

Background: Transient receptor potential channel TRPP2 is a glycoprotein mutated in polycystic kidney disease.
Results: Five N-glycosylation sites in TRPP2 and glucosidase II-mediated glycan trimming determine TRPP2 protein abundance.
Conclusion: N-Glycosylation of TRPP2 in the first extracellular loop is required for efficient protein translation and stability.
Significance: The mechanisms controlling the biogenesis of TRPP2 may provide insights into the pathogenesis of polycystic kidney and polycystic liver disease.
Keywords: Genetic Diseases, Glycosylation, Kidney, Lysosomes, TRP Channels
Abstract
Glycosylation plays a critical role in the biogenesis and function of membrane proteins. Transient receptor potential channel TRPP2 is a nonselective cation channel that is mutated in autosomal dominant polycystic kidney disease. TRPP2 has been shown to be heavily N-glycosylated, but the glycosylation sites and the biological role of N-linked glycosylation have not been investigated. Here we show, using a combination of mass spectrometry and biochemical approaches, that native TRPP2 is glycosylated at five asparagines in the first extracellular loop. Glycosylation is required for the efficient biogenesis of TRPP2 because mutations of the glycosylated asparagines result in strongly decreased protein expression of the ion channel. Wild-type and N-glycosylation-deficient TRPP2 is degraded in lysosomes, as shown by increased TRPP2 protein levels upon chemical inhibition of lysosomal degradation. In addition, using pharmacological and genetic approaches, we demonstrate that glucosidase II (GII) mediates glycan trimming of TRPP2. The non-catalytic β subunit of glucosidase II (GIIβ) is encoded by PRKCSH, one of the genes causing autosomal dominant polycystic liver disease (ADPLD). The impaired GIIβ-dependent glucose trimming of TRPP2 glycosylation in ADPLD may explain the decreased TRPP2 protein expression in Prkcsh−/− mice and the genetic interaction observed between TRPP2 and PRKCSH in ADPLD. These results highlight the biological importance of N-linked glycosylation and GII-mediated glycan trimming in the control of biogenesis and stability of TRPP2.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD)4 is one of the most common potentially fatal monogenetic diseases in man, affecting one in 400 to one in 1000 live births (1). Mutations in Polycystic Kidney Disease 2 (PKD2) cause ADPKD type 2 (OMIM no. 613,095) (2). The PKD2 gene product polycystin 2 is a member of the transient receptor potential (TRP) channel superfamily. It is classified as transient receptor potential channel polycystin 2 (TRPP2), the prototypical member of the TRPP subfamily (3). TRP channels are involved in diverse physiological processes, including sensory physiology, male fertility, regulation of vascular tone, as well as Ca2+ and Mg2+ homeostasis (3). They share the leitmotif of permeability to cations and rather low sensitivity to membrane voltage (3). Studies on the architecture of TRP channels yielded considerable evidence that these channels function as tetramers (4–6). Monomeric TRPP2 channel subunits are integral membrane proteins with six transmembrane helices (S1-S6), framing a pore-forming loop between S5 and S6 (TRPP2634–659), and cytosolic amino and carboxyl termini (TRPP21–223 and TRPP2680–968, respectively) (7). A prominent feature of TRPP2 is the large extracellular loop between S1 and S2, consisting of 223 amino acids (TRPP2245–468) (Fig. 1A). Loss-of-function phenotypes in ADPKD patients and TRPP2-deficient animal models have revealed important biological roles of this ion channel, ranging from renal tubular morphogenesis to determination of embryonic left-right asymmetry (8). Yet, our knowledge about the molecular mechanisms translating ion channel activity into complex morphogenetic processes is still limited. An important step toward understanding the function of TRPP2 is a molecular dissection of its posttranslational modifications and the characterization of their biological relevance. Posttranslational phosphorylation and glycosylation have been shown to significantly impact the behavior of ion channels, including channel activity, conductance, and protein stability. Several studies have demonstrated a critical role of phosphorylation in TRPP2 function (9–14). Biochemical studies have revealed that TRPP2 is glycosylated, but a detailed mapping and characterization of glycosylated amino acid residues is lacking (15, 16).
FIGURE 1.

Native TRPP2 is an N-glycosylated protein. A, TRPP2 is a six-transmembrane (S1-S6) protein with a large extracellular loop between S1 and S2 (TRPP2245–468), a pore-forming loop between S5 and S6 (TRPP2634–659), as well as cytosolic amino and carboxyl termini (TRPP21–223 and TRPP2680–968). B, immunoprecipitation (IP) of native TRPP2 from wild-type C57BL/6 mouse kidneys were analyzed by anti-TRPP2 Western blot (WB) staining. Two distinct bands were observed, one at ∼120 kDa and one below 100 kDa. Endo H-mediated deglycosylation led to an electrophoretic mobility shift of both bands to ∼110 kDa and ∼80 kDa, respectively. C, enzymatic deglycosylation of endogenous TRPP2 immunoprecipitates with Endo H from M1 renal epithelial cells. Similar to mouse kidneys, two distinct bands were observed. D, immunoprecipitation of native TRPP2 from M1 cells. After SDS-PAGE, samples were stained with colloidal Coomassie and cut into eight pieces. TRPP2 peptides were identified in gel fragments corresponding to a molecular mass from 35–150 kDa. IHC, immunoglobulin heavy chain; ILC, immunoglobulin light chain. E, total nano-LC-MS/MS coverage of murine TRPP2 was 63%. aa, amino acids.
Glycosylation of proteins plays crucial roles in biology, from quality control of protein folding to involvement in a large number of biological recognition events. The addition and processing of asparagine-linked (N-linked) oligosaccharides in the endoplasmic reticulum (ER) is the most common form of glycosylation. N-Linked glycosylation promotes proper folding and assembly of glycoproteins and increases their stability. Glycosylation begins with the cotranslational addition of a preformed, 14-residue glycan to asparagine residues at the NX[ST] consensus sequences, where N is asparagine, X can be any amino acid except proline, followed by either serine or threonine ([ST]), respectively. For all N-glycans, processing of the core glycan continues with trimming of terminal glucose residues by ER glucosidases I and II (17, 18). Glucosidase II-mediated trimming of carbohydrates eventually facilitates glycoprotein trafficking to the Golgi apparatus. Persistent glucosylation of unfolded or misfolded proteins inhibits ER exit and triggers degradation (19). Glucosidase II (GII) is of particular interest to glycan processing of TRPP2 because in vivo studies have placed TRPP2 and the non-catalytic glucosidase II β (GIIβ) subunit of this enzyme in a common biogenetic pathway (20). Although the kidney-specific elimination of GIIβ causes mild cystic kidney disease in mice, a severe PKD phenotype manifests on a Pkd2+/−-sensitized background (20). Loss of GIIβ is accompanied by a reduction of TRPP2 protein expression (21, 20). In humans, mutations in Protein Kinase C Substrate 80K-H (PRKCSH), the gene encoding for GIIβ, cause autosomal dominant polycystic liver disease (ADPLD), which manifests with bile duct cysts that are indistinguishable from the liver phenotype in ADPKD (evident in 94% of ADPKD patients who are older than 35 years) (OMIM no.174,050) (22–25). GII is an ER enzyme with a key role in glycoprotein folding and quality control. It is comprised of two subunits: catalytic GIIα and accessory GIIβ (26). GIIβ has been shown to physically interact with TRPP2, and it has been suggested that GII may be involved in the processing of TRPP2 glycosylation (21, 20). However, direct experimental data supporting this model are missing.
We are only beginning to understand the importance of glycosylation of TRP channels. It has been shown for some TRP channels that N-glycosylation plays a critical role for cellular localization and function (27–32). Biochemical studies have revealed that TRPP2 is glycosylated but the glycosylation sites and relevance of glycosylation for TRPP2 biogenesis and stability have not been investigated (15, 16). Here we present a comprehensive mapping of N-glycosylated amino acid residues in TRPP2. Furthermore, we show that GIIβ is required for glycan trimming of TRPP2 and that proper N-linked glycosylation is essential for adequate TRPP2 protein expression.
EXPERIMENTAL PROCEDURES
Molecular Biology
Full-length human PKD2 (GenBankTM accession no. U50928) in pcDNA3 (Invitrogen) was provided by Feng Qian (University of Maryland) (33). Using this wild-type plasmid, PKD2 D511V, PKD2 N299G/N305G/N328G/N362G (TRPP2Δ4-Glyc), PKD2 N299Q/N305Q/N328Q/N362Q, PKD2 N299G/N305G/N328G/N362G/N375G (TRPP2Δ5-Glyc), and PKD2 N299Q/N305Q/N328Q/N362Q/N375Q were generated by site-directed mutagenesis. The asparagine-to-glutamine and asparagine-to-glycine mutations showed identical biochemical properties. All figures depict experiments with the asparagine-to-glycine mutations. TRPP2L703X-HA, TRPP2HA, and PRKCSHFLAG (GIIβFLAG) constructs were generated as described previously (15, 21).
