

Abstract
The role for the inhibitors of differentiation (Ids) proteins in melanomagenesis has been poorly explored. In other cell types, Ids have been shown to contribute to cell proliferation, migration and angiogenesis and, along with a number of other genes, are direct downstream targets of the transforming growth factor (TGF)-β pathway. Expression of Smad7, which suppress TGF-β signaling, or synthetic TGF-β inhibitors, was shown to potently suppress melanomagenesis. We found that endogenous Id2, Id3 and Id4 expression was elevated in 1205Lu versus 1205Lu cells constitutively expressing Smad7, indicating Ids may play a role in melanomagenesis. Therefore, the effects of Tet-inducible expression of Id2, Id3 or Id4 along with Smad7 in TGF-β-dependent 1205Lu human melanoma cells were explored in vitro and in vivo. 1205Lu cells formed subcutaneous tumors in athymic mice, whereas cells expressing Smad7 failed to form tumors. However, 1205Lu cells expressing Smad7 along with doxycycline-induced Id2, Id3 or Id4 were able to overcome the potent tumorigenic block mediated by S7, to varying degrees. Conversely, Id small interfering RNA knockdown suppressed anchorage-independent growth of melanoma. Histology of tumors from 1205Lu cells expressing Smad7 + Id4 revealed an average of 31% necrosis, compared with 5.2% in tumors from 1205Lu with vector only. Downstream, Ids suppressed cyclin-dependent kinase inhibitors, and re-upregulated invasion and metastasis-related genes matrix metalloproteinase 2 (MMP2), MMP9, CXCR4 and osteopontin, shown previously to be downregulated in response to Smad7. This study shows that Id2, Id3 and Id4 are each able to overcome TGF-β dependence, and establish a role for Ids as key mediators of TGF-β melanomagenesis.
Summary
Suppression of the cyclin-dependent kinase inhibitor p15Ink4b and upregulation of the tumor invasion and metastasis genes MMP2, MMP9, CXCR4 and osteopontin by Id2, Id3 or Id4 contribute to melanomagenesis.
Introduction
Different TGF-β family members are involved in diverse cellular functions (1). Smads 2 and 3 are receptor-regulated Smads and propagate the TGF-β signal through interaction with the co-Smad, Smad4. Smads 6 and 7 are inhibitor Smads and function to repress the TGF-β signal by competing with receptor-regulated Smads for the receptor (TβR) (1). In normal cells, TGF-β induces phosphorylation of Smads 2 and 3, which results in their association with Smad4 and, in turn, inhibits Id synthesis (2,3). It has been suggested that Id de-regulation is a contributing factor to cancer initiation and progression (3). In early-stage melanomagenesis, TGF-β potently inhibits growth, whereas in late-stage melanoma, cells may secrete high levels of TGF-β, to which cells become desensitized and eventually use as a positive growth signal (4,5). In malignant melanoma, TGF-β overproduction correlates with increased tumor thickness and disease progression (6). In late-stage disease, TGF-β is also associated with a significantly decreased survival time and suppression of the immune response (6,7). The switch from TGF-β growth inhibition to growth promotion may somehow be linked to inhibitors of differentiation (Ids), which are typically suppressed by TGF-β in normal cells, although not in all malignant melanomas.
Ids are a small family of helix-loop-helix factors, which lack a basic domain and the ability to associate directly with DNA. Ids are believed to function primarily by sequestration of other factors, including certain basic helix-loop-helix, E-twenty six and retinoblastoma proteins, thereby acting as dominant-negative transcription factors. In this way, Ids regulate a myriad of cellular functions including cell cycle progression and proliferation, migration, angiogenesis and invasion while simultaneously inhibiting differentiation (8,9). Id expression has been found upregulated in many types of cancer, including those of the breast, pancreas, ovaries, and head and neck; implicating Ids as cooperating oncogenes (3). Id2 shows some association with melanoma, whereas the roles of Id3 and Id4 have not been well examined, begging the question of the potential role of these Ids in this disease.
