

Background: The mechanism of coordination between LPS synthesis and translocation is unknown.
Results: Two new proteins, LapA and LapB, co-purify with LPS transport proteins. lapB mutants display defects in lipid A and core assembly.
Conclusion: lapB mutants accumulate precursor LPS core species and exhibit elevated levels of LpxC.
Significance: Coordinated assembly of LPS is a critical step for targeting to the outer membrane.
Keywords: Endotoxin, Glycobiology, Glycolipid Structure, Glycosyltransferases, Lipopolysaccharide (LPS), RpoH, Heptosyltransferase, Lipid A, Myristoyltransferase, Tetratricopeptide Repeat (TPR)
Abstract
Here, we describe two new heat shock proteins involved in the assembly of LPS in Escherichia coli, LapA and LapB (lipopolysaccharide assembly protein A and B). lapB mutants were identified based on an increased envelope stress response. Envelope stress-responsive pathways control key steps in LPS biogenesis and respond to defects in the LPS assembly. Accordingly, the LPS content in ΔlapB or Δ(lapA lapB) mutants was elevated, with an enrichment of LPS derivatives with truncations in the core region, some of which were pentaacylated and exhibited carbon chain polymorphism. Further, the levels of LpxC, the enzyme that catalyzes the first committed step of lipid A synthesis, were highly elevated in the Δ(lapA lapB) mutant. Δ(lapA lapB) mutant accumulated extragenic suppressors that mapped either to lpxC, waaC, and gmhA, or to the waaQ operon (LPS biosynthesis) and lpp (Braun's lipoprotein). Increased synthesis of either FabZ (3-R-hydroxymyristoyl acyl carrier protein dehydratase), slrA (novel RpoE-regulated non-coding sRNA), lipoprotein YceK, toxin HicA, or MurA (UDP-N-acetylglucosamine 1-carboxyvinyltransferase) suppressed some of the Δ(lapA lapB) defects. LapB contains six tetratricopeptide repeats and, at the C-terminal end, a rubredoxin-like domain that was found to be essential for its activity. In pull-down experiments, LapA and LapB co-purified with LPS, Lpt proteins, FtsH (protease), DnaK, and DnaJ (chaperones). A specific interaction was also observed between WaaC and LapB. Our data suggest that LapB coordinates assembly of proteins involved in LPS synthesis at the plasma membrane and regulates turnover of LpxC, thereby ensuring balanced biosynthesis of LPS and phospholipids consistent with its essentiality.
Introduction
Lipopolysaccharide (LPS) in the outer leaflet of the outer membrane (OM)2 constitutes the major amphiphilic component of the envelope of most Gram-negative bacteria, including Escherichia coli (1–3). It is known that LPS is highly heterogeneous (Fig. 1). This heterogeneity is controlled by various regulatory factors and growth conditions (4). However, LPSs in general share a common architecture composed of a membrane-anchored phosphorylated and acylated β(1→6)-linked GlcN disaccharide, termed lipid A, to which a carbohydrate moiety of varying size is attached (1, 2). The latter may be divided into a lipid A proximal core oligosaccharide and, in smooth-type bacteria, a distal O-antigen. The core oligosaccharide can be further structurally divided in the inner and outer core. The inner core usually contains a residue(s) of 3-deoxy-α-d-manno-oct-2-ulosonic acid (Kdo) and l-glycero-d-manno-heptose (Hep). Both lipid A and the inner core are generally conserved in structure but often carry nonstoichiometric modifications (1–3).
FIGURE 1.
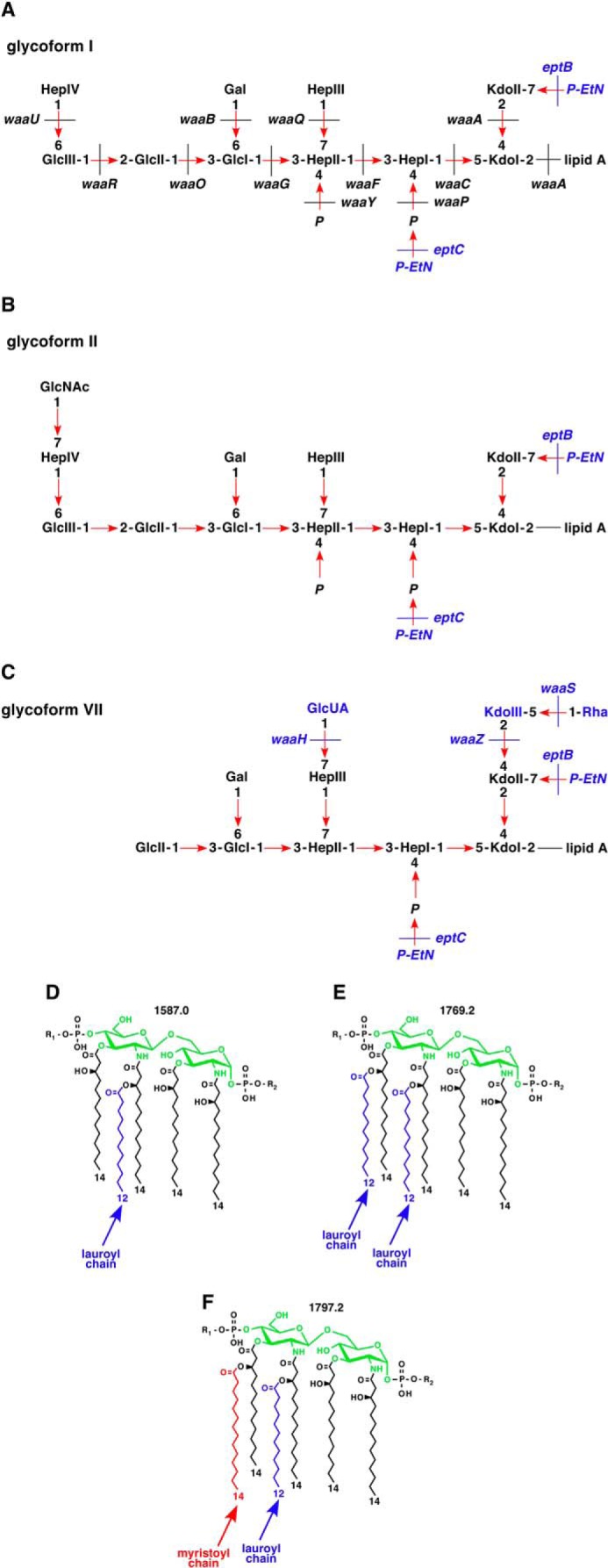
Proposed LPS structures of major glycoforms observed in E. coli K12. Shown is a schematic drawing of LPS glycoform I (A), II (B), and VII (C) with various nonstoichiometric substitutions. D–F, chemical structures of different lipid A variants: pentaacylated (D), hexaacylated with two lauroyl chains (E), and typical hexaacylated lipid A (F). Mass numbers of these lipid A species are indicated. R1 and R2, modification by Ara4N and P-EtN, respectively, observed in lipid A-modifying conditions.
In E. coli, the biosynthesis of LPS proceeds in a discontinuous manner. A tetraacylated lipid A (lipid IVA) precursor is synthesized first, in a pathway requiring six enzymes. Next, two Kdo residues are added on to the lipid IVA precursor (5). Every step involved in the synthesis of these intermediates is essential for the bacterial viability (1, 2). Kdo incorporation ensures the addition of secondary lauroyl and myristoyl chains, resulting in the synthesis of hexaacylated lipid A (Fig. 1). Following the synthesis of Kdo2-lipid A, additional sugars are sequentially transferred by a number of membrane-associated glycosyltransferases, using nucleotide sugars as donors (1, 2). The Kdo2-lipid A portion of LPS defines the minimal structure required to support the growth of E. coli up to 42 °C and is the most conserved part of the LPS (1, 2, 6).
Of nine enzymes involved in the lipid A biosynthesis, LpxA, LpxC, and LpxD are soluble proteins, whereas LpxB and LpxH are associated with the inner membrane (IM). The remaining enzymes (LpxK, WaaA, LpxL, and LpxM) are integral IM proteins (1). LpxC, the enzyme that catalyzes the first committed step of LPS biosynthesis, is tightly regulated (7, 8). LpxC is a substrate of the essential protease FtsH (7, 8). FtsH-dependent regulated proteolysis of LpxC sets a balance between phospholipid and LPS biosynthesis (7, 8).
After its synthesis, LPS is flipped across the IM by the essential ABC transporter MsbA (9) and translocated by the IM multiprotein complex LptBFGC and the periplasmic LptA (10–14). Last, LPS is delivered to LptD and LptE in the OM (10–14). MsbA displays higher specificity toward hexa- and pentaacylated lipid A substrates as compared with tetraacylated derivatives (9, 15), whereas Lpt proteins appear to interact with all LPS molecules regardless of the level of acylation (14, 16). It is unclear how bacteria coordinate LPS synthesis and translocation. Here, we speculate that a quality control-like system or a scaffold-like structure ensures that only completely synthesized LPS molecules are translocated. Ordered and rapid completion of LPS synthesis at the IM demands that all LPS biosynthetic enzymes must be fully active and present in stoichiometric amounts at the assembly site for LPS.
We reported earlier that synthesis of glycosylation-free LPS or Kdo2 + lipid IVA precursor induced the envelope stress response (6). Under such stress, bacteria can grow only in minimal medium at lower temperatures (6). Mutations in an uncharacterized locus, mapping at 28 min on the chromosome of E. coli, also caused induction of the envelope stress response (17). In this work, we constructed non-polar deletions of two genes of the 28 min locus and found that they exhibit defects in the LPS assembly. We designated these genes lapA and lapB (lipopolysaccharide assembly proteins). The lapB gene was found to be essential under laboratory growth conditions, and suppressor-free ΔlapB mutant could be constructed only on minimal medium at 30 °C. Such mutants accumulate early intermediates of LPS biosynthesis and exhibit elevated levels of LpxC. Consistent with a role in the LPS assembly, during pull-down experiments, LapA and LapB co-purified with LPS, FtsH, and Lpt proteins. Extragenic suppressors of ΔlapB mapped to genes whose products either control the lipid A biosynthesis (lpxC) or the LPS core biogenesis or factors that dampen the envelope stress response.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Media
The bacterial strains and plasmids used in this study are described in supplemental Table S1. Luria-Bertani (LB) broth, M9 (Difco), and 121 phosphate-limiting minimal media were prepared as described (4, 18).
Generation of Null Mutations and Construction of Their Combinations
Non-polar deletions of lapA, lapB, (lapA lapB), lpp, yceK, and slrA genes were constructed by using the λ Red recombinase/FLP-mediated recombination system as described previously (19, 20). The coding sequence of each gene was replaced with either a kanamycin (aph) or chloramphenicol (cat) resistance cassette flanked by FRT (FLP recognition target) recognition sequences, using plasmids pKD13 and pKD3 as templates (19). To replace the coding sequence by a spectinomycin resistance cassette (ada), the pLC1921 plasmid was used as the template (21). PCR products were used for recombineering on the chromosome of E. coli K12 strain GK1942, a derivative of BW25113 containing the λ Red recombinase-encoding plasmid pKD46. Gene replacements and their exact chromosomal locations were verified by PCR and sequencing of PCR products and then transduced into BW25113. Multiple null combinations were made through a series of bacteriophage P1- or T4-mediated transductions, followed by the removal of the antibiotic cassette when required. To avoid accumulation of suppressors, all of the transductions were carried out in M9 minimal medium at 30 °C. When required, the Δ(lapA lapB) or ΔlapB mutation was introduced in the presence or absence of a plasmid carrying the cloned wild-type copy of the target gene. The chromosomal DNA of isogenic Δ(lapA lapB) or ΔlapB deletion derivatives was used to amplify waaC, gmhA, and lpxC genes with their flanking sequences, and PCR products were verified to contain the wild-type sequence. The strain carrying deletion of the dnaK dnaJ operon (PK101) (22) was used as a donor in bacteriophage T4-mediated transduction to construct a deletion derivative in BW25113 (GK3078) and combined with Δ(lapA lapB), resulting in strain SR17161. This strain was verified to have correct deletion by the absence of DnaK and DnaJ antigens and resistance to bacteriophage λ. The sfhC21 mutation was transduced into BW25113 from A8926 ΔftsH3::Kan sfhC21 zad220::Tn10 using linked Tet marker (23). This resulted in strain GK3576, which served as the host strain for the introduction of ΔftsH3::Kan and Δ(lapA lapB). Strain slrAC SR17675 with chromosomal constitutive expression of the slrA RNA was constructed by the replacement of the processed RNA region upstream of mature slrA using appropriate oligonucleotides (supplemental Table S2). The construction of non-polar deletion derivatives of surA, waaC, and rseA genes was described previously (6, 17, 24).
For the complementation analysis, the wild-type (lapA lapB) genes were cloned from the genomic DNA in low copy pSC101-based pWSK29 and pWSK30 vectors (25). This resulted in the construction of plasmids pSR16726 and pSR16730, respectively. Complementing clones were verified by DNA sequencing. For the complementation of ΔlapB derivatives, plasmid pSR16881 from pSR16726 was used. This plasmid expresses only the lapB gene due to the introduction of stop codon (TAG in the place of TGG) corresponding to amino acid residue 64 in the lapA gene.
For protein production, the minimal coding sequence of the lapA gene of E. coli was PCR-amplified and cloned in the expression vector pET28b (NdeI-XhoI), generating C-terminal hexa-His-tagged LapA (pGK3605). Similarly, the minimal coding sequence of the lapB gene was PCR-amplified and cloned in pET28b (NcoI-XhoI), generating C-terminal hexa-His-tagged LapB (pSR7815). The expression of the waaC gene was induced from the ptac-based expression vector pCA24N (26). The minimal coding sequence of the lptC gene was PCR-amplified with appropriate oligonucleotides (supplemental Table S2) and cloned into pET28b using (NdeI-XhoI) sites. This resulted in the pSR15978 (plptC+) plasmid.
Error-prone PCR random mutagenesis was employed using the GeneMorph mutagenesis kit (Agilent Technologies). For PCR amplification, plasmid pSR16726 carrying lapA and lapB genes was used as template using appropriate oligonucleotides (supplemental Table S2). PCR products were recloned in pWSK29 and pWSK30 vectors and used to transform Δ(lapA lapB) or ΔlapB mutants on M9 minimal medium. Transformants that were unable to grow on rich medium at 30 or 37 °C were retained, and their plasmids were analyzed by DNA sequencing.
RNA Purification and Mapping of 5′ Ends
For the examination of lapAB and slrA transcripts, RNA was extracted from the wild-type strain BW25113. The cultures were grown in M9 medium at 30 °C until an absorbance of 0.2 at 600 nm, and when required, an aliquot was subjected to heat shock at 42 °C for 15 min. Cultures were centrifuged (10,000 rpm, 3 min). Typically, pellet from a 5-ml culture was resuspended in 1 ml of cold TRIsure solution (Bioline UK). RNA was purified according to the manufacturer's protocol and digested with RQ1 DNase (Promega) to remove any chromosomal DNA, followed by phenol extraction and RNA precipitation.