Antibodies
Anti-TRPP2 (G-20 and D-3) antibodies were obtained from Santa Cruz Biotechnology. A monoclonal anti-TRPP2 antibody against amino acid residues 698–799 (anti-TRPP2698–799) was provided by Gerd Walz (University Medical Center Freiburg). Clone AC-15 anti-β-actin, DM1A anti-α-tubulin, and anti-FLAG M2 antibodies were purchased from Sigma-Aldrich. For HA epitope detection, polyclonal anti-HA antibodies (Zymed Laboratories Inc.) were used. Western blot detection was performed using either an anti-mouse (Dako) or anti-rabbit (GE Healthcare) horseradish peroxidase-coupled secondary antibody. For immunofluorescence-mediated antigen visualization, primary rabbit anti-LAMP1 (catalog no. ab24170, Abcam) and goat anti-PKD2 (G-20) together with secondary donkey anti-goat Alexa Fluor 488 (Invitrogen) and Cy3-coupled donkey anti-rabbit (Dianova) antibodies were used.
Cell Culture
HeLa, HEK293T, LLCPK1, IMCD3, and murine cortical collecting duct M1 cells were cultivated as adherent monolayers in DMEM (Lonza) supplemented with 10% heat-inactivated FBS (Biochrom). Cell lines were maintained in a humidified 10% CO2 incubator at 36.5 °C. Cells were passaged every 3–4 days using 0.05% trypsin-EDTA (Invitrogen). HeLa and HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen) and calcium phosphate, respectively. Stable LLCPK1 cell lines expressing TRPP2L703X-HA were obtained as described previously (34). Prkcsh wild-type and null cells were isolated by tubule microdissection (20).
Mice
C57BL/6 mice were used as the wild type in Fig. 1B. PRKCSH experiments were performed on a C57BL/6–129 mixed background (Fig. 8, B and C). The conditional Prkcshflox mice have been described previously (20). Deletion of exons 6 and 7 by Cre recombinase results in a functional null allele (20). Ksp-Cre mice with constitutive Cre recombinase expression in the thick ascending limb of the loop of Henle, distal convoluted tubule, and collecting duct starting at 9.5 days after fertilization have been described previously (35).
FIGURE 8.

Inactivation of glucosidase II results in defects in TRPP2 N-glycan trimming. A, TRPP2 and GIIβ interact physically. A HA epitope-tagged TRPP2 fusion protein (TRPP2HA) was coexpressed with either FLAG-tagged GIIβ (GIIβFLAG) or with the control protein GFPFLAG in HEK293T cells. After affinity purification with immobilized anti-FLAG antibodies, TRPP2HA was detected by Western blotting (WB) in the immunoprecipitates (IP) of GIIβFLAG. Expression of TRPP2HA in the cell lysates was confirmed with anti-HA antibody. B, Ksp-Cre-mediated kidney-specific deletion of GIIβ results in significantly reduced TRPP2 protein levels in comparison with wild-type TRPP2 in 2-month-old mice. Western blot signal intensities were measured using ImageJ. Samples were normalized for α-tubulin. C, the mean reduction of relative TRPP2 protein levels was 66.75% (n = 4, p = 0.04). D, NB-DNJ-mediated inhibition of endoplasmic reticulum GII in vitro. LLCPK cell lysates were incubated with 8–80 μm for 24–96 h prior to experiments. Cell lysates were then incubated with the GII substrate 4MUG (1 mm), and fluorescence (excitation 365 nm, emission 455 nm) was recorded immediately after stopping the reaction at the indicated time points. NB-DNJ (80 μm) provided potent in vitro inhibition with a ∼95% reduction in GII enzyme activity. DMSO, dimethyl sulfoxide. E, GII inhibition was assayed in vivo in live cells by application of 2 mm NB-DNJ to cell culture medium for 24–96 h prior to experiments. Cells were lysed and assayed for GII activity using 4MUG (1 mm) at the indicated time points. GII activity was reduced by ∼70%. F, autoradiogram of metabolically labeled anti-HA immunoprecipitates from cells following inhibition of GII by NB-DNJ treatment (NBDN, 2 mm for 96 h). The electrophoretic mobility pattern of TRPP2L703X-HA at a higher molecular weight is the result of resistance to jack bean mannosidase digestion compared with TRPP2L703X-HA of untreated cells. Tunicamycin (TUN, 5 μg/ml), an inhibitor of N-linked glycosylation, served as a control, shifting TRPP2L703X-HA toward a lower molecular weight product, indicative of the unglycosylated native protein. G, TRPP2L703X-HA was transiently expressed in mouse kidney epithelial Prkcshflox/flox (wt) and Prkcsh−/− (GIIβ−/−) cells. Cells were labeled metabolically, lysed, immunoprecipitated with anti-HA, treated with jack bean mannosidase, and resolved by SDS-PAGE, followed by autoradiography. The absence of the non-catalytic GIIβ subunit resulted in an electrophoretic mobility shift to a larger molecular weight similar to the inhibition of the GII by NB-DNJ, indicating that loss of GIIβ, as occurs in ADPLD, results in a glucose trimming defect in N-glycans.
Protein Isolation
Native TRPP2 was isolated from confluent M1 cells or from the kidneys of 8-day-old mice. Transiently transfected cells were harvested 24 h after transfection if not stated otherwise. Samples were lysed in ice-cold IP buffer (1% Triton X-100, 20 mm Tris-HCl (pH 7.5), 50 mm NaCl, 50 mm NaF, 15 mm Na4P2O7, and 0.1 mm EDTA (pH 8)) supplemented with 2 mm Na3VO4 and complete protease inhibitor mixture (Roche). Lysates were first centrifuged at 4 °C for 15 min at 20,000 × g and subsequently subjected to ultracentrifugation at 4 °C for 30 min at 100,000 × g. Supernatants were either directly denatured 30 min at 42 °C with 2× Laemmli sample buffer and 100 mm DTT or used for immunoprecipitation and glycosylation studies.
TRPP2 Immunoprecipitation
For TRPP2 immunoprecipitation, protein G-Sepharose beads (GE Healthcare) were incubated with 2 μg of anti-TRPP2 (G-20, Santa Cruz Biotechnology) antibody in PBS (Biochrom) for 90 min at 4 °C. Antibody-coupled beads were washed three times for 10 min at 4 °C with PBS and then combined with the respective lysate (>5 mg of total protein). Immunoprecipitation took place overnight on a rotating wheel at 4 °C. Subsequently, beads were washed three times for 10 min at 4 °C with IP buffer. Proteins were eluted for 30 min at 42 °C with 2× Laemmli sample buffer and 100 mm DTT or, for glycosylation assays, with 0.5% SDS and 40 mm DTT.
Glycosylation Assays of TRPP2 Immunoprecipitates
Endoglycosidase H (Endo H, New England Biolabs) sample digestion was performed for 24 h at 37 °C with 5000 units of Endo H in G5 reaction buffer (New England Biolabs). For peptide-N-glycosidase F (PNGase F, New England Biolabs) cleavage, samples were incubated in G7 reaction buffer (New England Biolabs) with 2.5% Nonidet P-40 (New England Biolabs) and 500 units of PNGaseF for 24 h at 37 °C.
SDS-PAGE, Coomassie Staining, Western Blot Analysis, and Charge-coupled Device Camera-based ECL Detection
Protein samples (∼20 μg of total protein for cell lysates) were separated by SDS-PAGE using Mini-PROTEAN TGX 4–15% precast gels (Bio-Rad). For MS, gels were stained with colloidal Coomassie (36). Otherwise, proteins were Western-blotted semidry on Immobilon-P PVDF (Millipore) membranes. Membranes were blocked with 5% BSA (Serva) and probed with anti-TRPP2698–799 diluted 1/400 in PBS (Biochrom). Immobilized antigens conjugated to horseradish peroxidase-labeled antibodies were detected using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). A 16-bit ChemoCam (Intas) was used for light detection. Images were taken within the dynamic range of the charge-coupled device sensor. None of the depicted bands were saturated.
