

Background: Mitochondrial proteins are controlled by glutaredoxin-2 (Grx2)-mediated deglutathionylation reactions.
Results: Grx2 deficiency compromises cardiac mitochondrial functions leading to hypertrophy and fibrosis in male mice. This is associated with deregulated glutathionylation reactions and mitochondrial dysfunction.
Conclusion: Through deglutathionylation, Grx2 controls mitochondrial oxidative phosphorylation in cardiac muscle.
Significance: Deregulated glutathionylation in heart can have pathological consequences.
Keywords: Bioenergetics, Cardiac Hypertrophy, Cardiac Metabolism, Glutathionylation, Mitochondria, Redox, Glutaredoxin-2, Glutathione, Oxidative Phosphorylation
Abstract
Glutaredoxin-2 (Grx2) modulates the activity of several mitochondrial proteins in cardiac tissue by catalyzing deglutathionylation reactions. However, it remains uncertain whether Grx2 is required to control mitochondrial ATP output in heart. Here, we report that Grx2 plays a vital role modulating mitochondrial energetics and heart physiology by mediating the deglutathionylation of mitochondrial proteins. Deletion of Grx2 (Grx2−/−) decreased ATP production by complex I-linked substrates to half that in wild type (WT) mitochondria. Decreased respiration was associated with increased complex I glutathionylation diminishing its activity. Tissue glucose uptake was concomitantly increased. Mitochondrial ATP output and complex I activity could be recovered by restoring the redox environment to that favoring the deglutathionylated states of proteins. Grx2−/− hearts also developed left ventricular hypertrophy and fibrosis, and mice became hypertensive. Mitochondrial energetics from Grx2 heterozygotes (Grx2+/−) were also dysfunctional, and hearts were hypertrophic. Intriguingly, Grx2+/− mice were far less hypertensive than Grx2−/− mice. Thus, Grx2 plays a vital role in modulating mitochondrial metabolism in cardiac muscle, and Grx2 deficiency leads to pathology. As mitochondrial ATP production was restored by the addition of reductants, these findings may be relevant to novel redox-related therapies in cardiac disease.
Introduction
Glutathionylation reactions have emerged as key post-translational modifications required to control protein function in response to changes in redox (1). These modifications involve the formation of a disulfide bridge between glutathione (GSH) and an available protein cysteine thiol. Reactions can either proceed nonenzymatically or enzymatically, depending on cell conditions (2). For instance, the former occurs mostly during oxidative stress when 2GSH/GSSG is ∼1, and levels of reactive oxygen species (ROS)2 are high. Nonenzymatic glutathionylation reactions are nonspecific and can lead to the hyper-glutathionylation of proteins, altering their activity (3–5). Enzymatic glutathionylation reactions, however, are tightly controlled and highly specific post-translational modifications that occur normally in response to fluctuations in local redox environments (6–8). The glutaredoxin (Grx) proteins are currently thought to be the chief enzymes that catalyze these reactions (9). Specifically the Grx proteins catalyze the deglutathionylation of protein glutathione disulfide (PSSG) mixtures. In the cytosol, Grx1 is required to catalyze the deglutathionylation of target proteins (10). Grx1 catalyzes the deglutathionylation of protein targets in two steps as follows: 1) N-terminal cysteine on Grx1 deglutathionylates PSSG via a thiol disulfide exchange reaction yielding the deglutathionylated protein (PSH) and a Grx1-glutathionylated protein (SSG) intermediate; 2) Grx1-SSG binds GSH, and the glutathionyl moiety is removed regenerating Grx1 and producing GSSG (11). Although the mechanistic details of the catalytic reaction still require clarification (12), several models for the characteristic nucleophilic displacement ping-pong mechanism of Grx1 have been proposed and include a glutathione scaffold, glutathione activator, and an oxidative half-reaction (12). However, emerging evidence has also suggested that the Grx enzymes can also catalyze protein glutathionylation (13). This can be mediated via the stabilization of a glutathione thiyl radical that is then subsequently transferred to an available protein thiol (13).
Mitochondrial processes are particularly susceptible to regulation by glutathionylation. This is due to the unique physical properties of the matrix environment. The slight alkalinity of the matrix ionizes protein thiols increasing reactivity toward glutathione (14). Mitochondria also contain high amounts of protein thiols (60–90 mm), reduced glutathione (GSH; ∼5 mm), and are a major source of ROS, factors that render protein glutathionylation reactions more favorable (14, 15). Mitochondrial redox environment fluctuations also occur during fuel oxidation, electron transfer, and the formation of ROS. The thiol oxidoreductase glutaredoxin-2 (Grx2), which has ∼34% homology to Grx1, was recently identified as the enzyme required to catalyze deglutathionylation reactions in mitochondria (16–19). The catalytic cycle of Grx2 is also quite similar to Grx1 except that the Grx2-SSG intermediate can be reduced by NADPH and thioredoxin reductase (20). It is also important to point out that unlike Grx1, Grx2 complexes iron (Fe), which is required to modulate its activity (21) (Grx2 regulation by coordination of 2Fe-2S cluster was recently reviewed in detail in Refs. 22, 23). Intriguingly, Grx2 has been shown to catalyze both the deglutathionylation and glutathionylation of target proteins in mitochondria. The reversible nature of Grx2 is associated with its sensitivity to changes in 2GSH/GSSG as follows: a high 2GSH/GSSG promotes protein deglutathionylation and a low 2GSH/GSSG activates Grx2 glutathionylase activity (24, 25). The main target for Grx2 in mitochondria is complex I (24). Increases and decreases in 2GSH/GSSG prompt Grx2-mediated deglutathionylation and glutathionylation of complex I, which then alters its activity and superoxide production (24). Complex I has also been shown to be a Grx2 target in lens epithelia and liver mitochondria (26–28). UCP3 has also been shown to be targeted by Grx2 in skeletal muscle (28). 2-Oxoglutarate dehydrogenase may also be targeted by Grx2 because it can be deglutathionylated in vitro by exogenously added Grx1 (29).
The heart muscle turns over ∼30 kg of ATP daily (30). Clearly, mitochondrial ATP production is central to heart function. A variety of control mechanisms converge on mitochondria to adjust ATP output. Changes in redox due to temporal differences in mitochondrial metabolism (ROS production, NAD(P)H levels, and GSH and GSSG levels) can feedback to modulate oxidative phosphorylation. A decrease in 2GSH/GSSG could prompt glutathionylation of cardiac mitochondrial proteins, although a reductive glutathione ratio has the opposite effect (24, 31). A number of glutathionylation targets in cardiac mitochondria have been identified, including complex I subunit NDUSF1, complex II subunit succinate dehydrogenase B, 2-oxoglutarate dehydrogenase, and ATP synthase α subunit (25, 29, 32, 33). These proteins fulfill important energetic roles in cardiac mitochondria, including energy substrate metabolism and oxidative phosphorylation. Alterations in the control of mitochondrial glutathionylation can have profound physiological effects. Grx2 overexpression prevents doxorubicin-induced cardiac injury, and this is associated with recovery of mitochondrial respiration (34). Irregularities in the glutathionylation of the ATP synthase α subunit are associated with cardiac dysfunction (33). In addition, glutathionylation of complex I compromises aerobic metabolism in the heart, contributing to the development of cardiac dysfunction (31). Ischemic reperfusion injury also alters the glutathionylation state of complex II (32). Thus, the disruption of controlled protein glutathionylation reactions can compromise ATP output by cardiac mitochondria, potentially leading to disease.
Despite the number of glutathionylation targets in cardiac mitochondria, it still remains unknown if reversible glutathionylation can regulate oxidative phosphorylation (OXPHOS; ATP output). It is also unclear if deregulation of protein deglutathionylation reactions (e.g. loss of Grx2 deglutathionylase activity) in mitochondria can contribute to the development of cardiac dysfunction. In this study, we examine the importance of Grx2 function in cardiac physiology and metabolism. We used germ line Grx2 whole body knock-out (Grx2−/−, Grx2-deficient) mice, which we have recently characterized (28). In our previous study, we found that Grx2 deficiency did not induce any major defects (linear growth, food intake, and liver physiology), but we did note that hearts weighed significantly more (28). In this study, we examined the impact of Grx2 deficiency on cardiac metabolism and function. We found that OXPHOS was substantially decreased in cardiac mitochondria from Grx2-deficient mice. The decrease in OXPHOS was due to the glutathionylation of complex I resulting in decreased activity. These biochemical effects were reversed with dithiothreitol (DTT), a reductant known to prompt protein deglutathionylation (35, 36), thereby favoring deglutathionylation. The biochemical changes in OXPHOS were associated with development of cardiac abnormalities, including left ventricular hypertrophy, fibrosis, and a metabolic shift toward increased glucose uptake. We conclude that Grx2-mediated reversible glutathionylation is critical in modulating mitochondrial ATP output in cardiac muscle, and disruption of this process is pathological.
EXPERIMENTAL PROCEDURES
Animals
Studies were conducted on male C57BL/6 wild type (WT) and Grx2 whole body knock-out (Grx2−/−) mice aged 9–12 weeks that were backcrossed for over 10 generations into the C57BL/6 strain. Mice were fed ad libitum a standard diet (44.2% carbohydrate, 6.2% fat, 18.6% crude protein; diet T.2018, Harlan, Indianapolis, IN). All experiments were performed according to the principles and guidelines of the Canadian Council of Animal Care, and the study was approved by the Animal Care Committee of the University of Ottawa. Mice were genotyped for Grx2 prior to experimentation. Body and heart weights along with femur lengths were measured. Serum hemoglobin levels were measured using a kit purchased from Sigma, and assays were conducted as described by the manufacturer. For time profile experiments, mice were aged either 6 or 10 weeks.
Echocardiography
For M-mode and pulsed-wave Doppler echocardiography, mice were anesthetized (2.0% isoflurane, 80 ml/min 100% O2); anterior chests were shaved, and two-dimensional guided M-mode and pulsed-wave Doppler echocardiography were performed using a VEVO 2100 system (Visual Sonics, Amsterdam) with a 30-MHz linear array transducer. Images and videos were analyzed using the VEVO 2100 analysis software.
