

Background: WIN 18,446, one of the class of compounds known as bisdichloroacetyldiamines, inhibits spermatogenesis.
Results: WIN 18,446 strongly and irreversibly inhibits the retinal dehydrogenase ALDH1A2 in vitro, alters vitamin A status, and blocks spermatogenesis in mice.
Conclusion: Inhibition of spermatogenesis by WIN 18,446 cannot be reversed by oral retinoic acid supplementation.
Significance: WIN 18,446 is a useful tool to study retinoid metabolism and spermatogenesis in vivo.
Keywords: Adipose Tissue, Animal Models, Metabolism, Obesity, Vitamin A, ALDH1A2, Bisdichloroacetyldiamine, Fertility, Retinoic Acid
Abstract
Knowledge of the regulation of testicular retinoic acid synthesis is crucial for understanding its role in spermatogenesis. Bisdichloroacetyldiamines strongly inhibit spermatogenesis. We reported previously that one of these compounds, WIN 18,446, potently inhibited spermatogenesis in rabbits by inhibiting retinoic acid synthesis. To understand how WIN 18,446 inhibits retinoic acid synthesis, we characterized its effects on human retinal dehydrogenase ALDH1A2 in vitro as well as its effects on retinoid metabolism in vivo using mice. WIN 18,446 strongly and irreversibly inhibited ALDH1A2 in vitro. In vivo, WIN 18,446 treatment completely abolished spermatogenesis after 4 weeks of treatment and modestly reduced adiposity in mice fed a chow diet. Effects of WIN 18,446 on retinoid concentrations were tissue-dependent. Although lung and liver retinyl ester concentrations were lower in WIN 18,446-treated animals, adipose retinyl ester levels were increased following the treatment. Interestingly, animals treated with WIN 18,446 had significantly higher circulating retinol concentrations compared with control mice. The effect on spermatogenesis by WIN 18,446 was not prevented by simultaneous treatment with retinoic acid, whereas effects on other tissues were partially or completely reversed. Cessation of WIN 18,446 treatment for 4 weeks reversed most retinoid-related phenotypes except for inhibition of spermatogenesis. Our data suggest that WIN 18,446 may be a useful model of systemic acquired retinoic acid deficiency. Given the effects observed in our study, inhibition of retinoic acid biosynthesis may have relevance for the treatment of obesity and in the development of novel male contraceptives.
Introduction
The importance of vitamin A in reproductive health in both males and females has been well documented (1, 2). In males, either vitamin A deficiency (3) or mutations in genes involved in retinoid signaling pathways leads to perturbations in meiosis and spermatogenesis (1, 2). Because animals lack enzymes to synthesize vitamin A de novo, it has to be derived from dietary sources as preformed vitamin A from animal products and provitamin A carotenoids from fruits and vegetables (4). However, the liver can store a large amount of vitamin A as retinyl esters, thus relieving animals from the daily acquirement of vitamin A from food (5). Testes as well as other extrahepatic tissues can either take up retinyl esters from chylomicrons after a meal containing vitamin A or from retinol released from the liver in between meals. Inside tissues, vitamin A is metabolized to retinoic acid, its biologically active metabolite, which regulates the expression of many genes through its cognate receptors, retinoic acid receptors and retinoid X receptors (6). The majority of functions attributed to vitamin A are thought to be carried out by retinoic acid with the exception being visual functions, which require retinaldehydes. Thus, most symptoms of vitamin A deficiency can be relieved by the administration of retinoic acid and can be mimicked by mutations in retinoid receptors. However, the one vitamin A deficiency symptom that cannot be readily rescued by retinoic acid but can be mimicked by retinoic acid receptor/retinoid X receptor mutations (7) is spermatogenesis. The precise reason for this is not fully understood. However, it is generally thought that a blood-testis barrier may exist that is formed by high expression of retinoic acid-metabolizing cytochrome P450 enzymes (Cyp26) in peritubular myoid cells (8, 9). Studies with radiolabeled retinoic acid have demonstrated that the testes take up almost no circulating retinoic acid (10). In contrast, a majority of the retinoic acid in liver and brain originates from the circulation (10). This may explain why dietary retinoic acid is unable to substitute for retinol in the maintenance of spermatogenesis in the setting of vitamin A deficiency (11, 12) and suggests that in situ retinoic acid production is required for this function in the testes.
Previously, we have reported that the administration of the bisdichloroacetyldiamine WIN 18,446 abrogates spermatogenesis through its action as an inhibitor of aldehyde dehydrogenase 1A2 (ALDH1A22; also known as RALDH2), one of the enzymes responsible for the oxidation of retinaldehyde to retinoic acid in tissues (13). The effect of WIN 18,446 on spermatogenesis is reversible in that cessation of drug administration results in complete recovery of spermatogenesis (13). Although the reversible nature of WIN 18,446 inhibition of spermatogenesis is promising for its use as an oral contraceptive (14), its broad inhibitory effects on other isozymes of aldehyde dehydrogenase, such as ALDH2, the enzyme important for the metabolism of alcohol, prevents it from being used for such a purpose (13). However, strong inhibitory effects of WIN 18,446 on ALDH1A2 enzymes provide an opportunity to address some of the questions regarding effects of retinoic acid synthesis on various biological functions, including spermatogenesis and adipogenesis. Thus, we examined characteristics of WIN 18,446 inhibition on ALDH1A2 using in vitro enzyme assays and determined its effects on retinoid metabolism in vivo using a mouse model.
EXPERIMENTAL PROCEDURES
Chemical Reagents
Retinol, retinyl acetate, retinyl palmitate, and disulfiram were purchased from Sigma. Retinyl stearate, retinyl linoleate, and retinyl oleate were synthesized from retinol and corresponding fatty acids following a reported method (15). WIN 18,446 was obtained from Acros Organics (Geel, Belgium).
Cloning and Purification of ALDH1A2
Full-length human ALDH1A2 cDNA was cloned from human testis RNA using RT-PCR. An open reading frame was subcloned into a pET28 bacterial expression vector containing a 3′ histidine tag, and the resulting sequence was verified through DNA sequencing (University of Washington DNA sequencing facility). Expression of the enzyme was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside, and purification was performed using His·Bind resin (Novagen, San Diego, CA) as described previously (16). Purified enzyme was dialyzed against 20 mm Hepes buffer (pH 8.5) containing 150 mm KCl and 1 mm EDTA. We found that enzyme activity became stable when reducing agents were present, and thus, we stored enzyme with 1 mm TCEP (Bond-Breaker TCEP solution, Thermo Scientific, Rockford, IL) at 4 °C after purification and dialysis. Protein purity was confirmed by SDS-PAGE followed by Coomassie staining, and concentrations were determined using a BCA protein assay kit (Thermo Scientific).
Enzyme Assays
The characterization of ALDH1A2 activity was determined as reported previously (13) with purified enzyme. Briefly, ALDH1A2 was incubated in an assay buffer containing 20 mm Hepes, 150 mm KCl, and 1 mm EDTA at 37 °C with varying concentrations of retinal and 2 mm NAD+. For inhibition assays, WIN 18,446 was dissolved in DMSO and added to assay reactions (the concentration of DMSO was less than 1%). Concentrations of retinoic acid were determined by HPLC following the method reported previously (17).
Animals and Diets
Two studies were performed. The first study was designed to determine effects of WIN 18,446 on spermatogenesis as well as its potential toxicity on other organs due to changes in retinoid physiology. For this study, C57BL/6J male mice (n = 60) were purchased from The Jackson Laboratory at 4 weeks of age and acclimated for 2 weeks in a specific pathogen-free facility at the University of Washington. During the acclimation period, mice were fed a regular chow diet (5053, Purina). At 6 weeks of age, mice were assigned to one of the three dietary treatment groups and continued on the diet for 4 weeks: AIN93M (control diet; 5D3L, Test Diet) and AIN93M containing WIN 18,446 or disulfiram (2 mg/g of diet; 9G0V and 9G0U, Test Diet). Disulfiram is an inhibitor of ALDH1A2 in vitro, but it does not inhibit spermatogenesis in vivo and thus serves as another negative control. During the dietary treatment, mice were weighed weekly, and fasting blood glucose levels were determined from tail blood using a glucometer (OneTouch Ultra, LifeScan). All mice were euthanized after the 4 weeks of dietary treatment after overnight fasting, and tissues from half of the mice were collected after perfusion with PBS, weighed, snap frozen in liquid nitrogen, stored at −80 °C until retinoid analyses (n = 30). Tissues from the other half of the mice were fixed in 10% neutral buffered formalin for histopathological analyses (n = 30). Blood was also collected from these mice for retinoid and lipid analyses.