The GeneRacer from Invitrogen was used to obtain 5′ ends of cDNA according to the manufacturer's protocol. Two μg of total RNA, obtained from cultures grown at either 30 °C or after 15-min heat shock at 42 °C, was ligated to the GeneRacer RNA oligonucleotide and was reverse transcribed with SuperScript III RT. In the case of slrA, RNA with and without treatment with calf intestinal phosphatase was used to identify primary and processed products. The cDNA was amplified by PCR using 2 pmol of reverse gene-specific primers (supplemental Table S2) and GeneRacer 5′ primer. An additional round of nested PCR was employed, using GeneRacer nested and gene-specific oligonucleotides. PCR products from the first amplification and the second round nested PCR were purified and cloned into pCR-4-TOPO vector. From each reaction, at least six independent plasmids were isolated and sequenced using M13 forward primer.
β-Galactosidase Assays
To measure the activity of rpoEP2, rpoHP3, groESL, and cpxP promoters, single-copy chromosomal promoter fusions to the lacZ gene were used, whose construction has been described previously (27, 28). The regions covering the P2hs and P3 promoters of the lapA lapB operon were amplified by PCR. To quantify heat shock regulation, the minimal lapABP2hs promoter region was cloned using specific oligonucleotides (supplemental Table S2). Similarly, the promoter region of the slrA gene was amplified by PCR. PCR products were digested with EcoRI and BamHI and cloned into the promoter probe vectors pRS415 or pRS551. All of the cloned promoters were transferred to the chromosome in single copy by recombination with λRS45, selecting for lysogens as described previously (27–29). β-Galactosidase activity was determined as described previously (4).
Construction of Chromosomal Single-copy LapA, LapB, LptD, WaaC, and WaaO FLAG Derivatives
To construct chromosomal C-terminal 1× or 3× FLAG-tagged derivatives, plasmids pSUB312 and pSUB11 (30) were used as templates for PCR amplification as described previously (4). PCRs were carried out using appropriate oligonucleotides. Verification of non-disturbed function of the concerned gene was done on the basis of the presence of the wild-type phenotype. The construction of chromosomal LpxM-3×FLAG was described previously (4).
Because the translation of LapA and LapB was found to be coupled, an additional LapA-FLAG derivative was constructed. This was achieved by inserting the P2hs and P3 promoter region of the lapA lapB operon and by providing the ribosome-binding site upstream of the lapB coding sequence. This was achieved by a two-step overlap PCR using the appropriate oligonucleotides (supplemental Table S2). The PCR product from the second round of amplification was used for λ Red-mediated recombineering. The resulting construct (SR16430) was also verified to have normal LapA and LapB function in terms of synthesis of the wild type-like LPS.
Protein Purification
Expression of hexa-His-tagged LapA and LapB was induced in the strain E. coli BL21 at an optical density of 0.1 at 600 nm in a 3-liter culture by the addition of 0.6 mm isopropyl 1-thio-β-d-galactopyranoside (IPTG) at 28 °C. When required, rifampicin (200 μg ml−1) was added after a 1-h incubation with IPTG to block the host protein synthesis. Similarly, expression of hexa-His-tagged WaaC was induced in the strain E. coli BW25113 by the addition of 0.7 mm IPTG. To validate the in vivo interaction of WaaC and LapB, WaaC expression was induced in the strain GK2642, carrying chromosomal LapB-FLAG. Compatible plasmids expressing His-tagged LptC and non-His-tagged LapB in pWSK29 (lapB) were co-expressed in E. coli BL21 strain. For their co-expression, the mild inducing conditions with 0.15 mm IPTG were used. After 4 h of induction at 28 °C, cells were harvested by centrifugation at 7,000 rpm for 20 min. The pellet was resuspended in B-PER reagent (Pierce) (1:500 volume of culture), supplemented by 200 μg ml−1 of lysozyme, 300 units of benzonase, and 500 μg ml−1 of a protease inhibitor mixture (Sigma), and incubated with gentle stirring for 30 min on ice. The lysates were centrifuged at 45,000 × g for 30 min at 4 °C. The pellet fraction containing IM, OM proteins, and aggregates was resuspended in B-PER reagent and 2 volumes of buffer A (50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazole) supplemented with 1% octyl-β-d-glucoside and a mixture of protease inhibitors. Pellet was stirred at 4 °C for 4 h and centrifuged at 45,000 × g for 30 min at 4 °C. Supernatant was applied over nickel-nitrilotriacetic acid beads (Qiagen), pre-equilibrated with buffer A. The column was extensively washed with 20 mm imidazole supplemented with 1% octyl-β-d-glucoside. Proteins were eluted with buffer A, using a step gradient ranging from 50, 100, 250, and 500 mm imidazole and analyzed on 12.5% SDS-PAGE.
Multicopy and Extragenic Suppressor Isolation
A plasmid library containing a complete genomic set of E. coli ORFs (26), except for lapA and lapB genes, was used to transform either ΔlapB or Δ(lapA lapB). Transformants were plated in parallel under non-permissive growth conditions in the presence of either 25 or 75 μm IPTG. Individual colonies that grew at 40 or 42 °C on LA medium or on M9 medium at 42 °C (Ts+) or MacConkey-resistant transformants were selected. Plasmid DNA was isolated from such suppressors. This DNA was used to retransform original suppressor-free ΔlapB and Δ(lapA lapB) mutants to confirm the suppression. The identity of the multicopy-suppressing gene was obtained by DNA sequencing.
An additional multicopy library was constructed in p15A-based vector as described previously (31), using chromosomal DNA isolated from the Δ(lapA lapB) mutant. DNA from such a library was used to transform suppressor-free Δ(lapA lapB) or ΔlapB mutants to isolate multicopy suppressors of various growth defects as described above. Plasmids that confirmed the suppression were used to identify the suppressing gene(s) upon standard subcloning.
For the isolation and characterization of extragenic suppressors, several independent single-colony cultures of suppressor-free Δ(lapA lapB) BW25113 derivative were grown at 30 °C in M9 minimal medium. Aliquots were plated on LA medium at 37 and 42 °C or streaked on M9 minimal agar at 42 °C. Temperature-resistant colonies were grown, and suppressor mutation was marked with Tn10 as described previously (27). The position of Tn10 was determined by the inverse PCR. To characterize suppressor mutation(s) in W3110 and BW30207 backgrounds, ΔlapB transductants obtained in the absence of complementing plasmid were analyzed. The suppressor mutation was marked by Tn10 and further transduced in the wild-type strain. Such derivatives were phenotypically characterized, and the position of Tn10 was determined, followed by the complementation analysis using the plasmid DNA from the library of E. coli ORFs (26). The position of mutation was determined by DNA sequencing of PCR-amplified products.
Isolation of Aggregated Proteins
The aggregation and/or folding status of various LPS biosynthetic enzymes was analyzed using isogenic strains with a chromosomal FLAG epitope-encoding tail appended to the 3′ end of the individual gene. The protocol for quantification of aggregation was followed (32) with the following modifications. Bacterial cultures (25–30 ml) were grown in M9 minimal medium at 30 °C up to an A600 of 0.2. Half of the culture was heat-shocked at 42 °C and incubated with shaking for another 90 min. Heat-shocked cultures and the control (30 °C) were harvested by centrifugation. Pellets were resuspended in 600 μl of B-PER reagent; supplemented with 1 mg ml−1 lysozyme, a mixture of protease inhibitors (Sigma), and 30 units of benzonase; and incubated for 90 min on ice with gentle mixing. Intact cells were removed by centrifugation at 2,000 × g for 15 min at 4 °C. Soluble and insoluble cell fractions (containing membrane and aggregated proteins) were isolated by subsequent centrifugation at 15,000 × g for 20 min at 4 °C. The pellet fractions were resuspended in 300 μl of B-PER reagent, supplemented with 2% octyl-β-d-glucoside, a mixture of protease inhibitors (Sigma), and 30 units of benzonase, followed by brief sonification. The mixture was incubated with mixing for 4 h and centrifuged (15,000 × g, 90 min, 4 °C) to obtain IM fractions solubilized by octyl-β-d-glucoside. The pellet fraction, containing OM and aggregated proteins, was resuspended in 40 μl of 10 mm Tris buffer supplemented by a mixture of protease inhibitors. Soluble, IM fractions and aggregates were analyzed by gel electrophoresis.
LPS Extraction
Typically, 400-ml cultures of isogenic bacteria were grown in either M9 medium or in phosphate-limiting 121 medium until reaching an optical density of 0.8–1.0 at 600 nm at 30 °C. Cultures were harvested by centrifugation (8,671 × g, 30 min, 4 °C), and LPS was extracted by the phenol-chloroform-petroleum ether procedure (33) and lyophilized. For the LPS analysis, lyophilized material was dispersed in water by sonication and resuspended at a concentration of 2 mg ml−1. For detection of the chemotype, an equivalent portion of whole cell lysate treated with proteinase K was applied to a 16.5% Tricine gel. Gels were silver-stained for LPS analysis.
Mass Spectrometry
Electrospray ionization (ESI) Fourier transform ion cyclotron (FT-ICR) mass spectrometry was performed on intact LPS in the negative ion mode, using an APEX QE (Bruker Daltonics) equipped with a 7-tesla actively shielded magnet and dual ESI-MALDI. LPS samples were dissolved at a concentration of ∼10 ng μl−1 and analyzed as described previously (6). For nonspecific fragmentation, the DC offset (collision voltage) of the quadruple interface was set from 5 to 30 V. Under these conditions, the labile linkage between lipid A and the core oligosaccharide is cleaved. Mass spectra were charge-deconvoluted, and mass numbers given refer to the monoisotopic peaks. Mass calibration was done externally using well characterized similar compounds of known structure.
Western Blot Analysis
Routinely, cultures were grown at 30 °C for 20–24 h in 30 ml of M9 minimal medium, harvested by centrifugation at 14,000 rpm for 10 min, and resuspended in SDS lysis buffer. For heat shock induction, overnight bacterial cultures were grown at 30 °C. Cultures were diluted to an optical density of 0.02 at 600 nm and allowed to grow up to an optical density of 0.2 at 600 nm. Half of the sample was shifted to a prewarmed flask at 42 °C. Samples were withdrawn after every 5-min interval, and proteins were immediately precipitated by the addition of 10% TCA and resolved by 12% SDS-PAGE. After electrophoresis, proteins were blotted to a PVDF membrane and detected with specific antibodies as described previously (27, 34).
RESULTS
LapB Is Essential for Bacterial Growth in Rich Medium
We previously described the isolation of mutants with elevated levels of envelope stress response during a screen to identify periplasmic protein folding factors (17). However, no function could be assigned to the genes affected by these mutations, all of which mapped at 28 min on the chromosome of E. coli. These mutations mapped to two uncharacterized genes, yciS and yciM. We speculated that these mutations could affect the synthesis or translocation of LPS because genetic variants synthesizing minimal LPS composed of Kdo2-lipid IVA also exhibit similar phenotypes (6). In depth analysis revealed that products of yciS and yciM genes indeed play an important role in the assembly of LPS, and they were therefore designated lipopolysaccharide assembly proteins LapA and LapB. Several non-polar deletions, ΔlapA, ΔlapB, and Δ(lapA lapB), were constructed and examined in detail for the LPS content. Such deletions were transduced in well characterized E. coli K12 backgrounds, such as W3110, MG1655 derivative BW30207, and BW25113 on M9 minimal and LA-rich medium at 23, 30, 37, and 42 °C. Δ(lapA lapB) or ΔlapB suppressor-free mutants could be obtained in the BW25113 parental strain but only on M9 medium at 30 °C, and these mutants formed very small size colonies (Table 1). Transductants were obtained at 23 °C only after prolonged incubation (72 h). Δ(lapA lapB) and ΔlapB transductants were only obtained in the presence of plasmid carrying lapA and lapB genes in the parent strain W3110 or BW30207 (Table 1). However, ΔlapA deletion was tolerated in all backgrounds, unless when such strains were grown on MacConkey agar at 43 °C (Table 1). The growth defects of BW25113 Δ(lapA lapB) and ΔlapB derivatives were quantified by measuring growth in liquid cultures (Fig. 2, A and B). After a shift to 42 °C, the Δ(lapA lapB) and ΔlapB mutants ceased to grow after 2 h even in M9 minimal medium. A growth defect in minimal and LB medium was also observed for Δ(lapA lapB) and ΔlapB mutants even at 30 °C (Fig. 2, A and B). Together, these results show that the lapB gene is essential under standard laboratory growth conditions, and its deletion results in a severe growth defect.
TABLE 1.
The essentiality of lapB gene and suppressors that bypass lethality of ΔlapB; colony-forming ability in transductional assays
| M9 |
LA (37 °C) | MacConkey |
|||
|---|---|---|---|---|---|
| 30 °C | 37 °C | 42 °C | 43 °C | ||
| BW25113 + pWSK29 lapAB+ | ++a | ++ | ++ | ++ | ++ |
| BW25113 ΔlapA | ++ | ++ | ++ | ±b | −c |
| BW25113 ΔlapB | +d | − | − | − | − |
| BW25113 ΔlapB + pWSK29 lapB+ | ++ | ++ | ++ | ++ | ++ |
| BW25113 Δ(lapA lapB) | + | − | − | − | − |
| BW25113 Δ(lapA lapB) + pWSK29 lapAB+ | ++ | ++ | ++ | ++ | ++ |
| W3110 + pWSK29 lapAB+ | ++ | ++ | ++ | ++ | ++ |
| W3110 ΔlapB | −e | − | − | − | − |
| W3110 ΔlapB waaCT187K | + | + | ± | − | − |
| BW30207 + pWSK29 lapAB+ | ++ | ++ | ++ | ++ | ++ |
| BW30207 ΔlapB | −e | − | − | − | − |
| BW30207 ΔlapB gmhAIS1 | + | ± | ± | − | − |
| BW25113 Δ(lapA lapB) + plapBArg371 | + | − | − | − | NTf |
| BW25113 Δ(lapA lapB) + plapBPro378 | + | − | − | − | NT |
| BW25113 Δ(lapA lapB) + plapBLeu372 | + | − | − | − | NT |
| BW25113 Δ(lapA lapB) lptD4213 + pWSK29 | − | − | − | − | NT |
| BW25113 Δ(lapA lapB) lptD4213 + pWSK29 lapAB+ | ++ | ++ | ++ | NT | NT |
| BW25113 Δ(lapA lapB surA) | − | − | − | − | NT |
| BW25113 Δ(lapA lapB surA) + pWSK29 | − | − | − | − | NT |
| BW25113 Δ(lapA lapB surA) + pWSK29 lapAB+ | ++ | ++ | ++ | NT | NT |
| BW25113 Δ(lapA lapB) lpxC186 | ++ | + | + | − | − |
| BW25113 Δ(lapA lapB) lpxCA60g | ++ | + | + | ± | − |
| BW25113 sfhC21 Δ(lapA lapB) | ++ | ++ | ++ | NT | NT |
| BW25113 Δ(lapA lapB) lpxA2 | ++ | ± | ± | NT | − |
| BW25113 Δ(lapA lapB) lpxD36 | ++ | + | ± | NT | − |
| BW25113 Δ(lapA lapB) lpxD201 | ++ | ± | ± | NT | − |
| BW25113 Δ(lapA lapB) lpp::Tn10 | ++ | + | + | − | − |
| BW25113 Δ(lapA lapB lpp) | ++ | + | + | − | − |
| BW25113 Δ(lapA lapB) waaQ::Tn10 | ++ | + | + | − | − |
| BW25113 Δ(lapA lapB waaG) | ++ | + | + | − | − |
a ++, ≥1,000 colonies normal size.
b ±, small colonies.
c −, inability to support colony forming capacity.
d +, 200–1000 colonies but small in size.
e −, suppressors mostly mapping to the waaC gene or the gmhA gene.
f NT, not tested.
g Single nt deletion at position 60 upstream of ATG of the lpxC gene.