Nano-LC-MS/MS
Native TRPP2 was immunoprecipitated from a total of 9.4 m2 M1 cells for MS. After SDS-PAGE, gel lanes were cut into 8–12 pieces (Fig. 1D). Gel slices were destained (10 min of shaking at room temperature, 100 mm ammonium bicarbonate (pH 7.8), 100% acetonitrile, both from Sigma-Aldrich), equilibrated (10 min of shaking at room temperature, 100 mm ammonium bicarbonate (pH 7.8), dehydrated (5 min of shaking at room temperature, 100% acetonitrile), and finally dried using a vacuum centrifuge. To increase tandem MS coverage, in-gel digestion was performed with either trypsin (Promega) or elastase (Promega) overnight at 37 °C or thermolysin (R&D Systems) for 2 h at 60 °C. Peptides were recovered by 5% methanoic acid (Sigma-Aldrich). For fractionation, samples were subjected to high-performance liquid chromatography (bonded stationary phase, ZORBAX RRHD 300SB-C18 (Agilent Technologies), mobile phase, water/acetonitrile mixture (97:3)). After electrospray ionization, samples were analyzed on an Agilent Technologies 6520 Quadrupole time of flight or an Agilent Technologies 6340 ion trap hybrid mass spectrometer. Spectra were evaluated for murine TRPP2 (UniProtKB, O35245) using Mascot Server 2.3 (Matrix Science).
Immunofluorescence and Live Cell Imaging
HeLa cells were cultivated in 6 wells (Greiner Bio-One) on cover glasses (Carl Roth) until ∼80% confluence for transient transfection. 24 h after transfection, HeLa cells were either fixed or used for live cell imaging. For immunofluorescence staining, cells were rinsed twice with PBS (Biochrom) and fixed for 15 min at 21 °C using 3% paraformaldehyde (Electron Microscopy Sciences) and 5% sucrose (Sigma-Aldrich) in PBS. After washing twice for 3 min with PBS, cells were blocked for 30 min at 21 °C with 5% horse serum (Invitrogen) and 0.1% Triton X-100 (Merck) in PBS. Subsequently, samples were incubated for 3 min with PBS and for 1 h with primary antibodies (1/500 in PBS) at 21 °C. Cells were then washed for 3 min with PBS and incubated with secondary antibodies (1/1000 in PBS) for 30 min at 21 °C. After an additional 3 min at 21 °C with PBS, Prolong Gold and DAPI (Invitrogen) antifade reagent were added. Finally, samples were transferred to microscope slides (R. Langenbrinck) and sealed with nail varnish (L'Oréal). Acidic organelles in transfected HeLa cells were visualized in live cells with Lysotracker Red DND-99 (Invitrogen) according to the instructions of the manufacturer. Microscopy images were recorded using an Axio Observer microscope (Zeiss).
RNA Isolation, RT-PCR, and Real-time Quantitative PCR (qPCR)
RNA of transiently transfected HeLa cells was isolated (RNeasy Plus mini kit, Qiagen) and retrotranscribed to complementary DNA (Quantitect, Qiagen) according to the protocols of the manufacturer. Subsequently, qPCR (LightCycler 480 SYBR Green I Master, Roche) was performed using the following oligonucleotides: human PKD2, 5′-AGCGGACATAGCTCCAGAAGGAG and 5′-GATGGAATGCTCCATCCGGTC; human HSPCB, 5′-TCTGGGTATCGGAAAGCAAGCC and 5′-GTGCACTTCCTCAGGCATCTTG. Samples were analyzed on a LightCycler 480 (Roche), and relative quantification was calculated: r = 2−(ΔCP PKD2 − ΔCP HSPCB) (37).
Metabolic Labeling
Cells were cultured until ∼80% confluent in DMEM minus-Met/Cys (Invitrogen) with 10% heat-inactivated FBS (Gemini Bioproducts). Subsequently, cells were incubated in medium plus 100–200 μCi/ml [35S]Met/[35S]Cys (PerkinElmer Life Sciences), washed with PBS (Invitrogen), and then maintained in chase medium (DMEM (Invitrogen) with 10% FBS (Gemini Bioproducts)). Cells were then lysed, and the protein of interest was immunoprecipitated, followed by SDS-PAGE and Western blot analysis. Depending on the experiment, the beads were incubated with jack bean mannosidase (20 units/mg of protein, Sigma-Aldrich) prior to SDS-PAGE. Wherever specified, cells were preincubated with 2 mm N-butyldeoxynojirimycin (NB-DNJ, Toronto Research Chemicals) for 96 h or 5 μg/ml tunicamycin (Sigma-Aldrich) for 2 h before and during labeling.
Small Molecule Inhibitors
Drug incubation of HeLa cells was started 24 h after transfection. Small molecule inhibitors were added to DMEM (Lonza) supplemented with 10% heat-inactivated FBS (Biochrom). Cells were maintained in a humidified 10% CO2 incubator at 36.5 °C. Drug concentrations and incubation times were as follows: 200 μm chloroquine (Sigma-Aldrich) or 6 mm ammonium chloride (Sigma-Aldrich) for 24 h, 210 μm MG-132 (Enzo Life Sciences) for 6 h, and 18 μm cycloheximide (Sigma-Aldrich) for 2–10 h. Cells were counted after harvesting using a TC10 automated cell counter (Bio-Rad). Sample volumes were normalized for cell count.
Glucosidase II Inhibition Assay
NB-DNJ (Toronto Research Chemicals) was added either to the cell lysate (8–80 μm) or to the cell medium (2 mm) for 24–96 h prior to experiments. Glucosidase II activity was measured in the lysate of LLCPK cells. The substrate used was 4-methyl-umbelliferyl α-d-glucopyranoside (4MUG, Toronto Research Chemicals). Cell lysates were incubated with 1 mm 4MUG in a total reaction volume of 100 μl for 30 min at 37 °C. The reactions were stopped with 100 μl of 2 m Tris, and the fluorescence was recorded immediately (excitation 365 nm and emission 455 nm).
Statistical Analysis
For data quantitation, at least three independent experiments were evaluated. Samples were normalized for β-actin or α-tubulin Western blot signals. Relative expression was calculated for samples on the same gel. Western blot signal intensities were measured using ImageJ. Student's t test was performed to assess statistical significance.
RESULTS
Native TRPP2 Is N-glycosylated
TRPP2 is a six-transmembrane (S1-S6) protein with a large extracellular loop between S1 and S2 (TRPP2245–468), a pore-forming loop between S5 and S6 (TRPP2634–659), and cytosolic amino and carboxyl termini (TRPP21–223 and TRPP2680–968, respectively) (Fig. 1A) (7). Endogenous human TRPP2 has been shown to be heavily N-glycosylated (15, 16). TRPP2 contains seven potential NX[ST] glycosylation sites: 3, 299, 305, 328, 362, 375, and 580 (NetNGlyc 1.0 Server) (2). To further study the N-glycosylation of TRPP2 in a native context, we immunoprecipitated TRPP2 from wild-type mouse kidneys and analyzed the immunoreactive profile to TRPP2 in a Western blot analysis. Invariably, two distinct bands were observed, one at ∼120 kDa and one below 100 kDa. Enzymatic deglycosylation of TRPP2 immunoprecipitates with Endo H, which cleaves within the chitobiose core of high mannose glycans, led to an electrophoretic mobility shift of both bands to ∼110 and ∼80 kDa, respectively (Fig. 1B). N-Linked glycosylation contributes ∼15 kDa to the molecular mass of mature TRPP2. Similar results were obtained for endogenous TRPP2 from murine cortical collecting duct M1 cells (Fig. 1C). Interestingly, the size of both TRPP2-reactive bands was decreased after deglycosylation, and the faster migrating band was significantly smaller than the predicted size of 109 kDa for unmodified murine TRPP2 (966 amino acids). The high similarity of the Endo H-mediated glycosylation pattern of TRPP2 from mouse kidney and M1 renal epithelial cells make the latter an attractive model system to investigate N-linked glycosylation in detail.