The second study was conducted to determine whether the phenotype associated with WIN 18,446 treatment can be reversed by simultaneous treatment with retinoic acids and to determine whether the treatment effects can be reversed by cessation of the drug. For this study, C57BL/6J male mice (n = 96) were purchased from The Jackson Laboratory at 5 weeks of age and acclimated for 1 week in our facility with control diet (AIN93M). Mice were then assigned to one of the six treatment groups: 1) AIN93M, 2) AIN93M with WIN 18,446, 3) AIN93M with WIN 18,446 + all-trans-retinoic acid (5WOT; 2 mg of WIN 18,446/g of diet + 5 μg of all-trans-retinoic acid/g of diet), 4) AIN93M with WIN 18,446 + 13-cis-retinoic acid (5W0U; 2 mg of WIN 18,446/g of diet + 5 μg of 13-cis-retinoic acid/g of diet), 5) AIN93M, and 6) AIN93M with WIN 18,446 (4 weeks) followed by AIN93M (4 weeks). Groups 1–4 were fed the respective diets for 4 weeks, whereas groups 5 and 6 were fed the diets for 8 weeks. All mice were euthanized by CO2 asphyxiation followed by cardiac puncture after 4 or 8 weeks of diet treatment after overnight fasting, and tissues were collected for histological (liver, lung, adipose tissues, and testes) and biochemical (serum, liver, lung, testes, and adipose tissues) analyses. All animal study procedures were reviewed and approved by the University of Washington Institutional Animal Care and Use Committee.
Analysis of Spermatogenesis
Using techniques reported previously (13), testes and epididymides were dissected, cleaned of fat, and processed for histologic and quantitative analysis of spermatogenesis. In each experiment, five mice per group were assigned for this analysis. A portion of one testis was dissected, fixed in Bouin's solution, and stained with hematoxylin-periodic acid-Schiff for histological analysis. For testicular spermatid counts (the primary end point), 15–40 mg of testis was individually weighed and homogenized in 0.1 m sodium phosphate buffer (pH 7.4) containing 0.1% Triton X-100 using eight strokes in an all-glass Kontes 15-ml homogenizer. Ten microliters of the homogenate was counted using a Neubauer phase-contrast hemocytometer. Total homogenization-resistant spermatids per gram and per organ were calculated by correcting for the number of squares counted, dilution, volume, and weight (18, 19).
Lipid Analysis
Liver lipids were extracted following the Folch method (20, 21). Total cholesterol and triglyceride concentrations from serum and liver were determined using colorimetric assays following the manufacturers' instructions (Genzyme, Charlottetown, Prince Edward Island, Canada and Wako Chemicals, Richmond, VA).
Retinol and Retinyl Ester Analysis
Retinoid extractions and detections were carried out as described previously (16, 22). Briefly, tissue samples were weighed, homogenized in PBS, and extracted with hexane. The retinoid-containing hexane phase was dried under a gentle stream of nitrogen and injected onto a Symmetry C18 column (5 μm, 4.6 × 250 mm; Waters, Milford, MA) with a running solvent (acetonitrile/methanol/methylene chloride, 70:15:15, v/v) at 1.8 ml/min. The loss during the extraction was accounted for with an internal standard, retinyl acetate.
Measurement of Serum Retinol-binding Protein (RBP)
Serum RBP concentrations were determined using an ELISA kit (Aviva Systems Biology, San Diego, CA) following the manufacturer's instructions.
Retinoic Acid Analysis from Testes
Intratesticular retinoic acid (RA) concentrations were measured by LC-MS/MS as described previously with a few modifications to the sample preparation (23). All sample processing, preparation, and extraction were conducted on ice under red light to minimize degradation of retinoic acids. For standard curve generation, RA-depleted canine testicular tissue (obtained from Dr. Mary Ellen Zoulas at the Seattle Animal Shelter) was prepared by homogenizing decapsulated testicular tissue in a 1:1 volume of 0.9% NaCl with a hand-held homogenizer (Omni International, Kennesaw, GA) and exposing the homogenate to UV light for 12 h. Standard curves were generated with 13-cis-RA and all-trans-RA spiked into 200 μl of UV light-exposed testicular homogenate (0–15 nm). Aliquots of each standard (60 μl) were extracted along with the mouse testicular tissue samples. Mouse testicular tissue was decapsulated and homogenized in a 1:1 volume of 0.9% NaCl with a 2-ml Kontes glass Dounce homogenizer (Kimble Chase, Vineland, NJ). Both mouse testicular homogenates and RA standards were spiked with 13-cis-RA-d5, an internal standard. An equal volume of methanol/water (v/v, 50:50) + 1% formic acid was added to each homogenate and vortexed. Samples were transferred to a 96-well 200-μl solid-supported liquid extraction plate (Biotage, Charlotte, NC.) The sample was allowed to sit for 5 min followed by application of 6 p.s.i. dried nitrogen for 10 s using a positive pressure manifold (Biotage). To extract RA from the tissue loaded on the plate, 2 ml of hexanes was added to each well and allowed to flow through under gravity. After 10 min, 6 p.s.i. nitrogen was used to complete the elution. The eluent was evaporated to dryness under a nitrogen stream at 32 °C, reconstituted in 60 μl of 60:40 ACN/H2O, and injected (10 μl) onto an Ascentis Express RP-Amide column (2.7 μm, 150 × 2.1 mm; Sigma). Gradient elution with a flow rate of 0.5 ml/min using water (A) and acetonitrile (B) with 40% methanol and 0.1% formic acid in A and B was used. The gradient was from an initial 60% A for 2 min to 45% A over 8 min and then to 10% A over 7 min. The column was then washed with 95% B for 3 min and returned to initial conditions. The column heater was set to 40 °C, and samples were kept in the autosampler at 4 °C in the dark. Retinoic acids were detected using an AB SCIEX 5500 qTrap Q-LIT mass spectrometer (Foster City, CA) operated in positive ion atmospheric pressure chemical ionization mode (Fig. 1).
FIGURE 1.

Representative chromatograms of RA. Testicular retinoic acids were determined as described under “Experimental Procedures,” and the representative multiple reaction monitoring chromatograms are shown. A, testes depleted of RA. B, RA-depleted testes spiked with all-trans-retinoic acids. C, a representative mouse testicular sample. Arrows point to the all-trans-retinoic acid peak. CPS, counts/s.
Immunohistochemistry and Histopathology
Tissues taken for histological analyses were fixed in 10% phosphate-buffered formalin and routinely processed and stained with hematoxylin and eosin (H&E). Comprehensive histopathology was performed and included all major organs and decalcified cross-sections of the head. The following tissues were examined: head (rostral and caudal section with brain in situ), lungs, trachea, esophagus, gall bladder, kidney, adrenal gland, heart, spleen, pancreas, mesenteric lymph nodes, salivary gland, epididymis, seminal vesicle, bulbourethral gland, prostate, penis, urethra, bladder, small intestine, large intestine, stomach, skin, and adipose tissues (epididymal, retroperitoneal, mesenteric, and inguinal). White adipose tissues (epididymal fat pad) and brown adipose tissues from the second study were immunohistologically stained for uncoupling protein-1 (UCP-1; ab23841, Abcam), and the signal was detected with 3,3′-diaminobenzidine using polymer poly-HRP anti-rabbit IgG in the Leica Bond Polymer Refine Detection kit (Leica Biosystems). UCP-1 signal (brown staining) was scored using a subjective 0–2 scale for intensity as follows: 0, negative; 1, patchy and faint; 2, positive.