FIGURE 2.

LapB is essential for the bacterial growth in rich medium. Isogenic bacterial cultures of the wild type and ΔlapB and Δ(lapA lapB) mutants were grown overnight in M9 medium at 30 °C. Cultures were adjusted to A600 of 0.05 in 12 ml of prewarmed M9 medium at 30 and 42 °C (A). Aliquots of samples were drawn at different intervals, and the bacterial growth was monitored by measuring A600. In parallel, growth of the wild type and its Δ(lapA lapB) derivative was monitored after shift at A600 of 0.05 in prewarmed LB medium (B). An equivalent amount of bacterial cells obtained from the wild type and its ΔlapB derivative grown on M9 agar at 30 °C were used to prepare whole cell lysates. Samples were digested with proteinase K and applied on a 16.5% SDS-Tricine gel, and LPS was revealed by silver staining (C). The asterisk in the lane corresponding to the LPS from ΔlapB mutant indicates faster migrating species.
lapA and lapB Are Heat Shock Genes
Our previous studies of transcriptional profiling revealed increased lapB transcription at high temperature (35). Further independent studies suggested that the lapB gene belongs to the RpoH regulon (36, 37). To confirm these observations, we decided to map the 5′ ends of the RNA by 5′ RACE from RNA extracted from wild-type cultures grown at 30 °C and upon shift for 15 min to 42 °C. These experiments identified three transcription initiation sites designated P1, P2hs, and P3 (Fig. 3A). The transcription initiation start site corresponding to the P2hs promoter was found to be located 108 nt upstream of the translational initiation codon of the lapA gene. Plasmids bearing this 5′ end were enriched from clones based on RNA extracted upon shift to 42 °C. Examination of DNA sequence upstream of the P2hs initiation site revealed a remarkable similarity to RpoH-regulated heat shock promoters. Sequence conservation could be seen both in the −10 and −35 boxes of well characterized RpoH-regulated promoters (Fig. 3B). The other major initiation site P3 was mapped at either 56 or 57 nt upstream of the initiation codon from clones derived from RNA extracted from 30 °C growth conditions (Fig. 3A). The sequence of the predicted −10 region of the P3 promoter suggested that it is a housekeeping promoter. However, the lack of −35 consensus sequence and the presence of TGG adjacent to the −10 sequence suggests extended −10 promoter (38). The P1 initiation site was found to be located upstream of the pgpB gene with a good match to the −10 consensus of housekeeping promoters (Fig. 3A). Highly conserved TG dinucleotide (39) is present in the putative −35 region (Fig. 3A). Transcription initiating from the P1 promoter suggested coupling of the lapAB transcription with phospholipid synthesis as the pgpP gene encodes phosphatidylglycerophosphate phosphatase (40).
FIGURE 3.

Transcriptional regulation of lapA and lapB genes. A, nucleotide sequence of the promoter region of the lap operon. Transcriptional start sites were identified from RNA obtained from the wild-type bacteria grown in M9 medium at 30 °C and after a 15-min shift to 42 °C. The arrows indicate the position of transcription start sites. The site marked as P2hs corresponds to the heat shock promoter. The corresponding −10 and −35 elements are shown. The P1 and P3 start sites represent initiation sites under non-heat shock conditions. The P1 start is located upstream of the pgpB gene as indicated. The highly conserved T and G nucleotides present in the putative −35 region of the P1 promoter are indicated. The TG dinucleotide upstream of the −10 region of the P3 promoter, corresponding to the presence of putative extended −10 promoter, is shown in orange. B, alignment of −10 and −35 regions of the lapABP2hs promoter with well characterized heat shock promoters. C, the activity of lapABP2hs and groESL promoters was measured using strains carrying single-copy chromosomal promoter fusions. Cultures were grown as described above; heat-shocked at 42 °C for 5, 10, 15, 20, and 30 min; and analyzed for β-galactosidase activity. Shown is Western blot analysis of whole cell extracts from strains expressing LapA-FLAG (D) and LapB-FLAG (E). Cultures were grown in M9 medium at 30 °C up to an A600 of 0.2. One-ml aliquots were shifted to prewarmed tubes held at 42 °C and incubated for 5, 10, 15, or 20 min. Proteins were precipitated by TCA (10%) and analyzed on 12% SDS-PAGE, followed by immunoblotting with anti-FLAG antibody.
To confirm heat shock regulation of the P2hs promoter, the activity of a single-copy chromosomal promoter fusion lapABP2hs-lacZ was analyzed. Examination of its activity revealed a heat shock induction comparable with that of a well characterized heat shock promoter of the groESL operon (Fig. 3C). Heat shock induction of LapA and LapB was validated by examining levels of chromosomally encoded LapA-FLAG and LapB-FLAG. Levels of LapA and LapB follow a classical heat shock induction, followed by a typical shut-off (Fig. 3, D and E). Taken together, these results show that the lapA lapB operon is subjected to heat shock induction consistent with the promoter sequence element upstream of the P2hs transcription start site.
ΔlapB and ΔrpoH Mutants Have Defects in LPS
Analyses of whole cell lysates revealed that ΔlapB mutant accumulated elevated levels of LPS with a few faint faster migrating species (Fig. 2C). However, it is known that heptose-deficient LPS and underacylated LPS stain less intensely than when the LPS is with the full-length core (41) and thus may not reflect complete LPS alterations. To determine the LPS composition, LPS was purified and analyzed by mass spectrometric analysis. Cultures were grown under permissive suppressor-free growth conditions at 30 °C in either M9 or phosphate-limiting 121 medium. The phosphate-limiting growth condition was used because it allows us to study lipid A and LPS core modifications not found in the LPS obtained from M9 medium (4). Modifications by P-EtN and Ara4N served as indicators of LPS translocation (9). Mass spectra of LPS from the ΔlapB mutant grown in M9 medium revealed a considerable degree of heterogeneity. For example, the spectra of LPS from the ΔlapB mutant contained the mass peak at 2,237.3 Da and its derivatives (Fig. 4B). The mass peak at 2,237.3 Da corresponds to the minimal LPS structure composed of only LAhexa + Kdo2. Further mass peaks at 2,429.5 and 2,621.7 Da suggested the incorporation of one and two additional Hep residues, respectively, derived from LAhexa + Kdo2 (Fig. 4B). Additionally, mass peaks corresponding to pentaacylated derivatives were also present, as revealed by the mass peak at 2,219.2 Da and its derivatives (Fig. 4B). Further, a range of mass peaks depicting incomplete synthesis could be observed for spectra of LPS extracted from the ΔlapB mutant but not in the LPS of the wild type. Mass peaks corresponding to complete core synthesis were also present in spectra of LPS from the ΔlapB mutant.
FIGURE 4.

ΔlapB mutant accumulates precursor forms of LPS and restoration of normal LPS composition upon complementation. Shown are charge-deconvoluted ESI FT-ICR mass spectra in the negative ion mode of native LPS obtained from the wild-type strain (A) and its ΔlapB derivative (B) grown in M9 medium at 30 °C. C–G, spectra of LPS obtained from cultures grown in 121 medium at 30 °C. The relevant genotype is indicated. Mass numbers refer to monoisotopic peaks. In B, mass peaks, mostly corresponding to the substitution by phosphate or sodium adducts or due to carbon change length polymorphism, are not labeled. Rectangular boxes, mass peaks corresponding to the glycoform containing the third Kdo. Ovals, derivatives with two Kdo residues with either complete core or incomplete core.
Mass spectrometric analyses of LPS from ΔlapB mutant obtained from phosphate-limiting growth conditions also revealed the presence of several mass peaks corresponding to early intermediates and to mature LPS (Fig. 4D). Further, some of the mass peaks indicated the presence of pentaacylated lipid A in the LPS of the ΔlapB mutant (Fig. 4D). Thus, the mass peak at 2,815.4 Da in the LPS of ΔlapB mutant corresponds to LApenta + Kdo2 + Hep2 + Hex2 + P composition. Mass peaks at 3,279.6 and 3,402.6 Da correspond to hexaacylated LPS, composed of only 2 Hep and 2 Hex residues in the core. Interestingly, mass peaks corresponding to the complete glycoform I with additional modifications by GlcUA, Ara4N, and P-EtN (the mass peak at 4,040.9 Da and its derivatives) were also present. Mass peaks revealing substitutions with P-EtN and Ara4N suggested that the defects in LPS of ΔlapB mutant do not hinder LPS translocation.
Normal LPS composition was restored when the ΔlapB mutant was transformed with plasmid pSR16881 encoding the wild-type copy of the lapB gene (Fig. 4E). Distribution of mass peaks, corresponding to mature glycoforms and their derivatives, was found to be nearly identical to that of the LPS obtained from the wild type (Fig. 4C). Thus, the observed accumulation of LPS precursors in the ΔlapB mutant is the direct consequence of a loss of LapB function and not due to the accumulation of extragenic suppressors.
Mass spectra of LPS from ΔlapA mutant also revealed accumulation of a few incomplete LPS precursors. However, the majority of mass peaks corresponded to the complete glycoform I or glycoforms with a third Kdo and rhamnose (Figs. 1 and 4F). These results are consistent with minor growth defects observed for the ΔlapA mutant. Because one of the promoters of lapAB genes is RpoH-regulated, we analyzed LPS of a ΔrpoH derivative as well. This analysis revealed that premature termination of LPS biosynthesis occurred in this mutant (Fig. 4G). For example, the mass peak at 3,402.6 Da, which is also present in the LPS of ΔlapB mutant, is predicted to contain only 2 Kdo, 2 Hep, and 2 Hex residues in the truncated core (Fig. 4, D and G). A defect in the LPS assembly linked to a loss of the rpoH gene has not been reported before. However, this defect is not as severe as in the case of ΔlapB mutant, presumably because transcription from the P1 and P3 promoters is RpoH-independent.
Analysis of the lipid A portion of LPS from the ΔlapB mutant confirmed the accumulation of pentaacylated species, as revealed by the mass peak at 1,587.0 Da in addition to hexaacylated lipid A derivative (mass peak at 1,797.2 Da) (Fig. 5B). Further, mass peaks at 1,769.2 Da and derivatives with P-EtN (1,892.5 Da) and Ara4N (1,900.3 Da) substitutions were present in the lipid A of ΔlapB mutant but were absent in the wild type (Fig. 5B). The mass peak at 1,769.2 Da suggested the incorporation of additional lauroyl chain at the place where myristoyl chain is usually incorporated. Such lipid A alterations have been observed in lpxD mutants or mutations mapping to the fabZ gene, which confer resistance to LpxC inhibitors (42, 43).
FIGURE 5.

Defects in the lipid A biogenesis of ΔlapB derivatives. Shown are charge-deconvoluted ESI FT-ICR mass spectra in the negative ion mode depicting the lipid A composition and its modifications from the LPS obtained under permissive growth conditions. The relevant genotype corresponding to panels A–D is indicated. Part of the negative ion mass spectra of the native LPS after nonspecific fragmentation, leading to the cleavage of the labile lipid A-Kdo linkage, is presented. The mass peaks corresponding to the penta- and hexaacylated lipid A part and substitutions with P-EtN and/or Ara4N are indicated. Insets, predicted chemical composition of the hexa- and pentaacylated lipid A part.
LapA and LapB Co-purify with Lpt Proteins
To further understand the function of LapA and LapB, we purified these proteins. LapA and LapB were found to be present in IM fractions and could be solubilized by a mild detergent like octyl-β-d-glucoside. LapA and LapB were found to co-purify with each other and some other proteins (Fig. 6, A and B). MALDI-TOF analyses of co-purifying proteins revealed that LapA co-purifies with LptE/D, LptBFGC, LptA, and DnaK/J proteins (Fig. 6A). Similarly, independently expressed His-tagged LapB was found to co-elute with the same set of Lpt proteins, FtsH as well as WaaC (Fig. 6B). Quite surprisingly, LptE was enriched in LapA preparations.
FIGURE 6.

Co-purification of LapA and LapB with Lpt proteins. Purification of LapA and LapB and co-purification with Lpt proteins upon co-overexpression were performed as described under “Experimental Procedures.” Samples after the induction of LapA (A) and LapB (B), after washing columns and after elution with indicated concentrations of imidazole, were resolved on 12.5% SDS-PAGE. The identity of co-purifying proteins is indicated by numbers in A and B. Lanes 5 (A and B), purified samples of LapA and LapB, respectively, obtained after blocking host protein synthesis by the addition of rifampicin. Lanes 6 (A and B), complex after co-expression of LapB with LptC. Purified samples corresponding to lanes 5 and 7 in the A representing LapA and LapB, respectively, were digested with proteinase K and applied onto a 16.5% SDS-Tricine gel, followed by silver staining to reveal LPS (C). D, as a negative control, 300 μg of purified LPS from the wild-type bacteria was applied over nickel-nitrilotriacetic acid beads, pre-equilibrated with the same buffer used in the purification of LapA. One μg of purified LPS, a portion of the flow-through (FT), and LPS after elution were digested with proteinase K and analyzed as described above. E and F, WaaC heptosyltransferase I co-purifies with LapB. His-tagged WaaC was overproduced in the strain expressing LapB-FLAG from the chromosome. WaaC purification profile was examined by the analysis of WaaC elutions on 12% SDS-PAGE (E). The arrows indicate the position of LapB and WaaC. The same samples as used in E were analyzed on 12% SDS-PAGE and tested for the presence of LapB-FLAG, using FLAG antibody after immunoblotting (F).
To validate LapA and LapB interaction with LPS transport proteins, LapB and His-tagged LptC were co-induced and used for purification. In these experiments, LptC and LapB co-eluted, and these fractions were also enriched in LapA, LptBFG, DnaJ, and LptE/D (Fig. 6, A and B). Co-elution of LapB and Lpt proteins suggested that LapA and LapB could be part of the Lpt transenvelope complex. Further, the IM localization and co-purification of LapB with heptosyltransferase I and Lpt proteins support a role in coupling LPS synthesis and translocation. In this process, LapA and LapB could provide a scaffold-like structure for LPS assembly. Co-purification of FtsH with LapB could be part of the mechanism that controls levels of LpxC.