In some melanomas, unlike primary cells, Id2 is not downregulated by TGF-β, a mechanism proposed to explain loss of melanoma growth inhibition in response to TGF-β (10). Id2, but not Id1 or Id3, was found to interact physically and genetically with hypophosphorylated retinoblastoma family members, important in cell cycle progression and melanomagenesis (11,12). Consensus on the correlation between Id3 and Id4 expression and prognosis in human cancer has been mixed. Id3 is upregulated in some cancers including prostate (13), ovarian (14) and non-small-cell lung carcinoma (15), but, conversely, carries potentially inactivating mutations in 36 of 53 cases of Burkitt’s lymphoma (16). In breast and prostate cancer, as well as leukemia, the Id4 gene has been shown to be silenced by hypermethylation, suggesting its role as a putative tumor suppressor; however, Id4 silencing correlates with improved clinical outcome in glioblastoma (17–19) suggesting that Id3 and Id4 can function either as cooperating oncogenes or tumor suppressors, depending on the type of cancer. Their exact roles in melanoma are as yet undetermined.
Expression of Smad7 in 1205Lu metastatic melanoma cells results in a tumorigenic block through both TGF-β-dependent and -independent mechanisms. Using multiple approaches including subcutaneous injection, intra-cardiac injection and human skin grafts, others and we found that Smad7 not only blocked melanoma formation but also mitigated metastasis in highly aggressive, TGF-β-dependent 1205Lu cells. Mechanisms for these observations include downregulation of metastasis-related genes such as matrix metalloproteinase 2 (MMP2), MMP9, osteopontin and CXCR4, as well as stabilization of cell-adhesion-related proteins β-catenin and N-cadherin (20–23).
This study evaluates the role of Id2–4 during melanomagenesis in the presence of a functioning or blocked TGF-β pathway, via Smad7. We show that Id2–4 are each able to overcome the potent tumorigenic block imposed by Smad7. This lends confirmation that Id2, Id3 or Id4 alone can promote tumorigenesis independently, to varying degrees, following TGF-β inhibition (24,25).
Materials and methods
Cell culture, retroviral transductions and expression vectors
1205Lu, 1205Lu/Smad7, WM852, Sk-28 and 501-Mel were kindly provided by Dr A.Mauviel (Institut Curie, Orsay, France). Cells have been cultured and characterized as described previously (20,23). Pigmented MNT1 melanoma cells were kindly provided by Dr V.Hearing (NIH, Bethesda, MD) and maintained as described previously (26). Primary human foreskin keratinocytes and human foreskin melanocytes (HFM) were derived from neonatal foreskin and cultured as described with approval from Georgetown IRB (27). 1205Lu cells stably expressing Smad7 (1205Lu/S7) or vector only (1205Lu/Vc) were transduced with pLHCX-DsRed retroviral vector (Clontech, Mountain View, CA) as described previously (28). Id2, Id3 and Id4 were each separately cloned into pcDNA4/TO Tet-on expression vector (Invitrogen, Carlsbad, CA). 1205Lu cells were transfected with pcDNA6/Tet repressor, selected in 10 µg/ml of blasticidin; then transfected with pcDNA4/TO-Id constructs and further selected with 1200 µg/ml of Zeocin™ (Invitrogen) for >2 weeks. Highly expressing Tet-inducible clones were isolated, confirmed by immunoblot and used for subsequent studies.
Immunoblot analysis, immunoprecipitation and antibodies
Protein analyses were performed according to standard protocols. For most immunoblots, 40 μg of total protein was resolved on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane and blocked in 5% non-fat milk. Corresponding blots with primary antibody were incubated overnight, and membranes were then washed with 3× phosphate-buffered saline–Tween. Secondary donkey anti-mouse-horseradish peroxidase or sheep anti-rabbit-horseradish peroxidase was used at 1:8000 dilution. Enhanced chemiluminescence (Thermo, Rockford, IL) was used to detect proteins. Smad7 (N19), Id2 (C20), Id3 (C20) and Id4 (L20) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-Smad3 was a generous gift from Dr Edward Leof (Mayo Clinic, Rochester, MN). Glyceraldehyde 3-phosphate dehydrogenase was used as a loading control using antibody from Millipore (Billerica, MA).
In vivo subcutaneous injection
Subcutaneous injections were performed using athymic NCr-nu/nu mice (Harlan Laboratories, Indianapolis, IN). Animals were anesthetized using 2% isoflurane. A total of 1 × 106 cells of each type were injected into hind flanks using a 20 gauge syringe. At least eight animals were used for each condition. Animals were monitored for tumor growth for 60 days via Maestro II™ live-animal fluorescence imaging (Caliper Life Sciences, Hopkinton, MA). Animals were euthanized, tumor tissue was harvested and fixed in 10% neutral-buffered formalin; fresh 5 µm sections were cut using a microtome. All animal protocols were approved and performed according to guidelines established by the Georgetown University Animal Care and Use Committee.