Adipocyte Size Analysis
White adipocyte size was determined by image analysis. H&E-stained slides of retroperitoneal, mesenteric, and inguinal fat pads were digitally scanned (NanoZoomer Digital Imaging System). Three areas of interest per slide (regions of adipocytes without sectioning artifacts or large blood vessels) were determined, and the average area of adipocyte size was calculated using automated image analysis (Visiopharm® image analysis software).
Quantitative RT-PCR
Total RNA was extracted from liver, lung, and adipose tissue (mesenteric fat) following the manufacturer's protocol (RNeasy Tissue Mini kit and RNeasy Lipid Tissue Mini kit, Qiagen, Valencia, CA). cDNA was synthesized using 1 μg of RNA (SuperScript III first strand synthesis system, Invitrogen). Quantitative RT-PCR was carried out to determine expression of lecithin-retinol acyltransferase (Lrat), stimulated by retinoic acid 6 (Stra6), and Cyp26a1 using SYBR Green Master Mix (Invitrogen) and gene-specific primers (Lrat: forward, 5′-CTTACTGCAGATATGGCTCTCG; reverse, 5′-CTAATCCCAAGACAGCCGAAG; Stra6: forward, 5′-GGGTGACAGATGACTACAGC; reverse, 5′-AACCAGGAACGACAGTGAAG; Cyp26a1: forward, 5′-ACTTACCTAGGACTCTACCCAC; reverse, 5′-GCTGTTCCAAAGTTTCCATGTC).
Statistical Analysis
All data were analyzed using a statistical software, Prism (GraphPad, La Jolla, CA). One-way analysis of variance with a post hoc test (Bonferroni correction) was used for comparisons between three groups, and t test was used for comparison between two groups. Statistical significance was defined as having a p value of 0.05 or less.
RESULTS
Characterization of Human ALDH1A2 Enzyme
Initially DTT was added for enzyme reactions as DTT strongly increased the enzyme activity. A more stable reducing agent, TCEP, was also effective for optimal enzyme activity. Interestingly, the purified enzyme lost its activity if it was kept without reducing agent for an extended period at 4 °C. Thus, we tested the stability of the enzyme with various additives: 1 mm DTT, 1 mm TCEP, and 20% glycerol. Enzyme activity was similar when it was stored with either DTT or TCEP after 1 week at 4 °C. However, 20% glycerol was not sufficient to protect enzyme activity when stored at −20 °C. Enzyme activity remained stable after 3 weeks when TCEP was added at 1 mm concentration, whereas the enzyme with DTT lost its activity by this time. Thus, all our enzyme reactions were carried out using purified and dialyzed enzyme stored with 1 mm TCEP.
Production of retinoic acid by purified ALDH1A2 was dependent upon protein concentration, incubation time, and substrate concentration (Fig. 2, A–C). The rate of retinoic acid production was saturated with increasing concentrations of retinal (Fig. 2C). The relationship between retinal and reaction velocity was sigmoidal and fitted best using the Hill equation. Under our experimental conditions, ALDH1A2 has a Hill coefficient of 1.736, K0.5 for retinal of 0.296 μm, and Vmax of 82.37 nmol/mg of protein/min.
FIGURE 2.

Effects of protein, time, and substrate concentrations on all-trans-retinoic acid production by recombinant human ALDH1A2. Production of retinoic acids was determined with increasing concentrations of purified ALDH1A2 (0–5 μg) (A), variable incubation time (0–60 min) (B), and variable retinal concentrations (0–2 μm) (C) under the conditions described under “Experimental Procedures.” Each data point represents mean ± S.E. of triplicate samples. Errors bars are contained within data points in some instances.
We have reported previously that WIN 18,446 strongly inhibits ALDH1A2 in vitro (13). It is known that WIN 18,446 also inhibits another aldehyde dehydrogenase, ALDH2 (the main enzyme involved in alcohol metabolism), causing the “disulfiram reaction” when a subject consumes alcohol while taking WIN 18,446 that is the basis for aversion therapy in treatment of alcoholism (14). To determine whether the mechanism of inhibition of ALDHs by WIN 18,446 is similar to that of disulfiram, we compared disulfiram and WIN 18,446 for their inhibitory effects on ALDH1A2. Both WIN 18,446 and disulfiram strongly inhibited retinoic acid production by ALDH1A2 (Fig. 3, A and B). However, only disulfiram-induced inhibition was reversed by addition of DTT, suggesting that the mechanism of inhibition for these two compounds is distinct.
FIGURE 3.

WIN 18,446 is an irreversible inhibitor of ALDH1A2. Retinoic acid production by ALDH1A2 was determined using 1 μg of ALDH1A2, 5 μm all-trans-retinal and 10 μm disulfiram (A) or 10 μm WIN 18,446 (B) with and without DTT. Each bar represents mean of triplicate samples, and error bars denote S.E. C, time-dependent inhibition of ALDH1A2 by WIN 18,446 was determined by preincubating the enzyme with WIN 18,446 (WIN) for predetermined times in the presence and absence of all-trans-retinal (at-RAL) before initiating the enzyme reaction by addition of substrate and cofactor. Enzyme reactions were carried out at 37 °C for 5 min. Duplicate samples are shown for each data point. D, substrate concentration-dependent ALDH1A2 activity in the presence of increasing concentrations of WIN 18,446. Reactions were carried out at 37 °C for 15 min with 0.2 μg of enzyme. Each data point with error bars represents mean ± S.E. of triplicate samples.
After incubation with WIN 18,446, ALDH1A2 was dialyzed extensively against dialysis buffer to determine whether the inhibition could be reversed. After overnight incubation with WIN 18,446 at 4 °C, ALDH1A2 lost almost all activity (2–3% activity remaining compared with control). Extensive dialysis did not restore ALDH1A2 activity (4–6% of control), suggesting that the inhibition is likely irreversible. To further characterize the inhibitory effects of WIN 18,446, the enzyme was preincubated with the inhibitor for 0–40 min at 37 °C before the enzyme reaction was initiated by adding NAD+ and substrate (Fig. 3C). Most of the inhibition occurred within 5 min of the preincubation period. Although some protection was seen by the presence of the substrate at a concentration twice that of WIN 18,446 during the preincubation period, protection became less pronounced with a prolonged preincubation period. Increasing the concentration of WIN 18,446 in the enzyme reaction decreased both K0.5 and Vmax (Fig. 3D). However, a dose-dependent decrease was seen only for Vmax (82.43, 42.71, and 36.09 versus 19.81 nmol/mg of protein/min) but not for K0.5 (0.296, 0.196, 0.153, and 0.197 μm).
WIN 18,446 Treatment Does Not Cause Significant Toxicity in Mice
To determine effects of WIN 18,446 on spermatogenesis and retinoid metabolism, mice were fed a diet with or without WIN 18,446 or disulfiram. Diets containing WIN 18,446 and disulfiram were well tolerated throughout the 4-week treatment period. No significant clinical symptoms associated with toxicity (hunched posture, poor grooming, weight loss, etc.) were observed. All mice steadily gained weight during the treatment period, although mice fed a WIN 18,446-containing diet showed slower weight gain compared with mice fed control diet (significant differences were seen at 3 and 4 weeks after the diet initiation; data not shown). Fasting blood glucose levels were within the normal range for all treatment groups during the treatment period (data not shown). No gross abnormalities of tissues were detected at the necropsy except that slight paleness of liver was noted in mice treated with WIN 18,446. These mice were found to have increased hepatocellular vacuolation during histological analysis (see below).
Histological Examination of Potential Toxicity Associated with WIN 18,446 Treatment
With the exception of the liver and testes, there were no significant treatment-related lesions noted in the toxicologic histology screen. As expected, the testes were depleted of sperm and precursors (discussed below). In the liver, there was mild microvesicular vacuolation of centrilobular to midzonal hepatocytes in the WIN 18,446-treated animals. In this study, the descriptive diagnosis of hepatic vacuolation (favoring lipid) was scored. To determine whether this vacuolation was associated with increased liver lipids, we measured triglyceride levels and found them to be similar among the three groups (38.2 ± 1.6 mg/g of liver, WIN 18,446 treatment; 33.60 ± 2.9 mg/g of liver, control; 37.5 ± 3.5 mg/g of liver, disulfiram treatment). In contrast, serum triglyceride levels were significantly lower in mice fed a diet containing WIN 18,446 compared with those fed the control or disulfiram diet (100 ± 8.9 versus 164.4 ± 4.3 and 142 ± 12.1 mg/dl). After completion of the tissue retinol and retinyl ester level analysis (see below), the lungs were closely reexamined histologically again for any subtle lesions that may suggest dysfunction secondary to retinoid depletion; none were noted.