Purified LapA and LapB Contain LPS
We wondered whether LapA and LapB might also interact with LPS. To obtain relatively pure LapA and LapB proteins, the expression of their cognate genes was induced from the T7 promoter in the presence of rifampicin to block the host protein synthesis, and proteins were purified as described above. Under these conditions, relatively pure LapA preparations could be obtained (Fig. 6A, lane 5). However, LapB still contained LptBFG proteins, although their amounts were much lower when rifampicin was used. It has been reported that free LPS does not directly bind to nickel affinity resins in the presence of imidazole (14). Such LapA and LapB protein preparations were digested with proteinase K, and LPS was revealed by silver staining of SDS-Tricine gels. Both LapA and LapB preparations were found to contain LPS with relatively higher amounts in LapA preparations (Fig. 6C). Further, as a negative control, free LPS did not bind to the nickel affinity resin, and most of it came out in the flow-through (Fig. 6D). However, it is likely that LapA and LapB bind LPS in a complex. These results support a model whereby LapA and LapB serve as docking sites in the IM for the assembly of LPS.
Co-purification of Heptosyltransferase I and LapB
Because WaaC was found to co-purify with LapB, we performed additional experiments to confirm this co-purification. Expression of hexa-His-tagged WaaC was induced in the strain carrying the single-copy LapB-FLAG tag on the chromosome. WaaC was purified by affinity chromatography (Fig. 6E). WaaC eluted in an identical manner whether purified from a strain encoding or not encoding the LapB-FLAG. Fractions containing WaaC were analyzed by Western blotting using anti-FLAG antibody, which revealed the presence of LapB-FLAG (Fig. 6F). It should be noted that, as reported earlier, WaaC associates with the IM (44). Taken together, these results suggest that LapB could recruit WaaC to the site of LPS synthesis. This would ensure heptose transfer because it is the first step in the LPS core assembly after the Kdo attachment.
LpxC Accumulates in Δ(lapA lapB) Mutants
Extragenic suppressor mutations of ΔlapB or Δ(lapA lapB) mutants that allow growth at non-permissive growth conditions were isolated and characterized. Three independent suppressor mutations mapping to the lpxC gene were identified. Two of them contained a single nucleotide deletion (loss of A at position 60 nt upstream of the ATG initiation codon). This missing nucleotide was mapped to the −10 promoter region of the lpxC gene (45), thus presumably decreasing the transcription of the lpxC gene. The third suppressor mutation was a single nt change at the position 557 (T to A, leading to Ile-186 to Asn-186 substitution) and was designated lpxC186. LpxC is an unstable protein that is rapidly turned over by the essential protease FtsH (7, 8). Thus, ftsH mutants accumulate high amounts of LpxC, leading to an increase in LPS amounts and toxicity (7, 8). However, the ftsH gene can be deleted in the presence of mutations in the fabZ gene, such as sfhC21, which restore a balance between phospholipids and LPS (7). Levels of LpxC were examined by Western blot in extracts prepared from the wild-type, Δ(lapA lapB), ΔftsH sfhC21, and Δ(lapA lapB) lpxC186 bacterial cultures. This analysis revealed that Δ(lapA lapB) and ΔftsH sfhC21 derivatives accumulate high levels of LpxC that is otherwise barely detectable in the wild type (Fig. 7A). Consistent with the isolation of lpxC186 as suppressor mutation, the Δ(lapA lapB) lpxC186 mutant had a much reduced level of LpxC as compared with a Δ(lapA lapB) or ΔftsH sfhC21 mutant. Interestingly, a Δ(lapA lapB ftsH) sfhC21 or Δ(lapA lapB) ftsH+ derivatives contained similar elevated levels of LpxC. These enhanced levels of LpxC in the Δ(lapA lapB) mutant did not change when extracts were prepared from culture grown under permissive or non-permissive growth conditions (shift to LB medium at 37 or 42 °C). These results establish that FtsH-mediated degradation of LpxC requires LapB, and this may explain the genetic requirement for the lapB gene. Consistent with toxicity caused by elevated levels of LpxC, mutations that reduced the lipid A synthesis like lpxD36, lpxD201, and lpxA2 conferred a modest suppression of growth defects of Δ(lapA lapB) mutants (Table 1).
FIGURE 7.
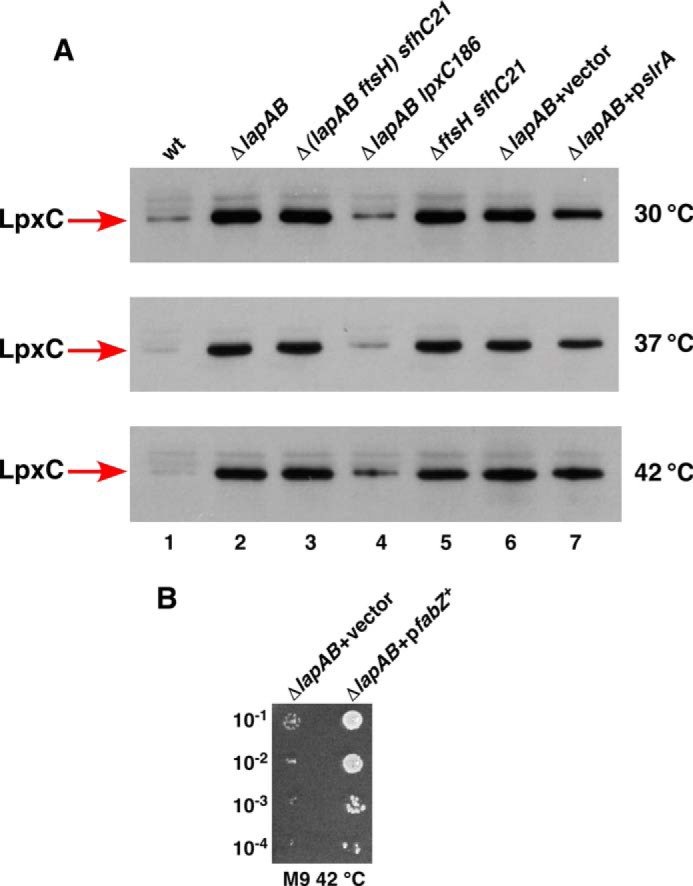
The accumulation of LpxC in Δ(lapA lapB) mutant and suppression of its growth defects by the overexpression of the fabZ gene. A, cultures were grown at 30 °C in M9 medium up to an A600 of 0.2, and aliquots were shifted to 37 and 42 °C for 30 min in LB medium. An equivalent amount of total protein was analyzed on 12% SDS-PAGE, followed by immunoblotting using LpxC antibodies. Note the elevated amounts of LpxC in Δ(lapA lapB) mutant and its reduction in lpxC186 background and upon overexpression of the slrA sRNA. B, cultures of Δ(lapA lapB) mutant carrying the vector alone or expressing the fabZ gene from plasmid under the control of the ptac promoter were grown in M9 medium at 30 °C, adjusted to an A600 of 0.1, and serially diluted. Five-μl aliquots were spotted on M9 plate supplemented with 25 μm IPTG and incubated at 42 °C.
It has been shown that the essential gene ftsH can be deleted in a background carrying the sfhC21 allele of the fabZ gene (7). This prompted us to examine whether introduction of the sfhC21 allele would also bypass the lethality of Δ(lapA lapB) and ΔlapB mutants under laboratory growth conditions. Thus, a BW25113 derivative with sfhC21 allele was constructed and found to tolerate a Δ(lapA lapB) mutation at 37 °C on M9 and LA medium (Table 1). These results are consistent with the notion that toxicity due to high amounts of LpxC in the absence of the lapB gene can be relieved when a gain of function mutation in the fabZ gene is introduced.
Consistent with our overall model, the fabZ overexpression to a modest level also suppressed the lethality of Δ(lapA lapB) mutation and restored growth up to 42 °C on M9 medium (Fig. 7B). Because the gain of function fabZ mutation (sfhC21) allows deletion of the ftsH gene as well as the lapB gene, we can conclude a similar restoration of a balance between phospholipid and LPS upon the fabZ overexpression.
Characterization of Additional ΔlapB Suppressors
Mapping of additional suppressor mutations identified loss of function mutations in waaQ/G operon, gmhA, waaC, or lpp in addition to the lpxC gene. GmhA is required for the isomerization of d-sedoheptulose 7-phosphate into d-glycero-d-manno-heptose 7-phosphate, and the waaC gene encodes heptosyltransferase I (1). It should be noted that suppressor mutation in waaC or gmhA genes allowed a limited growth on LA medium up to 37 °C only (Table 1). However, such suppressors were quite preponderant in ΔlapB derivatives in the W3110 or BW30207 backgrounds (Table 1). The suppressor mutation in the waaC gene corresponded to a single nucleotide change at position 560 (C to A). This resulted in the substitution of the highly conserved amino acid Thr-187 to Lys-187 (Fig. 8B). Amino acid Thr-187 is located within the sugar-nucleotide binding site of WaaC (46). In the crystal structure of the WaaC-ADP complex, the β-phosphate interacts with Thr-187, Thr-188, and Lys-192. The suppressor mutation in the gmhA gene was due to an insertion sequence (IS1) in the Shine-Dalgarno sequence.
FIGURE 8.

A single point mutation in the waaC gene suppresses ΔlapB. Charge-deconvoluted ESI FT-ICR mass spectra in the negative ion mode of LPS isolated from strains with ΔwaaC deletion (A) and ΔlapB with waaCT187K suppressor mutation (B). Cultures were grown in permissive conditions at 30 °C in M9 medium. Mass peaks corresponding to pentaacylated lipid A species lacking the myristoyl chain, and mass peaks representing carbon chain polymorphism are drawn schematically. Mass numbers refer to monoisotopic peaks, and the predicted compositions of mass peaks are indicated. C, equivalent amount of bacterial cells obtained from isogenic strains carrying the ΔwaaC deletion (lane 1) and ΔlapB waaCT187K derivative (lane 2) grown on LA agar at 30 °C and processed to obtain whole cell lysates. Samples were digested with proteinase K and applied on a 16.5% SDS-Tricine gel, and LPS was revealed by silver staining. Isogenic bacterial cultures were grown at 30 °C in M9 medium up to an A600 of 0.2 and harvested by centrifugation. An equivalent amount of total protein was analyzed on 12% SDS-PAGE, followed by immunoblotting using LpxC antibodies (D), and as a loading control, the same samples were tested by immunoblotting using antibodies to the α subunit of RNA polymerase, in the bottom panel (D). The relevant genotype is indicated.
Mass spectrometry analysis of LPS from ΔlapB waaC187 or ΔlapB gmhAIS1 mutants revealed LPS composition corresponding to LAhexa + Kdo2 (the mass peak at 2,237.3 Da) resembling the spectra of LPS obtained from ΔwaaC mutant (Fig. 8, A and B). However, mass spectra of ΔlapB waaC187 derivative revealed significant differences, as manifested by the presence of several additional mass peaks that were not present in the spectra of LPS obtained from isogenic ΔwaaC mutants. The mass peak at 2,027.1 Da corresponding to LApenta + Kdo2 was more intense in the spectra of LPS from ΔlapB waaC187 mutant as compared with the corresponding peak in the spectra of LPS from the ΔwaaC mutant (Fig. 8B). Further, several mass peaks with a mass difference of 14 Da were present in Δ(lapB waaC187) derivative (Fig. 8B). Mass peaks at 2013.1, 1999.1, and 1985.1 Da seem to arise from LApenta + Kdo2 derivative with the mass peak at 2027.1 Da, following successive loss of 14 mass units. Similarly, mass peaks with loss of 14 mass units also seemed to originate from LAhexa + Kdo2 mass peak 2,237.3 Da (Fig. 8B). Further mass peaks with an additional 14 mass units were also present, which originate from LApenta + Kdo2. It should be noted that Δ(lapB waaC) mutant exhibits the same carbon chain polymorphism. This carbon chain polymorphism can be explained if Δ(lapB waaC) mutant accumulates odd chain fatty acids because they can also be substrates for LpxA and LpxD (47). Also, underacylation has been reported to cause accumulation of mass peaks with similar 14-mass unit differences (48).
In order to address the molecular basis of suppression by a loss of function mutation in the waaC gene, LPS was examined from whole cell lysates prepared from isogenic ΔlapB waaC187 and ΔwaaC derivatives. This analysis revealed a reduction in the total amount of LPS in ΔlapB waaC187 mutant as compared with the LPS amount in the isogenic ΔwaaC mutant (Fig. 8C). A partial explanation for such results could be ascribed to the observed reduction in the elevated levels of LpxC in ΔlapB waaC187 mutant when compared with the LpxC amount in Δ(lapA lapB) mutant (Fig. 8D). A similar reduction in the elevated amounts of LpxC was observed in Δ(lapA lapB) waaQ::Tn10 derivative (Fig. 8D). This reduction might partly restore the balance between LPS and phospholipids. It should be noted that reduction in the amounts of LpxC does not restore it to wild-type levels. Thus, more in-depth studies are needed to fully explain the mechanism of suppression and molecular basis of carbon chain polymorphism.
Multicopy Suppression Reveals That Components of LPS/Phospholipid Biosynthesis Require LapB
To further understand the function of LapA and LapB, multicopy suppressors of Δ(lapA lapB) or ΔlapB mutants were isolated. This approach can identify substrates and/or limiting factors in Δ(lapA lapB) mutants. As described earlier, Δ(lapA lapB) mutants do not grow on LA or M9 at 37 and 42 °C or on MacConkey agar even at 30 or 37 °C. We used these phenotypes to obtain suppressor clones, which upon mild induction allowed growth of Δ(lapA lapB) mutants at such non-permissive growth conditions. This approach identified genes whose products are involved in phospholipid biosynthesis (fabB, fabZ), predicted to be involved in LPS assembly (yceK) or LPS synthesis (yeaD), peptidoglycan biogenesis (murA, ydhA, yffH), envelope stress response regulators (small non-coding sRNA designated slrA, hicA, and rcsF), and chaperones (dnaK and dnaJ) (Table 2). The degree of suppression varied among these suppressors (Table 2). Thus, we further analyzed suppression by the multicopy fabZ and dnaK dnaJ genes, the role and function of non-coding RNA slrA, and defects in Δ(dnaK dnaJ), and ΔyceK derivatives.
TABLE 2.