Transient transfection
Equal cell numbers were plated into six-well or 10 cm2 dishes, 24 h before transfection. A total of 2.5 or 5 µg of either Id4 or pcDNA4-empty vector control plasmid were used per well using Lipofectamine LTX (Invitrogen) and cells were transfected for 4 h. Transfection was removed; cells were washed in 1× phosphate-buffered saline and replaced with growth media.
Quantitative reverse transcription–PCR and primer sequences
Quantitative reverse transcription–PCR was performed by standard protocols using two-step reverse transcription–PCR (Invitrogen), 0.75 µg of RNA and specific primers: MMP2 forward-5ʹ-CTGGCTTTTCACTGCTGGCT-3ʹ; reverse-5ʹ-TGCTAAGTAGAGTGAACAGGG-3ʹ; MMP9 forward-5ʹ-CATTCAGGGAGACGCCCA-3ʹ; reverse-5ʹ-AACCACGACGCCCTTGC-3ʹ; CXCR4 forward-5ʹ-CAGTGGCCGACCTCCTCTT-3ʹ; reverse-5ʹ-CAGTTT GCCACGGCATCA-3ʹ; osteopontin forward-5ʹ-AGGCAGAGCACAGCAT CGT-3ʹ; reverse-5ʹ-TTGGCTGAGAAGGCTGCAA-3ʹ; β-actin forward-5ʹ-GCACTCTTCCAGCCTTCCTT-3ʹ; reverse-5ʹ-AATGCCAGGGTACATGGT GG-3ʹ.
Id knockdown
Knockdown experiments were performed as reported previously (20) using small interfering RNAs (siRNAs) specific for Id2, Id3, Id4 or scrambled siRNA controls (Santa Cruz).
Colony-forming assays
The colony assay was performed with modification as described previously (22,29). Briefly, 1205Lu, 1205Lu/Smad7, WM852 or Sk-28 melanoma cells expressing DsRed were seeded into 12-well plates at low density (5 × 103 cells per well) following knockdown of Id2, Id3 or Id4. Scrambled sequences were used as control for knockdown. Cells were mixed with growth media containing 0.3% low-melting agarose then layered onto a 0.6% solid agarose base layer. Cell colonies were counted using fluorescence microscopy for ease in accurately identifying small colonies. Graphs are shown for each cell line examined.
Statistical analyses
Statistical analyses were performed using JMP10 Pro (SAS Institute, Cary, NC) and SigmaPlot (Systat, San Jose, CA). Results show the mean ± standard error (SE) of three independent experiments (30), where each experiment was performed in triplicate. A Student’s t-test was used to calculate P-values using the data from the three independent experiments; values <0.05 was considered statistically significant. * indicates a P-value of <0.05, ** indicates <0.01 and *** indicates <0.001.
Results
Upregulation of Id2, 3 and 4 in 1205Lu metastatic melanoma cells
Endogenous Id protein expression was examined in TGF-β-dependent and -independent melanoma cell lines (Figure 1). Immunoblots were probed for Id2, Id3 or Id4. In general, Id protein expression was variable across the multiple melanoma cell lines examined, this was not unexpected as certain Ids share some overlapping functions, e.g. Id2 with Id4 (31–33). Id2 was weakly expressed in HFM and increased in melanoma. Id3, was not observed in HFM, yet was expressed in MNT1 and 1205Lu, and at higher levels in the more proliferative Mel501, Sk-28 and WM852 melanoma cells. Interestingly, Id4 was expressed in pigmented HFMs and at low levels in 1205Lu/Vc, but not detected in other melanoma lines. The loss of Id4 protein expression may be due to promoter methylation, as has been observed in other cancer types (19). Importantly, Id2, Id3 and Id4 each appeared to be TGF-β-dependent, as they were observed elevated in 1205Lu/Vc when compared with 1205Lu cells expressing Smad7 (1205Lu/S7; Figure 1, lanes 6 and 7). These results suggested that Ids may revert 1205Lu/S7 cells back to the tumorigenic phenotype observed with 1205Lu/Vc cells.