MicroPET Imaging
Small animal PET FDG imaging was conducted with the InveonTM DPET small animal scanner (Siemens, Knoxville, TN). Mice were scanned under anesthesia (2–2.5% isoflurane, 2–2.5 ml/min oxygen) in a fed state. A 60-min list-mode acquisition was started together with a 10–20-s tail vein injection of FDG (25.9 ± 6.94 MBq in 150 μl). List data were sorted into 26 dynamic frames (12 × 10, 3 × 60, and 11 × 300s) and reconstructed using OSEM3D with 10 iterations, 16 subsets, zoom of 2.5 with a 128 × 128 matrix, resulting in a 0.35-mm transaxial pixel size. Images were corrected for radioactive decay, random coincidences, and dead-time losses using the vendor software (IAW version 1.5). Blood glucose was measured prior to FDG injection with Advantage blood glucose strips (AccuChek, Roche Diagnostics). Myocardial time activity curves were compared using standard uptake values calculated by dividing the activity concentration in the region of interest (Bq/ml) by the activity concentration per body weight injected into the animal (Bq/g) (Siemens IRW software). Patlak kinetic analysis of reconstructed images was performed on the myocardium at 10–40 min as described previously with FlowQuant semi-automated software (63). Regional rate of myocardial glucose uptake (rMGU) was determined for each animal using the Equation 1,
where Ki is the Patlak uptake rate constant; BG is the blood glucose concentration, and LC is the lumped constant equal to 0.67, which accounts for differences in the uptake and phosphorylation of FDG versus glucose.
Fibrosis Determinations
Hearts were placed in 10% formalin for 48 h and then in 70% ethanol prior to paraffin embedding. Transverse sections were taken at the vertical midpoint of the heart (4 μm each), and deparaffinized sections were stained with Sirius Red, which stains fibrillar types 1 and 3 collagen. Analysis of the extent of fibrosis was determined using Zen software (Carl Zeiss Microscopy) whereby thresholds were set for collagen-stained areas, and identified areas were expressed as a percentage of the total tissue area.
Blood Pressure
Systolic blood pressure was determined using a BP-2000 Blood Pressure Analysis SystemTM (Visitech Systems; Apex, NC). This is a noninvasive computerized tail-cuff system. Mice (age of 10 weeks) were placed on the BP-200 platform (pre-warmed to 30 °C), and tails were passed through the tail cuffs. Mice were acclimated to the system daily for 5 days. After acclimation, blood pressure measurements were conducted between 9:00 and 11:00 a.m. daily for 5 days. Each day, mice were further acclimatized for 10 min prior to the 20-min measurement period.
Mitochondrial Ultrastructure
Electron microscopic imaging was performed on fixed and stained sections of the left ventricle. For heart fixation, mice were fully anesthetized using isofluorane (∼5%, O2 at ∼1 liter/min). The left ventricle near the apex was punctured with a 25-gauge needle, and a small incision was made in right atrium for fluid efflux. Hearts were then perfused slowly with heparinized saline until the efflux was clear and then perfused with fixative (2.5% glutaraldehyde + 2% formaldehyde in PBS). Hearts were excised, placed in fixative, and stored at 4 °C. A Leica Ultracut E ultramicrotome was used to section the tissue, and sections were counterstained with lead citrate and uranyl acetate. Images were collected using JEOL 1230 transmission electron microscopy at 60 kV.
Mitochondrial Isolation
Mitochondria were isolated as described previously (37). All steps were performed on ice or at 4 °C. Hearts were excised and placed immediately in homogenizing buffer (HM: 220 mm mannitol, 70 mm sucrose, 5 mm HEPES, 1 mm EGTA, 10 mm pyruvate, 2 mm malate or 0.5 mm l-carnitine, 2 mm malate, pH 7.2). On the day of experiments, HM was supplemented with 1% (w/v) defatted BSA + 2 units of subtilisin A. Tissue was cut into small pieces and washed four times in HM. Pieces were minced and then homogenized using the Potter-Elvehjem method. Homogenates were placed in centrifugation tubes and spun at 18,000 × g for 9 min. Pellets were resuspended in fresh HM and centrifuged at 800 × g for 9 min. Supernatants were carefully transferred into new centrifugation tubes and spun at 10,000 × g for 9 min. The mitochondrial pellets were resuspended in a 100 μl of HM devoid of BSA and subtilisin A. Protein content was determined by a Bradford assay. Mitochondria were then diluted according to protein equivalents (micrograms of mitochondrial protein/ml) for various assays.
Mitochondrial Bioenergetics
The bioenergetics of isolated mitochondria were studied in a Seahorse XF24 extracellular flux analyzer (Seahorse Biosciences; North Billerica, MA) using a method adapted from Refs. 28, 37. Mitochondria were diluted to 200 μg/ml in BSA-free HM, and 50 μl (∼10 μg of mitochondria) was loaded into each Seahorse TC plate well. Mitochondria were attached to the plate surface by centrifugation at 2000 × g for 20 min at 4 °C, incubated for 10 min in reaction buffer (HM containing 10 mm KH2PO4, 5 mm MgCl2, 0.2% w/v defatted BSA, with/without 10 mm DTT + 10 mm pyruvate, 2 mm malate or 0.1 mm palmitoylcarnitine, 0.1 mm palmitate-BSA complex, 2 mm malate), and loaded into the XF24 Analyzer.
Bioenergetic parameters were measured as described previously (28). Following measurement of state 2 respiration (presence of substrate only), mitochondria were sequentially treated with ADP (0.1 mm), oligomycin (2.5 μg/ml), FCCP (8 μm), and antimycin A (4 μm) to test state 3 (phosphorylating respiration), state 4O (proton leak-dependent respiration), maximal respiration (uncoupled), and respiration independent of the respiratory chain, respectively. Respiratory control ratio (RCR) values were calculated by dividing the absolute rate of state 3 respiration by state 4O respiration. ADP titration and cytochrome c titration experiments were performed as described previously (28) to determine whether mitochondrial outer membranes were intact. All values were corrected for background O2 consumption and any O2 consumption not associated with the respiratory chain (antimycin A).
2GSH/GSSG
The potential of the 2GSH/GSSG pair was determined as described previously (38). Isolated mitochondria were diluted to 0.5 mg/ml in ice-cold 0.5% (v/v) perchloric acid solution and incubated on ice for 10 min. Samples were then centrifuged at 10,000 × g for 10 min at 4 °C to remove any precipitate. The supernatant was injected into an Agilent HPLC system equipped with a Pursuit C18 column (150 × 4.6 mm, 5 μm; Agilent Technologies) operating at a flow rate of 1 ml/min. The mobile phase consisted of 1% trifluoroacetic acid diluted in double distilled H2O and mixed with HPLC-grade methanol in a 90:10 ratio. Retention times for GSH and GSSG were estimated by injecting standard solutions. Absolute amounts of GSH and GSSG were determined by integrating the area under each peak using Agilent ChemStation software. Values were interpolated from standard curves to give nanomoles of GSH and GSSG. Values were used to calculate millimolar amounts by assuming a mitochondrial volume of 0.75 μl (28, 39). Resulting values were used to calculate 2GSH/GSSG and the Eh of the pair as shown in Equation 2,
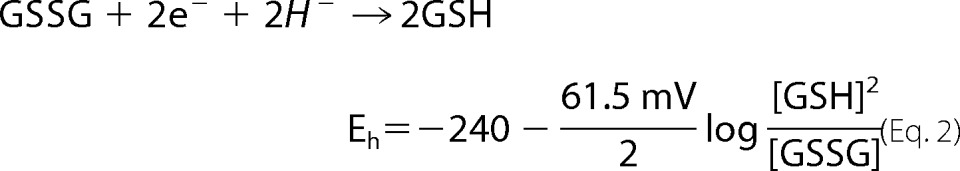 |
at 37 °C.
Glutathionylated Proteome
Protein glutathionylation levels were determined as described previously (28, 38). Freshly isolated mitochondria were treated with 25 mm N-ethylmaleimide to block unoccupied glutathionylation sites, and the suspension was diluted to 2 mg/ml in Laemmli buffer. Samples were electrophoresed on 12% SDS gels under nonreducing conditions. Upon completion, proteins were electroblotted to nitrocellulose membranes, blocked for nonspecific binding sites, and probed with PSSG antiserum (1:500, Virogen). Note that the antibody was diluted in 5% (w/v) nonfat skim milk dissolved in TBS-T (Tris-buffered saline + 0.1% (v/v) Tween 20) and incubated under constant agitation overnight at 4 °C. Membranes were then probed with anti-mouse HRP-conjugated secondary antibody diluted in 5% (w/v) nonfat skim milk in TBS-T for 1 h at room temperature (1:3000, Santa Cruz Biotechnology). Between each step, the membranes were washed two times for 5 min with TBS-T. Samples prepared in Laemmli buffer containing 2% (v/v) β-mercaptoethanol (β-MetOH) served as the control. β-Mercaptoethanol is a powerful reductant and is thus very effective at removing glutathionyl moieties from protein cysteine thiols. Thus, samples electrophoresed under reducing conditions ensure that the observed immunoreactivity of PSSG antiserum is due to the presence of glutathionylated proteins. Immunoreactive bands were detected using enhanced chemiluminescent substrate for visualization (ECL kit, Thermo Scientific). Ponceau S staining of membranes served as a loading control. The total amount of glutathionylated proteins was then quantified using ImageJ software.
Complex I Activity Assays
Mitochondria were treated with 1% (v/v) digitonin and incubated on ice for 30 min. Permeabilized mitochondria were then diluted to 0.5 mg/ml in assay buffer (70 mm sucrose, 220 mm mannitol, 1 mm EGTA, 2 mm HEPES, 10 mm KH2PO4, 5 mm MgCl2, 0.2% w/v defatted BSA, pH 7.2) containing 2 mm KCN, 1 μm antimycin A, with or without 10 mm DTT. Reactions were initiated by the addition of NADH (0.2 mm). The rate of NADH consumption was calculated for the first 30 s to 1 min of the reaction because this represents the time for most rapid consumption of NADH. Reactions performed in the absence of mitochondria or NADH served as controls. All values were corrected for background NADH consumption.
