sense oligonucleotide is 5′-CCCUGAUGGAGGAAGUGCUCCUGCU-3′, and the antisense oligonucleotide is 5′-AGCAGGAGCACUUCCUCCAUCAGGGUC-3′. For POMGnT1, the sense oligonucleotide is 5′-GACGUAGAGGUGUAUUCAAUU-3′, and the antisense oligonucleotide is 5′-UUGAAUACACCUCUACGUCCA-3′.
Flow Cytometry
For cell surface staining of α-dystroglycan with mAb IIH6 (Santa Cruz Biotechnology), HEK 293 cells were detached with enzyme-free dissociation solution (Sigma-Aldrich) and incubated with mAb IIH6 (1:100) in 1% BSA/PBS for 1 h on ice. Cells were then washed twice in PBS and labeled with goat anti-mouse conjugated with Alexa Fluor 488 (1:500) (Invitrogen) in 1% BSA/PBS for 45 min on ice in the dark. Cells were washed twice in PBS and analyzed with a FACSCalibur flow cytometry (BD Biosciences) using Cell Quest Software. Image analysis was done using FloJo Software (Tree Star, Inc.).
Quantitative Reverse Transcription (qRT) PCR Analyses
Total RNA was extracted from the siRNA-transfected HEK 293 cell lines using RNeasy kit (Qiagen). First strand cDNA was synthesized from 2 μg of total RNA using Moloney murine leukemia virus reverse transcriptase (MMLV-RT) (Promega) and oligo(dT(15)) (Promega). The following primers for GOLPH3 and POMGnT1 were designed: GOLPH3 primer, 5′-CAGCCACGTAATCCAGATGAT-3′ (forward) and 5′-ACCCTGATGGAGGAAGTGCT-3′ (reverse); POMGnT1 primer, 5′-CCTGGAACCGTGTTGAACTT-3′ (forward) and 5′-CGATCCTACCACTTTGGCAT-3′ (reverse). PCR analysis was performed using the ABI Prism7000qRT PCR Detection System (Applied Biosystems). For each reaction of 25 μl, 12.5 μl of 2× SYBR Green PCR Master Mix (Applied Biosystems) was mixed with 5 μl cDNA from 2 μg of total RNA) and 12.5 pmol each of forward and reverse primers and topped up with water in a MicroAmp optical 96-well reaction plate and sealed with optical adhesive covers (Applied Biosystems). PCR analysis was performed using the ABI Prism7000 qRT PCR Detection System (Applied Biosystems). The PCR reaction conditions were as follows: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 60 s. Threshold cycle (Ct) was determined using ABI Prism 7000 SDS program (Applied Biosystems). All reactions were run in triplicate. Relative transcript abundance was calculated by transforming the average Ct values to 2−Ct and normalizing to a housekeeping gene, GAPDH.
RESULTS
Identification of POMGnT1 as a Novel Binding Partner for the GOLPH3 Proteins
Full-length human GOLPH3 was fused to the C terminus of GST and used as the bait in a phage display screen against a human liver cDNA library. Five rounds of biopanning were performed and the enrichment after each round was ascertained by PCR. Specific DNA bands emerged in the fourth and fifth round of selection, suggesting that specific GOLPH3 binding phage was present in these phage pools (data not shown). Subsequently, the phage pool from the fourth round of biopanning was panned to harvest single clones. Sequencing analysis revealed that a dominant peptide, HLKESVLRAGWSHTW, was obtained as 50% of the clones contained this same peptide motif. A BLAST search revealed that this sequence is highly similar to a stretch of 27 amino acids located in the stem region of POMGnT1. Another potential binder of GOLPH3, identified in the phage display was N-acetylgalactosaminyltransferase 12, a glycosyltransferase implicated in the initiation of mammalian O-linked glycosylation. Alignment of the two glycoysltransferases identified in the phage display revealed the presence of a conserved motif (RXLRR) present in the cytoplasmic tail of the transferases. Further validation of the interaction between GOLPH3 and N-acetylgalactosaminyltransferase 12 will be important for defining the potential function of GOLPH3 in O-linked glycosylation.