Multicopy suppressors of ΔlapB or Δ(lapA lapB) mutants
Suppression on deoxycholate (DEO) and SDS was tested on LA at 30 °C. +, normal size of colonies; ±, small colonies; −, no growth.
| Gene(s) | Suppression on |
Function | |||
|---|---|---|---|---|---|
| M9/LA, 42 °C | MacConkey, 37 °C | 0.5% SDS LA, 30 °C | 0.5% DEO LA, 30 °C | ||
| slrA | + | + | + | + | Non-coding RNA, repress elevated LpxC and envelope stress, overexpression also represses synthesis of Lpp |
| yceK | + | + | ± | ± | LPS assembly |
| yeaD | + | + | + | + | Galactose mutarotase |
| lptBFG | + | + | ± | ± | LPS transport |
| yrbL | ± | + | + | − | PhoP/Q- and EvgA/R-regulated |
| fabZ | + | ± | ± | ± | 3-R-hydroxymyristoyl acyl carrier protein (ACP) dehydratase |
| dnaK/J | ± | + | ± | ± | Chaperones |
| fabB | ± | ± | + | + | β-Ketoacyl-ACP synthase I |
| murA | + | ++ | + | + | UDP-N-acetylglucosamine 1-carboxyvinyltransferase |
| ydhA | + | + | + | + | Lysozyme inhibitor (MliC) |
| yffH | + | + | ± | ± | Nudix hydrolase NudK (Rcs regulon) |
| yecF | + | + | + | + | Sigma 28-regulated |
| rcsF | − | + | + | + | Regulator of Rcs two-component system |
| hicA | + | + | + | ± | Toxin/antitoxin, down-regulation of RpoE and Cpx response |
| ydhM | ± | + | + | ± | NemR sensing of electrophile, maintenance of redox balance |
| ydhX | ± | ± | ± | ± | Oxidoreductase for anaerobic sulfur metabolism |
| citC | + | ± | + | + | Antibiotic resistance |
| hybG | + | ± | + | − | [NiFe]-hydrogenase accessory chaperone |
The New Small Non-coding RNA slrA as a Multicopy Suppressor
Subcloning of minimal DNA fragments that cause suppression led to the identification of the cutC gene and the downstream UTR. The cutC gene is a member of the RpoE regulon required for copper homeostasis (28). Deletion analysis showed that the suppression did not require the promoter region of the cutC gene and suggested a potential non-coding region within the cutC gene and its terminator region (Fig. 9A). This gene was further characterized by mapping 5′ and 3′ ends of RNA. This revealed expression of an ∼307-nt primary RNA transcript, which is processed to an 80-nt mature non-coding RNA (Fig. 9, A and B). This sRNA is located at the 3′ UTR of the cutC gene (Fig. 9A) and was designated slrA (suppressing lap RNA). The transcription initiation site revealed that its −35 and −10 regions and their spacing resembled the consensus sequence of RpoE-regulated promoters (Fig. 9, A and C). The −35 region perfectly matches the −35 consensus of promoters recognized by RpoE (Fig. 9C) (28). This regulation was confirmed by generating a single-copy promoter fusion of slrA. Such slrA promoter fusion behaved as predicted for RpoE-regulated promoters because its expression was highly increased in a ΔrseA mutant (Fig. 9D). RseA acts as anti-σ factor for RpoE and negatively regulates RpoE activity (24).
FIGURE 9.

RpoE-regulated novel sRNA slrA as a multicopy suppressor of Δ(lapA lapB) mutation and its role in modulating RpoE activity. A, nucleotide sequence of the gene encoding slrA RNA and mapping of 5′ and 3′ ends by RACE. The arrows indicate the beginning of the primary transcript or processing sites. Nucleotides in red correspond to the region encoding the mature form of slrA sRNA, which is modeled in B. The −10 and −35 promoter elements are underlined and aligned with conserved RpoE-regulated promoters, and consensus is shown (A and C). Shown are elevated levels of the slrA promoter activity monitored by single-copy slrA-lacZ promoter fusion in a ΔrseA derivative as compared with that in the wild type (D). Cultures of Δ(lapA lapB) derivative with vector alone or expressing the slrA gene from a plasmid were grown at 30 °C in the M9 medium up to an A600 of 0.2 and analyzed by serial spot dilution for the restoration of growth on MacConkey agar at 42 °C (E). Exponentially grown cultures of the wild type (wt), its slrAC derivative with constitutive expression of slrA, ΔdegP, and ΔdegP slrAC derivatives were grown in LB medium at 30 °C, adjusted to an A600 of 0.1, and serially spot-diluted on LA plates. Plates were incubated at 30 and 42 °C (F). Isogenic cultures were grown as described in the legend to Fig. 7A. A portion of the cultures were shifted to 37 and 42 °C for 30 min in LB medium. An equivalent amount of proteins were resolved on 12% SDS-PAGE and immunoblotted with DegP antibodies (G).
To address the function of slrA non-coding RNA and the molecular basis of its multicopy suppression, we examined specific defects of Δ(lapA lapB) mutants that could be suppressed. The minimal plasmid clone in a medium copy plasmid, carrying the gene encoding this non-coding RNA, can restore growth of a Δ(lapA lapB) mutant on rich medium and MacConkey agar (Fig. 9E and Table 2). As shown above, Δ(lapA lapB) mutants accumulate high amounts of LpxC, causing toxicity, and have limiting amounts of functional WaaC at high temperature. Immunoblotting of whole cell extracts with and without overexpression of the slrA RNA caused a modest decrease in the amounts of LpxC at 30 and 37 °C (Fig. 7A).
Further, it seems that the suppression upon overexpression of the slrA RNA could also be explained by the slrA-mediated negative feedback mechanism. This negative feedback mechanism could be ascribed to the down-regulation of elevated levels of the RpoE-dependent envelope stress response in Δ(lapA lapB) mutants upon slrA overexpression. Analysis of proteomic changes in the whole cell extracts from Δ(lapA lapB) mutant or the wild-type bacteria after overexpression of the slrA RNA revealed that the amounts of Lpp lipoprotein were specifically reduced based on its identification by MALDI-TOF (data not shown). Lpp is the most abundant protein in E. coli and carries three fatty acyl chains (49, 50). Overall, these results are consistent with the isolation of a loss of function mutation in the lpp gene as an extragenic suppressor (Table 1). The repression of Lpp synthesis could be due to the predicted base pairing of slrA RNA and lpp mRNA in the region around the AUG and Shine-Dalgarno sequence. Because mutations in the lpp gene or overexpression of the sohB gene have been shown to rescue the temperature-sensitive phenotype of degP mutants (51, 52), a strain with a constitutive elevated expression of slrA was constructed to verify if increased synthesis of slrA can suppress the temperature-sensitive phenotype of a ΔdegP mutant. Consistent with these predictions, an slrAC degP derivative did not exhibit the temperature-sensitive phenotype like its parental isogenic ΔdegP mutant at 42 °C (Fig. 9F). The rescue of the temperature-sensitive phenotype in degP lppA has been previously attributed to release of toxic substances from the periplasm (51). The negative feedback action of down-regulation of the RpoE-dependent stress response was validated by the examination of levels of DegP, when the slrA sRNA was overexpressed from a plasmid as compared with the Δ(lapA lapB) derivative carrying the vector alone. This analysis revealed a reduction in the levels of DegP, which are otherwise elevated in a Δ(lapA lapB) mutant (Fig. 9G). Taken together, reduction in LpxC and Lpp amounts and a decrease in the levels of elevated stress response can explain the identification of the slrA sRNA as a multicopy suppressor of a Δ(lapA lapB) mutant. The reduction in Lpp amounts could also restore a balance between phospholipid and LPS amount by making available more free fatty acids, given the known presence of three acyl chains in the Lpp lipoprotein (49).
Synergistic Effect of Δ(lapA lapB) and Δ(dnaK dnaJ) Mutations and Mutation in a New Potential LPS Assembly Factor
Because DnaK and DnaJ co-purify with LapA and LapB and a modest overexpression of the dnaKJ operon or yceK gene product(s) partially suppressed growth defects of Δ(lapA lapB), we further explored their roles. Thus, deletion derivatives of dnaK dnaJ and yceK genes were constructed and combined with a Δ(lapA lapB) mutation at 30 °C on M9 medium.
Mass spectrometric analysis of LPS from Δ(dnaK dnaJ) mutant revealed that it contains primarily the wild type-like LPS and does not accumulate any major early intermediates of the LPS core biogenesis as is in the case with Δ(lapA lapB) (Fig. 10). However, LPS of a Δ(lapA lapB dnaK dnaJ) derivative revealed differences in the distribution of mass peaks when compared with spectra of LPS from Δ(lapA lapB) mutant (Fig. 10C). Further, Δ(lapA lapB dnaK dnaJ) derivative also contained the mass peak at 2,237.3 Da corresponding to LPS composed of LAhexa + Kdo2. Interestingly, the intensity of pentaacylated lipid A in the LPS of Δ(lapA lapB dnaK dnaJ) derivative was increased, as revealed by the high resolution mass spectrometric analysis of native, non-derivatized LPS (Fig. 5). Such mass peaks corresponding to pentaacylated lipid A (1,587.0 and 1,710.0 Da) are absent in the spectra from the lipid A part of LPS from the wild-type and its Δ(dnaK dnaJ) derivative (Fig. 5D). Further, the lipid A part of LPS from Δ(lapA lapB dnaK dnaJ) derivative also contained mass peaks at 1892.5 and 1900.3 Da derived from the parental ion at 1,769.2 Da, as is the case in the isogenic derivative of Δ(lapA lapB) mutant (Fig. 5, B and D). As explained earlier, these mass peaks correspond to P-EtN and Ara4N derivatives of hexaacylated lipid A containing two lauroyl chains and lacking the myristoyl chain. Because a mild overexpression of dnaK dnaJ genes suppressed some of the growth defects of Δ(lapA lapB) mutants, LPS was also analyzed from Δ(lapA lapB) derivative carrying plasmid-born dnaK and dnaJ genes. Mass spectrometric analysis of LPS from such a strain revealed a partial restoration of accumulation of mature glycoforms (Fig. 10F). However, some of the early intermediates were also observed but with a reduced heterogeneity (Fig. 10F).
FIGURE 10.

Effect in the accumulation of LPS precursors of Δ(lapA lapB) derivatives lacking DnaK/J or YceK. Shown are charge-deconvoluted ESI FT-ICR mass spectra in the negative ion mode of LPS isolated from strains carrying different mutations: Δ(lapA lapB) (A), Δ(dnaK dnaJ) (B), Δ(lapA lapB dnaK dnaJ) (C), ΔyceK (D), Δ(lapA lapB yceK) (E), and Δ(lapA lapB) + pdnaK+dnaJ+ (F). Cultures were grown under permissive growth conditions at 30 °C in 121 medium. Mass numbers are the monoisotopic masses of major peaks, and the predicted compositions of mass peaks are indicated.
The presence of more intense mass peaks, corresponding to pentaacylated lipid A derivatives and changes in the LPS core composition of Δ(lapA lapB dnaK dnaJ) mutant, suggested a synergistic role of LapA/B proteins with cytosolic chaperones DnaK and DnaJ for folding/activity of various LPS-specific enzymes. Because DnaK and DnaJ are major cytoplasmic chaperones, it is likely that the folding of some LPS-specific enzymes is chaperone-mediated and could be limiting if they are not correctly delivered to the LPS assembly site, where LapA and LapB proteins function together with Lpt IM complex. This can also explain a partial suppression offered by the overexpression of dnaK and dnaJ gene products.
Next, the requirement for YceK was analyzed. The precise function of the yceK gene is not known. However, the yceK gene encodes an OM lipoprotein, which has been found to interact with components of the LPS transport complex (53). Examination of LPS from a strain with a single ΔyceK mutation revealed that the majority of mass peaks correspond to glycoforms present in the wild-type strain (Fig. 10D). However, LPS from Δ(lapA lapB yceK) derivative revealed more severe defects in the LPS maturation than in Δ(lapA lapB) mutant alone. Several mass peaks present in the spectra of LPS obtained from Δ(lapA lapB yceK) mutant were composed of either penta- or hexaacylated lipid A derivatives with only Kdo2 + Hep2 + Hex2 + P in the core region (Fig. 10E). Taken together, these results suggest that the product of the yceK gene could be required for proper LPS assembly in the OM. However, overexpression of the yceK gene did not alter LPS properties (data not shown). Because the YceK is an OM lipoprotein, known to interact with Lpt proteins, the absence of YceK could lead to such a synergistic defect in the final assembly of LPS in the OM.
Aggregation of Myristoyltransferase and Early Glycosyltransferases in Δ(lapA lapB) Derivatives
Accumulation of early intermediates and the presence of pentaacylated lipid A in the LPS of Δ(lapA lapB) or ΔlapB mutants can arise due to the requirement for LapB in the synthesis or the folding, localization, or the activity of the responsible enzymes. Thus, Δ(lapA lapB) mutation was transduced into strains expressing chromosomally encoded in-frame C-terminal FLAG tag epitopes fused to specific enzymes, and their cell lysates were subjected to fractionation. Cell lysates were obtained from permissive growth conditions (M9 at 30 °C) and after shift to non-permissive growth conditions. Consistent with the accumulation of pentaacylated lipid A, Δ(lapA lapB) mutant contained reduced amounts of total LpxM at all temperatures, and the protein migrated with a lower mobility on SDS-PAGE as compared with the wild type (Fig. 11A). Further, LpxM extracted from the mutant was more sensitive to proteolysis and prone to aggregation, particularly under non-permissive growth conditions (Fig. 11A). These observations suggested that presumably LpxM was unable to achieve proper folding in the Δ(lapA lapB) mutant. The reason(s) for slower migration needs further investigation.
FIGURE 11.

Defects in the accumulation of myristoyltransferase LpxM, early glycosyltransferases, and LptD in Δ(lapA lapB) mutant. Cultures of the wild type and its Δ(lapA lapB) derivatives carrying chromosomal single-copy C-terminal 3× FLAG epitope fused to various enzymes were grown at 30 °C in M9 medium up to an A600 of 0.2, shifted to 42 °C for 90 min in LB medium. An equivalent portion of total proteins was fractionated to obtain IM proteins and total protein aggregates. Samples obtained after fractionation into aggregates, IM, and samples without fractionation (total fractions) from 30 or 42 °C were analyzed on 12% SDS-PAGE and immunoblotted with FLAG antibody. Shown are Western blots of samples from LpxM-FLAG derivatives (A) and Western blots from samples with FLAG tag on different glycosyltransferases (B). B (lanes 9 and 10), samples from aggregate fractions obtained from a strain with the Δ(lapA lapB) mutation in WaaC-3×FLAG background carrying plasmid-borne slrA RNA and subjected to immunoblotting with FLAG antibody. The relevant genotype, temperature, and correspondence to samples without fractionation (total) and after fractionation are marked in each case. C, cultures of the wild type with LptD-3×FLAG and its Δ(lapA lapB) derivative were grown in M9 medium at 30 °C up to an A600 of 0.2. A portion of the culture was shifted for 90 min to 42 °C and fractionated to obtain OM fractions. Proteins were resolved on 12% SDS-PAGE and subjected to immunoblotting with FLAG antibody. Lanes 1–4, samples without fractionation, representing the total amount of LptD. Lanes 5–9, samples from OM fractions. In lane 9, a 5× volume of sample is applied from the OM fraction obtained from Δ(lapA lapB) mutant after the shift to 42 °C.