The impact of 2GSH/GSSG and exogenous Grx1 on complex I activity was tested as described previously (24) with a few modifications. Digitonized mitochondria were incubated at 37 °C in different amounts of reduced and oxidized glutathione (GSH and GSSG, respectively) to provide 2GSH/GSSG ratios of 100 and 1, respectively. For mitochondrial preparations that were incubated with a 2GSH/GSSG of ∼1, incubations were also performed in the absence or presence of 10 units of Grx1. Following a 5-min incubation, complex I activity was tested. The potential of the 2GSH/GSSG redox pair was calculated using Equation 2.
Mitochondrial ROS Emission
Mitochondrial ROS production kinetics were measured using dihydrodichlorofluorescein diacetate in a BioTek SynergyMX2 microplate reader (Winooski, VT) using Gen5 software, as described previously (38). Mitochondria were diluted to 0.5 mg/ml in reaction buffer (70 mm sucrose, 220 mm mannitol, 1 mm EGTA, 2 mm HEPES, 10 mm KH2PO4, 5 mm MgCl2, 0.2% w/v defatted BSA, pH 7.2) containing dihydrodichlorofluorescein diacetate (20 μm), 1 μg/ml oligomycin, with or without 10 mm DTT. Reactions were initiated by the addition of pyruvate (10 mm) and malate (2 mm). Reactions devoid of mitochondria served as the control. Values were corrected for background fluorescence.
Metabolite Analysis
Mitochondria were diluted to 0.5 mg/ml in perchloric acid solution and clarified as described above. Supernatants were then injected into an Agilent HPLC system equipped with a C18 reverse phase hydrophilic HPLC column (Synergy Hydro-RP, 4 μm, 250 × 4.6 mm; Phenomenex) and operated at a flow rate of 0.7 ml/min. The mobile phase consisted of 20 mm KH2PO4 dissolved in double distilled H2O, pH 2.9. TCA cycle intermediates and ATP were detected using an Agilent variable wavelength detector set to 210 and 254 nm. Retention times were confirmed by injecting standard solutions. Levels of metabolites were quantified using Agilent ChemStation software.
Oxidative Damage
4-Hydroxy-2-nonenal-histidine covalent adducts were measured using the OxiSelect ELISA kit provided by CellBio Labs (CellBio Labs, San Diego). Assays were conducted according to the manufacturer's instructions. 8-Hydroxy-2′-deoxyguanosine levels were measured by HPLC. Mitochondria were diluted to 0.5 mg/ml in perchloric solution and incubated on ice as described above. Following removal of precipitate, the clarified supernatant was injected into an Agilent HPLC system equipped with a C18 reverse phase hydrophilic HPLC column (Synergy Hydro-RP, 4 μm, 250 × 4.6 mm; Phenomenex). Run conditions were the same as described above. 8-Hydroxy-2′-deoxyguanosine was detected using an Agilent variable wavelength detector set to 254 nm. Retention times were confirmed by injecting standard solutions. We also injected 2′-deoxyguanosine standard solutions to ensure no overlap with the retention and detection of 8-hydroxy-2′-deoxyguanosine. Levels of metabolites were quantified using Agilent ChemStation software.
Cytochrome c + c1 Concentration
Mitochondria were diluted to 1 mg/ml in BSA-free HM containing either excess sodium dithionite (reducing) or ferricyanide (oxidizing), as described previously (40, 41). Concentrations were determined by reading the maximum absorbance of mitochondria at 550 nm. The difference in absorbance under reducing and oxidizing conditions was then calculated and normalized to isosbestic points measured at 535 nm. The concentration of cytochrome c + c1 was determined according to the Beer-Lambert law, as described previously (42), with a molar extinction coefficient of 18,500 m−1·cm−1.
Immunoblots
Mitochondria were diluted to 2 mg/ml, and protein was electrophoresed and electroblotted as described above. Ventricular heart tissue was mechanically homogenized in homogenization buffer (50 mm Tris, pH 7.5, 1 mm EDTA disodium salt, 1 mm EGTA, 50 mm NaF, 5 mm sodium glycerophosphate, 10% v/v glycerol, 1% v/v Triton X-100, 1 mm DTT, 1 mm PMSF), and protein content was determined by a BCA assay. Samples were electrophoresed on 15% isocratic, 12% isocratic, and 10% isocratic SDS gels to afford proper immunodetection of Grx2, thioredoxin-2 (Trx2), uncoupling protein 3 (UCP3), and manganese superoxide dismutase (MnSOD). In addition, for MnSOD determinations only 15 μg of protein was loaded into each well, whereas Trx2 and Grx2/UCP3 determinations required 45 and 60 μg of protein, respectively, for proper detection. Membranes were blocked and probed for 24 h at 4 °C with primary antibodies directed against Grx2 (1:500, Abcam), Trx2 (1:1000, Abcam), UCP3 (1:1000, Abcam), and MnSOD (1:3000, Santa Cruz Biotechnology). Membranes were then incubated for 1 h at room temperature in the requisite secondary HRP-conjugated antibody. Succinate dehydrogenase A or α-tubulin served as the loading control (1:2000, Santa Cruz Biotechnology) and (1:5000, Sigma) respectively. Immunoreactive bands were visualized as described above.
Simultaneous detection of different respiratory complex subunits was performed with the MitoProfile OXPHOS antibody mixture (Abcam). The mixture consisted of five different antibodies directed against a specific subunit associated with each complex (I to V) as follows: NDUFB8, succinate dehydrogenase B, UQCRC2, MTCO1, and ATP5A. In bovine heart mitochondria, the ratio of complexes I/II/III/IV/V is estimated to be 1.1:1.3:3:6.7:3.5 indicating that the respiratory complexes vary in abundance (41). To afford proper detection and resolution of each complex subunit, we electrophoresed 10 μg of mitochondrial protein for detection of UQCRC2, MTCO1, and ATP5A and 45 μg of mitochondrial protein for NDUFB8 and succinate dehydrogenase B. Electrophoresis and protein amounts along with antibody dilution were optimized prior to experimentation. After blocking, membranes were probed overnight at 4 °C under constant agitation. OXPHOS antibody was diluted by 1:800 in TBS-T supplemented with 5% (w/v) defatted BSA and 0.02% (w/v) NaN3. After three washes in TBS-T membranes were probed with anti-mouse HRP conjugate. Immunoreactive bands were identified using molecular weight markers and quantified with ImageJ software. Quantification values were adjusted for the final amount of protein used for detection.
NDUSF1 Glutathionylation
NDUSF1 glutathionylation was tested by immunoblot. Samples were loaded in duplicate on gels to allow alignment of NDUSF1 immunoreactive bands with PSSG immunoreactive bands that appear at the same molecular weight. Mitochondria were diluted to 2 mg/ml in Laemmli buffer. 30 μg of sample was electrophoresed in 12% isocratic SDS gels. Following transfer of proteins to nitrocellulose membranes, membranes were stained with Ponceau S and then cut in half vertically to be probed for NDUSF1 and PSSG. As NDUSF1 has a molecular mass of ∼75 kDa, blots were cut a second time horizontally below the 50-kDa marker to measure MnSOD, which served as the loading control. All blots were incubated overnight under constant agitation at 4 °C. NDUSF1 was diluted to 1:2000 in TBS-T, 5% (w/v) defatted BSA, 0.02% (w/v) NaN3, and PSSG and MnSOD were diluted as described above. Experiments were also performed on samples electrophoresed under reducing conditions to ensure that the immunoreactive bands in the 75-kDa range were due to interaction of PSSG antiserum with glutathionylated proteins.
Statistics
Statistical analysis was performed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA). Comparison of controls with treated groups was performed with Student's t test. For comparisons of multiple treatments in the same group with a control, one-way ANOVA with a post hoc Tukey's test was used.
RESULTS
Grx2 Deficiency Induces Left Ventricular Hypertrophy and Moderate Fibrosis but Does Not Compromise Heart Contractility
Hearts from Grx2-deficient mice aged 9–12 weeks weighed significantly more (Fig. 1a). No changes in serum hemoglobin were observed indicating Grx2−/− mice are not anemic (Fig. 1b). Increased heart weight was associated with an increase in cardiomyocyte size and moderate left ventricular fibrosis in mice at 9 weeks of age; no left ventricular fibrosis was observed at 6 weeks (Fig. 1c). Histological analyses indicated concentric hypertrophy; there was no evidence of myocytolysis or myocyte disarray. Echocardiographic measurements revealed that the left ventricular wall was significantly thicker in Grx2-deficient mice (Fig. 1, d and e). Measurement of the size of the interventricular septum, left ventricular internal dimension, and left ventricular wall in diastole and systole confirmed that the left ventricle in Grx2−/− mice is hypertrophied (Table 1). Thus, the increased heart size is associated with hypertrophying of the left ventricle and fibrosis. We then calculated fractional shortening and ejection fractions, indices for diastolic and systolic function, and measured blood flow through the mitral valve during diastole. No significant changes in fractional shortening or ejection volume were observed (Fig. 1e). At 16 weeks of age, left ventricles were still hypertrophied (Table 2), and a small but significant increase in fractional shortening and ejection volume was observed (Fig. 1f). Despite this small increase in fractional shortening, overall cardiac function (contraction and relaxation) was maintained.
FIGURE 1.

Grx2 deficiency induces cardiac hypertrophy and left ventricular fibrosis in 10-week-old mice. a, heart weight. Values were normalized to femur length (g/cm). n = 4, mean ± S.E., Student's t test. b, serum hemoglobin levels. n = 6, mean ± S.E. c, histological analysis of cell size (H&E) and fibrosis (Sirius Red) in left ventricle. d, echocardiographic images of left ventricle in M-mode of mice aged 10 weeks. Left ventricular wall parameters (Tables 1 and 2) calculate left ventricular wall mass, percent fractional contraction, and ejection efficiency in mice aged 10 weeks (e) and 16 weeks (f). n = 7, mean ± S.E., Student's t test.