We chose to focus our efforts on POMGnT1 as this peptide represented the dominant binder in our screen. Co-immunoprecipitation analyses were performed to confirm the physiological interaction of the GOLPH3 and POMGnT1 interaction in mammalian cells. Cells were co-transfected with GOLPH3 and HA-tagged POMGnT1 or GOLPH3 alone followed by IP analyses with anti-HA antibody. The immunoprecipitates were tested for the presence of GOLPH3 by immunoblotting with anti-GOLPH3 antibody. GOLPH3 was found in the immunoprecipitated complexes of POMGnT1-HA expressing cells but not in cells that did not express POMGnT1-HA (Fig. 1A). A reciprocal IP was also preformed to further verify this interaction. Cells were co-transfected with GOLPH3 and POMGnT1-HA, following which IP was carried out using an anti-GOLPH3 antibody. The immunoprecipitates were tested for the presence of POMGnT1 by immunoblotting with anti-HA antibody. POMGnT1-HA was detected in the immunoprecipitated complexes as shown in Fig. 1B. These data offer strong support for the physical association of GOLPH3 and POMGnT1 in cells.
FIGURE 1.

POMGnT1 interacts and co-localizes with GOLPH3 proteins. A, HeLa cells were transfected with an expression plasmid to ectopically express GOLPH3- and HA-tagged POMGnT1. IP was performed using the cell lysates prepared from the transfected cells and an anti-HA antibody. The immunoprecipitated complexes and the protein inputs were analyzed by Western blotting (Western) using anti-GOLPH3 antibodies. GOLPH3 displayed interactions with POMGnT1. B, the cell lysates of GOLPH3 and POMGnT1-HA expressing cells were used for IP with an anti-GOLPH3 antibody. Normal IgG (IgG) was used as a negative control. The presence of POMGnT1 in the immunoprecipitates was determined by Western blotting using anti-HA antibodies. C, POMGnT1-HA was co-expressed with GOLPH3 and fixed cells were stained with anti-HA (green) and anti-GOLPH3 (red) antibodies and DAPI. GOLPH3 co-localized with POMGnT1 at the Golgi.
Based on the results of the co-immunoprecipitation, we predicted that POMGnT1 would co-localize with GOLPH3. To determine this, we performed confocal fluorescence microscopy. As previously determined, we found that a HA-tagged POMGnT1 co-localized with the Golgi marker giantin (data not shown). Importantly, the co-transfection of POMGnT1 and GOLPH3 showed that both proteins co-localized at the Golgi (Fig. 1C).
GOLPH3 Mediates the Golgi Localization of POMGnT1
The amino acid residues within the stem domain of POMGnT1 identified in the T7 phage display may play an important role in interacting with GOLPH3. To determine the functional significance of these residues, we deleted amino acids 184 to 210 of POMGnT1 (Fig. 2A) and assessed its GOLPH3-binding capabilities. However, we found that the deletion of this region did not affect its binding to GOLPH3 (Fig. 2B). Due to prior knowledge that GOLPH3 and its homolog Vps74p binds to the cytoplasmic tails of C2GnT1 (10) and yeast mannosyltransferases (8, 9), we investigated the role of the cytoplasmic tail of POMGnT1 in mediating its interaction with GOLPH3. A truncated form of POMGnT1 was generated by deleting the cytoplasmic tail of POMGnT1 (Fig. 2A). HeLa cells were co-transfected with constructs expressing a HA-tagged POMGnT1 with its C-terminal tail removed and GOLPH3. This was followed by co-immunoprecipitation using anti-HA antibodies. The immunoprecipitated complexes were then immunoblotted with anti-GOLPH3 antibodies to detect for the presence of GOLPH3. As shown in left panel of Fig. 2C, the deletion of the cytoplasmic tail of POMGnT1 led to the reduction in the binding capacity of GOLPH3 when compared with that of the full-length POMGnT1 protein. Quantification of the GOLPH3 bands from two separate co-immunoprecipitation experiments showed that there was an approximate 3-fold decrease in GOLPH3 binding to the truncated POMGnT1 protein (Fig. 2C, right panel).
FIGURE 2.

GOLPH3 influences the subcellular localization of POMGnT1. A, illustration of truncated POMGnT1 proteins. B, HeLa cells were transfected with the indicated plasmids. IP was performed using an anti-HA antibody followed by Western blotting with anti-GOLPH3 antibody. Deletion of amino acids within the stem domain did not affect GOLPH3 binding. C, HeLa cells were transfected with the indicated plasmids. IP was performed using an anti-HA antibody followed by Western blotting with anti-GOLPH3 antibody (left panel). The deletion of the POMGnT1 cytoplasmic tail (CT) resulted in reduced interactions with GOLPH3. Shown is quantification of the reduction in binding capacity of GOLPH3 to truncated POMGnT1 (n = 2, p < 0.01) (right panel). D, the indicated plasmids were transiently expressed in HeLa cells following which fixed cells were stained with anti-HA-specific (green) and anti-giantin-specific (red) antibodies. The deletion of the POMGnT1 cytoplasmic tail resulted in failure of the protein to localize precisely to the Golgi. E, HeLa cells were transfected with control siRNA or GOLPH3 siRNA, followed by a vector expressing HA-tagged POMGnT1. Cells were fixed and stained with an anti-giantin (left panel) or anti-PDI (right panel) antibody (red), HA (green), and DAPI (blue). POMGnT1 localizes to the ER in GOLPH3-depleted cells. TMD, transmembrane domain.