WIN 18,446 Treatment Inhibits Spermatogenesis and Reduces Retinoic Acid Synthesis in Mice
Testes were significantly smaller in mice fed WIN 18,446 compared with control, whereas disulfiram treatment did not affect the size of testes (Fig. 4A). Spermatid numbers were significantly lower in WIN 18,446-treated mice compared with control or disulfiram-treated mice (Fig. 4B). As reported previously in rabbits (13), epididymal weight in mice treated with WIN 18,446 was not different from that of control mice (Fig. 4C). Histological analysis showed that WIN 18,446 treatment completely blocked spermatogenesis in mice (Fig. 5A). Testis histology in WIN 18,446-treated mice demonstrated mostly Sertoli cells in the seminiferous tubules with about 5% of tubules containing germ cells or late stage spermatids in a disrupted epithelium (Fig. 5A, right panel). In contrast, mice fed control or disulfiram diets had normal spermatogenesis (Fig. 5A, left panel, and data not shown). In addition, WIN 18,446 treatment significantly reduced all-trans-retinoic acid concentrations in testes compared with control or disulfiram treatment (Fig. 5B). Because of very low levels of retinoic acids found in WIN 18,446-treated mice, each data point for this group was obtained from pooled testes of three to four mice.
FIGURE 4.

WIN 18,446 treatment inhibits spermatogenesis in mice after 4 weeks of treatment. Mice were fed a control diet, WIN 18,446-, or disulfiram-containing diet for 4 weeks, and testis and epididymis were analyzed for weight and spermatid counts. A, weight of testes per mouse. B, spermatid counts per testis. C, weight of epididymis. Mean and S.E. are shown as horizontal bars. ****, p < 0.0001.
FIGURE 5.

WIN 18,446 treatment abolishes mature spermatocytes and decreases intratesticular retinoic acids. Testes were histologically analyzed for spermatogenesis after 4 weeks of treatment (control, WIN 18,446, or disulfiram). A, histology of testis from control (left) and WIN 18,446-treated (right) mice. Original magnification, 200×. Testes were stained with hematoxylin-periodic acid-Schiff. Normal spermatogenesis is seen in control testis, and tubules of WIN 18,446-treated mice exhibited an apparent complete depletion of spermatocytes. B, intratesticular retinoic acids were determined from five to seven mice per treatment group. Because of very low levels of retinoic acids in testis of mice treated with WIN 18,446, intratesticular retinoic acids of mice treated with WIN 18,446 were obtained from pooled testes of three to four mice. Retinoic acid concentrations were calculated by dividing by the number of pooled testes. Horizontal bars represent mean.
WIN 18,446 Treatment Alters Adiposity in Mice
Because WIN 18,446-fed mice gained weight more slowly, we determined whether adiposity differed in these mice compared with control or disulfiram-treated mice. At necropsy, three visceral adipose depots were removed and weighed individually, and visceral adiposity was calculated (Sum of adipose tissue weights/Body weight × 100; Fig. 6A). Individual adipose depots (Fig. 6, B–D) were significantly smaller in the WIN 18,446-treated group compared with either the control and/or the disulfiram-treated group.
FIGURE 6.

WIN 18,446 decreases adipose tissue weight in mice after 4 weeks of treatment. Visceral adipose depots were dissected and weighed. A, visceral adiposity was determined by the Sum of three adipose depots (epididymal, retroperitoneal, and mesenteric fat mass)/Body weight × 100. B, retroperitoneal adipose weight. C, mesenteric adipose weight. D, epididymal adipose weight. Horizontal bars represent mean ± S.E. *, p < 0.05; **, p < 0.01.
During routine examination of histopathology of adipose tissues, the pathologist noted morphological differences (adipocyte size) in some adipose tissue sections. Thus, we determined adipocyte size (retroperitoneal and mesenteric adipose tissue) using computer-assisted morphometry. Adipocyte sizes tended to be larger in control diet-fed mice compared with WIN 18,446- or disulfiram-treated mice, although the differences did not reach a statistical significance (data not shown).
WIN 18,446 Alters Whole Body Retinoid Physiology
To determine whether inhibition of endogenous retinoic acid synthesis by WIN 18,446 influences storage and metabolism of upstream retinoids, we determined retinol and retinyl ester levels in five retinoid target tissues, i.e. liver (storage), lung, kidney (catabolism), adipose tissue, and serum. Liver retinol levels were significantly higher in WIN 18,446-treated mice compared with the control group (Fig. 7A). In contrast, total retinyl ester levels in the liver tended to be lower in WIN 18,446 mice, but the difference was not statistically significant (Fig. 7B). Serum retinol levels were significantly elevated in mice fed a WIN 18,446-containing diet (Fig. 7C). Similarly, serum RBP levels were significantly elevated in mice treated with WIN 18,446 compared with controls (Fig. 7D). Retinyl esters were not detected in any of the serum samples as these were collected from mice fasted overnight. Both retinol and retinyl ester levels were significantly higher in kidney and adipose tissues of mice treated with WIN 18,446 (Table 1). In contrast, both retinol and retinyl ester levels were significantly lower in lungs of WIN 18,446-treated mice compared with those of control or disulfiram-treated mice (Table 1).
FIGURE 7.

WIN 18,446 alters retinoid levels in serum and liver. Retinyl ester and retinol levels were determined at the end of the study period (4 weeks of dietary treatment). A, retinol levels in liver. B, total retinyl ester levels in the liver. C, serum retinol levels. D, serum RBP levels. Horizontal bars represent mean ± S.E. *, < 0.05; **, p < 0.01; ****, p < 0.0001.
TABLE 1.
Summary of tissue retinoid levels from experiment 1
Values are mean ± S.E. WIN, WIN 18,446.
| Tissue | Retinol |
Total retinyl esters |
||||
|---|---|---|---|---|---|---|
| Control | WIN | Disulfiram | Control | WIN | Disulfiram | |
| μg/g tissue | nmol/g tissue | |||||
| Lung | 2.86 ± 0.7a | 0.41 ± 0.1b | 2.19 ± 0.5a | 308.1 ± 36.8a | 10.1 ± 0.8b | 139.8 ± 15.2c |
| Kidneys | 0.20 ± 0.01a | 0.24 ± 0.01b | 0.22 ± 0.01a,b | 0.1 ± 0.02a | 0.2 ± 0.03b | 0.1 ± 0.01a |
| Adipose (retroperitoneal) | 0.20 ± 0.02a | 0.33 ± 0.03b | 0.17 ± 0.01a | 2.1 ± 0.2a | 3.2 ± 0.3b | 2.0 ± 0.2a |
a,b,c Values with different superscript characters represent statistically significant differences between them based on a post hoc test with adjustment with multiple comparisons (p < 0.05). Comparisons were made among the treatment groups of the same duration (4 or 8 weeks).
Because WIN 18,446 inhibits retinoic acid biosynthesis, we examined whether tissue retinoic acid levels differ among treatment groups by examining expression levels of two retinoic acid-responsive genes that are involved in retinoid metabolism, Lrat and Cyp26a1, by quantitative RT-PCR in liver and lung (Fig. 8). Both Lrat and Cyp26a1 levels were significantly lower in mice fed a WIN 18,446-containing diet.
FIGURE 8.

Expression of retinoic acid-responsive genes was reduced in WIN 18,446-treated mice. Relative expression of retinoic acid-responsive genes, Lrat and Cyp26a1, was determined using quantitative RT-PCR and normalized by hypoxanthine phosphoribosyltransferase (HPRT). Normalized expression levels were then compared with that of control. Expression of Lrat (A) and Cyp26a1 (B) in the liver is shown. C, Lrat expression in lung. Each bar represents mean and error bars denote S.E. *, p < 0.05; ***, p < 0.001.