TABLE 1.
Echocardiographic measurement of left ventricular size and function in diastole and systole in male mice 10 weeks of age
All measurements are in millimeters and were performed on M-mode images acquired with conscious mice. n = 7, mean ± S.E., Student's t test. IVS is interventricular septum; LVID is left ventricular internal dimension; LVPW is left ventricular posterior wall; d is diastole; s is systole.
| IVS, d | LVID, d | LVPW, d | IVS, s | LVID, s | LVPW, s | |
|---|---|---|---|---|---|---|
| WT | 0.68 ± 0.03 | 4.38 ± 0.18 | 0.65 ± 0.03 | 1.09 ± 0.04 | 3.12 ± 0.15 | 1.01 ± 0.03 |
| Grx2−/− | 0.83 ± 0.02a | 4.92 ± 0.19a | 0.80 ± 0.03b | 1.18 ± 0.02a | 3.61 ± 0.17a | 1.14 ± 0.05a |
a p < 0.05.
b p < 0.01.
TABLE 2.
Echocardiographic measurement of left ventricular size and function in diastole and systole in male mice 16 weeks of age
All measurements are in millimeters and were performed on M-mode images acquired with conscious mice. n = 4, mean ± S.E., Student's t test. IVS is interventricular septum; LVID is left ventricular internal dimension; LVPW is left ventricular posterior wall; d is diastole; s is systole.
| IVS, d | LVID, d | LVPW, d | IVS, s | LVID, s | LVPW, s | |
|---|---|---|---|---|---|---|
| WT | 0.62 ± 0.02 | 4.46 ± 0.10 | 0.66 ± 0.04 | 0.95 ± 0.03 | 3.11 ± 0.10 | 1.06 ± 0.06 |
| Grx2−/− | 0.78 ± 0.02a | 4.09 ± 0.11a | 0.82 ± 0.05b | 1.08 ± 0.04b | 2.87 ± 0.08b | 1.24 ± 0.06a |
a p < 0.01.
b p < 0.05.
We next examined heart glucose uptake in vivo using the glucose radiotracer analog [18F]FDG and microPET. Analysis of representative images showed stark visual differences in glucose uptake in the heart of Grx2−/− mice compared with WT mice (Fig. 2a). Three-dimensional polar mapping of the left ventricle revealed significantly higher [18F]FDG uptake in this region of heart in Grx2−/− mice (Fig. 2a). The rate of [18F]FDG uptake by the left ventricle and septum was also higher in Grx2−/− mice (Fig. 2c).
FIGURE 2.

Glucose uptake is increased in the left ventricle of Grx2-eficient mice. a, [18F]FDG images collected from WT and Grx2−/− mice. Static images were collected after 60 min of imaging. White arrow represents location of heart. Asterisk denotes location of bladder. b, three-dimensional polar map of glucose uptake in left ventricle. Maps were generated after 60 min of imaging. The rate of myocardial glucose uptake (rMGU) by the left ventricle was then calculated. n = 4–5, mean ± S.E., Student's t test. c, dynamic measurement of the rate of [18F]FDG uptake by left ventricle and septum. n = 4–5, mean ± S.E., Student's t test. * denotes p < 0.05.
Impact of Grx2 Deficiency on Mitochondrial OXPHOS Proteins and Redox Environment
Electron microscopic analysis of sections from the left ventricle revealed no alterations in cristae structure (Fig. 3a). However, sharp changes in the relative amounts of the various complexes were observed (Fig. 3b). Significant decreases in complex I accessory protein NDUSB8, complex II subunit succinate dehydrogenase B, and complex IV subunit MTCO1 were evident in Grx2-deficient mitochondria. Intriguingly, Grx2 deficiency significantly increased the amounts of complex III subunit UQCRC2 and complex V subunit ATP5A (Fig. 3b). Grx2 deficiency also significantly increased the concentration of cytochrome c + c1 in mitochondria (Fig. 3c).
FIGURE 3.

Grx2 deficiency alters content of respiratory complexes and redox environment. a, electron microscopic images of mitochondrial cristae structure in left ventricle. Scale bar, 500 nm. n = 3. b, immunoblot analysis of respiratory complex subunits. Protein bands were identified and quantified to determine respiratory complex subunit amounts. n = 3, mean ± S.E., Student's t test. SDHB, succinate dehydrogenase B. c, absolute content of cytochromes c and c1 in cardiac mitochondria. The absolute amounts were calculated from the difference in absorbance values when mitochondria were fully reduced (dithiothreitol) or fully oxidized (ferrocyanide). n = 4, mean ± S.E., Student's t test. d, measurement of the mitochondrial 2GSH/GSSG ratio. Absolute amounts of reduced glutathione (GSH) and glutathione disulfide (GSSG) in mitochondria were measured by HPLC and then used to calculate the 2GSH/GSSG. The Eh of the ratio was then calculated using the Nernst equation. n = 4, mean ± S.E., Student's t test. * denotes p < 0.05. e, immunodetection of PSSG in cardiac mitochondria. Electrophoresis was conducted under nonreducing (left) and reducing (right; + 2% v/v βMetOH). Bands were quantified using ImageJ software. n = 4, mean ± S.E., Student's t test.
We next measured the impact of Grx2 deficiency on the redox potential of mitochondria and the total glutathionylated proteome. Grx2−/− cardiac mitochondria contained significantly more GSSG than WT mitochondria (Table 3). In addition the total glutathione pool was significantly larger in Grx2−/− cardiac mitochondria suggesting that loss of Grx2 induces glutathione biosynthesis in the cytosol (Table 3). Although Grx2−/− mitochondria also contained more GSH, no significance was recorded in comparison with WT. In Grx2-deficient mitochondria, 2GSH/GSSG was approximately a quarter of the WT ratio (Fig. 3d). The lower 2GSH/GSSG had a significant effect on the potential (Eh) of the redox pair (Fig. 3d). Previous reports have estimated that the Eh value of the 2GSH/GSSG varies between −280 and −340mV (43). From our calculations, the Eh value in WT mitochondria falls within this range. Grx2 deficiency increases the Eh value of this pair creating a more oxidizing matrix environment. Grx2 is required to catalyze reversible glutathionylation, but changes in 2GSH/GSSG can also alter the glutathionylation state of proteins as well. Grx2 deficiency significantly increased the total amount of glutathionylated protein in cardiac mitochondria (Fig. 3e). The specificity of the anti-PSSG antibody was confirmed by performing electrophoresis under reducing conditions (+βMetOH). As shown in Fig. 3e, addition of βMetOH abolished anti-PSSG antibody binding, except for a faint band at ∼40 kDa.
TABLE 3.
Absolute GSH and GSSG levels in WT and Grx2−/− cardiac mitochondria
All values reported as mm/liter. Calculations were performed as described under “Experimental Procedures.” n = 4, mean ± S.E., Student's t test.
| GSH | GSSG | Total glutathione (GSH + GSSG) | |
|---|---|---|---|
| WT | 2.39 ± 0.065 | 0.081 ± 0.014 | 2.47 ± 0.069 |
| Grx2−/− | 3.24 ± 0.39 | 0.27 ± 0.037a | 3.51 ± 0.41b |
a p < 0.01.
b p < 0.05.
The increase in the Eh value of 2GSH/GSSG prompted us to determine whether Grx2 deficiency induced oxidative damage or changes in the levels of antioxidative defense and redox enzymes. No differences in the levels of 4-hydroxy-2-nonenal-histidine adducts or 8-hydroxy-2′-deoxyguanosine, two end products of ROS damage, were observed in Grx2-deficient heart mitochondria when compared with WT (Fig. 4, a and b). Thioredoxin-2 (Trx2) is required to reduce intra- or intermolecular disulfide bridges that may form during oxidative stress. No changes in Trx2 protein levels were recorded (Fig. 4c). Also, no changes in the protein levels of MnSOD were observed (Fig. 4c). Collectively, our results indicate that Grx2 deficiency changes mitochondrial redox, increases the glutathionylated proteome, and alters the composition of the respiratory complexes. However, these changes occurred in the absence of oxidative damage.
FIGURE 4.
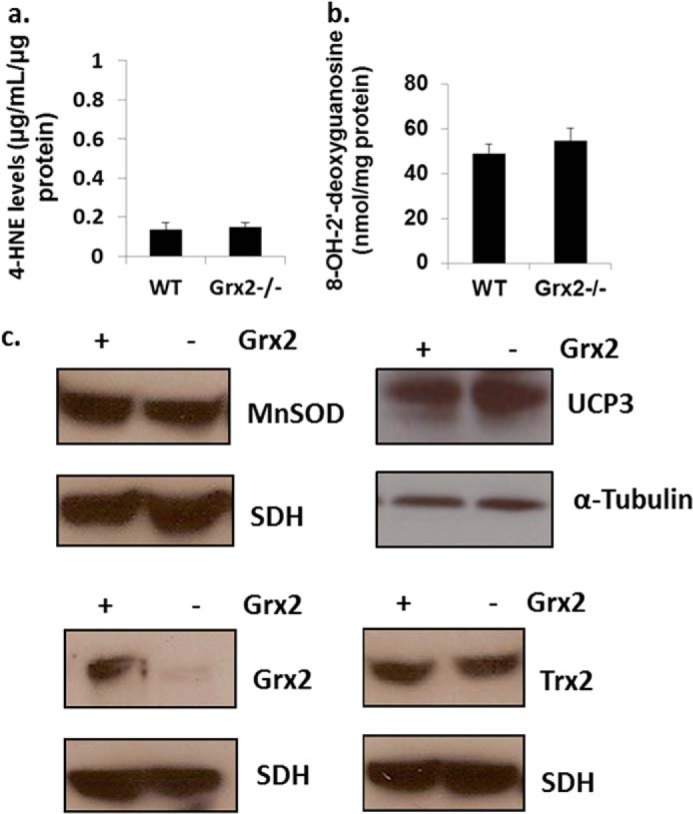
Grx2 deficiency does not induce oxidative damage in cardiac mitochondria. a, measurement of 4-hydroxy-2-nonenal (4-HNE) histidine adduct levels in isolated mitochondria. n = 4, mean ± S.E., Student's t test. b, assessment of 8-hydroxy-2′-deoxyguanosine (8-OH-2′-deoxyguanosine) levels in isolated mitochondria. n = 4, mean ± S.E., Student's t test. c, immunoblot analysis of MnSOD, uncoupling protein-3 (UCP3) protein levels, Grx2, and Trx2. Succinate dehydrogenase (SDH) or α-tubulin served as loading controls.