Due to the reduction in interaction between GOLPH3 and POMGnT1, we speculated that the localization of POMGnT1 would be affected. As shown by confocal fluorescence microscopy, we observed that the removal of the cytoplasmic tail of POMGnT1 led to the inability of the protein to precisely co-localize with the Golgi marker (Fig. 2D, middle panel). Further analysis demonstrated that the truncated protein mislocalized to endoplasmic reticulum (ER) as shown by the co-localization with the ER marker, protein disulfide isomerase (PDI) (Fig. 2D, lower panel). We have thus mapped the GOLPH3 interaction region to the cytoplasmic tail of on POMGnT1 and further demonstrated that this interaction is important in maintaining the Golgi localization of POMGnT1.
In an effort to further evaluate the finding that GOLPH3 is critical in maintaining the Golgi localization of POMGnT1, we employed RNAi-based interference to reduce the expression level of endogenous GOLPH3 and assessed the subcellular localization of POMGnT1-HA. Due to the existence of a GOLPH3 variant (GOLPH3V), and with the knowledge that GOLPH3V exists in human cell lines such as HeLa (27), we utilized an siRNA to target both the variant and full-length form of the GOLPH3 protein. The knockdown of GOLPH3 resulted in the inability of POMGnT1 to precisely co-localize with the Golgi marker (Fig. 2E, left panel). On further analysis, we show POMGnT1 localized to the ER in GOLPH3-depleted cells as observed by the co-localization with anti-PDI (Fig. 2E, right panel). Taken together, the in vivo data further demonstrate that GOLPH3 proteins determine the Golgi localization of POMGnT1.
Down-regulation of GOLPH3 Reduces Functional Glycosylation of α-Dystroglycan
To investigate the biological significance of the novel interaction between GOLPH3 and POMGnT1 identified in this study, we used RNAi-based gene silencing to reduce endogenous levels of GOLPH3 and assessed the functional glycosylation of cell-surface α-dystroglycan. The glycosylation of α-dystroglycan was examined based on the immunoreactivity of the glycoprotein to the IIH6 monoclonal antibody, which is known to recognize the laminin-binding glyco-epitope (28). HeLa cells were not used in this assay as glycosylation of α-dystroglycan is regulated in a cell type-specific manner, and studies have shown that epithelium-derived cancer cell lines, including HeLa, are unable to functionally glycosylate α-dystroglycan (29). We instead used HEK293 cells to analyze the impact of GOLPH3 knockdown on α-dystroglycan glycosylation. siRNA targeting POMGnT1 was included in our study as a positive control, and cells treated with this siRNA clearly demonstrated a reduction in IIH6 immunoreactivity (Fig. 3A), in comparison with cells treated with the control siRNA. To knock down endogenous levels of the GOLPH3 proteins, siRNA-targeting GOLPH3 was transfected into HEK293 cells. FACS analysis on these cells showed that the knockdown of GOLPH3 reduced IIH6 immunoreactivity when compared with cells transfected with the control siRNA (Fig. 3B). This reduction is indicative of an alteration in the glycosylation of α-dystroglycan. The knockdown of POMGnT1 and GOLPH3 following RNAi-mediated silencing was confirmed at the mRNA level using qRT-PCR. The results show that the POMGnT1 and GOLPH3 siRNA resulted in ∼80% knockdown of POMGnT1 and GOLPH3 mRNA levels, respectively (Fig. 3, C and D).
FIGURE 3.

GOLPH3 knockdown reduces functional glycosylation of α-dystroglycan. A, HEK293 cells were transfected with POMGnT1 siRNA, followed by FACS analysis with IIH6 antibody. B, HEK293 cells were transfected with GOLPH3 siRNA, followed by FACS analysis with IIH6 antibody. Control (Ctrl) siRNA indicates non-targeting siRNA control. NC, negative control without the primary antibody. Cell surface α-dystroglycan produced in cells treated with POMGnT1 and GOLPH3 siRNA displayed reduced IIH6 immunoreactivity. The relative POMGnT1 (C) and GOLPH3 mRNA levels (D) following transfection with the corresponding siRNAs were quantified by qRT-PCR. Bar charts summarize data obtained from three replicates and was normalized against endogenous GAPDH. Error bars indicate S.E. or S.D. A significant reduction in the expression of both genes was observed.