Identification of Five N-glycosylation Sites in the First Extracellular Loop of TRPP2
To determine the individual asparagine residues occupied with N-glycans, endogenous TRPP2 was immunoprecipitated and deglycosylated with Endo H. This generated truncated carbohydrate molecules with one N-acetylglucosamine residue remaining on the respective asparagine, i.e. a definite mass for N-linked glycosylation (asparagine + N-acetylglucosamine). After SDS-PAGE and colloidal Coomassie staining, samples were subjected to MS analyses. TRPP2 peptides were identified in gel fragments corresponding to a molecular weight from 35–150 kDa (Fig. 1D). The obtained data represent 63% of TRPP2 amino acids, covering almost all of the protein except for hydrophobic parts (Fig. 1E). The computational analysis of MS/MS spectra identified four glycosylated asparagine residues in murine TRPP2: 297, 303, 326, and 360 (Fig. 2, A–F). The sites are evolutionarily conserved in vertebrates and correspond in human TRPP2 to amino acid positions 299, 305, 328, and 362. To validate N-linked glycosylation in TRPP2, two constructs were generated by site-directed mutagenesis, replacing the four asparagines with either glycine or glutamine: N299G/N305G/N328G/N362G (TRPP2Δ4-Glyc) and N299Q/N305Q/N328Q/N362Q. However, upon heterologous expression in HEK293T and HeLa cells, these were still sensitive to Endo H-mediated deglycosylation, as seen by an electrophoretic mobility shift of TRPP2 bands in SDS-PAGE (∼3 kDa) (Fig. 3). A fifth N-glycosylation site was identified by taking advantage of the in silico predictions for TRPP2 (2). The additional mutation of asparagine 375 in TRPP2 (TRPP2Δ5-Glyc), which is partially conserved in vertebrates, abrogates any size shift after enzyme-mediated deglycosylation of the protein (Fig. 4A).
FIGURE 2.

Mass spectrometry identifies four N-glycosylated asparagine residues in murine TRPP2. A, MS precursor spectrum N297HTQADN303RSFI after elastase digestion (charge state +3). B, MS/MS spectrum of N297HTQADN303RSFI after fragmentation and reduction to charge state +1. C, MS precursor spectrum N326GSCSIPQDLR after trypsin digestion (charge state +2). 13C caused a shift of 1 unit of the monoisotopic peptide. D, MS/MS spectrum of N326GSCSIPQDLR after fragmentation and reduction to charge state +1. E, MS precursor spectrum SSEDRAPFGPRN360GT after elastase digestion (charge state +3). F, MS/MS spectrum of SSEDRAPFGPRN360GT after fragmentation and reduction to charge state +1.
FIGURE 3.
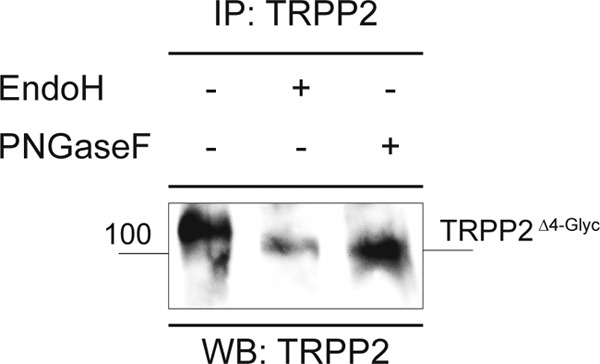
TRPP2 with mutations in four N-glycosylation sites is still sensitive to enzymatic deglycosylation. TRPP2Δ4-Glyc was expressed heterologously in HeLa cells, immunoprecipitated (IP), and analyzed by SDS-PAGE and subsequent anti-TRPP2 Western blot (WB) staining. Enzymatic deglycosylation of TRPP2Δ4-Glyc with Endo H or PNGase F caused a shift in electrophoretic mobility (∼3 kDa), indicating that TRPP2Δ4-Glyc is N-glycosylated.
FIGURE 4.

TRPP2 comprises five N-linked glycosylation sites that are required for adequate TRPP2 protein expression. A, Endo H treatment (24 h at 37 °C) of wild-type TRPP2 causes a size decrease, that was slightly more pronounced after PNGase F treatment (24 h at 37 °C). No electrophoretic mobility shift is observed after Endo H or PNGase F treatment of TRPP2Δ5-Glyc, suggesting that TRPP2Δ5-Glyc was not N-glycosylated. After immunoprecipitation (IP), normalized amounts of protein were used to visualize SDS-PAGE electrophoresis by anti-TRPP2 Western blot (WB) staining. B, illustrative schematic of TRPP2 N-glycosylation. All five N-linked glycosylation sites are located in the first extracellular loop (amino acids 244–469) of human TRPP2: 299, 305, 328, 362, and 375 (corresponding to the conserved asparagines 297, 303, 326, 360, and 373 in murine TRPP2). C, heterologous expression of wild-type TRPP2 and TRPP2Δ5-Glyc in HeLa cells. Cells were counted to normalize for cell number. After lysis and SDS-PAGE, anti-TRPP2 and anti-β-actin Western blot signal intensities were measured using ImageJ. Sample intensities were normalized for β-actin. Wild-type TRPP2 protein expression was set to 100%, and relative TRPP2Δ5-Glyc amounts were calculated. TRPP2Δ5-Glyc shows a significant reduction in protein levels by 86.2% compared with wild-type TRPP2 (n = 3, p = 0.003). D, overexpression of TRPP2Δ5-Glyc in HEK293T cells. Similar to C, TRPP2Δ5-Glyc shows a significant reduction in protein levels compared with wild-type TRPP2 (−74.4%, n = 3, p = 0.016).
The comprehensive analysis of N-linked glycosylation identified five widely conserved glycosylated asparagines in human TRPP2: 299, 305, 328, 362, and 375. Notably, all glycosylation sites are located in the first extracellular loop (Fig. 4B). These asparagines appear to be the only N-glycosylation sites of the protein because neither mass spectrometry nor enzymatic digestion with Endo H or PNGase F detected evidence for additional glycosidic bonds. Further in vitro analysis was facilitated by the recapitulation of native glycosylation patterns with high-mannose glycans by heterologously expressed TRPP2 (Figs. 1B and 4A).
N-Glycosylation of TRPP2 Is Required for Adequate TRPP2 Protein Levels
It has been shown that N-linked glycans determine the homeostasis of secretory proteins and membrane proteins in eukaryotic cells (19). Therefore, we hypothesized that aberrant N-glycosylation of TRPP2 might impair its protein biogenesis. To test this hypothesis, TRPP2Δ5-Glyc was expressed heterologously in HeLa and HEK293T cells. Indeed, TRPP2Δ5-Glyc shows a significant reduction in protein levels by 86.2% compared with wild-type TRPP2 in HeLa cells (n = 3, p = 0.003) (Fig. 4C). This effect is not cell type-specific because a similar reduction is observed in HEK293T cells (mean, −74.4%; n = 3; p = 0.016) (Fig. 4D). To ensure that this decrease is not solely caused by a change in kinetics of TRPP2 protein processing, we measured protein levels after 96 h in HEK293T cells. TRPP2Δ5-Glyc protein levels compared with wild-type TRPP2 remain significantly reduced after 96 h (mean, −79%; n = 4; p = 0.00002).
Lower protein levels may be caused by either transcriptional down-regulation, impaired translation, or decreased protein stability. To evaluate a possible impact on mRNA transcription or stability, RNA of transiently transfected HeLa cells was isolated. TRPP2 wild-type and TRPP2Δ5-Glyc mRNA abundance was similar, as assessed by qPCR (Fig. 5A). Protein stability of TRPP2 was investigated using the Streptomyces griseus metabolite cycloheximide, which inhibits protein translation by blocking elongation factor 2-mediated ribosomal translocation (38). Cells were incubated with 18 μm cycloheximide for 2–10 h, and TRPP2 expression was evaluated by Western blot analysis (Fig. 5, B and C). These experiments show a reduced stability of TRPP2Δ5-Glyc compared with wild-type TRPP2 after 10 h (mean, −46.8%; n = 3; p = 0.006) (Fig. 5D).
FIGURE 5.

TRPP2Δ5-Glyc protein stability is decreased significantly compared with wild-type TRPP2. A, in transiently transfected HeLa cells, mRNA expression of TRPP2Δ5-Glyc is similar to wild-type TRPP2, as assessed by qPCR. B, wild-type TRPP2 protein stability was probed using cycloheximide, which blocks protein translation. Cells were incubated with 18 μm cycloheximide for 2–10 h, samples were normalized for cell count, and TRPP2 protein persistence was evaluated by Western blot (WB) analysis. β-Actin was used as loading control. C, TRPP2Δ5-Glyc protein stability was probed as described in B. D, group data from B and C. Western blot signal intensities at 0 and 10 h were measured using ImageJ. After 10 h of 18 μm cycloheximide treatment, 95.5% of wild-type TRPP2 was still detected (n = 3). TRPP2Δ5-Glyc, in contrast, shows a significant reduction of protein levels to 44.72% (n = 3, p = 0.006).