Grx2 Deficiency Causes Dysfunctional Oxidative Phosphorylation
We next measured the impact of Grx2 deficiency on cardiac mitochondrial energetics using a high throughput multiwell technique (28). Mitochondrial di-oxygen consumption was measured sequentially under state 2 (substrate only), state 3 (substrate + ADP), oligomycin-induced state 4 (proton leak-dependent respiration), and following treatment with protonophore FCCP (Fig. 5, a and b). No significant differences in proton leak-dependent respiration (state 4) were observed (Fig. 5a). No significant change in UCP3 protein was observed (Fig. 4c). Remarkable, however, was the significantly lower rate of ADP-stimulated (state 3) respiration (Fig. 5a). Addition of DTT to reaction mixtures restored state 3 respiration in Grx2-deficient cardiac mitochondria to WT levels (Fig. 5b). Similarly, uncoupled respiration was significantly lower in mitochondria isolated from Grx2−/− (Fig. 5a) but could be recovered to WT levels by the inclusion of DTT (Fig. 5b). The observation that uncoupled respiration in Grx2−/− cardiac mitochondria was also significantly decreased indicates that the decrease in OXPHOS is likely associated with a decrease in the activity of a respiratory complex or complexes rather than a decrease in ATP synthase activity.
FIGURE 5.

Grx2 deficiency decreases complex I-supported oxidative phosphorylation, which can be restored with DTT. Measurement of O2 consumption in isolated cardiac mitochondria under different bioenergetic states was conducted using the Seahorse XF24. a and b, states 3, state 4, and FCCP-induced uncoupled respiration rates in the absence or presence of 10 mm dithiothreitol (DTT). Pyruvate and malate were used as substrates. Seahorse traces detailing the experiment are included in a and b. c, calculated respiratory control ratios for a and b. n = 4, mean ± S.E., Student's t test. d, calculated respiratory control ratios for mitochondria respiring on palmitoylcarnitine. e, ADP titration assay; f, mitochondrial quality test. n = 3, mean ± S.E. Cyt c, cytochrome c; Ant A, antimycin A.
The decrease in state 3 respiration resulted in an RCR that was less than a half of the WT RCR (Fig. 5c). Inclusion of DTT in reaction mixtures restored RCR values in Grx2−/− cardiac mitochondria to WT levels (Fig. 5c). RCR was also significantly decreased (∼50%) when palmitoylcarnitine was the respiratory substrate, and this was readily recovered by the addition of DTT (Fig. 5c). The observed decrease in OXPHOS was confirmed by measuring absolute amounts of ATP in mitochondria respiring on pyruvate and malate (Fig. 6). A significant decrease in ATP synthesis was observed in Grx2-deficient mitochondria. This was matched by significant decreases in various TCA cycle metabolites (Fig. 6).
FIGURE 6.
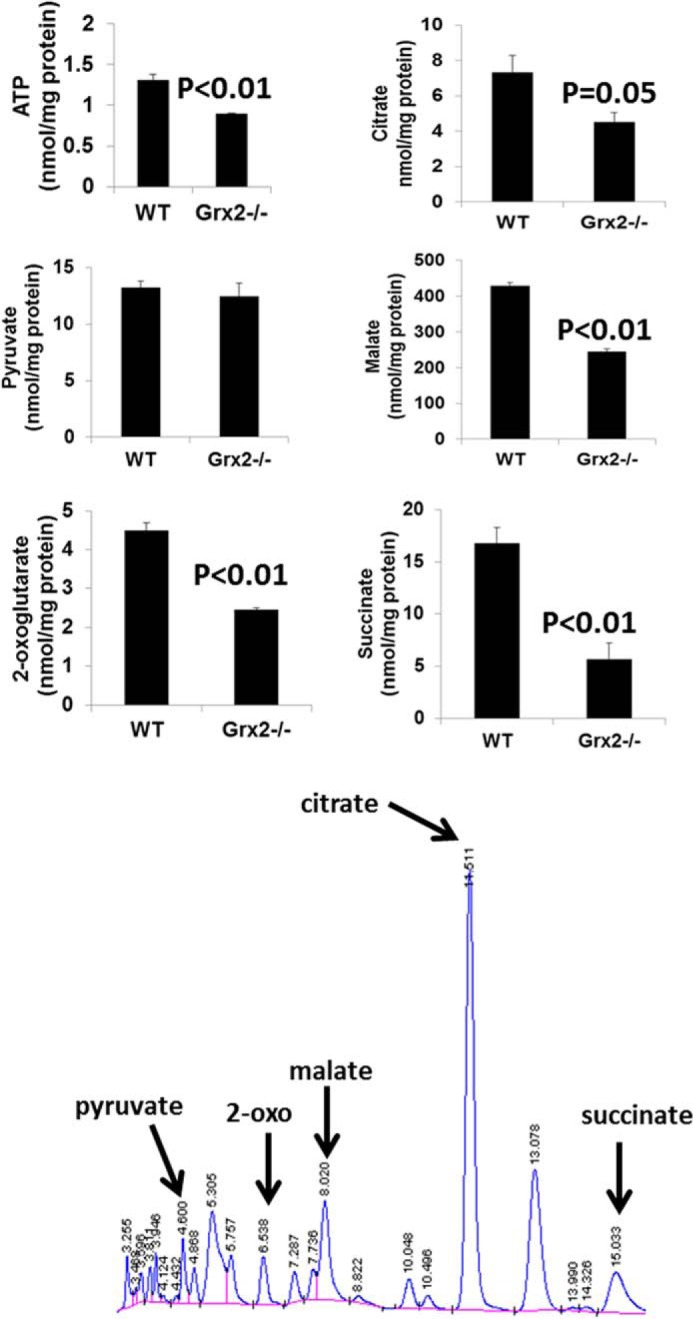
ATP and tricarboxylic acid intermediate levels in cardiac mitochondria are altered by Grx2 deficiency. Levels of various TCA cycle metabolites and ATP were measured by HPLC as described under “Experimental Procedures.” n = 4, mean ± S.E., Student's t test.
ADP titration experiments revealed that Grx2 deficiency induced a modest decrease in the affinity of ATP synthase for ADP (Fig. 5e). We also examined whether changes in respiration were due to damage to the outer membrane (e.g. during mitochondrial isolation and attachment to the surface of Seahorse TC plates). As shown in Fig. 5f, no changes in respiration were recorded following treatment of mitochondria with increasing concentrations of cytochrome c after induction of state 3 respiration.
Impact of Grx2−/− and Redox Environment on Complex I Activity and Glutathionylation
The specific activity of complex I was significantly lower in Grx2-deficient cardiac mitochondria when compared with WT (Fig. 7a). Incubation in 10 mm DTT restored the activity of complex I in Grx2-deficient mitochondria to levels that were not significantly different from those in WT mitochondria. We also measured ROS production in isolated mitochondria. Grx2-deficient mitochondria respiring on complex I-linked substrates pyruvate and malate generated more ROS under state 4O conditions than WT cardiac mitochondria (Fig. 7b). Performing the reactions in the presence of 10 mm DTT lowered ROS emission by Grx2-deficient cardiac mitochondria to WT levels.
FIGURE 7.

Complex I activity is compromised but is recovered by restoration of redox environment. Measurement of complex I activity (a) and mitochondrial ROS production (b) in the absence or presence of 10 mm dithiothreitol (DTT). n = 4, mean ± S.E., Student's t test. c, complex I activity can be recovered by restoration of redox environment or the addition of exogenous glutaredoxin-1 (Grx). Mitochondria were permeabilized and then incubated for 5 min in various combinations of reduced glutathione (GSH), oxidized glutathione (GSSG) in the absence or presence of 10 units Grx. Complex I activity was then measured. The Eh values were estimated according to the Nernst equation (see Equation 1). n = 5, mean ± S.E., 1-way ANOVA with a post hoc Tukey's test. * denotes p < 0.05. d, estimation of the glutathionylation state of complex I subunit NDUSF1 (∼75 kDa) by immunoblot. Glutathionylation state was preserved by treating mitochondria with 25 mm N-ethylmaleimide. To afford a proper electrophoretic estimation of NDUSF1 glutathionylation, NDUSF1 and PSSG levels were detected on the same gels. Samples were also electrophoresed under reducing conditions (+2% v/v βMetOH) as a control for PSSG immunodetection. Bands were quantified using ImageJ. n = 3, mean ± S.E., Student's t test. Mice were age 9–12 weeks.
Complex I has previously been reported to be modulated by glutathionylation (24, 27, 28). Thus, we next tested whether complex I activity can be recovered with exogenously added Grx1 or GSH. We also wanted to determine whether complex I activity in mitochondria from WT mice could be inhibited by adding excess GSSG. Addition of 10 mm GSH and 0.1 mm GSSG did not alter complex I activity in WT cardiac mitochondria (Fig. 7c). However, adjustment of the 2GSH/GSSG to ∼1 induced an ∼50% decrease in complex I activity in WT mitochondria relative to control (Fig. 7c). Addition of exogenous Grx1 restored the activity of complex I to control levels in mitochondria exposed to a 2GSH/GSSG of ∼1 (Fig. 7c). We next performed similar determinations on Grx2−/− mitochondria. As shown in Fig. 7c, complex I activity was lower in Grx2−/− mitochondria. This activity increased ∼2.1-fold when 10 mm GSH was added (Fig. 7c). Adjustment of the 2GSH/GSSG to ∼1 did not decrease the activity of complex I any further, relative to control. Addition of exogenous Grx1 induced an ∼2.3-fold increase in complex I activity relative to control even when the 2GSH/GSSG was set to ∼1 (Fig. 7c). These results illustrate that Grx plays a central role in reversing the deactivation of complex I by glutathionylation. In addition, our results indicate that redox potential plays an important role in modulating complex I activity. For instance, if the redox potential of 2GSH/GSSG is highly oxidizing, it can lead to spontaneous deactivation of complex I by glutathionylation. The opposite is also true for a highly reducing 2GSH/GSSG, which spontaneously recovers complex I activity by deglutathionylation.