Clinically Relevant Mutations in POMGnT1 Failed to Localize to the Golgi
We next attempted to gain further insights into three clinically relevant mutations in POMGnT1 represented by E223K (30), C269Y (30), and R265H (31). These mutations occurred within the stem domain of POMGnT1. In vitro data showed that E223K displayed complete inactivity (32), whereas R265H and C269Y resulted in a 5-fold decrease in enzymatic activity (32). The subcellular localization of these mutant proteins, however, has not been examined. Thus, we performed immunofluorescence microscopy to determine this.
Single point mutations of POMGnT1 representing E223K, R265H, and C269Y were generated via site-directed mutagenesis. The intracellular distribution of these mutant proteins was assessed. As depicted in Fig. 4A, the mutants failed to localize to the Golgi as demonstrated by their inability to co-localize with giantin. Upon further analysis, we demonstrated that these mutant proteins were mislocalized to ER as shown by the co-localization with the PDI (Fig. 4B). These data represents the first to demonstrate the mislocalization of these clinically relevant mutations in POMGnT1 and thus demonstrates the importance of Golgi localization of glycosyltransferases.
FIGURE 4.
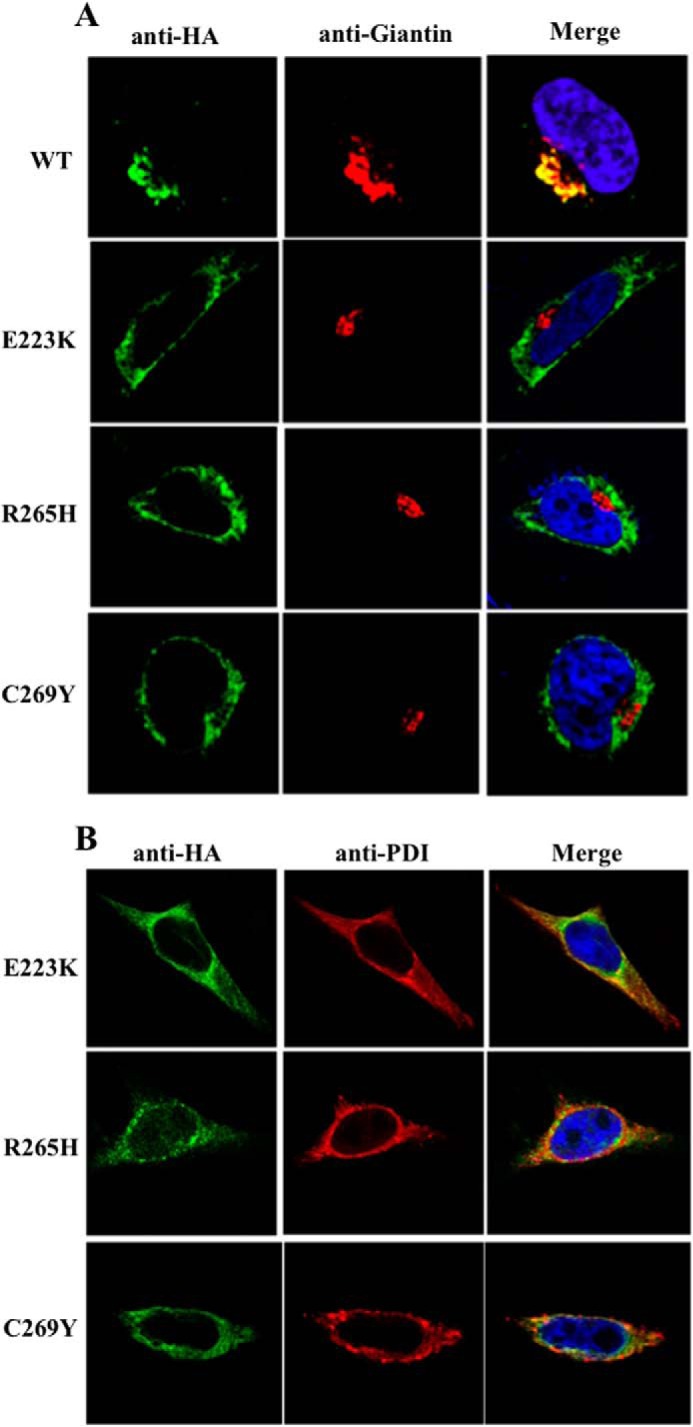
Three clinically relevant POMGnT1 mutations failed to localize to the Golgi. A, the indicated HA-tagged POMGnT1 mutants were transiently expressed in HeLa cells. Fixed cells were stained for HA (green), giantin (red), and DAPI (blue). E223K, R265H, and C269Y failed to localize to the Golgi. B, HA-tagged POMGnT1 mutants were transiently expressed in HeLa cells. Fixed cells were stained for HA (green), PDI (red), and DAPI (blue). E223K, R265H, and C269Y localized to the ER.