TRPP2 Is Subject to Lysosomal Degradation
Nascent polypeptides transit between a significant number of intermediate folding states before attaining their native state. It has been demonstrated that the fidelity of this process may be compromised by the absence of posttranslational glycosylation. Missing glycosylation may cause newly synthesized proteins to be degraded or to attain their native states more slowly (39). To investigate the cause for decreased levels of glycosylation-deficient TRPP2, small molecule inhibitors were employed.
Two major proteolytic pathways have been implicated in protein turnover in intact cells: lysosome- and proteasome-mediated degradation. Chloroquine is a weak base that accumulates in acidic cellular organelles, raises the pH, and, thereby, blocks lysosomal function. The peptidyl aldehyde MG-132 is a specific and potent inhibitor of the proteolytic activity of the 26 S proteasome complex (40). Wild-type TRPP2 is significantly more abundant in cells treated with lysosomotropic chloroquine (mean, 575.36%; n = 4; p = 0.0003). MG-132 shows no effect (Fig. 6A). Similarly, MG-132 has no effect on TRPP2Δ5-Glyc, but chloroquine also significantly increases TRPP2Δ5-Glyc protein levels (mean, 329.64%; n = 4; p = 0.048) (Fig. 6B). The observation that chloroquine increases TRPP2 protein levels suggests lysosome-mediated degradation as a pathway for TRPP2 degradation. However, chloroquine is known to have pleiotropic effects on cellular metabolism. To test the hypothesis that lysosomes degrade TRPP2, we inhibited lysosomal function using another structurally different compound, ammonium chloride, which neutralizes the acidic pH value of lysosomes. Both wild-type TRPP2 (mean, 203.07%; n = 3; p = 0.04) and TRPP2Δ5-Glyc (mean, 218.69%; n = 3; p = 0.02) protein levels rise under ammonium chloride treatment (Fig. 6C). Indirect immunofluorescence suggests a partial colocalization of TRPP2 and lysosomal LAMP1 (Fig. 7, A and B). However, the majority of TRPP2 is detected in a reticular pattern, consistent with the published ER localization of the overexpressed channel (41). The gross distribution, number, and appearance of lysosomes are not affected by transfection of TRPP2 or TRPP2Δ5-Glyc (Fig. 7, A–C).
FIGURE 6.

TRPP2 is subject to lysosomal degradation. A, drug incubation of HeLa cells started 24 h after wild-type TRPP2 transfection. Small molecule inhibitors were added to cell medium, 200 μm chloroquine (C) for 24 h, 210 μm MG-132 (M) for 6 h, or 6 mm ammonium chloride (A) for 24 h. As a negative control, untreated (ø) cells were processed in parallel. Cells were counted after harvesting, and sample volume was normalized for cell count. β-Actin was used as the Western blot (WB) loading control. Wild-type TRPP2 protein expression in cells without drug treatment was set to 100%, and relative TRPP2 amounts after drug treatment were calculated. Wild-type TRPP2 was significantly more abundant in cells treated with chloroquine compared with control cells (mean = 575.36%, n = 4, p = 0.0003). B, likewise, TRPP2Δ5-Glyc protein levels increased in the presence of chloroquine (mean, 329.64%; n = 4; p = 0.048). C, incubation of wild-type TRPP2 (mean, 203.07%; n = 3; p = 0.04) and TRPP2Δ5-Glyc (mean, 218.69%; n = 3; p = 0.02) with ammonium chloride significantly increased protein levels compared with untreated controls.
FIGURE 7.

Cellular localization of TRPP2 in HeLa cells. A, HeLa cells were transiently transfected with wild-type TRPP2. Cells were stained for DNA (DAPI, blue), TRPP2 (green), and lysosomes (LAMP1, red) 24 h after transfection. Scale bar = 20 μm. B, likewise, DNA, TRPP2, and lysosomes were visualized in TRPP2Δ5-Glyc-transfected cells. Scale bar = 20 μm. C, LysoTracker-mediated labeling of acidic organelles in live HeLa cells, which were transfected with wild-type TRPP2 and TRPP2Δ5-Glyc, respectively. Scale bar = 20 μm.
Protein turnover is determined by the dynamic balance between protein synthesis and degradation. Our data support a critical role for lysosomes in the degradation of TRPP2. The relatively modest increase of TRPP2Δ5-Glyc protein levels upon lysosomal inhibition together with the insensitivity of TRPP2Δ5-Glyc expression to proteasomal inhibition suggests reduced translation efficiency as an important additional mechanism for reduced expression of TRPP2Δ5-Glyc.
Inactivation of Glucosidase II Results in Defects in N-Glycan Trimming of TRPP2
N-linked glycans are processed and monitored in the ER by enzymes and molecular chaperones (19). Because addition of N-linked oligosaccharides is required for normal protein levels and mutations in at least one protein involved in processing N-linked glycans, GIIβ, result in reduced TRPP2 levels and polycystic kidney and liver disease, we next studied the effects of GIIβ on processing of the precursor glycan of TRPP2. TRPP2 and GIIβ interact physically in cells expressing tagged versions of both proteins (Fig. 8A) (21, 20). Kidney-specific conditional deletion of GIIβ in mice using Ksp-Cre results in significantly reduced tissue levels of the TRPP2 protein in 2-month-old mice (mean, 66.75%; n = 4; p = 0.04) (Fig. 8, B and C). Because the Ksp-Cre is active in a portion of kidney tubule cells primarily in the distal nephron, the actual reduction in TRPP2 in cells lacking GIIβ is likely more pronounced than what can be detected at the whole organ tissue level.
We next wanted to directly examine the role of GII on the trimming of N-linked glycans in TRPP2. Because this required the sensitive determination of differences in the migration of TRPP2 on the basis of variable presence or absence of two glucose residues per N-linked glycan, we used a carboxyl terminal-truncated form of TRPP2, truncated at codon 703 (TRPP2L703X-HA), to allow us to better resolve migration differences on the basis of loss of GII activity. We first used a potent inhibitor of α-glucosidases, NB-DNJ, to establish our ability to reliably detect the lack of N-glycan deglycosylation in TRPP2L703X-HA. NB-DNJ provides potent inhibitory activity when applied in vitro to cell lysates or in vivo to intact cells (Fig. 8, D and E). GII catalytic activity was measured by the fluorescence units released through the cleavage of the specific substrate 4MUG. The studies with the synthetic substrate establish 2 mm NB-DNJ as sufficient to provide at least 70% GII inhibition in intact LLCPK1 cells. To examine the GII-dependent glycosylation status of TRPP2L703X-HA, LLCPK1 cells stably expressing TRPP2L703X-HA were incubated with 2 mm NB-DNJ for 96 h. Cells were pulsed for 30 min with medium containing [35S]Met/[35S]Cys and chased with non-radioactive medium for 30 min. After lysis, TRPP2L703X-HA was immunoprecipitated using anti-HA antibodies and digested with jack bean mannosidase, which cleaves mannose residues from deglycosylated N-glycans but not from N-glycan moieties that were not deglycosylated because of inhibition of GII activity. Thus, the jack bean mannosidase treatment improves the resolution of the migrating protein species to differentiate deglycosylated forms from non-deglycosylated forms. Inhibition of GII by NB-DNJ resulted in a distinct electrophoretic mobility pattern for TRPP2L703X-HA, migrating at a higher molecular weight compared with TRPP2L703X-HA from untreated cells, in which GII was active to permit deglycosylation of the protein (Fig. 8F). This showed the ability to detect a defect in glucose trimming by this method.
GII function in vivo is dependent on GIIβ (42). We therefore tested the hypothesis whether GIIβ deficiency as seen in ADPLD results in defective glucose trimming of TRPP2. TRPP2L703X-HA was transiently expressed in mouse kidney epithelial cells either wild-type (Prkcshflox/flox) or null (Prkcsh−/−) for GIIβ. Cells were labeled metabolically, lysed, immunoprecipitated with anti-HA, digested with jack bean mannosidase, and resolved by SDS-PAGE prior to autoradiography. TRPP2L703X-HA expressed in Prkcsh−/− cells showed slower electrophoretic mobility compared with TRPP2L703X-HA expressed in wild-type cells, indicating impaired glucose trimming in the absence of the non-catalytic GIIβ subunit (Fig. 8G). These data show that the enzymatic activity of GII is indeed impaired in the absence of GIIβ in ADPLD cells and leads to the accumulation of di- or monoglycosylated TRPP2L703X-HA intermediates. Because all five N-linked glycosylation sites are located in the first extracellular loop (TRPP2245–468) of TRPP2 and should not be affected by the TRPP2L703X-HA truncation, it is likely that the observed impaired glycan trimming is analogous in wild-type TRPP2.