Examination of WaaC and WaaO amounts revealed that their overall synthesis is nearly similar in the wild-type and its Δ(lapA lapB) derivative (Fig. 10B). However, WaaC and WaaO were found to be aggregation-prone in Δ(lapA lapB) derivatives upon a shift to non-permissive growth conditions (Fig. 11B). Interestingly, the aggregation of WaaC at non-permissive growth conditions was dampened upon overexpression of the slrA RNA (Fig. 11B). These aggregation defects and a proposed scaffold-like function for LapB for LPS-specific enzymes seem to correlate with a specific requirement for individual glycosyltransferases in the cell envelope integrity.
Reduction in LptD Amounts in the OM in Δ(lapA lapB) Mutants at High Temperature
Because LapA and LapB co-purify with Lpt proteins, we expect a synergistic effect of Δ(lapA lapB) when the Lpt system is compromised. Folding of LptD requires the SurA periplasmic folding catalyst, and partial loss of function alleles of the lptD gene exhibit permeability defects (54, 55). Thus, Δ(lapA lapB) was introduced in the wild-type strain and its derivative carrying lptD mutation imp4213 in the presence or absence of a plasmid carrying wild-type lapA lapB genes under permissive growth conditions. Interestingly, no viable transductants were obtained in imp4213 background unless wild-type lapA lapB genes were present (Table 1). Similarly, Δ(lapA lapB surA) multiple deletion derivatives could be constructed only when either SurA or LapB was provided in trans from a plasmid (Table 1). These results show a requirement for functional LptD and SurA in Δ(lapA lapB) mutant. To further support such a role, we examined the amounts of LptD-FLAG in the wild type and its Δ(lapA lapB) derivative. Immunoblotting with FLAG antibody revealed that the total amount of LptD was nearly similar in the extracts from Δ(lapA lapB) mutant and its parental wild type. However, a 4–5-fold reduction in LptD levels was observed in the OM fractions at non-permissive temperature in Δ(lapA lapB) mutant (Fig. 11C). However, this reduction could be a consequence of the requirement of proper LPS for the folding/insertion of LptD in the OM and needs more investigation. However, the overexpression of the lptD gene alone was not found to suppress growth defects of a Δ(lapA lapB) mutant (data not shown). The lack of suppression by lptD overexpression could be due to a simultaneous requirement of other partners of LptD.
Constitutive Induction of Envelope Stress-responsive Pathways
Because the initial screen that identified lapA and lapB genes was based on the elevated envelope stress response, such results were quantified. Examination of the RpoE-dependent promoter activity in Δ(lapA lapB) derivatives revealed an ∼9-fold induction of rpoEP2-lacZ and a nearly 4-fold increase of rpoHP3-lacZ even under permissive growth conditions at 30 °C (Fig. 12A). The Cpx-dependent cpxP promoter activity was also elevated by ∼3-fold (Fig. 12A). These results were further validated by the examination of DegP levels, which are jointly controlled by the RpoE and Cpx pathways (27, 28). Confirming these results, the amount of DegP was found to be elevated in Δ(lapA lapB) derivative at the permissive temperature and was further increased upon shift to higher temperature (Fig. 12B). Thus, we can conclude that Δ(lapA lapB) or ΔlapB mutants exhibit constitutive high expression of major envelope stress regulons (RpoE and Cpx) consistent with their initial identification.
FIGURE 12.
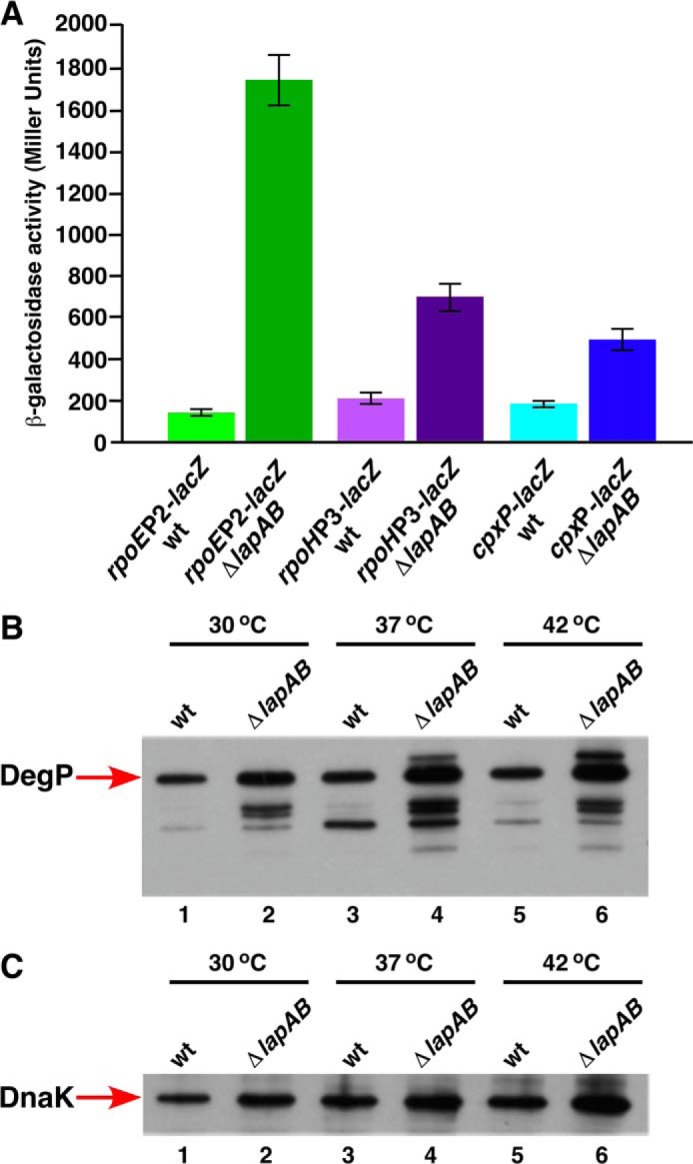
Induction of stress response pathways in Δ(lapA lapB) derivatives. Isogenic cultures of the wild type and its Δ(lapA lapB) derivative, carrying a specific single-copy chromosomal promoter fusion, were grown to early log phase at 30 °C in M9 medium and adjusted to an A600 of 0.02 and allowed to grow further. Samples were analyzed for β-galactosidase activity after different intervals. Data are shown after a 90-min incubation (A). B and C, cultures of the wild type and its Δ(lapA lapB) derivative were grown in M9 medium up to A600 of 0.2 at 30 °C. Aliquots were shifted to 37 and 42 °C and allowed to grow for 30 min in prewarmed flasks. Cultures were harvested by centrifugation, and an equivalent amount of total protein was applied on 12% SDS-PAGE. The relative abundance of DegP (B) and DnaK (C) was revealed by immunoblotting using DegP and DnaK antibodies, respectively. The relevant genotype and temperature are indicated.
Constitutive Induction of the RpoH-dependent Heat Shock Response
RpoH is known to sense misfolding of cytoplasmic and IM proteins (37). Until now, defects in LPS have not been documented to impact RpoH activity, although LpxC and WaaA are subjected to proteolysis by the RpoH-regulated FtsH protease (7, 8, 56). Thus, relative amounts of highly conserved heat shock protein DnaK were analyzed. Levels of DnaK were found to be elevated in the Δ(lapA lapB) mutant even in permissive growth conditions (Fig. 12C). Amounts of DnaK were further elevated upon shift to higher temperatures. If LapB functions as a scaffold for the LPS biosynthetic enzymes, such enzymes might not be correctly located at the site of LPS synthesis in the cytoplasm or in the IM. This in turn could trigger the RpoH-dependent heat shock induction.
LapA and LapB Are Highly Conserved
Modeling of LapB and LapA was performed with the Phyre server. LapB was predicted to share structural similarity to the TPR-containing N-terminal domain of eukaryotic O-linked GlcNAc transferases, eukaryotic APC/C subunit Cdc/Cut9, MamA protein required for magnetosome assembly, and several other TPR-containing proteins (Fig. 13). The addition of GlcNAc is a ubiquitous form of intracellular glycosylation catalyzed by the conserved O-linked GlcNAc transferase in eukaryotes. TPR repeats of the O-linked GlcNAc transferase in the N terminus are required for the recognition of a broad range of target proteins (57). However, such post-translational modification is not known to occur in E. coli K12. The presence of TPR repeats suggested that LapB could be involved in mediating protein-protein interactions. The C-terminal domain of LapB contains a rubredoxin-like domain (RYCQKCGXXWHCPSCXP). This motif is also highly conserved among LapB homologs. Error-prone mutagenesis was used to identify some residues critical for the LapB function. This identified several residues, including some mapping to the rubredoxin-like domain (Fig. 13), indicating its essential role for the LapB function. Thus, Cys-371 → Arg, Ser-378 → Pro, and Pro-372 → Leu mutations rendered LapB non-functional (Table 1). Further, Cys-371 → Arg conferred a dominant negative phenotype, suggesting that LapB could be a multimeric protein. The C-terminal domain (amino acids 61–89) of LapA is predicted to have a structural fold resembling connector region LβH in the C-terminal domain of LpxD (42). This connector loop is located in proximity to the active site residues in the crystal structure of LpxD (42). In almost all cases, lapA and lapB genes seem to be organized in a presumed operon and are present in all members of Enterobacteriaceae. However, a LapB-like protein was not found in the genome of Pseudomonas, where LpxC is not a substrate for FtsH and slrA sRNA is also absent. The control by slrA RNA in response to elevated LpxC could be specific because this sRNA presence is correlated with LpxC stability and was not found to be present in bacterial species when LpxC stability is independent of FtsH. Consistent with our overall results, LapB seems to act like other TPR-containing proteins and could thus serve as a docking site in the IM for LPS assembly.
FIGURE 13.
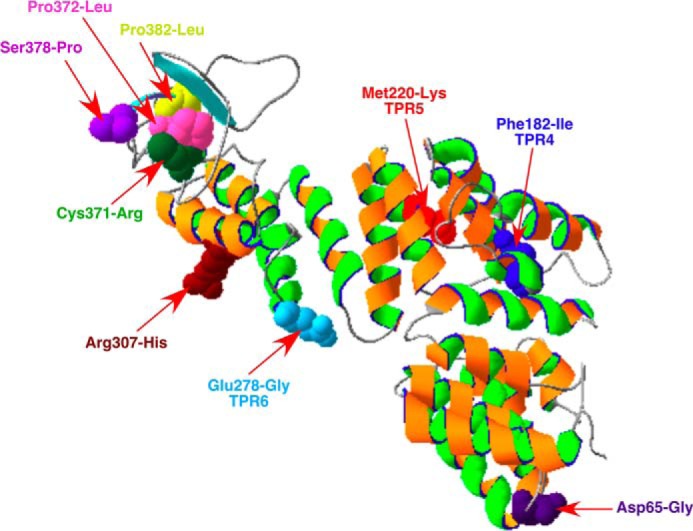
Modeling of LapB from E. coli. The primary amino acid sequence was analyzed for the best fitting model on the Phyre 2 server. Mutations identified in this work are indicated.
DISCUSSION
In this work, we characterized two proteins of previously unknown function. These new proteins were named LapA and LapB based on their contribution to the LPS assembly. Disruption of the lapB gene caused a constitutive high expression of RpoE and Cpx envelope stress-responsive pathways and severe defects in LPS. The lapB gene was found to be essential under normal laboratory growth conditions. Suppressor-free ΔlapB mutants could be obtained only when cells were grown on minimal medium at 30 °C in some but not in all genetic backgrounds. These phenotypes are reminiscent of phenotypes observed in strains with minimal LPS composed of either Kdo2 + lipid IVA or, in the case of ΔwaaA mutants, synthesizing glycosylation-free LPS (6). The lapB gene is co-transcribed with the lapA gene, and their transcription is directed from three promoters, one of which (P2) is heat shock-regulated. A non-polar deletion in the lapA gene confers mild permeability defects and temperature sensitivity when grown on bile salt-containing medium. To address the molecular basis of essentiality of LapB and causes of envelope stress response, we analyzed the LPS composition of various mutants, isolated various multicopy and extragenic suppressors, and purified LapA and LapB proteins.
Based on the biochemical analyses of Lap proteins, properties of LPS from Δ(lapA lapB) mutant, and genetic analyses of various suppressors, our results suggest a pivotal role for LapB protein in the coupling of LPS synthesis and translocation. LapB protein seems to act in the pathway of lipid A assembly by regulating amounts of LpxC and in either the delivery or maintenance of the folding competence of early glycosyltransferases. Such a key role is consistent with the essentiality of LapB and with the presence of modules in LapB like TPR, which could mediate protein-protein interaction and also serve as a scaffold for LPS and LPS-specific enzymes.
Purification of LapA and LapB provided important clues concerning their function in the interaction with Lpt proteins that mediate LPS translocation. When used in pull-down experiments, LapA and LapB co-purify with LPS, Lpt proteins, and FtsH protease. LptE and LptD together with LptBFGC and LptA proteins constitute a large transenvelope LPS transport complex (10, 11). LptE is an OM lipoprotein and forms a complex with LptD in the OM. LptD and LptE are required in the final steps of LPS insertion at the OM (58, 59). LptB is presumed to provide energy to drive LPS translocation for delivery to LptC and LptA (60). Indeed, in the pull-down experiments using the co-overexpression system of LapB with LptC-expressing plasmid, we were able to purify a complex consisting of LapB and Lpt proteins. Co-purification of LapA and LapB with LPS supports these conclusions. However, this co-purification with LPS can also arise due to co-purification of Lap proteins with Lpt proteins. It has been shown that LptA, LptC, and LptE bind LPS (13, 14, 58). However, LapA samples that were tested for LPS presence do not contain detectable amounts of Lpt proteins.
Consistent with LPS defects and interaction of LapA and LapB with Lpt proteins, a partially functional lptD4213 allele causes lethality in the Δ(lapA lapB) background. Further, the amounts of LptD in the OM were found to be 4–5-fold diminished in a Δ(lapA lapB) mutant at non-permissive growth conditions. However, this decrease can also arise due to the known requirement of LPS for folding of OM proteins (17) and needs more investigation. Overall, these results can explain synthetic lethality of Δ(surA lapA lapB). SurA is known to be required for the folding of LptD (55) and can explain previous report of Δ(surA lapB) lethality (61).
Co-purification of LapB and WaaC suggested that LapB protein could couple LPS synthesis with LPS translocation. However, it seems that LapA and LapB might function together with some chaperones in this pathway. Support for such a conclusion is based on co-purification of LapA and LapB with DnaK and DnaJ and partial suppression of growth and LPS defects upon the overexpression of these chaperones. There seems to be a parallel in the function of LapB with eukaryotic TPR-containing protein at the organellar surfaces serving as docking proteins for chaperone-bound preproteins (62). Thus, LapB may act in a similar manner in the pathway of folding with common substrates with Hsp40 (DnaJ) and Hsp70 (DnaK).