The above results indicate that glutathionylation reactions mediated by Grx2 regulate complex I activity. We also observed that excess GSSG can inhibit complex I. In a previous report, it was found that complex I subunit NDUSF1 is glutathionylated by Grx2 (25). We next used comparative electrophoresis and immunoblotting to indirectly determine whether Grx2 deficiency alters the glutathionylation state of NDUSF1. Immunoblotting for protein glutathione disulfide adducts (PSSG) revealed protein bands at ∼72–75 kDa that matched the electrophoretic mobility of NDUSF1 (Fig. 7d). The PSSG bands at this molecular weight were quantified and normalized to the intensity of NDUSF1 immunoreactive bands. These results suggest that NDUSF1 was more glutathionylated in mitochondria from the Grx2-deficient heart (Fig. 7d). Intriguingly, NDUSF1 levels were ∼10% higher in WT mitochondria. Immunoreactivity toward PSSG antiserum was abolished when samples were electrophoresed under reductive conditions (+βMetOH) confirming that the protein band detected at ∼72–75 kDa was a glutathionylated protein (Fig. 7d). These results suggest that NDUSF1 may be the site for glutathionylation of complex I.
OXPHOS and Heart Physiology in Younger Grx2-deficient Mice
Our above results demonstrate that mitochondria from hearts of Grx2-deficient male mice aged 9–12 weeks have decreased OXPHOS efficiency, which can be reversed by adding DTT. We next decided to test the impact of Grx2 deficiency on mitochondrial energetics and heart physiology in mice aged 6 weeks. To afford a comparison, we also measured OXPHOS and heart weights in mice that were 10 weeks of age. In mice aged 10 weeks, hearts from Grx2-deficient mice weighed significantly more than WT hearts (Fig. 8a). These physiological changes coincided with a decrease in OXPHOS. Incubation of Grx2-deficient cardiac mitochondria in 10 mm DTT restored the efficiency of ATP production (Fig. 8a). These observations are consistent with the results generated above. Intriguingly, hearts from 6-week-old WT and Grx2−/− mice weighed the same, and there were no signs of hypertrophy in the Grx2−/− mice (Fig. 8b). In addition, no differences in the efficiency of OXPHOS were observed between WT and Grx2-deficient cardiac mitochondria (Fig. 8b).
FIGURE 8.

Effect of age on OXPHOS and heart weight. a and b, measurement of RCR in mitochondria isolated from WT and Grx2−/− mice aged 10 and 6 weeks. RCR values were calculated by dividing absolute state 3 respiration rates by state 4O respiration rates. Hearts were weighed prior to experimentation and normalized to femur length (g/cm). n = 4, mean ± S.E., Student's t test.
Heart Physiology and OXPHOS in Grx2 Heterozygotes
We next tested whether mice heterozygous for Grx2 (Grx2+/−) also presented with abnormalities in OXPHOS. Weights of hearts from Grx2+/− and Grx2−/− mice were similar and were heavier than hearts from WT mice (Fig. 9a). The amount of Grx2 protein in Grx2+/− mitochondria was approximately half that in WT mitochondria (Fig. 9b). We then tested the impact of Grx2+/− on OXPHOS efficiency. As shown in Fig. 9c, mitochondria from Grx2+/− mice had decreased RCRs, similar to values from Grx2−/− mice (Fig. 9c). Treatment of mitochondria with 10 mm DTT recovered RCRs in mitochondria from Grx2+/− and Grx2−/− mice (Fig. 9c). Left ventricular hypertrophy is often induced by hemodynamic stress. Thus, we assessed diastolic and systolic blood pressure to discern if hypertension and hemodynamic stress were evident. At 9 weeks of age, systolic blood pressure in Grx2-deficient mice was the highest (Fig. 9d). Systolic pressure was also elevated in mice heterozygous for Grx2 but not to the same magnitude as Grx2-deficient mice. No differences in diastolic pressure were observed at 9 weeks of age (Fig. 9d). Intriguingly, at 6 weeks the systolic blood pressure in Grx2 heterozygous mice was comparable with WT (Fig. 9d). By contrast, systolic blood pressure in Grx2-deficient mice was still significantly elevated. At 6 weeks of age, diastolic blood pressure was significantly lower in Grx2-deficient mice and mice heterozygous for Grx2 compared with WT mice (Fig. 9d).
FIGURE 9.

Grx2 heterozygotes. a, heart weight. Values were normalized to femur length (g/cm). b, Grx2 protein levels. c, calculated RCR for isolated cardiac mitochondria. Pyruvate and malate served as the respiratory substrate. n = 3, mean ± S.E., one-way ANOVA with Tukey's post hoc test. d, measurement of systolic and diastolic blood pressure. n = 6, mean ± S.E., one-way ANOVA with a post hoc Tukey's test, * and ** denote p < 0.05 and p < 0.01. Mice were aged 9–12 weeks.
DISCUSSION
Mitochondrial dysfunction contributes to the pathogenesis of a number of diseases, including neurological disorders, obesity, type 2 diabetes, and cardiovascular disorders. Perturbations in mitochondrial function have also been associated with cardiac injury and heart failure (44). Studies have established that defects in mitochondrial bioenergetics are associated with development of heart disease. Dysfunctional mitochondria in cardiac tissue can lead to energy starvation, which is associated with defects in the conversion of fuels into ATP (45). Recent work has shown that perturbations in post-translational modifications that regulate OXPHOS, like acetylation, can result in heart disease (44). In particular, a recent study established that regulation of complex I by acetylation in cardiac tissue is essential for responding to chronic increases in workload (44). In this study, we have established that Grx2 plays a central role in regulating OXPHOS efficiency in cardiac mitochondria. The importance of Grx2 in maintaining efficient mitochondrial respiration in cardiac tissue stems from its capacity to deglutathionylate target proteins like complex I. Loss of Grx2 led to the hyper-glutathionylation of the mitochondrial proteome and decreased OXPHOS efficiency. Grx2 deficiency was associated with cardiac hypertrophy and hypertension. Intriguingly, Grx2 heterozygous mice also had cardiac hypertrophy and a decrease in mitochondrial ATP production associated with defective complex I activity. However, Grx2 heterozygotes presented with a milder degree of hypertension suggesting that perturbations in mitochondria can contribute to development of heart disease. To our knowledge, this is the first study to show that controlled glutathionylation reactions mediated by Grx2 are key for modulating cardiac mitochondrial energetics. In addition, we provide evidence that intrinsic factors, such as deregulation of redox signaling, can contribute to development of heart disease.
Complex I is a major site for respiratory chain control in the heart (46). Indeed, complex I can serve as a major therapeutic target for prevention of cardiac ischemic-reperfusion injury (46). Thus, a need for a better understanding of the mechanisms that control complex I activity in cardiac tissue is required. We confirmed that complex I is a major target for Grx2-mediated deglutathionylation and demonstrated that complex I activity was decreased with Grx2 deficiency and that this could be reversed with DTT, exogenous Grx1, or GSH. Importantly, similar observations have been made in previous studies where addition of DTT, exogenous Grx1, or simply adding GSH to reactions reversed glutathionylation and activated mitochondrial proteins like complex I or carnitine/acyl-carnitine carrier (25, 31, 47). We also provided evidence that NDUSF1 subunit of complex I may be the site for glutathionylation of complex I. In addition, the band at ∼72–75 kDa was more glutathionylated mitochondria from Grx2-deficient mice. β-MetOH abolished band reactivity at ∼72–75 kDa confirming that NDUSF1 is glutathionylated. This observation is in line with previous studies that showed that Cys531 and Cys704 on NDUSF1 are sites for glutathionylation (25). In addition, recent work has shown that NDUSF1 is a target for S-nitrosylation and S-oxidation reactions (25, 46). We also noted that NDUSF1 levels were ∼11% higher in WT mitochondria indicating Grx2 deficiency can also affect the expression of mitochondrial proteins. It is important to point out that other subunits for complex I, including Ndufv1 (∼51 kDa), have been shown to be glutathionylated in perfused isolated mouse hearts (48). Another subunit with a mass of ∼40 kDa have also been shown to be glutathionylated (31). This coincides with the mass of the ND3 subunit of complex I that harbors Cys39, which can be modified by S-oxidation reactions (49). It has been reported that complex I transitions between activated (A-form) and deactivated (D-form) states that regulate its activity (50). In the transition to the D-form, Cys39 on ND3 becomes accessible and thus amenable to S-oxidation. Thus, ND3 subunit could serve as an important site for regulation of complex I activity by glutathionylation (51). Simultaneous immunoblotting for specific subunits for all respiratory complexes revealed that Grx2 deficiency decreased the amount of complex I subunit NDUFB8. This subunit is an accessory protein that plays no catalytic role in complex I. However, NDUFB8 has been suggested to be required for supercomplex assembly in mitochondria (52). According to our results, complex I activity is influenced by its “state of glutathionylation,” which is directly modulated by Grx2 and not by the decrease in NDUFB8. Indeed, inclusion of DTT, restoration of 2GSH/GSSG, or addition of exogenous Grx1 fully recovered complex I activity in Grx2-deficient mitochondria. We also observed that high GSSG inhibited complex I activity in WT mitochondria, which is consistent with previous observations that if GSSG levels are high enough then glutathionylation of complex I can proceed via a simple thiol exchange reaction (53, 54). Inclusion of exogenous Grx1 with GSSG prevented the deactivation of complex I by GSSG in WT mitochondria. It is important to consider though that inclusion of GSSG at high concentrations may influence complex I activity by altering the redox state of other redox couples. Addition of GSSG can deplete NADPH pools and result in the oxidation of thioredoxin-2. However, based on our results and work published by others (24, 31), we can conclude that GSSG lowers complex I activity by glutathionylation and that this can be reversed by either restoring the mitochondrial redox environment or in the presence of Grx2.