We identified five N-glycosylation sites in TRPP2 and showed that their glycosylation is required for adequate TRPP2 protein expression. Processing of these N-linked glycans is dependent on GII, highlighting the molecular association of the ADPKD protein TRPP2 and its ADPLD partner GIIβ.
DISCUSSION
The addition and processing of N-linked oligosaccharides in the ER play a pivotal role in the biogenesis and quality control of many membrane proteins (43, 18). It has been shown that TRPP2 is a glycoprotein, but the glycosylation sites and the importance of glycosylation for TRPP2 biogenesis and stability have not been investigated (15, 41). In this study, we identified five N-glycosylation sites in TRPP2. All glycosylated asparagines in TRPP2 are found in the first extracellular loop (TRPP2245–468) between the first two transmembrane domains (Asn-299, Asn-305, Asn-328, Asn-362, and Asn-375 in human TRPP2). These asparagines are probably the only N-linked glycosylation sites in TRPP2 because enzyme-mediated deglycosylation of TRPP2 with mutations in all five residues does not cause any additional electrophoretic mobility shift of the protein. Our data indicate that glycosylation of TRPP2 regulates the abundance of the channel complex and that deglycosylation results in inefficient translation and decreased stability. TRPP2 appears to be degraded through lysosomes. Incubation of cells with the lysosomal inhibitor chloroquine leads to a significant increase of both wild-type and glycosylation-deficient TRPP2, whereas proteasomal inhibition with MG-132 shows no effect. In addition, we show that glucosidase II mediates trimming of TRPP2 glycosylation. This is required for efficient maintenance of steady-state levels of the protein. These combined results provide a mechanism for the decrease of TRPP2 levels in PRKCSH-deficient cells, which may contribute to the pathogenesis of ADPLD.
There is still rather limited information on the role of N-linked glycosylation of TRP channels. For a subset of TRP channels it has been shown that N-glycosylation plays an important role in cellular localization and ion channel function (31). In TRPV5, a channel critical for Ca2+ reabsorption in the distal tubule of the kidney, cleavage of a single N-linked oligosaccharide by Klotho, a glucuronidase enzyme, stabilizes the channel at the apical plasma membrane (29). In TRPC6 and TRPC3, two closely related cation channels, differences in the N-glycosylation patterns are responsible for their distinct functional properties, switching the channel from a tightly receptor-regulated mode to a constitutively active mode (27). In TRPV4, an osmosensitive TRP channel, mutation of a single N-glycosylation site within the pore-forming loop increases the expression of plasma membrane-bound channels compared with the wild type (30). The temperature-sensitive TRP channels TRPV1 and TRPM8 are also N-glycosylated at a position near the pore loop. Glycosylation of these asparagines affects the biophysical properties and response to canonical activators of both channels (44, 45, 32). All these TRP channels display glycosylation at one or two asparagines. TRPP2, however, is more heavily glycosylated akin to TRPML1, the channel mutated in mucolipidosis type IV (46, 47). One hallmark of the TRPP and the TRPML subfamilies of TRP channels is the prominent first extracellular loop, which harbors the glycosylation sites (Fig. 4B) (48). Interestingly, TRPML1 is cleaved within this first extracellular loop, and there is evidence that TRPP2 undergoes proteolytic cleavage as well (46, 47, 49). In addition to full-length TRPP2, we invariably detect TRPP2 antibody-reactive bands on Western blot analyses that are considerably smaller than the calculated size of the full-length protein. This applies to native and overexpressed protein (Figs. 1, B and C, and 4, C and D). The biological function of cleavage and of the particularly big first extracellular loop of these TRP channels is not known. However, on the basis of data from other ion channels and membrane proteins, it is tempting to speculate that cleavage may be a regulatory mechanism that extends beyond mere inactivation of TRPP2 channels (50, 51). Whether the biological roles of cleavage and glycosylation in the first extracellular loop intersect remains to be determined. Unfortunately, the impact of glycosylation on channel function and cellular localization of TRPP2 cannot be studied easily using heterologous expression systems because overexpressed wild-type TRPP2 is trapped in the ER and does not traffic to the plasma membrane (15, 41). Although this precludes the straightforward investigation of the effect of glycosylation on the biophysical properties of TRPP2 using electrophysiological approaches, the biogenesis and degradation of the channel can be studied using heterologously expressed TRPP2.
The proteasome and lysosome are the main sites of cellular proteolysis (52). The majority of regulated degradation occurs through the ubiquitin-proteasome system in the cytoplasm. Proteins are marked for degradation by covalent attachment of multiple copies of ubiquitin molecules. The polyubiquitinated protein is recognized by the proteasome, a multisubunit protease that degrades the substrate into short polypeptides (53, 54). In contrast, selective lysosomal protein degradation is achieved by controlling the trafficking of proteins into the acidic lysosome, where they are exposed to resident proteases. Transmembrane proteins require special mechanisms for their degradation because different parts of such proteins are on opposite sides of a membrane. Eukaryotic cells have devised several solutions to this problem. Membrane proteins in the ER and in mitochondria are removed from the lipid bilayer by retrotranslocation into the cytoplasm, where they are ubiquitinated and degraded by the proteasome (43). In contrast, integral membrane proteins from the plasma membrane and the Golgi apparatus are collected in vesicles and sent to multivesicular bodies via endosomes. Fusion of multivesicular bodies with lysosomes leads to degradation of proteins and lipids by lysosomal proteases and lipases (55).
The pathways responsible for the degradation of proteins can be studied by incubating cells with specific and selective inhibitors of proteasomes or lysosomes, such as MG-132 or chloroquine, respectively. We used these inhibitors to investigate where wild-type and non-glycosylated TRPP2 are degraded. Because overexpressed TRPP2 is an ER-resident protein and because glycosylation has been shown to be important for folding and quality control of membrane proteins, we speculated that TRPP2 is likely degraded by the ubiquitin-proteasome system via ER-associated degradation (43). Indeed, previous studies have shown that TRPP2 is ubiquitinated (56, 21). Surprisingly, inhibition of the proteasome with MG-132 did not show any effect on wild-type or glycosylation-deficient TRPP2 levels. Inhibition of lysosomal degradation with chloroquine or ammonium chloride, however, resulted in a significant increase of wild-type and glycosylation-deficient TRPP2 levels. Because overexpressed TRPP2 has been shown to be fully Endo H-sensitive and because immunofluorescence shows exclusive ER localization of the channel, lysosomal degradation of the protein appears to be rather counterintuitive (15, 41). It is conceivable, though, that transient trafficking of TRPP2 to the Golgi apparatus and plasma membrane as well as to the lysosomal compartment cannot be detected under steady-state conditions because the retrieval of the channel to the ER profoundly exceeds its forward transport. It has been reported that wild-type TRPP2 can be found at the plasma membrane under certain circumstances (57, 58). Furthermore, truncation mutations of the carboxyl terminus that delete an ER retention/retrieval motif in TRPP2 can be detected at the plasma membrane (15, 10, 59). A recent study has shown that TRPP2 takes different routes to the plasma membrane and the membrane of the primary cilium (59). Notably, this study showed that TRPP2 does not traverse the Golgi apparatus on its way to the primary cilium, raising the possibility that similar mechanisms apply to lysosomal targeting. The stability of TRPP2Δ5-Glyc is decreased significantly, as shown in the experiments with cycloheximide-treated cells (Fig. 5D). However, it should be noted that lysosomal degradation may not explain the strong decrease of glycosylation-deficient TRPP2 because the increase in protein levels upon lysosomal inhibition is not increased for TRPP2Δ5-Glyc compared with wild-type TRPP2. The relatively modest increase of TRPP2Δ5-Glyc protein levels upon inhibition of lysosomes suggests reduced efficiency of translation and folding of TRPP2 as an important additional mechanism for low-level expression of TRPP2Δ5-Glyc.