Fig. 1.

Expression of Id2, Id3 and Id4 in melanocytes and melanoma cells. Endogenous Id2 (top; 15kDa), Id3 (middle; 15kDa) and Id4 (bottom; 19kDa) protein were probed across multiple melanoma cell lines using immunoblot analysis of total cell lysate and specific anti-sera against Id proteins. Id expression was variable across melanoma cell lines with Id2 and Id3 expression more prominent in the more proliferative MNT1, Sk-28, 501-Mel and WM852 cells. In 1205Lu cells, increased endogenous expression of Id2, Id3 and Id4 were detected when 1205Lu was compared with 1205Lu cells expressing Smad7. Endogenous Id4 was observed only in primary HFM and 1205Lu cells lacking Smad7 expression. Glyceraldehyde 3-phosphate dehydrogenase (34kDa) is used as a loading control.
Role of Ids in proliferation
Effects of Id2–4 on cell cycle were first assessed in normal HFM (Figure 2A and B). To determine if Id mediated changes in cell proliferation in vitro, HFM expressing green fluorescent protein were compared with cells expressing Id2 (top), Id3 (middle) or Id4 (bottom). In HFM expressing either Id2 or Id3, S-phase was significantly increased, indicating an alteration in cell proliferation, with a more robust increase (1.98-fold; P < 0.01) detected in cells expressing Id3. Id3 also induced a concomitant drop in G1-phase (P < 0.001) and increased in G2/M-phase (P < 0.001). HFM cells expressing Id4 did not result in statistically significant changes to the percentage of cells in each phase of the cell cycle, suggesting that Id4 does not significantly alter proliferation in melanocytes.
Fig. 2.
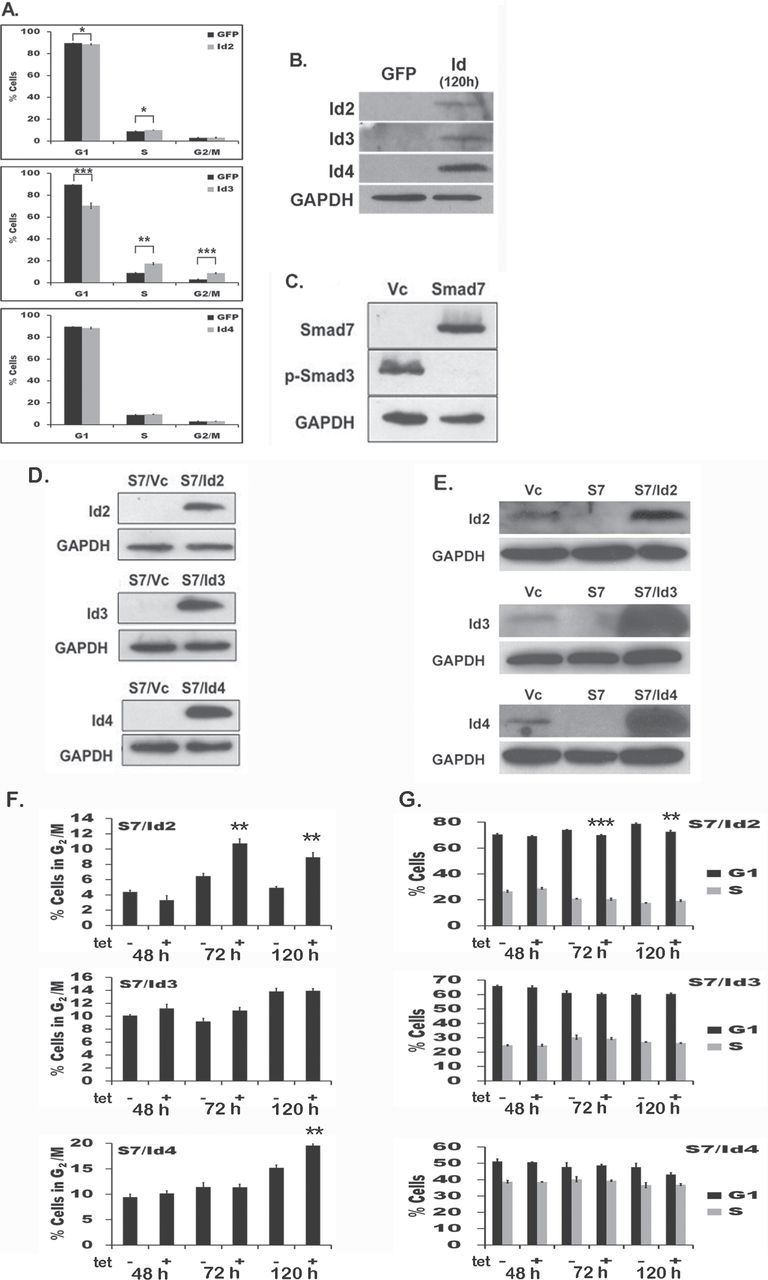
Id2 and Id3 increase S-phase in primary HFM, but not in 1205Lu in cell culture. (A) HFM were transiently transfected with Id2 (top), Id3 (middle) or Id4 (bottom) and subjected to cell cycle analysis after 48h; the percentage of cells in each cell cycle phase are plotted. HFM cells expressing either Id2 or Id3 showed a statistically significant increase in percentage of cells in S-phase of the cell cycle, whereas Id4 resulted in no significant changes. (B) Ectopic expression of Id2, Id3 or Id4 in HFM after 120h. (C) Smad7 blocks TGF-β expression in 1205Lu cells. Cells were subjected to immunoblot with antibodies to Smad7 (upper panel; 51kDa) or p-Smad3 (middle panel; 52kDa) to confirm Smad7 expression and inhibition of TGF-β signaling. (D) Stable clones of 1205Lu/S7 melanoma cells expressing Ids were subjected to immunoblot with antibodies to Id2 (top panel), Id3 (middle panel) or Id4 (bottom panel) to confirm Id protein expression. (E) Endogenous expression of Id2, Id3 or Id4 in 1205Lu is shown alongside induced Id clones. (F and G) S7/Id cell cycle analysis. No changes in S-phase were detected in S7 cells expressing Id2, Id3 or Id4. Stable S7/Id2 (top), S7/Id3 (middle) or S7/Id4 (bottom) clones were grown in presence or absence of tetracycline over a 48, 72 and 120h time course and showed only an increased percentage of cells in G2/M phase in both Id2- and Id4-expressing cells. Statistically significant changes are depicted with asterisk(s) and were compared with matched un-induced populations. *P < 0.05, **P < 0.01 and ***P < 0.001. Results show the mean ± SE of three independent experiments, where each experiment was performed in triplicate.
To investigate if Id2, Id3 or Id4 expression alters proliferation or tumorigenesis in Smad7-expressing 1205Lu melanoma cells, stable transfectants of each Id in a tetracycline-on (Tet-on) expression system were generated in 1205Lu/S7 cells (see Materials and methods). Individual cell colonies were isolated following the introduction of each construct (S7/Id2, S7/Id3 and S7/Id4) and assayed for expression of Ids, as well as Smad7 and p-Smad3. TGF-β signaling was completely inhibited (Figure 2C and D). 1205Lu cells expressing Smad7 plus Id2, Id3 or Id4 are also shown alongside 1205Lu/Vc in order to compare the level of Id overexpression achieved in Smad7 cells (Figure 2E). As we previously showed Id2 increases both S-phase and contributes to increased tumorigenesis in mouse fibroblasts (28), we investigated whether a similar effect could be observed in melanoma cells and therefore performed cell cycle analysis of 1205Lu/S7/Id-expressing cells in the presence or absence of Tet. However, no changes in S-phase were observed in S7/Id2, S7/Id3 or S7/Id4. This result was perhaps not surprising as previous work showing the same 1205Lu cells expressing Smad7, which mitigated tumorigenesis in vivo, also did induce changes in melanoma proliferation in vitro (22). Yet, an increase (P < 0.01) in G2/M was observed in S7/Id2 cells at 72 (1.67-fold) and 120h (1.79-fold) after Tet induction (Figure 2F and G). S7/Id3 cells did not exhibit any significant changes in the cell cycle. However, similar to the S7/Id2-expressing cells, S7/Id4 cells showed a 1.27-fold increase (P < 0.05) in the G2/M population 120h after Tet induction (Figure 2F), suggesting an overlapping relationship in cell division between Id2 and Id4 expression in 1205Lu melanoma cells.