WIN 18,446-induced Inhibition of Spermatogenesis Is Not Prevented by Simultaneous Treatment with Retinoic Acids
In a second experiment, we tested whether 1) WIN 18,446-induced “inhibition” of spermatogenesis can be prevented by concomitant treatment with retinoic acids and 2) WIN 18,446 cessation would reverse blockage in spermatogenesis. In addition to all-trans-retinoic acid, 13-cis-retinoic acid was chosen for this purpose because this isomer was reported to be a poor substrate for CYP26 enzymes (24, 25) that may create a blood-testis barrier for retinoic acid uptake (10). We hypothesized that 13-cis-retinoic acid might cross this barrier more efficiently, providing all-trans-retinoic acids through isomerization within the testes. Histological analysis showed that the simultaneous administration of retinoic acids (all-trans or 13-cis) with WIN 18,446 (Fig. 9A, panels 1 and 2) did not prevent inhibition of spermatogenesis by WIN 18,446. Testes lacked any mature spermatocytes after 4 weeks of WIN 18,446 treatment or concomitant retinoic acid treatment. The sizes of testes of retinoic acid + WIN 18,446-treated mice were similar to those of WIN 18,446-treated mice (Fig. 9, B and C). When WIN 18,446 treatment was followed by a control diet for 4 weeks, the testes became larger, but spermatogenesis did not recover completely (Fig. 9C). Histological analysis showed some recovery of spermatogenesis based on the presence of occasional round spermatids. However, in most tubules, mature elongated spermatids were not observed (Fig. 9A, panel 3).
FIGURE 9.

Simultaneous retinoic acid treatment does not rescue inhibition of spermatogenesis by WIN 18,446. Mice were fed control diet or control diet containing WIN 18,446, WIN 18,446 + all-trans-retinoic acid, or WIN 18,446 + 13-cis-retinoic acid for 4 weeks. For the reversibility study, mice were fed either control diet for 8 weeks or a diet containing WIN 18,446 for 4 weeks followed by a control diet for 4 weeks. A, histology of testes from WIN 18,446-treated mice given all-trans-retinoic acid (panel 1) or 13-cis-retinoic acid (panel 2), WIN 18,446 for 4 weeks followed by control diet for 4 weeks (panel 3), or control diet for 8 weeks (panel 4; control of panel 3). In panels 1 and 2, seminiferous tubules are depleted of mitotic and meiotic germ cells; Sertoli cells (SC) are common with occasional spermatogonia (Sg). In panel 3, spermatogenesis is recovering, although a few mature elongating spermatozoa are present in some tubules (not seen in this image); round spermatids (RS) are most often the most differentiated germ cell present. In panel 4, complete spermatogenesis is demonstrated, including elongating spermatids (ES). Original magnification, 400× for panels 1 and 2 and 250× for panels 3 and 4. B, relative size of testis of mice fed a control diet or a control diet containing WIN 18,446 or WIN 18,446 (WIN) + all-trans-retinoic acid. C, testes were weighed at the end of the treatment period (4 or 8 weeks (wks)). D, serum retinol levels were determined at the end of the study period (4 or 8 weeks). Horizontal bars represent mean ± S.E. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001. atRA, all-trans-retinoic acid; 13cRA, 13-cis-retinoic acid.
WIN 18,446-induced Changes in Retinoid Metabolism Are Partially Prevented by Concomitant Treatment with Retinoic Acids
Retinoid levels of animals treated with retinoic acids were altered during the treatment period in a tissue-dependent manner. Serum retinol levels of retinoic acid-treated animals were intermediate between WIN 18,446 treatment and control, whereas the cessation of WIN 18,446 treatment normalized serum retinol levels to those of control (Fig. 9D). Interestingly, although liver retinyl ester levels of retinoic acid + WIN 18,446-treated animals were similar to those of WIN 18,446-treated animals, lung retinyl ester levels of retinoic acid-treated animals were similar to those of control, i.e. significantly higher than that of WIN 18,446-treated animals (Table 2). This is in contrast to the observation that after cessation of WIN 18,446 treatment liver retinyl ester levels of the recovery group were similar to those of the control group, whereas lung retinyl esters were significantly lower in the recovery group compared with the control group (Table 2).
TABLE 2.
Summary of tissue retinoid levels from experiment 2
Values are mean ± S.E. WIN, WIN 18,446; atRA, all-trans-retinoic acid; 13cRA, 13-cis-retinoic acid; wk, weeks.
| Tissue | Total retinyl esters |
|||||
|---|---|---|---|---|---|---|
| Control | WIN 18,446 | WIN + atRA | WIN + 13cRA | Control (8 wk) | Recovery (WIN, 4 wk → control, 4 wk) | |
| nmol/g tissue | ||||||
| Liver | 601 ± 28.9a | 398 ± 24.2b | 364 ± 29.9b | 422 ± 41.1b | 743 ± 72.2 | 670 ± 50.6 |
| Lung | 212 ± 19.6a | 7 ± 3.8b | 417 ± 62.8c | 278 ± 30.0a,c | 121.3 ± 13.1a | 71 ± 6.3b |
a,b,c Values with different superscript characters represent statistically significant differences between them based on a post hoc test with adjustment with multiple comparisons (p < 0.05). Comparisons were made among the treatment groups of the same duration (4 or 8 weeks).
WIN 18,446-treated animals had lower weight gain compared with controls (Fig. 10A), and the weights of animals fed either 13-cis- or all-trans-retinoic acid were in between those of control and WIN 18,446-treated animals, suggestive of partial rescue from the WIN 18,446 effect. This trend was also reflected in visceral adipose tissue weight except that 13-cis-retinoic acid treatment was not as effective in normalizing adiposity as all-trans-retinoic acid (Fig. 11A). Serum triglyceride levels were lower in WIN 18,446-treated animals compared with control, whereas simultaneous treatment with all-trans-retinoic acid increased the levels closer to those of control (Fig. 10B). Compared with all-trans-retinoic acid treatment, 13-cis-retinoic acid treatment was not as effective in increasing serum triglyceride levels to those of control. Because mice lacking Aldh1a1, one of the retinal dehydrogenases found in adipose tissue, were reported to resist diet-induced obesity partly due to increased thermogenesis in white adipose tissue (26), we examined epididymal adipose tissue, one of the main visceral white adipose tissues, for UCP-1 expression by immunohistochemistry. UCP-1 is a marker of brown adipose tissue and was shown to be increased in white adipose tissue of Aldh1a1−/− animals (26). Although no UCP-1 protein was detected in mice fed a control diet, mice treated with WIN 18,446 showed significant UCP-1 expression (Fig. 11, B and C). Interestingly, concomitant all-trans-retinoic acid treatment abolished UCP-1 expression, whereas 13-cis-retinoic acid treatment slightly reduced the UCP-1 expression compared with the WIN 18,446-only treatment group.
FIGURE 10.
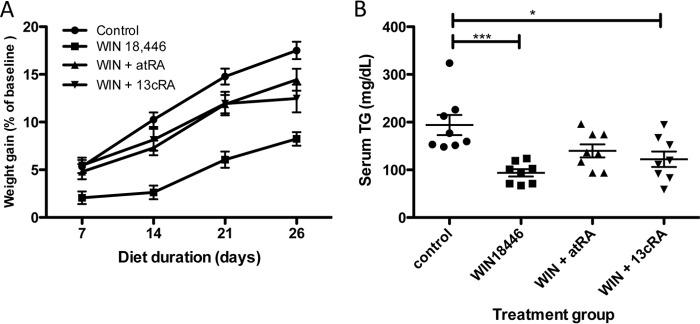
Concomitant treatment with retinoic acids partially reverses WIN 18,446 effects. Mice were fed a control diet or a control diet containing WIN 18,446 or WIN 18,446 (WIN) + all-trans-retinoic acid or WIN 18,446 + 13-cis-retinoic acid for 4 weeks. A, weight gain was determined by weekly measurements. B, serum triglyceride (TG) levels at the end of the study period. Horizontal bars represent mean ± S.E. *, p < 0.05; ***, p < 0.001. atRA, all-trans-retinoic acid; 13cRA, 13-cis-retinoic acid.