DISCUSSION
The yeast ortholog of GOLPH3, Vps74p, functions by interacting with and anchoring multiple mannosyltransferases at the Golgi. Here, we have identified a novel interaction between GOLPH3 and POMGnT1, thus highlighting the conserved nature of the GOLPH3 family of proteins in mediating interactions with glycosyltransferases. It is reasonable to speculate that these interactions are direct as the characteristic feature of phage display is its ability to identify binary interactions between proteins. In the same screen, alongside POMGnT1, we also identified ppGalNAc-12, which is implicated in the initiation of mammalian O-linked glycosylation. Interestingly, a conserved motif (RXLRR), exists within the cytoplasmic domain of both glycosyltransferases. Sequence alignments revealed that the conserved motif is also present in ppGalNAc-6. The significance of the observation that GOLPH3 may interact with specific members of the ppGalNAc transferase family remains to be established. It is clear that the identified motif in POMGnT1 does not match the consensus Vps74p recognition sequence (F/L)(L/I/V)XX(R/K) present in the cytoplasmic tails of yeast mannosyltransferases (8). Thus, our result potentially expands the repertoire of possible GOLPH3 recognition sequences.
POMGnT1 catalyzes the second step in the O-mannosylation of α-dystroglycan by mediating the transfer of GlcNAc from UDP-GlcNAc onto mannose in the 2-OH position. The localization of POMGnT1 at its Golgi compartment is a prerequisite for the enzyme to perform its designated role in glycosylation. However, the molecular mechanism responsible for the Golgi localization of POMGnT1 is not clearly defined. Several reports have indicated that the localization of glycosyltransferases at the Golgi is maintained via the interaction of its cytoplasmic tails with a glycosyltransferase specific cytoplasmic protein (8, 33–35). However, only a few of such proteins have been identified.
Here, we demonstrate that GOLPH3, a peripheral Golgi membrane protein mediates the Golgi localization of POMGnT1 by interacting with the cytoplasmic tail of the glycosyltransferase. This was demonstrated by the substantial reduction in binding capacity of GOLPH3 following the deletion of the cytoplasmic tail of POMGnT1 and the subsequent mislocalization of the glycosyltransferase. The finding is consistent with previous reports demonstrating that the GOLPH3 family of proteins indeed binds to the cytoplasmic tails of C2GnT1 (10) and yeast mannosyltransferases (8, 9). We did not observe a complete abrogation of GOLPH3 binding following the deletion of the POMGnT1 cytoplasmic tail. However, we speculate that this could be due to the fact that POMGnT1, similar to many other glycosyltransferases, form homodimers in vivo. Thus, the observed GOLPH3 band could be due to interactions with endogenous POMGnT1.
It is clear that the results from the phage display that identified the stem of POMGnT1 as the domain that interacts with GOLPH3 is incongruent with that of the co-immunoprecipitation data. Although this remains puzzling, we speculate that this discrepancy could be due to the fundamental differences inherent in the systems used to assess the interaction between POMGnT1 and GOLPH3. In the T7 phage display, protein interactions are assessed in a cellular environment that is not native to the respective proteins. Although we do not doubt that GOLPH3 binds to the stem domain of POMGnT1 in the phage system, this domain is unavailable for binding to GOLPH3 in mammalian system due to the type II membrane topology of glycosyltransferases.
The role of the GOLPH3 proteins in maintaining the Golgi localization of POMGnT1 is further supported by our observation that the transferase localized to the ER following RNAi-mediated knockdown of endogenous levels of GOLPH3. We speculate that this inability of POMGnT1 to localize to the Golgi represents a failure in the mechanism to retain the enzyme in the Golgi rather than failure in the targeting of proteins to the Golgi. This is based on results by Ali et al. (10), demonstrating that the processing of glycans on C2GnT1 precedes retaining of C2GnT1 in the Golgi by GOLPH3 as C2GnT1 in GOLPH3-depleted cells mislocalized to the ER and displayed complex-type N-glycans terminating with GlcNAc.