We also observed that Grx2 deficiency drastically altered the protein levels of all other measured respiratory complex subunits and cytochrome c. We also observed that the TCA cycle metabolite pool was diminished. The loss of a functional TCA cycle and changes in the protein levels of the respiratory complexes would inevitably also impact OXPHOS capacity. Deletion of Grx2 resulted in the glutathionylation of a number of mitochondrial proteins. Thus, Grx2 may have many mitochondrial protein targets, including TCA cycle enzymes. Indeed, previous reports have shown that various TCA cycle enzymes can be glutathionylated (22, 55, 56) and that cardiac ATP synthase is deactivated by glutathionylation (57). In our study, we observed that Grx2 deficiency induced a modest decrease in the sensitivity of ATP synthase toward ADP. However, it is also important to point out that adenine nucleotide translocator is also a target for glutathionylation, which may affect its capacity to exchange adenine nucleotides and would affect ATP synthase activity (58). Previous reports have shown that glutathionylation of the α subunit of ATP synthase diminishes ATP production (33, 35). Although our results do indicate that ATP synthase could potentially be affected by Grx2 deficiency, further analyses must be conducted to properly evaluate the sensitivity of ATP synthase in Grx2−/− mitochondria toward ADP.
Our analyses also revealed defective cardiac mitochondrial energetics in Grx2-deficient mitochondria respiring on either complex I-linked substrates or fatty acids, consistent with a recent report that carnitine/acylcarnitine carrier is inhibited by glutathionylation (47). Importantly, glutathionylation of carnitine/acylcarnitine carrier can be reversed by addition of exogenous Grx1 or DTT in vitro (47). It is important to acknowledge that fatty acid oxidation in mitochondria serves as the main source of ATP in cardiac tissue. Indeed, this inability to use fatty acids or TCA cycle substrates was associated with increased glucose uptake and reliance on glycolysis to produce ATP. Decreased OXPHOS in cardiac mitochondria induces a shift toward glycolysis to preserve ATP pools (59). Notably, we have demonstrated that the addition of DTT to Grx2-deficient cardiac mitochondria restored OXPHOS, thus indicating that the primary cause of the observed defects in mitochondrial function were due to perturbed redox and glutathionylation reactions.
Hearts from 9- to 12-week-old Grx2-deficient mice were heavier and presented with left ventricular hypertrophy and fibrosis. Grx2-deficient mice were also hypertensive indicating that hemodynamic stress may be responsible for the hypertrophying of the left ventricle. In Grx2+/− mice, hearts were also hypertrophied, and ATP production was decreased to values half those of WT controls. As in Grx2−/− mitochondria, OXPHOS could be fully recovered by the addition of DTT to reaction mixtures. Intriguingly though, systolic blood pressure in 6-week-old Grx2+/− mice was not significantly different compared with WT mice. At 9 weeks, increased blood pressure was observed in Grx2+/− mice, but the levels were not as high as in Grx2−/− mice. Left ventricular hypertrophy is often associated with increased hemodynamic stress associated with hypertension (60). However, deregulated ATP production by mitochondria has also been implicated in left ventricular hypertrophy, which would indicate that cardiac disease can be caused by intrinsic (cardiac mitochondrial dysfunction) and extrinsic stress (hemodynamic stress) (61). Thus, the cardiac hypertrophy in mice heterozygous for Grx2 is likely more directly associated with deregulated mitochondrial glutathionylation and inefficient ATP production, whereas in Grx2−/− mice left ventricular hypertrophy and fibrosis are due to a mixture of extrinsic and intrinsic stressors (hemodynamic stress and inadequate mitochondrial ATP production). It is important to note that Grx2−/− did not induce oxidative damage, oxidative stress, or changes in mitochondrial ultrastructure. Although we cannot exclude the possibility that the increased ROS production by mitochondria and decreased 2GSH/GSSG ratio may also contribute to the observed hypertrophy, it can be concluded that deregulated mitochondrial glutathionylation reactions are responsible, in part, for the observed cardiac hypertrophy. It has been previously reported that myocardial overexpression of Grx2 maintains mitochondrial OXPHOS in mice treated with doxorubicin, a chemotherapeutic that can induce cardiac dysfunction (34). Also, Brautigam et al. (62) observed that silencing Grx2 in zebrafish does not induce oxidative stress but does induce neuronal cell death due to changes in redox and possibly protein glutathionylation.
This is the first study to investigate the relationship between Grx2-mediated regulation of mitochondrial bioenergetics and heart function. We demonstrated that Grx2 is a key component of the regulatory machinery of cardiac mitochondria. Loss of Grx2 function leads to deregulated mitochondrial glutathionylation, changes in mitochondrial redox state, and decreased mitochondrial ATP output. Our work also revealed that OXPHOS can be recovered by restoring the reductive nature of the mitochondrial redox environment. Collectively our findings illustrate that redox signaling plays a vital role in regulating mitochondrial OXPHOS in the heart and that disruption of glutathionylation reactions can lead to heart disease. The observation that glutathionylation status and mitochondrial function can be restored with reducing agents also indicates that therapies could potentially be designed to restore the mitochondrial glutathiome and thereby curtail the progression of heart disease.
Acknowledgments
We are grateful for the expert advice and assistance of Dr. Jean Veinot and members of the University of Ottawa Pathology Laboratory, including Louise Pelletier and Alex Doré. We also thank Linda Jui and Shin Her Huang for help with histological analyses.
This work was supported by Canadian Institutes of Health Research Grant INMD MOP57810 (to M. E. H.).
- ROS
- reactive oxygen species
- Grx
- glutaredoxin
- OXPHOS
- oxidative phosphorylation
- RCR
- respiratory control ratio
- PSSG
- protein glutathione disulfide
- MnSOD
- manganese superoxide dismutase
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- ANOVA
- analysis of variance
- PET
- positron emission tomography
- β-MetOH
- β-mercaptoethanol.
REFERENCES
- 1. Mieyal J. J., Gallogly M. M., Qanungo S., Sabens E. A., Shelton M. D. (2008) Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 10, 1941–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xiong Y., Uys J. D., Tew K. D., Townsend D. M. (2011) S-Glutathionylation: from molecular mechanisms to health outcomes. Antioxid. Redox Signal. 15, 233–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gallogly M. M., Mieyal J. J. (2007) Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 7, 381–391 [DOI] [PubMed] [Google Scholar]
- 4. Fratelli M., Demol H., Puype M., Casagrande S., Eberini I., Salmona M., Bonetto V., Mengozzi M., Duffieux F., Miclet E., Bachi A., Vandekerckhove J., Gianazza E., Ghezzi P. (2002) Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 99, 3505–3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang Y., Shi W., Cui N., Wu Z., Jiang C. (2010) Oxidative stress inhibits vascular K(ATP) channels by S-glutathionylation. J. Biol. Chem. 285, 38641–38648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang J., Boja E. S., Tan W., Tekle E., Fales H. M., English S., Mieyal J. J., Chock P. B. (2001) Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem. 276, 47763–47766 [DOI] [PubMed] [Google Scholar]
- 7. Gallogly M. M., Shelton M. D., Qanungo S., Pai H. V., Starke D. W., Hoppel C. L., Lesnefsky E. J., Mieyal J. J. (2010) Glutaredoxin regulates apoptosis in cardiomyocytes via NFκB targets Bcl-2 and Bcl-xL: implications for cardiac aging. Antioxid. Redox Signal. 12, 1339–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reynaert N. L., van der Vliet A., Guala A. S., McGovern T., Hristova M., Pantano C., Heintz N. H., Heim J., Ho Y. S., Matthews D. E., Wouters E. F., Janssen-Heininger Y. M. (2006) Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β. Proc. Natl. Acad. Sci. U.S.A. 103, 13086–13091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lillig C. H., Berndt C., Holmgren A. (2008) Glutaredoxin systems. Biochim. Biophys. Acta 1780, 1304–1317 [DOI] [PubMed] [Google Scholar]
- 10. Pai H. V., Starke D. W., Lesnefsky E. J., Hoppel C. L., Mieyal J. J. (2007) What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid. Redox Signal. 9, 2027–2033 [DOI] [PubMed] [Google Scholar]
- 11. Gallogly M. M., Starke D. W., Leonberg A. K., Ospina S. M., Mieyal J. J. (2008) Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: implications for intracellular roles. Biochemistry 47, 11144–11157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deponte M. (2013) Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 1830, 3217–3266 [DOI] [PubMed] [Google Scholar]
- 13. Starke D. W., Chock P. B., Mieyal J. J. (2003) Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J. Biol. Chem. 278, 14607–14613 [DOI] [PubMed] [Google Scholar]
- 14. Hurd T. R., Costa N. J., Dahm C. C., Beer S. M., Brown S. E., Filipovska A., Murphy M. P. (2005) Glutathionylation of mitochondrial proteins. Antioxid. Redox Signal. 7, 999–1010 [DOI] [PubMed] [Google Scholar]
- 15. Murphy M. P. (2012) Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 16, 476–495 [DOI] [PubMed] [Google Scholar]
- 16. Gallogly M. M., Starke D. W., Mieyal J. J. (2009) Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid. Redox Signal. 11, 1059–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ströher E., Millar A. H. (2012) The biological roles of glutaredoxins. Biochem. J. 446, 333–348 [DOI] [PubMed] [Google Scholar]