Δ(lapA lapB) mutant was found to accumulate high amounts of LpxC. LpxC catalyzes the first committed step in the lipid A synthesis. LpxC is an unstable protein whose turnover is regulated by FtsH. LpxC together with FabZ (3-R-hydroxymyristoyl acyl carrier protein dehydratase) are known to be key factors in the balanced synthesis of lipid A and phospholipids (7, 8). That the amounts of LpxC are elevated to the same extent in the Δ(lapA lapB) mutant irrespective of the presence or the absence of FtsH argues that LapB is required in vivo for the FtsH-mediated proteolysis of LpxC. This elevated level of LpxC could be one of the reasons for the essentiality of LapB. In agreement with this notion, we also observed that FtsH and LapB co-purify, and suppressor mutations, which restore growth of ΔlapB mutants, mapped to either the lpxC gene or genes, which reduce LpxC accumulation or LPS biosynthesis. Two suppressor mutations mapping to the lpxC gene dampened the elevated levels of LpxC. Interestingly, the sfhC21 suppressor mutation of a defined ftsH mutation also suppresses the growth defect of Δ(lapA lapB) mutants. Similarly, overexpression of a new non-coding sRNA, slrA, identified in this work as a multicopy suppressor, also caused a modest decrease in the accumulation of LpxC. This in turn could restore a balance between LPS and phospholipids arising due to an increase in LpxC amounts, consistent with previous reports of suppressors of ftsH mutations mapping to the fabZ gene (7). The biosynthesis of phospholipids and lipid A use the same precursor R-3-hydroxymyristoyl-ACP (7, 43). However, the LPS defects that are observed in Δ(lapA lapB) or ΔlapB mutants cannot be solely due to increased LpxC. It has been reported that ftsH mutants, accumulating a similar high amount of LpxC, have a normal LPS core chain length (7). There also seems to be a built-in mechanism that helps to maintain the balance in the levels of LapAB-dependent LPS and phospholipid synthesis, because one of the promoters of lapAB genes is located upstream of the pgpB gene. Consistent with imbalanced synthesis of LPS and other envelope components, overexpression of the murA gene product allowed Δ(lapA lapB) or ΔlapB mutants to grow under non-permissive growth conditions. MurA and LpxA use the common UDP-GlcNAc as a substrate. This explains the isolation of the murA gene as a multicopy suppressor. Overexpression of MurA could shift the balance toward peptidoglycan biosynthesis and prevent toxic buildup of LPS.
Mass spectrometric analyses of LPS from Δ(lapA lapB) or ΔlapB mutants revealed that their LPS is composed of a mixture of complete LPS and its precursor forms. A range of species corresponding to incomplete core with the lipid A part comprised of a mixture of penta- and hexaacylated derivatives were observed. Because the LPS of the ΔlapA mutant was nearly normal, it seems that in this process, the main LPS defect is due to a specific requirement of LapB. The accumulation of LPS intermediates implies that various glycosyltransferases are either limiting in the absence of LapB or LapB is needed to recruit such glycosyltransferases to sites of LPS assembly. This is consistent with the observed co-purification of WaaC with LapB. In line with these arguments, some of the key early glycosyltransferases were observed to be aggregation-prone at non-permissive growth conditions.
LPS from Δ(lapA lapB) or ΔlapB mutants accumulate significant amounts of lipid A species, which are either pentaacylated or species with an additional secondary lauroyl chain replacing the myristoyl chain. This can be explained by enhanced proteolysis and reduction in the overall amount of LpxM in the Δ(lapA lapB) background. Another explanation can be that LpxM is not delivered to the site of LPS assembly, where LapB could function as a scaffold for LPS assembly. The accumulation of pentaacylated lipid A and lipid A with two lauroyl chains can also be due to reduction in the availability of acyl-ACP substrates when LpxC levels are elevated and a concomitant reduction in FabZ activity. FabZ and LpxC activity are known to be co-regulated (43). Mutants with reduced FabZ activity also accumulate pentaacylated lipid A and lipid A derivatives with two lauroyl chains (43). Thus, a combination of a reduced amount of functional LpxM in the IM and reduction in the levels of acyl-ACP pools could together contribute to this defect. These results are consistent with co-immunoprecipitation of FabZ with LapB (see below) and suppression of lethality of the ΔlapB mutation in sfhC21 background or when the fabZ gene was overexpressed.
Another glaring defect observed in the LPS of Δ(lapA lapB) derivatives is the carbon chain length polymorphism of lipid A. This was most obvious in the LPS of Δ(lapA lapB) derivative carrying the WaaC T187K mutation. This waaC variant or a deletion of the waaC gene conferred a limited suppression onto Δ(lapA lapB) when grown on rich medium below 37 °C. The carbon chain polymorphism may be due to the changes in the composition of fatty acid odd chain versus even chain in Δ(lapA lapB) mutants. Such a polymorphism has also been reported in the lipid A of lpxD mutants (42) or when E. coli is grown in the presence of 1% propionic acid (47). Similar to the lipid A of Δ(lapA lapB) derivatives, lpxD mutant generated multiple lipid A species containing one or two longer hydroxy fatty acids in place of the usual R-3-hydroxymyristate at positions 2 and 2′. Consistent with these results, a partial suppression was also observed when a known variant lpxD36 was introduced in the Δ(lapA lapB) background. LpxD catalyzes the third step of lipid A biosynthesis, the R-3-hydroxyacyl-ACP-dependent N-acylation of UDP-3-O-(acyl)-R-d-glucosamine (42). Taken together, LapB seems to have a function related to maintaining a balance in various enzymes involved in the biosynthesis of LPS core and lipid A.
In a comprehensive approach to understanding the essentiality of LapB and its functional partners, multicopy and extragenic suppressors were isolated and characterized. The multicopy suppressor approach identified enzymes involved in the phospholipid and fatty acid biosynthesis (fabZ and fabB), peptidoglycan synthesis (murA), and envelope stress response regulators (rcsF, a new sRNA designated slrA, and toxin/antitoxin system hicA). The fabB gene encodes β-ketoacyl-ACP synthase I (63). FabB overexpression may suppress growth defects by maintaining a balance in saturated versus unsaturated fatty acids.
As mentioned above, balanced synthesis of phospholipids and LPS is crucial to bacterial viability. The gene cluster comprising lpxD, fabZ, lpxA, and lpxB is co-transcribed from RpoE and Cpx promoters (28). ΔlapB mutants have up-regulated RpoE and Cpx activity, and this could cause an increase in the synthesis of enzymes like FabZ. Thus, systems that will modulate RpoE and/or Cpx pathways and restore a balance in phospholipids and lipid A should suppress to some extent a ΔlapB mutation. The isolation of fabZ, non-coding sRNA slrA, and hicA support this model of suppression. Overexpression of the slrA sRNA dampens RpoE-dependent envelope stress response and reduces LpxC accumulation. This resetting of balanced cell envelope biogenesis, upon mild overexpression of the slrA sRNA, also caused a reduction in the WaaC aggregation. We also found that its overexpression causes reduction in the amounts of the most abundant lipoprotein Lpp. Interestingly, a null mutation in the lpp gene was isolated as an extragenic suppressor of growth defect of Δ(lapA lapB) mutants. A similar explanation can be provided to account for the multicopy suppressor effect of the hicA gene. It has been suggested that the HicA toxin acts by enhanced degradation of target mRNAs, which include RpoE and CpxR/A regulators (64). Characterization of Δ(lapA lapB lpp) mutants revealed that such bacteria can grow up to 40 °C. The Lpp murein lipoprotein is numerically the most abundant protein in E. coli and carries three fatty acyl chains (49, 50). Thus, the absence of Lpp or reduction of its amounts can lead to the increased availability of fatty acids and restore a balance between phospholipids and LPS in Δ(lapA lapB) mutants. This can provide a rational explanation for the isolation of suppressor mutation as a loss of function mutation in the lpp gene and multicopy suppression by overexpression of the slrA sRNA.
Additional multicopy suppressors identified four genes of unknown function: yceK, yeaD, yffH, and ydhA. Most of these genes have a predicted function in the envelope biogenesis. Indeed, analysis of LPS by mass spectrometry revealed that defects in LPS composition were exacerbated in the Δ(lapA lapB yceK) mutant. Thus, taken together, it seems that YceK is a genuine protein with a role in LPS translocation or assembly.
In addition to the suppressors in the lpxC gene, additional extragenic suppressors were mapped to genes involved either in the heptose biosynthesis (gmhA) or heptosyltransferases (waaC and waaQ) or glycosyltransferase I (waaG). Examination of LPS of the ΔlapB waaCT187K derivative revealed that it accumulated a lower amount of LPS as compared with a ΔwaaC mutant. This could be due to defects in the translocation of LPS present in ΔlapB waaCT187K derivative because it exhibited altered carbon chain polymorphism. Another explanation could be a partial reduction in the elevated amount of LpxC in a ΔlapB waaCT187K mutant. A similar explanation could be provided for suppressor mutation in the waaQ operon. A Δ(lapA lapB) waaQ::Tn10 derivative also accumulated a slightly lower amount of LpxC as compared with the parental strain carrying the Δ(lapA lapB) mutation. Thus, reduction in the LpxC and LPS amount could be a part of the feedback mechanism and could thus contribute to restoration of the balance between the amount of LPS and phospholipids. Further detailed studies are required to fully understand the mode of such a suppression mechanism.
During the revision process of this manuscript, three groups described characterization of the lapB gene (yciM) in E. coli and its homolog in Neisseria meningitidis (65–67). In one study, the lapB (yciM) gene was identified by serendipity, whereby the lapB deletion strain in the ASKA library carried a suppressor mutation in the lpxC gene (65). The lapB gene was found to be essential at 37 °C, and depletion of LapB was shown to cause accumulation of LpxC and increase in LPS amounts, which supports our results. In another study, the lapB mutant was identified in a screen of an E. coli genomic knock-out library (ASKA library), on the basis of its sensitivity to various antibiotics (66). The sensitivity of lapB (yciM) mutants toward antibiotics had already been shown in an earlier study using the same deletion library (68). In N. meningitidis, a mutation in the homologous gene ght was found to cause reduction in LPS amounts, and a suppressor mutation was found to synthesize more LpxC (67). These authors suggested that LpxC could be tethered to the IM by Ght (67).
Model of LapA and LapB Function and the Essentiality of the lapB Gene
In summary, we show that the essential function of LapB is to maintain balanced amounts of LPS biosynthetic enzymes, such as LpxC. In the absence of LapB, the elevated accumulation of LpxC would perturb the balance between phospholipids and LPS, leading to toxicity. Thus, defects in LpxC proteolysis support an acute requirement for LapB for maintaining cellular homeostasis of these important enzymes. This model (Fig. 14) is supported by restoration of growth by the overexpression of either the fabZ gene product or in the presence of the sfhC21 mutation. Indeed in our preliminary immunoprecipitation experiments, LapB was found to co-immunoprecipitate with FabZ. Another reason for the essentiality of the lapB gene could be a specific requirement for LapB-mediated coupling of LPS synthesis with its translocation, as revealed by the co-purification of WaaC with LapB and LapB with Lpt proteins (Figs. 6 and 14). However, in this process, an interaction with MsbA was not observed, implying that MsbA could translocate LPS without a specific requirement for either LapA or LapB. This is consistent with known properties of MsbA and the lack of its co-purification with either Lpt proteins or LapA or LapB. Another reason for the essentiality of the lapB gene could be toxic buildup of elevated RpoE, Cpx, and RpoH stress-responsive pathways. This is consistent with the isolation of some of the extragenic suppressors or multicopy suppressors that either dampen the elevated envelope stress response or cause a reduction in LPS biosynthesis.
FIGURE 14.

Proposed model of LapA and LapB function in the assembly of LPS. LapB functions at key steps, including the presentation of LpxC to FtsH protease, serving as a docking site for the LPS assembly by various IM-associated or IM-anchored enzymes, ensuring that only the completely synthesized LPS is translocated. In this process, LapB could couple LPS synthesis with its translocation. This mature LPS would be flipped by MsbA, and in subsequent steps LapA and LapB could function together with transenvelope Lpt complex components because such proteins were found to co-purify. In the absence of LapB, LpxC accumulates, causing toxicity due to the imbalance between LPS and phospholipids. Overexpression of the fabZ gene product can restore this balance between phospholipids and LPS.
Thus, in addition to regulating LpxC turnover, LapB could act as a docking site for various LPS biosynthetic enzymes, such as glycosyltransferases, accounting for the extreme heterogeneity of LPS that was observed in the absence of LapB. Finally, LapA and LapB appear to interact with proteins involved in the LPS translocation process. In this role, LapA and LapB could deliver LPS to the Lpt complex in the IM, a function that is supported by observations demonstrating that the Lap proteins interact with both LPS and Lpt factors (Fig. 14). The presence of TPR repeats in LapB is consistent with the notion that this protein functions as an organizing complex at the IM for the assembly of LPS, mediates delivery of various glycosyltransferases, and assists in the rapid turnover of LpxC by FtsH.
Supplementary Material
Acknowledgments
We gratefully acknowledge the generous help of U. Mamat, O. Holst, H. Brade, and their laboratory members (Research Center Borstel). We also thank F. Narberhaus for the kind gift of LpxC antibodies, A. Rokney for the gift of the ftsH mutant, S. Narita for the lptBGF plasmid, and D. Missiakas and the anonymous reviewer for helpful suggestions. Inputs of several members of the laboratory of S. R., particularly A. Kur and P. Gorzelak, at different stages of this work are acknowledged.
This work was supported by National Science Grant 2011/03/B/NZ1/02825 (to S. R.).

This article contains supplemental Tables S1 and S2.
- OM
- outer membrane
- IM
- inner membrane
- Kdo
- 3-deoxy-α-d-manno-oct-2-ulosonic acid
- Hep
- l-glycero-d-manno-heptose
- Hex
- hexose
- TPR
- tetratricopeptide repeat
- P-EtN
- phosphoethanolamine
- Ara4N
- 4-amino-4-deoxy-l-arabinose
- ESI
- electrospray ionization
- FT-ICR
- Fourier transform-ion cyclotron
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- nt
- nucleotide(s).