Role of Id2–4 in melanomagenesis
We then sought to examine whether Id2, Id3 or Id4 can bypass the TGF-β requirement for tumorigenesis in 1205Lu cells expressing Smad7. Therefore, 1205Lu/S7/Id cells were subcutaneously injected into athymic mice and maintained ad libitum with doxycycline-supplemented feed to induce Id expression. 1205Lu/Vc formed vigorously growing tumors, whereas 1205Lu/S7 did not, as we previously reported (20). However, 1205Lu cells harboring Smad7 along with Id2, Id3 or Id4 (S7/Id2, S7/Id3 or S7/Id4) were each able to establish subcutaneous tumors, though only in the presence of doxycycline. The ability of these cells to form tumors in the absence of TGF-β signaling (Figure 3A) demonstrates that Ids can completely bypass the previously established requirement for the TGF-β pathway for melanomagenesis in 1205Lu cells (22,23). Live fluorescence imaging (see Materials and methods) revealed that animals xenografted with S7/Id3 or S7/Id4 had the largest tumors when compared with either S7 or S7/Id2 (Figure 3B and Table I). In the absence of doxycycline, animals xenografted with the same cells did not develop a single tumor, showing the absolute requirement for Ids in these cells (Figure 3A, right panel and C). These results support the hypothesis that these Ids are able to overcome the Smad7-mediated block in tumorigenesis and support a role for Ids in melanomagenesis.
Fig. 3.
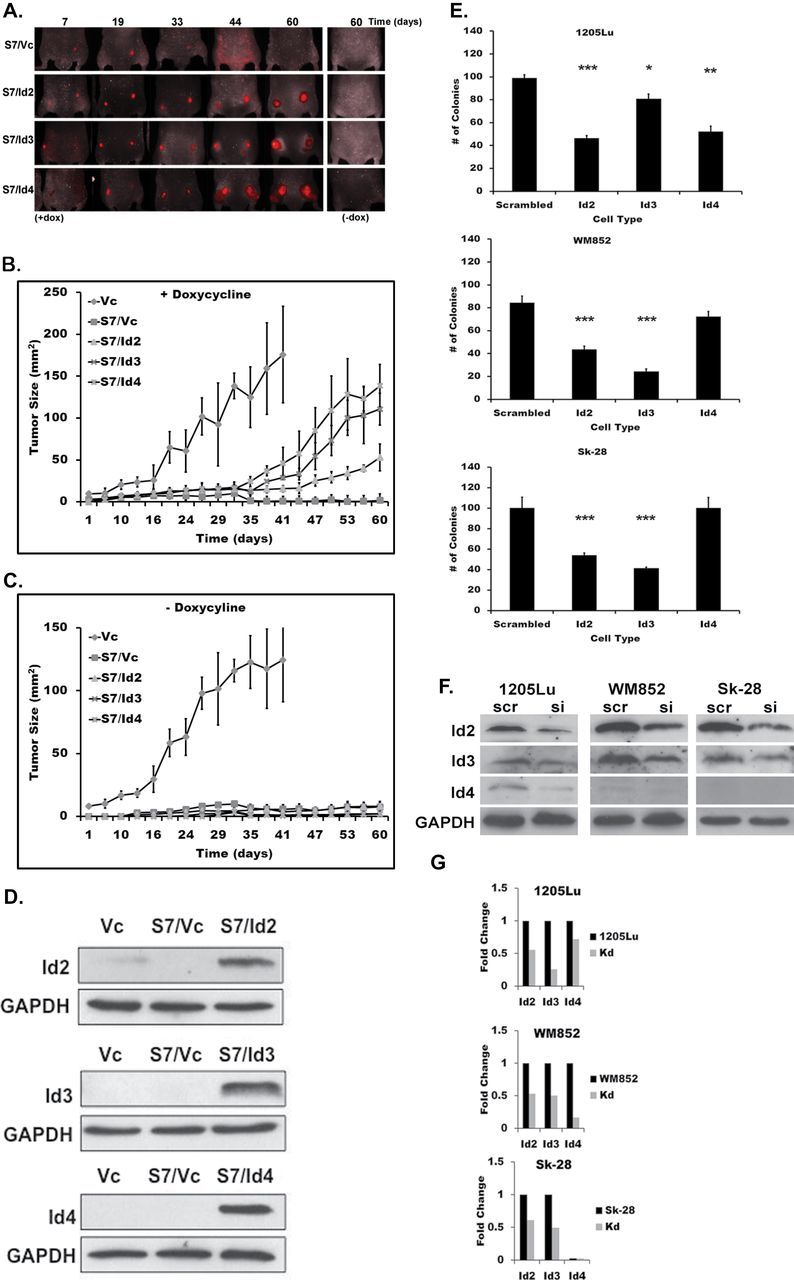
Id2, Id3 or Id4 expression bypasses Smad7-mediated block in tumorigenesis. (A) S7/Id cells were xenografted into athymic mice (n = 8) continuously fed with doxycycline-supplemented feed to induce Ids and were subjected to live-animal fluorescence imaging. Representative images of animals taken at days 7, 19, 33, 44 and 60 are shown; no tumor growth in S7/Vc animals (top) and prominent tumor formation in all 1205Lu/S7 cells expressing Ids (lower panels). Right panel shows representative images of animals lacking tumor formation after being injected with the same S7/Id clones grown in the absence of doxycycline. (B) Tumor size was monitored over the time course and plotted for mice fed with doxycycline. (C) No tumor growth was observed in animals maintained with feed lacking doxycycline, 1205Lu/Vc cells are shown for comparison. (D) Tumor lysates were probed for Id2, Id3 or Id4 expression. Protein expression indicates Ids were expressed in vivo and glyceraldehyde 3-phosphate dehydrogenase was used as a loading control. (E) Colony formation assay. 