- 18. Gladyshev V. N., Liu A., Novoselov S. V., Krysan K., Sun Q. A., Kryukov V. M., Kryukov G. V., Lou M. F. (2001) Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J. Biol. Chem. 276, 30374–30380 [DOI] [PubMed] [Google Scholar]
- 19. Lundberg M., Johansson C., Chandra J., Enoksson M., Jacobsson G., Ljung J., Johansson M., Holmgren A. (2001) Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J. Biol. Chem. 276, 26269–26275 [DOI] [PubMed] [Google Scholar]
- 20. Johansson C., Lillig C. H., Holmgren A. (2004) Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J. Biol. Chem. 279, 7537–7543 [DOI] [PubMed] [Google Scholar]
- 21. Lillig C. H., Berndt C., Vergnolle O., Lönn M. E., Hudemann C., Bill E., Holmgren A. (2005) Characterization of human glutaredoxin 2 as iron-sulfur protein: a possible role as redox sensor. Proc. Natl. Acad. Sci. U.S.A. 102, 8168–8173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mailloux R. J., Jin X., Willmore W. G. (2013) Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2, 123–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berndt C., Hudemann C., Hanschmann E. M., Axelsson R., Holmgren A., Lillig C. H. (2007) How does iron-sulfur cluster coordination regulate the activity of human glutaredoxin 2? Antioxid. Redox Signal. 9, 151–157 [DOI] [PubMed] [Google Scholar]
- 24. Beer S. M., Taylor E. R., Brown S. E., Dahm C. C., Costa N. J., Runswick M. J., Murphy M. P. (2004) Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J. Biol. Chem. 279, 47939–47951 [DOI] [PubMed] [Google Scholar]
- 25. Hurd T. R., Requejo R., Filipovska A., Brown S., Prime T. A., Robinson A. J., Fearnley I. M., Murphy M. P. (2008) Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of Cys residues in decreasing oxidative damage. J. Biol. Chem. 283, 24801–24815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu H., Lin L., Giblin F., Ho Y. S., Lou M. F. (2011) Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic. Biol. Med. 51, 2108–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu H., Xing K., Lou M. F. (2010) Glutaredoxin 2 prevents H(2)O(2)-induced cell apoptosis by protecting complex I activity in the mitochondria. Biochim. Biophys. Acta 1797, 1705–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mailloux R. J., Xuan J. Y., Beauchamp B., Jui L., Lou M., Harper M. E. (2013) Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. J. Biol. Chem. 288, 8365–8379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Applegate M. A., Humphries K. M., Szweda L. I. (2008) Reversible inhibition of α-ketoglutarate dehydrogenase by hydrogen peroxide: glutathionylation and protection of lipoic acid. Biochemistry 47, 473–478 [DOI] [PubMed] [Google Scholar]
- 30. Rosca M. G., Hoppel C. L. (2010) Mitochondria in heart failure. Cardiovasc. Res. 88, 40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Passarelli C., Tozzi G., Pastore A., Bertini E., Piemonte F. (2010) GSSG-mediated complex I defect in isolated cardiac mitochondria. Int. J. Mol. Med. 26, 95–99 [DOI] [PubMed] [Google Scholar]
- 32. Chen Y. R., Chen C. L., Pfeiffer D. R., Zweier J. L. (2007) Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J. Biol. Chem. 282, 32640–32654 [DOI] [PubMed] [Google Scholar]
- 33. Wang S. B., Foster D. B., Rucker J., O'Rourke B., Kass D. A., Van Eyk J. E. (2011) Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circ. Res. 109, 750–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Diotte N. M., Xiong Y., Gao J., Chua B. H., Ho Y. S. (2009) Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim. Biophys. Acta 1793, 427–438 [DOI] [PubMed] [Google Scholar]
- 35. Garcia J., Han D., Sancheti H., Yap L. P., Kaplowitz N., Cadenas E. (2010) Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 285, 39646–39654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mailloux R. J., Seifert E. L., Bouillaud F., Aguer C., Collins S., Harper M. E. (2011) Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J. Biol. Chem. 286, 21865–21875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rogers G. W., Brand M. D., Petrosyan S., Ashok D., Elorza A. A., Ferrick D. A., Murphy A. N. (2011) High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One 6, e21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mailloux R. J., Adjeitey C. N., Xuan J. Y., Harper M. E. (2012) Crucial yet divergent roles of mitochondrial redox state in skeletal muscle vs. brown adipose tissue energetics. FASEB J. 26, 363–375 [DOI] [PubMed] [Google Scholar]
- 39. Brand M. D., Nicholls D. G. (2011) Assessing mitochondrial dysfunction in cells. Biochem. J. 435, 297–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rossignol R., Letellier T., Malgat M., Rocher C., Mazat J. P. (2000) Tissue variation in the control of oxidative phosphorylation: implication for mitochondrial diseases. Biochem. J. 347, 45–53 [PMC free article] [PubMed] [Google Scholar]
- 41. Schägger H., Pfeiffer K. (2001) The ratio of oxidative phosphorylation complexes I-V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 276, 37861–37867 [DOI] [PubMed] [Google Scholar]
- 42. Benard G., Faustin B., Passerieux E., Galinier A., Rocher C., Bellance N., Delage J. P., Casteilla L., Letellier T., Rossignol R. (2006) Physiological diversity of mitochondrial oxidative phosphorylation. Am. J. Physiol. Cell Physiol. 291, C1172–C1182 [DOI] [PubMed] [Google Scholar]
- 43. Kemp M., Go Y. M., Jones D. P. (2008) Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic. Biol. Med. 44, 921–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Karamanlidis G., Lee C. F., Garcia-Menendez L., Kolwicz S. C., Jr., Suthammarak W., Gong G., Sedensky M. M., Morgan P. G., Wang W., Tian R. (2013) Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 18, 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neubauer S. (2007) The failing heart–an engine out of fuel. N. Engl. J. Med. 356, 1140–1151 [DOI] [PubMed] [Google Scholar]
- 46. Chouchani E. T., Methner C., Nadtochiy S. M., Logan A., Pell V. R., Ding S., James A. M., Cochemé H. M., Reinhold J., Lilley K. S., Partridge L., Fearnley I. M., Robinson A. J., Hartley R. C., Smith R. A., Krieg T., Brookes P. S., Murphy M. P. (2013) Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 19, 753–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Giangregorio N., Palmieri F., Indiveri C. (2013) Glutathione controls the redox state of the mitochondrial carnitine/acylcarnitine carrier Cys residues by glutathionylation. Biochim. Biophys. Acta 1830, 5299–5304 [DOI] [PubMed] [Google Scholar]
- 48. Kumar V., Kleffmann T., Hampton M. B., Cannell M. B., Winterbourn C. C. (2013) Redox proteomics of thiol proteins in mouse heart during ischemia/reperfusion using ICAT reagents and mass spectrometry. Free Radic. Biol. Med. 58, 109–117 [DOI] [PubMed] [Google Scholar]
- 49. Babot M., Labarbuta P., Birch A., Kee S., Fuszard M., Botting C. H., Wittig I., Heide H., Galkin A. (2014) ND3, ND1 and 39-kDa subunits are more exposed in the de-active form of bovine mitochondrial complex I. Biochim. Biophys. Acta 1837, 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grivennikova V. G., Kapustin A. N., Vinogradov A. D. (2001) Catalytic activity of NADH-ubiquinone oxidoreductase (complex I) in intact mitochondria. evidence for the slow active/inactive transition. J. Biol. Chem. 276, 9038–9044 [DOI] [PubMed] [Google Scholar]
- 51. Drose S., Brandt U., Wittig I. (2014) Mitochondrial respiratory chain complexes as sources and targets of thiol-based redox-regulation. Biochim. Biophys. Acta 10.1016/j.bbapap.2014.02.006 [DOI] [PubMed] [Google Scholar]
- 52. Davis C. W., Hawkins B. J., Ramasamy S., Irrinki K. M., Cameron B. A., Islam K., Daswani V. P., Doonan P. J., Manevich Y., Madesh M. (2010) Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic. Biol. Med. 48, 306–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Taylor E. R., Hurrell F., Shannon R. J., Lin T. K., Hirst J., Murphy M. P. (2003) Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 278, 19603–19610 [DOI] [PubMed] [Google Scholar]
- 54. Kang P. T., Zhang L., Chen C. L., Chen J., Green K. B., Chen Y. R. (2012) Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic. Biol. Med. 53, 962–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee D. W., Kaur D., Chinta S. J., Rajagopalan S., Andersen J. K. (2009) A disruption in iron-sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: implications for Parkinson's disease. Antioxid. Redox Signal. 11, 2083–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kil I. S., Park J. W. (2005) Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 280, 10846–10854 [DOI] [PubMed] [Google Scholar]
- 57. Wang S. B., Murray C. I., Chung H. S., Van Eyk J. E. (2013) Redox regulation of mitochondrial ATP synthase. Trends Cardiovasc. Med. 23, 14–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Queiroga C. S., Almeida A. S., Martel C., Brenner C., Alves P. M., Vieira H. L. (2010) Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J. Biol. Chem. 285, 17077–17088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Taegtmeyer H. (2002) Switching metabolic genes to build a better heart. Circulation 106, 2043–2045 [DOI] [PubMed] [Google Scholar]
- 60. Rosca M. G., Hoppel C. L. (2013) Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 18, 607–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rosca M. G., Tandler B., Hoppel C. L. (2013) Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 55, 31–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bräutigam L., Schütte L. D., Godoy J. R., Prozorovski T., Gellert M., Hauptmann G., Holmgren A., Lillig C. H., Berndt C. (2011) Vertebrate-specific glutaredoxin is essential for brain development. Proc. Natl. Acad. Sci. U.S.A. 108, 20532–20537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Thorn S. L., deKemp R. A., Dumouchel T., Klein R., Renaud J. M., Wells R. G., Gollob M. H., Beanlands R. S., DaSilva J. N. (2013) Repeatable noninvasive measurement of mouse myocardial glucose uptake with 18F-FDG: evaluation of tracer kinetics in a type 1 diabetes model. J. Nucl. Med. 54, 1637–1644 [DOI] [PubMed] [Google Scholar]