The inability of POMGnT1 to maintain its Golgi localization following GOLPH3 knockdown is suggestive of the loss of its enzymatic activity as a glycosyltransferase. In line with this, we showed that the RNAi-mediated knockdown of GOLPH3 resulted in the reduction in IIH6 immunoreactivity, indicative of an alteration in the glycosylation status of α-dystroglycan. Taken together, these results suggest a novel role for the GOLPH3 proteins in glycosylation of α-dystroglycan. The critical glycan on α-dystroglycan required for laminin binding has been identified to be a novel phosphorylated O-mannosyl glycan consisting of a phosphate group that is linked to the 6-hydroxyl position of the core O-mannose residue on α-dystroglycan (36) and is further extended with repeating units of [-3-xylose-α1,3-glucoronic acid-β1-] (37). POMGnT1-deficient cells or tissues were shown to display defects in the postphosphoryl modification of the O-mannosyl glycan (36, 38). Based on the negative impacts of down-regulated GOLPH3 and POMGnT1 on IIH6 immunoreactivity, we speculate that GOLPH3 influences the formation of the IIH6-reacting glycoepitope, at least in part, via mediating POMGnT1 Golgi localization. Recent data has demonstrated that glycosytransferase-like domain containing 2 present in the ER, possesses protein O-mannose β1,4-N-acetylglucosmainyltransferase activity and is responsible for generating the phosphotrisaccharide that likely contains the LARGE-dependant glycan essential for laminin binding (39). In line with this, we speculate that our observed reduction in IIH6 immunoreactivity of α-dystroglycan following GOLPH3 knockdown could be due to the inability to form the 4-GlcNAc extension of key mannose residues as a result of improper 2-GlcNAc extension by mislocalized POMGnT1.
We have also shown that three mutations within the stem domain of POMGnT1 that exists in patients with muscle-eye-brain mislocalized to the ER. To our knowledge, these data represents the first to demonstrate the mislocalization of these clinically relevant mutations in POMGnT1. It further demonstrates the importance of Golgi localization of glycosyltransferases. Thus, elucidating the mechanisms responsible for the localization of this family of enzymes is critical.
In this study, we have determined the mechanism for Golgi targeting of POMGnT1. This glycosyltransferase is targeted to the Golgi via interaction of its N-terminal cytoplasmic tail with GOLPH3. Subsequently, we showed that GOLPH3 affects the processing and/or maturation of α-dystroglycan raising the possibility that the GOLPH3 gene could be implicated in the development of muscle-eye-brain disease. These data provide a further understanding of the role of GOLPH3 in mediating the Golgi localization glycosyltransferases in mammalian cells.
This work was supported by the Agency for Science, Technology, and Research (A*STAR), Singapore.
- GlcNAc
- N-acetylglucosamine
- qRT
- quantitative reverse transcription
- ER
- endoplasmic reticulum
- PDI
- protein disulfide isomerase.
REFERENCES
- 1. Wu C. C., Taylor R. S., Lane D. R., Ladinsky M. S., Weisz J. A., Howell K. E. (2000) GMx33: a novel family of trans-Golgi proteins identified by proteomics. Traffic 1, 963–975 [PubMed] [Google Scholar]
- 2. Bell A. W., Ward M. A., Blackstock W. P., Freeman H. N., Choudhary J. S., Lewis A. P., Chotai D., Fazel A., Gushue J. N., Paiement J., Palcy S., Chevet E., Lafrenière-Roula M., Solari R., Thomas D. Y., Rowley A., Bergeron J. J. (2001) Proteomics characterization of abundant Golgi membrane proteins. J. Biol. Chem. 276, 5152–5165 [DOI] [PubMed] [Google Scholar]
- 3. Scott K. L., Chin L. (2010) Signaling from the Golgi: mechanisms and models for Golgi phosphoprotein 3-mediated oncogenesis. Clin. Cancer Res. 16, 2229–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kunigou O., Nagao H., Kawabata N., Ishidou Y., Nagano S., Maeda S., Komiya S., Setoguchi T. (2011) Role of GOLPH3 and GOLPH3L in the proliferation of human rhabdomyosarcoma. Oncol. Rep. 26, 1337–1342 [DOI] [PubMed] [Google Scholar]