FIGURE 11.

Concomitant retinoic acid treatment partially reverses WIN 18,446-induced suppression of adiposity. Mice were fed a control diet or a diet containing WIN 18,446, WIN 18,446 (WIN) + all-trans-retinoic acid, or WIN 18,446 + 13-cis-retinoic acid for 4 weeks. At the end of diet period, adipose tissues were dissected, weighed, and analyzed for UCP-1 expression. A, total weight of visceral adipose tissue (mesenteric, epididymal, and retroperitoneal adipose tissue). Horizontal bars represent mean ± S.E. B, UCP-1 expression levels in epididymal adipose tissues were scored by a pathologist blinded to the treatment group. Horizontal bars represent mean ± S.E. C, immunochemical staining of UCP-1 in epididymal fat (white adipose tissue (WAT)) and brown adipose tissue (BAT). Brown adipose tissue from the same animal was used as a staining control. atRA, all-trans-retinoic acid; 13cRA, 13-cis-retinoic acid; AU, arbitrary units.
DISCUSSION
We have reported previously that the bisdichloroacetyldiamine WIN 18,446 inhibits retinoic acid biosynthesis by ALDH1A2 using a cell line overexpressing this enzyme and that this is the likely mechanism by which spermatogenesis is inhibited (13). In this report, we used purified enzyme to further characterize ALDH1A2 and its inhibition by WIN 18,446 in addition to determining the effects of WIN 18,446 on general health and retinoid metabolism in mice.
Human ALDH1A2 kinetics data fitted best with the Hill equation, suggesting possible cooperativity in substrate binding (Fig. 2C). A structural study of rat ALDH1A2 suggests that the enzyme exists as a homotetramer and that the substrate access channel is formed by two homodimers “embracing” each other (27). However, it is not known whether all four substrate binding sites can be occupied at the same time and how the binding of one site might affect conformation of the enzyme and thus other binding sites. The Hill coefficient calculated from our kinetics data is 1.7, suggesting that there is a positive cooperativity regarding the substrate binding to the enzyme. Human ALDH1A2 had similar kinetic activity to rat ALDH1A2, which was reported to have a Km of 0.7 μm and Vmax of 105 nmol/min/mg of protein, although the rat enzyme did not show an allosteric relationship to retinal (28).
We found that the reduction of sulfhydryl groups is essential for enzyme activity as evidenced by the fact that either DTT or TCEP is necessary for the enzyme activity. The ability of DTT to overcome inhibition of ALDH1A2 by disulfiram suggests that the inhibitory effect of disulfiram is likely related to an available sulfhydryl group in the enzyme. In contrast, the mechanism of WIN 18,446 inhibition is likely irreversible binding of this compound to the enzyme or the induction of an irreversible change to the enzyme that results in permanent or near permanent inhibition of enzyme activity. In support of this hypothesis, we found that inhibition by WIN 18,446 is time-dependent (Fig. 3C) and cannot be reversed by extensive dialysis. Thus, we speculate that the strong inhibitory effects of WIN 18,446 on spermatogenesis stem from its ability to irreversibly disable ALDH1A2 activity, necessitating new enzyme synthesis to catalyze retinoic acid formation in germ cells.
Although 4-week treatment with WIN 18,446 was sufficient to block spermatogenesis (Fig. 5), we did not observe other gross or histological abnormalities in treated animals except for mild lipidosis that was completely reversed following the cessation of WIN 18,446 treatment. This together with the fact that WIN 18,446-treated testis contained small amounts of retinoic acid suggests that WIN 18,446 does not completely inhibit all retinoic acid-synthesizing enzymes. In adult testes, ALDH1A1 is expressed in Leydig and Sertoli cells, whereas ALDH1A2 is expressed in germ cells (9). Thus, it would be interesting to find out how the residual retinoic acid detected in the testis is synthesized and whether WIN 18,446 can inhibit other known retinal dehydrogenases.
Although we did not detect any vitamin A deficiency symptoms in mice treated with WIN 18,446, we observed variable changes in retinoid metabolism in the target tissues, including serum, liver, lung, and adipose tissues (Tables 1 and 2). Vitamin A is unique in that a large quantity of it can be stored in a specialized storage organ, the liver (4, 6). In between meals, the liver supplies fairly constant amounts of vitamin A to extrahepatic tissues through secretion of retinol bound to RBP (6, 29). However, it is not known what extrahepatic signaling regulates this homeostatic retinol secretion from the liver. Both retinoic acids and apo-RBP have been suggested to be potential signals (30, 31). Compared with vitamin A-deficient rats, rats fed a vitamin A-deficient diet but supplemented with retinoic acids have lower circulating levels of retinol likely due to sparing of the liver vitamin A reserve (30). Similarly, injection of supraphysiological levels (100 μg) of retinoic acid suppresses circulating levels of retinol in rats with and without renal failure (31). However, a lower dose of retinoic acid (10 μg) did not induce changes in circulating retinol levels, whereas injection of apo-RBP increased serum retinol. Thus, it was suggested that physiological levels of serum retinoic acid concentrations do not influence retinol secretion from the liver and that instead circulating levels of apo-RBP likely supply the signal (31).
Our studies provide a unique opportunity to consider what effects the low levels of retinoic acids in liver and extrahepatic tissues might have on circulating retinol concentrations. We detected increased levels of circulating retinol and RBP in mice treated with WIN 18,446 compared with control (Figs. 7, C and D, and 9D). Four weeks after the cessation of the WIN 18,446 treatment, circulating retinol levels were decreased to the levels seen in control animals (Fig. 9D). Thus, we hypothesize that retinoic acid deficiency in liver and/or extrahepatic tissues increased mobilization of retinoid from the liver, which was manifested as increased concentrations of circulating retinol and RBP and decreased levels of retinyl esters in the liver of WIN 18,446-treated animals. Interestingly, serum retinol levels of animals treated with retinoic acids were in between those of the control and WIN 18,446 treatment groups, suggesting that 5 mg of retinoic acid/kg of diet was not sufficient to “spare” liver retinoid reserves when retinoic acid synthesis is actively suppressed. These results indicate that WIN 18,446 may be a useful tool to determine effects of retinoic acids on retinoid homeostasis in the body. In this regard, changes in retinoid metabolism in the lung and liver following various treatments are intriguing.
Although the liver is the major storage organ for vitamin A, the lung contains high levels of retinyl esters in mice (32, 33) as our data support. However, WIN 18,446 treatment nearly depleted retinyl esters from the lung but not from the liver (Fig. 7B and Tables 1 and 2). In addition, cessation of the treatment restored levels of liver retinyl esters to control levels but did not fully restore those in lung. Most interestingly, co-treatment with retinoic acids and WIN 18,446 increased (all-trans-retinoic acid co-treatment) or spared (13-cis-retinoic acid co-treatment) lung retinyl esters, whereas liver retinyl ester storage was still reduced under the same treatment (Table 2). We hypothesize that these differences may be due to the fact that retinyl esters stored in the liver are used by extrahepatic tissues, whereas those in the lung are mainly used to meet local pulmonary needs. It also appears that dietary retinoic acid is delivered to and utilized more efficiently by the lung, thus sparing retinyl esters when simultaneous treatment with WIN 18,446 and retinoic acid is given (control versus WIN 18,446 versus WIN + all-trans-retinoic acid in Table 2). Further studies are needed to determine why the lung stores such high concentrations of retinyl esters and what the consequences are of long term depletion of this storage. For both study 1 and study 2, we did not detect any morphologic changes in lung after 4 weeks of WIN 18,446 treatment. However, a longer term study with complete depletion of retinoid storage in the lung will be needed to answer these questions.