The general importance of N-glycosylation in protein function is highlighted by severe and pleiotropic clinical phenotypes of patients with impaired glycosylation. These disorders are caused by several known heritable defects in N-glycan synthesis and processing (60, 18). Glycosylation begins with the cotranslational addition of a preformed, 14-residue precursor glycan to asparagine residues that are part of the consensus sequence NX[ST]. Processing of the core glycan commences with trimming of two of the three terminal glucose residues by ER glucosidases I and II in the calnexin/calreticulin cycle (17, 18). Mutations in these glucosidases result in human disease. The non-catalytic β subunit of glucosidase II, encoded by the PRKCSH gene, is of particular interest to glycan processing of TRPP2 because mutations in this gene cause ADPLD (22–24). Mutations in PRKCSH manifest with bile duct cysts indistinguishable from the liver phenotype in ADPKD. It has been shown that PRKCSH is part of a genetic interaction network with several PKD genes, including PKD1 and PKD2 (20). This study established that polycystin 1 is the rate-limiting component in cyst formation in human polycystic kidney and liver disease. Furthermore, it has been shown that GIIβ is required for adequate expression of polycystin 1 and TRPP2 (21, 20). Whether TRPP2 is a substrate for glucosidase II has not been addressed previously. Here we show that GII is required for glycan trimming of TRPP2 using pharmacological and genetic approaches. This observation provides a mechanism for the reduction of TRPP2 expression in cells lacking GIIβ. It should be noted that the reduction of TRPP2 protein levels in GIIβ mutant kidneys was performed with Ksp-Cre-mediated deletion of GIIβ. This approach leads to deletion of the gene in less than 30% of the TRPP2-expressing cells in the kidney.5 Therefore, the reduction in TRPP2 expression depicted in Fig. 8, B and C, would probably be more profound in Prkcsh null kidneys. This is supported by the observation that shRNA-mediated knockdown of PRKCSH in renal epithelial cells leads to a considerably stronger reduction of TRPP2 expression (21). Although it has been shown convincingly that polycystin 1 is the central component for cyst formation, the reduction of TRPP2 expression in cells lacking GIIβ also contributes to cyst formation or modulates the severity of the disease.
In summary, we demonstrate that TRPP2 is glycosylated at five asparagines in the first extracellular loop. N-glycosylation and glycan trimming through glucosidase II regulate the abundance of TRPP2, highlighting the importance of these posttranslational modifications for the biogenesis and stability of the protein. These results provide a basis for further studies addressing additional roles of TRPP2 glycosylation in health and disease.
Acknowledgments
These studies utilized reagents provided by the NIDDK sponsored Baltimore Polycystic Kidney Disease Research and Clinical Core Center, P30 DK090868. We thank Feng Qian for providing plasmids, Gerd Walz for TRPP2 antibodies, and Simone Diederichsen and Julia Merkel for technical assistance.
This work was supported by National Institutes of Health Grant NIH DK51041 (to S. S.). This work was also supported by Deutsche Forschungsgemeinschaft Grant DFG KFO 201 (to M. K.), by the Excellence Initiative of the German Federal and State Governments Grant GSC-4, by the Spemann Graduate School, and by the Alfried Krupp von Bohlen und Halbach Foundation (to M. K.).
F. Grahammer, personal communication.
- ADPKD
- autosomal dominant polycystic kidney disease
- TRP
- transient receptor potential
- TRPP2
- TRP polycystin 2
- GII
- glucosidase II
- ADPLD
- autosomal dominant polycystic liver disease
- Endo H
- endoglycosidase H
- PNGase F
- peptide-N-glycosidase F
- qPCR
- quantitative PCR
- NB-DNJ
- N-butyldeoxynojirimycin
- ER
- endoplasmic reticulum.
REFERENCES
- 1. United States Renal Data System (2012) USRDS 2012 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, pp. 362–375, National Institutes of Health, Bethesda, MD [Google Scholar]
- 2. Mochizuki T., Wu G., Hayashi T., Xenophontos S. L., Veldhuisen B., Saris J. J., Reynolds D. M., Cai Y., Gabow P. A., Pierides A., Kimberling W. J., Breuning M. H., Deltas C. C., Peters D. J., Somlo S. (1996) PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272, 1339–1342 [DOI] [PubMed] [Google Scholar]
- 3. Nilius B., Owsianik G. (2011) The transient receptor potential family of ion channels. Genome Biol. 12, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jahnel R., Dreger M., Gillen C., Bender O., Kurreck J., Hucho F. (2001) Biochemical characterization of the vanilloid receptor 1 expressed in a dorsal root ganglia derived cell line. Eur. J. Biochem. 268, 5489–5496 [DOI] [PubMed] [Google Scholar]
- 5. Kedei N., Szabo T., Lile J. D., Treanor J. J., Olah Z., Iadarola M. J., Blumberg P. M. (2001) Analysis of the native quaternary structure of vanilloid receptor 1. J. Biol. Chem. 276, 28613–28619 [DOI] [PubMed] [Google Scholar]
- 6. Moiseenkova-Bell V. Y., Stanciu L. A., Serysheva I. I., Tobe B. J., Wensel T. G. (2008) Structure of TRPV1 channel revealed by electron cryomicroscopy. Proc. Natl. Acad. Sci. U.S.A. 105, 7451–7455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoffmeister H., Gallagher A.-R., Rascle A., Witzgall R. (2011) The human polycystin-2 protein represents an integral membrane protein with six membrane-spanning domains and intracellular N- and C-termini. Biochem. J. 433, 285–294 [DOI] [PubMed] [Google Scholar]
- 8. Hofherr A., Köttgen M. (2011) TRPP channels and polycystins. Adv. Exp. Med. Biol. 704, 287–313 [DOI] [PubMed] [Google Scholar]
- 9. Cai Y., Anyatonwu G., Okuhara D., Lee K.-B., Yu Z., Onoe T., Mei C.-L., Qian Q., Geng L., Wiztgall R., Ehrlich B. E., Somlo S. (2004) Calcium dependence of polycystin-2 channel activity is modulated by phosphorylation at Ser-812. J. Biol. Chem. 279, 19987–19995 [DOI] [PubMed] [Google Scholar]
- 10. Köttgen M., Benzing T., Simmen T., Tauber R., Buchholz B., Feliciangeli S., Huber T. B., Schermer B., Kramer-Zucker A., Höpker K., Simmen K. C., Tschucke C. C., Sandford R., Kim E., Thomas G., Walz G. (2005) Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J. 24, 705–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Streets A. J., Moon D. J., Kane M. E., Obara T., Ong A. C. (2006) Identification of an N-terminal glycogen synthase kinase 3 phosphorylation site which regulates the functional localization of polycystin-2 in vivo and in vitro. Hum. Mol. Genet. 15, 1465–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Streets A. J., Needham A. J., Gill S. K., Ong A. C. (2010) Protein kinase D-mediated phosphorylation of polycystin-2 (TRPP2) is essential for its effects on cell growth and calcium channel activity. Mol. Biol. Cell 21, 3853–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Plotnikova O. V., Pugacheva E. N., Golemis E. A. (2011) Aurora A kinase activity influences calcium signaling in kidney cells. J. Cell Biol. 193, 1021–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Streets A. J., Wessely O., Peters D. J., Ong A. C. (2013) Hyperphosphorylation of polycystin-2 at a critical residue in disease reveals an essential role for polycystin-1-regulated dephosphorylation. Hum. Mol. Genet. 22, 1924–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cai Y., Maeda Y., Cedzich A., Torres V. E., Wu G., Hayashi T., Mochizuki T., Park J. H., Witzgall R., Somlo S. (1999) Identification and characterization of polycystin-2, the PKD2 gene product. J. Biol. Chem. 274, 28557–28565 [DOI] [PubMed] [Google Scholar]
- 16. Newby L. J., Streets A. J., Zhao Y., Harris P. C., Ward C. J., Ong A. C. (2002) Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J. Biol. Chem. 277, 20763–20773 [DOI] [PubMed] [Google Scholar]
- 17. Helenius A., Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 18. Moremen K. W., Tiemeyer M., Nairn A. V. (2012) Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aebi M., Bernasconi R., Clerc S., Molinari M. (2010) N-glycan structures: recognition and processing in the ER. Trends Biochem. Sci. 35, 74–82 [DOI] [PubMed] [Google Scholar]
- 20. Fedeles S. V., Tian X., Gallagher A.-R., Mitobe M., Nishio S., Lee S. H., Cai Y., Geng L., Crews C. M., Somlo S. (2011) A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat. Genet. 43, 639–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao H., Wang Y., Wegierski T., Skouloudaki K., Pütz M., Fu X., Engel C., Boehlke C., Peng H., Kuehn E. W., Kim E., Kramer-Zucker A., Walz G. (2010) PRKCSH/80K-H, the protein mutated in polycystic liver disease, protects polycystin-2/TRPP2 against HERP-mediated degradation. Hum. Mol. Genet. 19, 16–24 [DOI] [PubMed] [Google Scholar]