- 5. Li X. Y., Liu W., Chen S. F., Zhang L. Q., Li X. G., Wang L. X. (2011) Expression of the Golgi phosphoprotein-3 gene in human gliomas: a pilot study. J. Neurooncol. 105, 159–163 [DOI] [PubMed] [Google Scholar]
- 6. Wullschleger S., Loewith R., Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 7. Dippold H. C., Ng M. M., Farber-Katz S. E., Lee S. K., Kerr M. L., Peterman M. C., Sim R., Wiharto P. A., Galbraith K. A., Madhavarapu S., Fuchs G. J., Meerloo T., Farquhar M. G., Zhou H., Field S. J. (2009) GOLPH3 bridges phosphatidylinositol-4-phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell 139, 337–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tu L., Tai W. C., Chen L., Banfield D. K. (2008) Signal-mediated dynamic retention of glycosyltransferases in the Golgi. Science 321, 404–407 [DOI] [PubMed] [Google Scholar]
- 9. Schmitz K. R., Liu J., Li S., Setty T. G., Wood C. S., Burd C. G., Ferguson K. M. (2008) Golgi localization of glycosyltransferases requires a Vps74p oligomer. Dev. Cell 14, 523–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ali M. F., Chachadi V. B., Petrosyan A., Cheng P. W. (2012) Golgi phosphoprotein 3 determines cell binding properties under dynamic flow by controlling Golgi localization of Core 2 N-acetylglucosaminyltransferase 1. J. Biol. Chem. 287, 39564–39577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blake D. J., Weir A., Newey S. E., Davies K. E. (2002) Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 82, 291–329 [DOI] [PubMed] [Google Scholar]
- 12. Henry M. D., Campbell K. P. (1999) Dystroglycan inside and out. Curr. Opin. Cell Biol. 11, 602–607 [DOI] [PubMed] [Google Scholar]
- 13. Durbeej M., Henry M. D., Campbell K. P. (1998) Dystroglycan in development and disease. Curr. Opin. Cell Biol. 10, 594–601 [DOI] [PubMed] [Google Scholar]
- 14. Michele D. E., Barresi R., Kanagawa M., Saito F., Cohn R. D., Satz J. S., Dollar J., Nishino I., Kelley R. I., Somer H., Straub V., Mathews K. D., Moore S. A., Campbell K. P. (2002) Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature 418, 417–422 [DOI] [PubMed] [Google Scholar]
- 15. Michele D. E., Campbell K. P. (2003) Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J. Biol. Chem. 278, 15457–15460 [DOI] [PubMed] [Google Scholar]
- 16. Muntoni F., Brockington M., Torelli S., Brown S. C. (2004) Defective glycosylation in congenital muscular dystrophies. Curr. Opin. Neurol. 17, 205–209 [DOI] [PubMed] [Google Scholar]
- 17. Holzfeind P. J., Grewal P. K., Reitsamer H. A., Kechvar J., Lassmann H., Hoeger H., Hewitt J. E., Bittner R. E. (2002) Skeletal, cardiac and tongue muscle pathology, defective retinal transmission, and neuronal migration defects in the Large(myd) mouse defines a natural model for glycosylation-deficient muscle - eye - brain disorders. Hum. Mol. Genet. 11, 2673–2687 [DOI] [PubMed] [Google Scholar]
- 18. Kanagawa M., Michele D. E., Satz J. S., Barresi R., Kusano H., Sasaki T., Timpl R., Henry M. D., Campbell K. P. (2005) Disruption of perlecan binding and matrix assembly by post-translational or genetic disruption of dystroglycan function. FEBS Lett. 579, 4792–4796 [DOI] [PubMed] [Google Scholar]
- 19. Barresi R., Campbell K. P. (2006) Dystroglycan: from biosynthesis to pathogenesis of human disease. J. Cell Sci. 119, 199–207 [DOI] [PubMed] [Google Scholar]
- 20. Yoshida A., Kobayashi K., Manya H., Taniguchi K., Kano H., Mizuno M., Inazu T., Mitsuhashi H., Takahashi S., Takeuchi M., Herrmann R., Straub V., Talim B., Voit T., Topaloglu H., Toda T., Endo T. (2001) Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev. Cell 1, 717–724 [DOI] [PubMed] [Google Scholar]
- 21. Smith G. P. (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315–1317 [DOI] [PubMed] [Google Scholar]
- 22. Winter G., Milstein C. (1991) Man-made antibodies. Nature 349, 293–299 [DOI] [PubMed] [Google Scholar]
- 23. Clackson T., Hoogenboom H. R., Griffiths A. D., Winter G. (1991) Making antibody fragments using phage display libraries. Nature 352, 624–628 [DOI] [PubMed] [Google Scholar]
- 24. Yamamoto M., Kominato Y., Yamamoto F. (1999) Phage display cDNA cloning of protein with carbohydrate affinity. Biochem. Biophys. Res. Commun. 255, 194–199 [DOI] [PubMed] [Google Scholar]