Perhaps the most dramatic effect of WIN 18,446 administration is the complete abrogation of spermatogenesis observed with this agent. Indeed, WIN 18,446 has been extensively tested as a potential male contraceptive because of its potent and reversible effect on sperm production. Likely, this effect is mediated by the inhibition of spermatogonial differentiation in particular the transition from A to A1 spermatogonia (2); however, other investigators have suggested additional roles for retinoic acid in spermiogenesis and spermiation (7). In our second experiment, we sought to determine whether the effect of WIN 18,446 treatment on spermatogenesis could be prevented by concomitant administration of either all-trans-retinoic acid or 13-cis-retinoic acid, both of which are present in the murine testis (34). Our result confirmed previous findings that dietary retinoic acids are not sufficient to support spermatogenesis in vitamin A deficiency (11, 12). This finding is likely due to the unusual ability of the testes to exclude retinoic acid delivered from the blood. Previous studies with radiolabeled retinoic acid demonstrated that, in contrast to most other tissues, less than 1% of intratesticular retinoic acid is derived from circulating retinoic acid (10) probably due to the high expression of Cyp26 in the peritubular myoid cells (9). This creates a barrier to retinoic acid uptake from the circulation into tubules that is potentially essential for the prevention of prepubertal spermatogenesis by allowing germ cells to develop based on in situ retinoic acid synthesis and degradation regulated by retinal dehydrogenases and CYP26 enzymes. This barrier would provide tight spatial and temporal regulation of retinoic acid within the seminiferous tubule during the various stages of the spermatogenic cycle. Our observation that 13-cis-retinoic acid supplementation is not able to reverse spermatogenesis inhibition is interesting as this isomer was reported to be a poor substrate for CYP26 enzymes (24, 25). We chose this isomer to potentially circumvent the blood-testis barrier. Further studies are needed to address whether 13-cis-retinoic acid can cross the blood-testis barrier and how efficiently 13-cis-retinoic acid is isomerized to all-trans-retinoic acid in the testes.
We did not observe histological evidence of full recovery in spermatogenesis after 4 weeks of WIN 18,446 cessation, although the testes became larger compared with WIN 18,446-treated animals (Fig. 9C). We believe that this is likely due to insufficient recovery time from the WIN 18,446 treatment as we and others have shown that the blockage in spermatogenesis is reversible given sufficient time (13, 14, 35). In our previous study with rabbits, we observed mature spermatozoa in ejaculates after 16 weeks of drug cessation (13), and other studies reported recovery times of greater than 4 weeks in various animals, including humans (14, 35, 36).
The observed effects of WIN 18,446 on lipid and adipose physiology are also notable with animals treated with WIN 18,446 displaying reductions in visceral adiposity in the mesenteric, retroperitoneal, and epididymal areas when fed a low fat control diet (AIN93M) for a relatively short period of time (4 weeks). These findings are consistent with data from the Aldh1a1−/− mouse model, which demonstrates decreased abdominal fat and body weight (26, 37). In addition, we observed increased brown adipose tissue-like conversion of white adipose tissue (defined by increased expression of UCP-1) in animals treated with WIN 18,446 (Fig. 11). The finding that these phenotypes are induced by inhibiting retinoic acid synthesis in adult wild type animals suggests that the effects seen in the Aldh1a1−/− mice are not solely developmentally driven. Indeed, our findings suggest that small molecule inhibitors directed at inhibiting retinoic acid synthesis may have potential benefit for the treatment of obesity. One concern about such an approach to the treatment of obesity, however, may be our observation of microvesicular lipidosis in some of the liver specimens in WIN 18,446-treated animals. Vacuolation of hepatocytes may be due to lipids, phospholipidosis, and glycogen accumulation, all of which are removed from the tissues during routine processing and embedding, leaving vacuoles behind (38). On H&E-stained sections, medium to large, clear, round cytoplasmic vacuoles that occasionally displaced the nucleus within centrilobular to midzonal hepatocytes were noted. These lipid-like vacuoles are in contrast to glycogen-like vacuoles where there is irregular lacy intracytoplasmic clearing and no nuclear displacement. Phospholipidosis in rodent livers can be induced by a number of cationic amphiphilic drugs, and affected hepatocytes have variably sized clear cytoplasmic vacuoles that cannot be unequivocally distinguished from lipid based on morphology alone (38, 39). Although we did not see elevated liver triglyceride levels in these animals, it is possible that this may have appeared with a longer duration of treatment. Interestingly, we saw a decrease in serum triglyceride levels in WIN 18,446-treated mice compared with control diet-fed mice. Thus, it is possible that WIN 18,446 treatment decreases adiposity by decreasing trafficking of lipid from the liver to adipose tissue. If this is the case, blocking retinoic acid synthesis to treat obesity may not be realistic as it would lead to fatty liver. Because our primary interest was in the effects of retinoic acid reduction on spermatogenesis, we did not thoroughly investigate the metabolic aspects of retinoic acid reduction or changes in retinoic acid levels in adipose tissues directly. Thus, further study will be needed to determine whether WIN 18,446 could be used to prevent obesity under high fat diet feeding in a long term study and whether the adipose phenotype seen in our study was due to inhibition of retinoic acid synthesis.
In summary, we present a novel paradigm for acquired retinoic acid deficiency using the bischloroacetyldiamine WIN 18,446. We demonstrate that WIN 18,446 potently suppresses retinoic acid synthesis by ALDH1A2 in vitro. In vivo, WIN 18,446 administration markedly reduced retinoic acid concentrations, leading to increased serum retinol, a cessation of spermatogenesis, and reductions in visceral adiposity. These findings shed new light onto basic retinoid physiology and may have implication for the development of drugs for the treatment of obesity and as male contraceptives.
Acknowledgments
We thank Brian Johnson and Cara Appel of the University of Washington Comparative Pathology/Histology and Imaging Core Research Laboratory for immunohistochemistry and image analysis.
This work was supported, in whole or in part, by National Institutes of Health Grants U01 HD060488 and U54 HD42454 from the Eunice Kennedy Shriver NICHD (to J. K. A.).
- ALDH
- aldehyde dehydrogenase
- RA
- retinoic acid
- RBP
- retinol-binding protein
- TCEP
- tris(2-carboxyethyl)phosphine
- UCP-1
- uncoupling protein-1
- Lrat
- lecithin-retinol acyltransferase.