- 22. Li A., Davila S., Furu L., Qian Q., Tian X., Kamath P. S., King B. F., Torres V. E., Somlo S. (2003) Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am. J. Hum. Genet. 72, 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Drenth J. P., te Morsche R. H., Smink R., Bonifacino J. S., Jansen J. B. (2003) Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 33, 345–347 [DOI] [PubMed] [Google Scholar]
- 24. Drenth J. P., Martina J. A., van de Kerkhof R., Bonifacino J. S., Jansen J. B. (2005) Polycystic liver disease is a disorder of cotranslational protein processing. Trends Mol. Med. 11, 37–42 [DOI] [PubMed] [Google Scholar]
- 25. Bae K. T., Zhu F., Chapman A. B., Torres V. E., Grantham J. J., Guay-Woodford L. M., Baumgarten D. A., King B. F., Jr., Wetzel L. H., Kenney P. J., Brummer M. E., Bennett W. M., Klahr S., Meyers C. M., Zhang X., Thompson P. A., Miller J. P., and Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) (2006) Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clinical Journal of the American Society of Nephrology 1, 64–69 [DOI] [PubMed] [Google Scholar]
- 26. Trombetta E. S., Simons J. F., Helenius A. (1996) Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound noncatalytic HDEL-containing subunit. J. Biol. Chem. 271, 27509–27516 [DOI] [PubMed] [Google Scholar]
- 27. Dietrich A., Mederos y Schnitzler M., Emmel J., Kalwa H., Hofmann T., Gudermann T. (2003) N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J. Biol. Chem. 278, 47842–47852 [DOI] [PubMed] [Google Scholar]
- 28. Wirkner K., Hognestad H., Jahnel R., Hucho F., Illes P. (2005) Characterization of rat transient receptor potential vanilloid 1 receptors lacking the N-glycosylation site N604. Neuroreport 16, 997–1001 [DOI] [PubMed] [Google Scholar]
- 29. Chang Q., Hoefs S., van der Kemp A. W., Topala C. N., Bindels R. J., Hoenderop J. G. (2005) The β-glucuronidase Klotho hydrolyzes and activates the TRPV5 channel. Science 310, 490–493 [DOI] [PubMed] [Google Scholar]
- 30. Xu H., Fu Y., Tian W., Cohen D. M. (2006) Glycosylation of the osmoresponsive transient receptor potential channel TRPV4 on Asn-651 influences membrane trafficking. Am. J. Physiol. Renal Physiol. 290, F1103–F1109 [DOI] [PubMed] [Google Scholar]
- 31. Cohen D. M. (2006) Regulation of TRP channels by N-linked glycosylation. Semin. Cell Dev. Biol. 17, 630–637 [DOI] [PubMed] [Google Scholar]
- 32. Pertusa M., Madrid R., Morenilla-Palao C., Belmonte C., Viana F. (2012) N-glycosylation of TRPM8 ion channels modulates temperature sensitivity of cold thermoreceptor neurons. J. Biol. Chem. 287, 18218–18229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hanaoka K., Qian F., Boletta A., Bhunia A. K., Piontek K., Tsiokas L., Sukhatme V. P., Guggino W. B., Germino G. G. (2000) Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408, 990–994 [DOI] [PubMed] [Google Scholar]
- 34. Geng L., Okuhara D., Yu Z., Tian X., Cai Y., Shibazaki S., Somlo S. (2006) Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J. Cell Sci. 119, 1383–1395 [DOI] [PubMed] [Google Scholar]
- 35. Shao X., Somlo S., Igarashi P. (2002) Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 13, 1837–1846 [DOI] [PubMed] [Google Scholar]
- 36. Dyballa N., Metzger S. (2009) Fast and sensitive colloidal Coomassie G-250 staining for proteins in polyacrylamide gels. J. Vis. Exp. 30, 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jacob F., Guertler R., Naim S., Nixdorf S., Fedier A., Hacker N. F., Heinzelmann-Schwarz V. (2013) Careful selection of reference genes is required for reliable performance of RT-qPCR in human normal and cancer cell lines. PLoS ONE 8, e59180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schneider-Poetsch T., Ju J., Eyler D. E., Dang Y., Bhat S., Merrick W. C., Green R., Shen B., Liu J. O. (2010) Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 6, 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brodsky J. L. (2012) Cleaning up: ER-associated degradation to the rescue. Cell 151, 1163–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee D. H., Goldberg A. L. (1998) Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397–403 [DOI] [PubMed] [Google Scholar]
- 41. Koulen P., Cai Y., Geng L., Maeda Y., Nishimura S., Witzgall R., Ehrlich B. E., Somlo S. (2002) Polycystin-2 is an intracellular calcium release channel. Nat. Cell Biol. 4, 191–197 [DOI] [PubMed] [Google Scholar]
- 42. Trombetta E. S., Fleming K. G., Helenius A. (2001) Quaternary and domain structure of glycoprotein processing glucosidase II. Biochemistry 40, 10717–10722 [DOI] [PubMed] [Google Scholar]
- 43. Hampton R. Y. (2002) ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 14, 476–482 [DOI] [PubMed] [Google Scholar]
- 44. Dragoni I., Guida E., McIntyre P. (2006) The cold and menthol receptor TRPM8 contains a functionally important double cysteine motif. J. Biol. Chem. 281, 37353–37360 [DOI] [PubMed] [Google Scholar]
- 45. Veldhuis N. A., Lew M. J., Abogadie F. C., Poole D. P., Jennings E. A., Ivanusic J. J., Eilers H., Bunnett N. W., McIntyre P. (2012) N-glycosylation determines ionic permeability and desensitization of the TRPV1 capsaicin receptor. J. Biol. Chem. 287, 21765–21772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kiselyov K., Chen J., Rbaibi Y., Oberdick D., Tjon-Kon-Sang S., Shcheynikov N., Muallem S., Soyombo A. (2005) TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J. Biol. Chem. 280, 43218–43223 [DOI] [PubMed] [Google Scholar]
- 47. Miedel M. T., Weixel K. M., Bruns J. R., Traub L. M., Weisz O. A. (2006) Posttranslational cleavage and adaptor protein complex-dependent trafficking of mucolipin-1. J. Biol. Chem. 281, 12751–12759 [DOI] [PubMed] [Google Scholar]
- 48. Qian F., Noben-Trauth K. (2005) Cellular and molecular function of mucolipins (TRPML) and polycystin 2 (TRPP2). Pflugers Arch. 451, 277–285 [DOI] [PubMed] [Google Scholar]
- 49. Huang K., Diener D. R., Mitchell A., Pazour G. J., Witman G. B., Rosenbaum J. L. (2007) Function and dynamics of PKD2 in Chlamydomonas reinhardtii flagella. J. Cell Biol. 179, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yu S., Hackmann K., Gao J., Gao J., He X., Piontek K., García-González M. A., García González M. A., Menezes L. F., Xu H., Germino G. G., Zuo J., Qian F. (2007) Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc. Natl. Acad. Sci. U.S.A. 104, 18688–18693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kleyman T. R., Carattino M. D., Hughey R. P. (2009) ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J. Biol. Chem. 284, 20447–20451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ciechanover A. (2012) Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Biochim. Biophys. Acta 1824, 3–13 [DOI] [PubMed] [Google Scholar]
- 53. Hochstrasser M. (1996) Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30, 405–439 [DOI] [PubMed] [Google Scholar]
- 54. Glickman M. H., Ciechanover A. (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 82, 373–428 [DOI] [PubMed] [Google Scholar]
- 55. Katzmann D. J., Odorizzi G., Emr S. D. (2002) Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 3, 893–905 [DOI] [PubMed] [Google Scholar]
- 56. Liang G., Li Q., Tang Y., Kokame K., Kikuchi T., Wu G., Chen X.-Z. (2008) Polycystin-2 is regulated by endoplasmic reticulum-associated degradation. Hum. Mol. Genet. 17, 1109–1119 [DOI] [PubMed] [Google Scholar]
- 57. Luo Y., Vassilev P. M., Li X., Kawanabe Y., Zhou J. (2003) Native polycystin 2 functions as a plasma membrane Ca2+-permeable cation channel in renal epithelia. Mol. Cell. Biol. 23, 2600–2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tsiokas L. (2009) Function and regulation of TRPP2 at the plasma membrane. Am. J. Physiol. Renal Physiol. 297, F1–F9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hoffmeister H., Babinger K., Gürster S., Cedzich A., Meese C., Schadendorf K., Osten L., de Vries U., Rascle A., Witzgall R. (2011) Polycystin-2 takes different routes to the somatic and ciliary plasma membrane. J. Cell Biol. 192, 631–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Freeze H. H. (2007) Congenital disorders of glycosylation: CDG-I, CDG-II, and beyond. Curr. Mol. Med. 7, 389–396 [DOI] [PubMed] [Google Scholar]