- 25. Danner S., Belasco J. G. (2001) T7 phage display: a novel genetic selection system for cloning RNA-binding proteins from cDNA libraries. Proc. Natl. Acad. Sci. U.S.A. 98, 12954–12959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qin H., Pu H. X., Li M., Ahmed S., Song J. (2008) Identification and structural mechanism for a novel interaction between a ubiquitin ligase WWP1 and Nogo-A, a key inhibitor for central nervous system regeneration. Biochemistry 47, 13647–13658 [DOI] [PubMed] [Google Scholar]
- 27. Nagano-Ito M., Yoshikawa S., Tamura M., Tsurumaki M., Ichikawa S. (2007) Identification and characterization of a novel alternative splice variant of mouse GMx33α/GPP34. Gene 400, 82–88 [DOI] [PubMed] [Google Scholar]
- 28. Ervasti J. M., Campbell K. P. (1993) A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 122, 809–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. de Bernabé D. B., Inamori K., Yoshida-Moriguchi T., Weydert C. J., Harper H. A., Willer T., Henry M. D., Campbell K. P. (2009) Loss of α-dystroglycan laminin binding in epithelium-derived cancers is caused by silencing of LARGE. J. Biol. Chem. 284, 11279–11284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taniguchi K., Kobayashi K., Saito K., Yamanouchi H., Ohnuma A., Hayashi Y. K., Manya H., Jin D. K., Lee M., Parano E., Falsaperla R., Pavone P., Van Coster R., Talim B., Steinbrecher A., Straub V., Nishino I., Topaloglu H., Voit T., Endo T., Toda T. (2003) Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum. Mol. Genet. 12, 527–534 [DOI] [PubMed] [Google Scholar]
- 31. Vervoort V. S., Holden K. R., Ukadike K. C., Collins J. S., Saul R. A., Srivastava A. K. (2004) POMGnT1 gene alterations in a family with neurological abnormalities. Ann. Neurol. 56, 143–148 [DOI] [PubMed] [Google Scholar]
- 32. Voglmeir J., Kaloo S., Laurent N., Meloni M. M., Bohlmann L., Wilson I. B., Flitsch S. L. (2011) Biochemical correlation of activity of the α-dystroglycan-modifying glycosyltransferase POMGnT1 with mutations in muscle-eye-brain disease. Biochem. J. 436, 447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Osman N., McKenzie I. F., Mouhtouris E., Sandrin M. S. (1996) Switching amino-terminal cytoplasmic domains of α-1,2-fucosyltransferase and α1,3-galactosyltransferase alters the expression of H substance and Galα1,3Gal. J. Biol. Chem. 271, 33105–33109 [DOI] [PubMed] [Google Scholar]
- 34. Uliana A. S., Giraudo C. G., Maccioni H. J. (2006) Cytoplasmic tails of SialT2 and GalNAcT impose their respective proximal and distal Golgi localization. Traffic 7, 604–612 [DOI] [PubMed] [Google Scholar]
- 35. Okamoto M., Yoko-o T., Miyakawa T., Jigami Y. (2008) The cytoplasmic region of 1,6-mannosyltransferase Mnn9p is crucial for retrograde transport from the Golgi apparatus to the endoplasmic reticulum in Saccharomyces cerevisiae. Eukaryot. Cell 7, 310–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshida-Moriguchi T., Yu L., Stalnaker S. H., Davis S., Kunz S., Madson M., Oldstone M. B., Schachter H., Wells L., Campbell K. P. (2010) O-Mannosyl phosphorylation of α-dystroglycan is required for laminin binding. Science 327, 88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inamori K., Yoshida-Moriguchi T., Hara Y., Anderson M. E., Yu L., Campbell K. P. (2012) Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 335, 93–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuga A., Kanagawa M., Sudo A., Chan Y. M., Tajiri M., Manya H., Kikkawa Y., Nomizu M., Kobayashi K., Endo T., Lu Q. L., Wada Y., Toda T. (2012) Absence of post-phosphoryl modification in dystroglycanopathy mouse models and wild-type tissues expressing non-laminin binding form of α-dystroglycan. J. Biol. Chem. 287, 9560–9567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yoshida-Moriguchi T., Willer T., Anderson M. E., Venzke D., Whyte T., Muntoni F., Lee H., Nelson S. F., Yu L., Campbell K. P. (2013) SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science 341, 896–899 [DOI] [PMC free article] [PubMed] [Google Scholar]