1205Lu (top), WM852 (middle) or Sk-28 melanoma cells (bottom) were transfected with siRNAs specific for Id2, Id3, Id4 or scrambled siRNA controls as described previously (20), then analyzed for colony formation in soft agar as described in Materials and methods. To confirm knockdown, cells were subjected to immunoblot analysis using antibodies specific for each Id (F) and blots were quantified by densitometry (G). Kd: Id siRNA knockdown. Results show the mean ± SE of three independent experiments, where each experiment was performed in triplicate. *P < 0.05, **P < 0.01 and ***P < 0.001.
Table I.
Comparison of tumor sizes induced by Id2, Id3 and Id4
| Cell line 1 | Cell line 2 | P-value |
|---|---|---|
| Day 41 ANOVA | ||
| 1205Lu/Vc | 1205Lu/S7/Vc | 0.006 |
| 1205Lu/Vc | 1205Lu/S7/Id2 | 0.008 |
| 1205Lu/Vc | 1205Lu/S7/Id3 | 0.024 |
| 1205Lu/Vc | 1205Lu/S7/Id4 | 0.021 |
| 1205Lu/S7/Id4 | 1205Lu/S7/Vc | 0.015 |
| 1205Lu/S7/Id3 | 1205Lu/S7/Vc | <0.001 |
| 1205Lu/S7/Id2 | 1205Lu/S7/Vc | 0.003 |
| 1205Lu/S7/Id4 | 1205Lu/S7/Id2 | 0.034 |
| 1205Lu/S7/Id3 | 1205Lu/S7/Id2 | 0.006 |
| 1205Lu/S7/Id4 | 1205Lu/S7/Id3 | 0.199 |
| Day 60 ANOVA | ||
| 1205Lu/S7/Id4 | 1205Lu/S7/Vc | <0.001 |
| 1205Lu/S7/Id3 | 1205Lu/S7/Vc | <0.001 |
| 1205Lu/S7/Id4 | 1205Lu/S7/Id2 | 0.008 |
| 1205Lu/S7/Id3 | 1205Lu/S7/Id2 | 0.015 |
| 1205Lu/S7/Id2 | 1205Lu/S7/Vc | 0.005 |
| 1205Lu/S7/Id4 | 1205Lu/S7/Id3 | 0.201 |
ANOVA, analysis of variance.
To further address the ability of Ids to modulate tumorigenicity, a colony-forming assay was performed using Id2, Id3 or Id4 siRNA-mediated knockdown. WM852 and Sk-28 cells have elevated levels of endogenous Id2 and Id3 expression when compared with 1205Lu and were therefore included in the assay (Figure 1). We observe a significant reduction in the ability of 1205Lu, WM852 and Sk-28 cells to form colonies in soft agar following reduced Id expression, especially for Id2 and Id3 knockdowns (Figure 3E). Knockdown of Id4 expression impaired colony formation in 1205Lu cells, though not in other cells examined, likely due to the low levels of endogenous Id4 expression observed in WM852 and Sk-28 cells (Figure 1). Immunoblot analysis revealed that Id levels were reduced following knockdown using specific siRNAs (Figure 3F and G).