REFERENCES
- 1. Clagett-Dame M., Knutson D. (2011) Vitamin A in reproduction and development. Nutrients 3, 385–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Griswold M. D., Hogarth C. A., Bowles J., Koopman P. (2012) Initiating meiosis: the case for retinoic acid. Biol. Reprod. 86, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wolbach S. B., Howe P. R. (1925) Tissue changes following deprivation of fat-soluble A vitamin. J. Exp. Med. 42, 753–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vogel S., Gamble M. V., Blaner W. S. (1999) in Retinoids. The Biochemical and Molecular Basis of Vitamin A and Retinoid Action (Nau H., Blaner W. S., eds) pp. 31–95, Springer, New York [Google Scholar]
- 5. Quadro L., Blaner W. S., Salchow D. J., Vogel S., Piantedosi R., Gouras P., Freeman S., Cosma M. P., Colantuoni V., Gottesman M. E. (1999) Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 18, 4633–4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blaner W. S., Olson J. A. (1994) in The Retinoids: Biology, Chemistry, and Medicine (Sporn M. B., Roberts A. B., Goodman D. S., eds) pp. 229–256, Raven Press, New York [Google Scholar]
- 7. Packer A. I., Wolgemuth D. J. (1999) in Retinoids: The Biochemical and Molecular Basis of Vitamin A and Retinoid Action (Nau H., Blaner W. S., eds) pp. 347–368, Springer, Berlin [Google Scholar]
- 8. Wu J. W., Wang R. Y., Guo Q. S., Xu C. (2008) Expression of the retinoic acid-metabolizing enzymes RALDH2 and CYP26b1 during mouse postnatal testis development. Asian J. Androl. 10, 569–576 [DOI] [PubMed] [Google Scholar]
- 9. Vernet N., Dennefeld C., Rochette-Egly C., Oulad-Abdelghani M., Chambon P., Ghyselinck N. B., Mark M. (2006) Retinoic acid metabolism and signaling pathways in the adult and developing mouse testis. Endocrinology 147, 96–110 [DOI] [PubMed] [Google Scholar]
- 10. Kurlandsky S. B., Gamble M. V., Ramakrishnan R., Blaner W. S. (1995) Plasma delivery of retinoic acid to tissues in the rat. J. Biol. Chem. 270, 17850–17857 [DOI] [PubMed] [Google Scholar]
- 11. Howell J. M., Thompson J. N., Pitt G. A. (1963) Histology of the lesions produced in the reproductive tract of animals fed a diet deficient in vitamin A alcohol but containing vitamin A acid. I. The male rat. J. Reprod. Fertil. 5, 159–167 [DOI] [PubMed] [Google Scholar]
- 12. Huang H. F., Hembree W. C. (1979) Spermatogenic response to vitamin A in vitamin A deficient rats. Biol. Reprod. 21, 891–904 [DOI] [PubMed] [Google Scholar]
- 13. Amory J. K., Muller C. H., Shimshoni J. A., Isoherranen N., Paik J., Moreb J. S., Amory D. W., Sr., Evanoff R., Goldstein A. S., Griswold M. D. (2011) Suppression of spermatogenesis by bisdichloroacetyldiamines is mediated by inhibition of testicular retinoic acid biosynthesis. J. Androl. 32, 111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heller C. G., Moore D. J., Paulsen C. A. (1961) Suppression of spermatogenesis and chronic toxicity in men by a new series of bis(dichloroacetyl) diamines. Toxicol. Appl. Pharmacol. 3, 1–11 [DOI] [PubMed] [Google Scholar]
- 15. Hartmann S., Froescheis O., Ringenbach F., Wyss R., Bucheli F., Bischof S., Bausch J., Wiegand U. W. (2001) Determination of retinol and retinyl esters in human plasma by high-performance liquid chromatography with automated column switching and ultraviolet detection. J. Chromatogr. B Biomed. Sci. Appl. 751, 265–275 [DOI] [PubMed] [Google Scholar]
- 16. Fierce Y., de Morais Vieira M., Piantedosi R., Wyss A., Blaner W. S., Paik J. (2008) In vitro and in vivo characterization of retinoid synthesis from β-carotene. Arch. Biochem. Biophys. 472, 126–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Paik J., Blaner W. S., Swisshelm K. (2005) Cis-retinol dehydrogenase: 9-cis-retinol metabolism and its effect on proliferation of human MCF7 breast cancer cells. Exp. Cell Res. 303, 183–196 [DOI] [PubMed] [Google Scholar]
- 18. Amann R. P. (1986) Detection of alterations in testicular and epididymal function in laboratory animals. Environ. Health Perspect. 70, 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petersen P. M., Giwercman A., Pakkenberg B. (1999) Stereological methods as efficient and unbiased tools to quantitate structures in the testis. Scand. J. Work Environ. Health 25, Suppl. 1, 31–33 [PubMed] [Google Scholar]
- 20. Folch J., Lees M., Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 21. Paik J., Fierce Y., Drivdahl R., Treuting P. M., Seamons A., Brabb T., Maggio-Price L. (2010) Effects of murine norovirus infection on a mouse model of diet-induced obesity and insulin resistance. Comp. Med. 60, 189–195 [PMC free article] [PubMed] [Google Scholar]
- 22. Paik J., Vogel S., Piantedosi R., Sykes A., Blaner W. S., Swisshelm K. (2000) 9-cis-Retinoids: biosynthesis of 9-cis-retinoic acid. Biochemistry 39, 8073–8084 [DOI] [PubMed] [Google Scholar]
- 23. Arnold S. L., Amory J. K., Walsh T. J., Isoherranen N. (2012) A sensitive and specific method for measurement of multiple retinoids in human serum with UHPLC-MS/MS. J. Lipid Res. 53, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taimi M., Helvig C., Wisniewski J., Ramshaw H., White J., Amad M., Korczak B., Petkovich M. (2004) A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J. Biol. Chem. 279, 77–85 [DOI] [PubMed] [Google Scholar]
- 25. White J. A., Ramshaw H., Taimi M., Stangle W., Zhang A., Everingham S., Creighton S., Tam S. P., Jones G., Petkovich M. (2000) Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc. Natl. Acad. Sci. U.S.A. 97, 6403–6408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kiefer F. W., Vernochet C., O'Brien P., Spoerl S., Brown J. D., Nallamshetty S., Zeyda M., Stulnig T. M., Cohen D. E., Kahn C. R., Plutzky J. (2012) Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat. Med. 18, 918–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamb A. L., Newcomer M. E. (1999) The structure of retinal dehydrogenase type II at 2.7 Å resolution: implications for retinal specificity. Biochemistry 38, 6003–6011 [DOI] [PubMed] [Google Scholar]
- 28. Wang X., Penzes P., Napoli J. L. (1996) Cloning of a cDNA encoding an aldehyde dehydrogenase and its expression in Escherichia coli. Recognition of retinal as substrate. J. Biol. Chem. 271, 16288–16293 [DOI] [PubMed] [Google Scholar]
- 29. Soprano D. R., Blaner W. S. (1994) in The Retinoids: Biology, Chemistry, and Medicine (Sporn M. B., Roberts A. B., Goodman D. S., eds) pp. 257–282, Raven Press, New York, NY [Google Scholar]
- 30. Keilson B., Underwood B. A., Loerch J. D. (1979) Effects of retinoic acid on the mobilization of vitamin A from the liver in rats. J. Nutr. 109, 787–795 [DOI] [PubMed] [Google Scholar]
- 31. Gerlach T. H., Zile M. H. (1991) Effect of retinoic acid and apo-RBP on serum retinol concentration in acute renal failure. FASEB J. 5, 86–92 [DOI] [PubMed] [Google Scholar]
- 32. Quadro L., Blaner W. S., Hamberger L., Van Gelder R. N., Vogel S., Piantedosi R., Gouras P., Colantuoni V., Gottesman M. E. (2002) Muscle expression of human retinol-binding protein (RBP). Suppression of the visual defect of RBP knockout mice. J. Biol. Chem. 277, 30191–30197 [DOI] [PubMed] [Google Scholar]
- 33. Clugston R. D., Jiang H., Lee M. X., Berk P. D., Goldberg I. J., Huang L. S., Blaner W. S. (2013) Altered hepatic retinyl ester concentration and acyl composition in response to alcohol consumption. Biochim. Biophys. Acta 1831, 1276–1286 [PubMed] [Google Scholar]
- 34. Kane M. A., Chen N., Sparks S., Napoli J. L. (2005) Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem. J. 388, 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Munson L., Chassy L. M., Asa C. (2004) Efficacy, safety and reversibility of bisdiamine as a male contraceptive in cats. Theriogenology 62, 81–92 [DOI] [PubMed] [Google Scholar]
- 36. Drobeck H. P., Coulston F. (1962) Inhibition and recovery of spermatogenesis in rats, monkeys, and dogs medicated with bis(dichloroacetyl)diamines. Exp. Mol. Pathol. 1, 251–274 [DOI] [PubMed] [Google Scholar]
- 37. Reichert B., Yasmeen R., Jeyakumar S. M., Yang F., Thomou T., Alder H., Duester G., Maiseyeu A., Mihai G., Harrison E. H., Rajagopalan S., Kirkland J. L., Ziouzenkova O. (2011) Concerted action of aldehyde dehydrogenases influences depot-specific fat formation. Mol. Endocrinol. 25, 799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thoolen B., Maronpot R. R., Harada T., Nyska A., Rousseaux C., Nolte T., Malarkey D. E., Kaufmann W., Küttler K., Deschl U., Nakae D., Gregson R., Vinlove M. P., Brix A. E., Singh B., Belpoggi F., Ward J. M. (2010) Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol. Pathol. 38, 5S–81S [DOI] [PubMed] [Google Scholar]
- 39. Asaoka Y., Togashi Y., Imura N., Sai T., Miyoshi T., Miyamoto Y. (2013) Immunohistochemistry of LAMP-2 and adipophilin for phospholipidosis in liver and kidney in ketoconazole-treated mice. Exp. Toxicol. Pathol. 65, 817–823 [DOI] [PubMed] [Google Scholar]