REFERENCES
- 1. Raetz C. R., Whitfield C. (2002) Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gronow S., Brade H. (2001) Lipopolysaccharide biosynthesis: which steps do bacteria need to survive? J. Endotoxin Res. 7, 3–23 [PubMed] [Google Scholar]
- 3. Frirdich E., Whitfield C. (2005) Lipopolysaccharide inner core oligosaccharide structure and outer membrane stability in human pathogens belonging to the Enterobacteriaceae. J. Endotoxin Res. 11, 133–144 [DOI] [PubMed] [Google Scholar]
- 4. Klein G., Lindner B., Brade H., Raina S. (2011) Molecular basis of lipopolysaccharide heterogeneity in Escherichia coli: envelope stress-responsive regulators control the incorporation of glycoforms with a third 3-deoxy-α-d-manno-oct-2-ulosonic acid and rhamnose. J. Biol. Chem. 286, 42787–42807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Clementz T., Raetz C. R. (1991) A gene coding for 3-deoxy-d-manno-octulosonic-acid transferase in Escherichia coli: identification, mapping, cloning, and sequencing. J. Biol. Chem. 266, 9687–9696 [PubMed] [Google Scholar]
- 6. Klein G., Lindner B., Brabetz W., Brade H., Raina S. (2009) Escherichia coli K-12 suppressor-free mutants lacking early glycosyltransferases and late acyltransferases: minimal lipopolysaccharide structure and induction of envelope stress response. J. Biol. Chem. 284, 15369–15389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ogura T., Inoue K., Tatsuta T., Suzaki T., Karata K., Young K., Su L. H., Fierke C. A., Jackman J. E., Raetz C. R., Coleman J., Tomoyasu T., Matsuzawa H. (1999) Balanced biosynthesis of major membrane components through regulated degradation of the committed enzyme of lipid A biosynthesis by the AAA protease FtsH (HflB) in Escherichia coli. Mol. Microbiol. 31, 833–844 [DOI] [PubMed] [Google Scholar]
- 8. Führer F., Langklotz S., Narberhaus F. (2006) The C-terminal end of LpxC is required for degradation by the FtsH protease. Mol. Microbiol. 59, 1025–1036 [DOI] [PubMed] [Google Scholar]
- 9. Doerrler W. T., Raetz C. R. (2002) ATPase activity of the MsbA lipid flippase of Escherichia coli. J. Biol. Chem. 277, 36697–36705 [DOI] [PubMed] [Google Scholar]
- 10. Ruiz N., Kahne D., Silhavy T. J. (2009) Transport of lipopolysaccharide across the cell envelope: the long road of discovery. Nat. Rev. Microbiol. 7, 677–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Narita S., Tokuda H. (2009) Biochemical characterization of an ABC transporter LptBFGC complex required for the outer membrane sorting of lipopolysaccharide. FEBS Lett. 583, 2160–2164 [DOI] [PubMed] [Google Scholar]
- 12. Sperandeo P., Lau F. K., Carpentieri A., De Castro C., Molinaro A., Dehò G., Silhavy T. J., Polissi A. (2008) Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli. J. Bacteriol. 190, 4460–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ma B., Reynolds C. M., Raetz C. R. (2008) Periplasmic orientation of nascent lipid A in the inner membrane of an Escherichia coli LptA mutant. Proc. Natl. Acad. Sci. U.S.A. 105, 13823–13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tran A. X., Trent M. S., Whitfield C. (2008) The LptA protein of Escherichia coli is a periplasmic lipid A-binding protein involved in the lipopolysaccharide export pathway. J. Biol. Chem. 283, 20342–20349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vorachek-Warren M. K., Ramirez S., Cotter R. J., Raetz C. R. (2002) A triple mutant of Escherichia coli lacking secondary acyl chains on lipid A. J. Biol. Chem. 277, 14194–14205 [DOI] [PubMed] [Google Scholar]
- 16. Tran A. X., Dong C., Whitfield C. (2010) Structure and functional analysis of LptC, a conserved membrane protein involved in the lipopolysaccharide export pathway in Escherichia coli. J. Biol. Chem. 285, 33529–33539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Missiakas D., Betton J.-M., Raina S. (1996) New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol. Microbiol. 21, 871–884 [DOI] [PubMed] [Google Scholar]
- 18. Torriani A. (1960) Influence of inorganic phosphate in the formation of phosphatases by Escherichia coli. Biochim. Biophys. Acta 38, 460–469 [DOI] [PubMed] [Google Scholar]
- 19. Datsenko K. A., Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klein G., Müller-Loennies S., Lindner B., Kobylak N., Brade H., Raina S. (2013) Molecular and structural basis of inner core lipopolysaccharide alterations in Escherichia coli: incorporation of glucuronic acid and phosphoethanolamine in the heptose region. J. Biol. Chem. 288, 8111–8127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lerner C. G., Inouye M. (1990) Low copy plasmids for regulated low-level expression of cloned genes in Escherichia coli with blue/white screening capability. Nucleic Acids Res. 18, 4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kang P. J., Craig E. A. (1990) Identification and characterization of a new Escherichia coli gene that is a dosage-dependent suppressor of a dnaK deletion mutation. J. Bacteriol. 172, 2055–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rokney A., Kobiler O., Amir A., Court D. L., Stavans J., Adhya S., Oppenheim A. B. (2008) Host responses influence on the induction of λ prophage. Mol. Microbiol. 68, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Missiakas D., Mayer M. P., Lemaire M., Georgopoulos C., Raina S. (1997) Modulation of the Escherichia coli σE (RpoE) heat-shock transcription factor activity by the RseA, RseB and RseC proteins. Mol. Microbiol. 24, 355–371 [DOI] [PubMed] [Google Scholar]
- 25. Wang R. F., Kushner S. R. (1991) Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100, 195–199 [PubMed] [Google Scholar]
- 26. Kitagawa M., Ara T., Arifuzzaman M., Ioka-Nakamichi T., Inamoto E., Toyonaga H., Mori H. (2005) Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12, 291–299 [DOI] [PubMed] [Google Scholar]
- 27. Raina S., Missiakas D., Georgopoulos C. (1995) The rpoE gene encoding the σE (σ24) heat shock sigma factor of Escherichia coli. EMBO J. 14, 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dartigalongue C., Missiakas D., Raina S. (2001) Characterization of the Escherichia coli σE regulon. J. Biol. Chem. 276, 20866–20875 [DOI] [PubMed] [Google Scholar]
- 29. Simons R. W., Houman F., Kleckner N. (1987) Improved single and multicopy lac-based cloning vectors for protein and operon fusion. Gene 53, 85–96 [DOI] [PubMed] [Google Scholar]
- 30. Uzzau S., Figueroa-Bossi N., Rubino S., Bossi L. (2001) Epitope tagging of chromosomal genes in Salmonella. Proc. Natl. Acad. Sci. U.S.A. 98, 15264–15269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Missiakas D., Georgopoulos C., Raina S. (1993) Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc. Natl. Acad. Sci. U.S.A. 90, 7084–7088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tomoyasu T., Mogk A., Langen H., Goloubinoff P., Bukau B. (2001) Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol. Microbiol. 40, 397–413 [DOI] [PubMed] [Google Scholar]
- 33. Galanos C., Lüderitz O., Westphal O. (1969) A new method for the extraction of R lipopolysaccharides. Eur. J. Biochem. 9, 245–249 [DOI] [PubMed] [Google Scholar]
- 34. Dartigalongue C., Raina S. (1998) A new heat-shock gene, ppiD, encodes a peptidyl-prolyl isomerase required for folding of outer membrane proteins in Escherichia coli. EMBO J. 17, 3968–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murata M., Fujimoto H., Nishimura K., Charoensuk K., Nagamitsu H., Raina S., Kosaka T., Oshima T., Ogasawara N., Yamada M. (2011) Molecular strategy for survival at a critical high temperature in Escherichia coli. PLoS One 6, e20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao K., Liu M., Burgess R. R. (2005) The global transcriptional response of Escherichia coli to induced σ32 protein involves σ32 regulon activation followed by inactivation and degradation of σ32 in vivo. J. Biol. Chem. 280, 17758–17768 [DOI] [PubMed] [Google Scholar]
- 37. Nonaka G., Blankschien M., Herman C., Gross C. A., Rhodius V. A. (2006) Regulon and promoter analysis of the Escherichia coli heat-shock factor, σ32, reveals a multifaceted cellular response to heat stress. Genes Dev. 20, 1776–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Keilty S., Rosenberg M. (1987) Constitutive function of a positively regulated promoter reveals new sequences essential for activity. J. Biol. Chem. 262, 6389–6395 [PubMed] [Google Scholar]
- 39. Gardella T., Moyle H., Susskind M. M. (1989) A mutant Escherichia coli σ70 subunit of RNA polymerase with altered promoter specificity. J. Mol. Biol. 206, 579–590 [DOI] [PubMed] [Google Scholar]
- 40. Dillon D. A., Wu W. I., Riedel B., Wissing J. B., Dowhan W., Carman G. M. (1996) The Escherichia coli pgpB gene encodes for a diacylglycerol pyrophosphate phosphatase activity. J. Biol. Chem. 271, 30548–30553 [DOI] [PubMed] [Google Scholar]
- 41. Schnaitman C. A., Klena J. D. (1993) Genetics of lipopolysaccharide biosynthesis in enteric bacteria. Microbiol. Rev. 57, 655–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bartling C. M., Raetz C. R. (2009) Crystal structure and acyl chain selectivity of Escherichia coli LpxD, the N-acyltransferase of lipid A biosynthesis. Biochemistry 48, 8672–8683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zeng D., Zhao J., Chung H. S., Guan Z., Raetz C. R., Zhou P. (2013) Mutants resistant to LpxC inhibitors by rebalancing cellular homeostasis. J. Biol. Chem. 288, 5475–5486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kadrmas J. L., Raetz C. R. (1998) Enzymatic synthesis of lipopolysaccharide in Escherichia coli: purification and properties of heptosyltransferase I. J. Biol. Chem. 273, 2799–2807 [DOI] [PubMed] [Google Scholar]
- 45. Beall B., Lutkenhaus J. (1987) Sequence analysis, transcriptional organization, and insertional mutagenesis of the envA gene of Escherichia coli. J. Bacteriol. 169, 5408–5415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grizot S., Salem M., Vongsouthi V., Durand L., Moreau F., Dohi H., Vincent S., Escaich S., Ducruix A. (2006) Structure of the Escherichia coli heptosyltransferase WaaC: binary complexes with ADP and ADP-2-deoxy-2-fluoro heptose. J. Mol. Biol. 363, 383–394 [DOI] [PubMed] [Google Scholar]
- 47. Bainbridge B. W., Karimi-Naser L., Reife R., Blethen F., Ernst R. K., Darveau R. P. (2008) Acyl chain specificity of the acyltransferases LpxA and LpxD and substrate availability contribute to lipid A fatty acid heterogeneity in Porphyromonas gingivalis. J. Bacteriol. 190, 4549–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mamat U., Schmidt H., Munoz E., Lindner B., Fukase K., Hanuszkiewicz A., Wu J., Meredith T. C., Woodard R. W., Hilgenfeld R., Mesters J. R., Holst O. (2009) WaaA of the hyperthermophilic bacterium Aquifex aelicus is a monofunctional 3-deoxy-d-manno-oct-2-ulosonic acid transferase invplved in lipopolysaccharide biosynthesis. J. Biol. Chem. 284, 22248–22262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hantke K., Braun V. (1973) Covalent binding of lipid to protein. Digliceride and amide-linked fatty acid at the N-terminal end of the murein-lipoprotein of the Escherichia coli outer membrane. Eur. J. Biochem. 34, 284–296 [DOI] [PubMed] [Google Scholar]
- 50. Nikaido H. (1996) Outer membrane. in Escherichia coli and Salmonella: Cellular and Molecular Biology (Neidhardt F. C., ed) pp. 29–47, American Society for Microbiology Press, Washington, D. C [Google Scholar]
- 51. Strauch K. L., Johnson K., Beckwith J. (1989) Characterization of degP, a gene required for proteolysis in the cell envelope and essential for growth of Escherichia coli at high temperature. J. Bacteriol. 171, 2689–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baird L., Lipinska B., Raina S., Georgopoulos C. (1991) Identification of the Escherichia coli sohB gene, a multicopy suppressor of the HtrA (DegP) null phenotype. J. Bacteriol. 173, 5763–5770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Babu M., Díaz-Mejía J. J., Vlasblom J., Gagarinova A., Phanse S., Graham C., Yousif F., Ding H., Xiong X., Nazarians-Armavil A., Alamgir M., Ali M., Pogoutse O., Pe'er A., Arnold R., Michaut M., Parkinson J., Golshani A., Whitfield C., Wodak S. J., Moreno-Hagelsieb G., Greenblatt J. F., Emili A. (2011) Genetic interaction maps in Escherichia coli reveal functional crosstalk among cell envelope biogenesis pathways. PLoS Genet. 7, e1002377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sampson B. A., Misra R., Benson S. A. (1989) Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122, 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schwalm J., Mahoney T. F., Soltes G. R., Silhavy T. J. (2013) Role for Skp in LptD assembly in Escherichia coli. J. Bacteriol. 195, 3734–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Katz C., Ron E. Z. (2008) Dual role of FtsH in regulating lipopolysaccharide biosynthesis in Escherichia coli. J. Bacteriol. 190, 7117–7122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Iyer S. P., Hart G. W. (2003) Roles of the tetratricopeptide repeat domain in O-GlcNAc transferase targeting and protein substrate specificity. J. Biol. Chem. 278, 24608–24616 [DOI] [PubMed] [Google Scholar]
- 58. Freinkman E., Chng S. S., Kahne D. (2011) The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc. Natl. Acad. Sci. U.S.A. 108, 2486–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bos M. P., Tommassen J. (2011) The LptD chaperone LptE is not directly involved in lipopolysaccharide transport in Neisseria meningitidis. J. Biol. Chem. 286, 28688–28696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Okuda S., Freinkman E., Kahne D. (2012) Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli. Science 338, 1214–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Niba E. T., Naka Y., Nagase M., Mori H., Kitakawa M. (2007) A genome-wide approach to identify the genes involved in biofilm formation in E. coli. DNA Res. 14, 237–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schweiger R., Soll J., Jung K., Heermann R., Schwenkert S. (2013) Quantification of interaction strengths between chaperones and tetratricopeptide repeat domain-containing membrane proteins. J. Biol. Chem. 288, 30614–30625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xiao X., Yu X., Khosla C. (2013) Metabolic flux between unsaturated and saturated fatty acids is controlled by the FabA:FabB ratio in the fully reconstituted fatty acid biosynthetic pathway of Escherichia coli. Biochemistry 52, 8304–8312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jørgensen M. G., Pandey D. P., Jaskolska M., Gerdes K. (2009) HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 191, 1191–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mahalakshmi S., Sunayana M. R., SaiSree L., Reddy M. (2014) yciM is an essential gene required for regulation of lipopolysaccharide synthesis in Escherichia coli. Mol. Microbiol. 91, 145–157 [DOI] [PubMed] [Google Scholar]
- 66. Nicolaes V., El Hajjaji H., Davis R. M., Van der Henst C., Depuydt M., Leverrier P., Aertsen A., Haufroid V., Ollagnier de Choudens S., De Bolle X., Ruiz N., Collet J. F. (2014) Insights into the function of YciM, a heat shock membrane protein required to maintain envelope integrity in Escherichia coli. J. Bacteriol. 196, 300–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Putker F., Grutsch A., Tommassen J., Bos M. P. (2014) Ght protein of Neisseria meningitidis is involved in the regulation of lipopolysaccharide biosynthesis. J. Bacteriol. 196, 780–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu A., Tran L., Becket E., Lee K., Chinn L., Park E., Tran K., Miller J. H. (2010) Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating anantibiotic bar code. Antimicrob. Agents Chemother. 54, 1393–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.