To assess tumor proliferative potential, tissues were harvested and stained with the Ki-67 proliferation marker. 1205Lu/Vc and S7/Id3 tissues had the highest percentage of Ki-67 cell positivity, with averages of 15.3% and 14.5%, respectively. Interestingly, S7/Id2 and S7/Id4 had fewer Ki-67 positive cells (6.1% and 6.9%, respectively; Figure 4B). This was unexpected as S7/Id4 tumors had substantial growth observed via fluorescence imaging. However, hematoxylin and eosin sections revealed extensive multifocal necrosis within S7/Id4 tumors, explaining the low number of Ki-67 positive cells observed. To quantify necrosis, 10 low-power (×20) images of hematoxylin and eosin sections were analyzed per tumor, and necrotic areas were analyzed using Image J analysis software (NIH). Necrotic areas were then expressed as a percentage of total area per image. S7/Id4 tumors had extensive necrosis, with an average of 31% (Figure 4C and D). By contrast, 1205Lu/Vc tumors exhibited an average of only 5.2% necrosis, whereas S7/Id2 and S7/Id3 showed minimal necrosis with 0.13% and 0.95%, respectively. Even the aggressive 1205Lu/Vc tumors, which had substantially more tumor growth than those expressing Id4, did not show the degree of necrosis observed in Id4 sections, suggesting an active process rather than one that arose due to an exhaustion of tumor resources.
Fig. 4.

Tumor proliferation and necrosis. Ki-67 immunofluorescence was performed to assess in vivo proliferation; Ki-67 (red), 4′,6-diamidino-2-phenylindole (blue). (A) Five high-power fields (×40) representing >300 cells per field were used to count the number of Ki-67-positive cells; % Ki-67-positive cells in tumors are shown. (B) Ki-67 expression is quantitated and compared with 1205Lu/S7. All Id expressing tumors were more proliferative than non-tumorigenic S7-expressing cells. (C) Examining necrosis in xenografts. Ten low-power (×20) images were randomly captured across each tumor section; necrosis was readily detected and encircled using Image J software (NIH). Representative images are shown. (D) Tumors expressing S7/Id4 had an average of 31% necrosis, whereas other S7/Id expressing tumors had little observed necrosis. *P < 0.05, **P < 0.01 and ***P < 0.001.
To investigate the mechanism by which Ids bypass TGF-β-mediated tumorigenesis, Id2, Id3 or Id4 were transiently expressed in both 1205Lu/Vc- and 1205Lu/S7-expressing cells; stable downregulation was not performed, as Ids have overlapping functions. Quantitative reverse transcription–PCR was then performed to examine four tumor-promoting genes shown previously to be downregulated in response to Smad7 in 1205Lu cells (23). Id2, Id3 and Id4 strikingly de-repressed MMP2, MMP9, CXCR4 and osteopontin (Figure 5A–C), suggesting that Id2, Id3 and Id4 play a role downstream of TGF-β-mediated tumorigenesis. In fact, Id2, Id3 and Id4 even upregulated MMP2, MMP9, osteopontin and CXCR4 in 1205Lu parental cells. CXCR4 was observed to have the most robust upregulation in response to Id expression across each of the different Ids examined. Furthermore, Ids have been shown to alter the expression of certain cyclin-dependent kinase inhibitors. To address whether Id2, Id3 or Id4 can alter expression of cyclin-dependent kinase inhibitors in vitro, 1205Lu cells expressing Smad7 were transiently transfected as above and total cell lysates were probed for p15Ink4b, p16Ink4a, p18Ink4c, p19Ink4d, p21CIP1, p27KIP1 and p57KIP2. We found that p15Ink4b protein expression was significantly reduced in response to Id2, Id3 and Id4 expression after 48 h (Figure 5D). This is in agreement with previous observations showing Id2 reduces p15Ink4b expression in melanoma (10). p27KIP1 was also somewhat reduced by Id2, whereas p19Ink4d was slightly reduced by both Id2 and Id4.
Fig. 5.
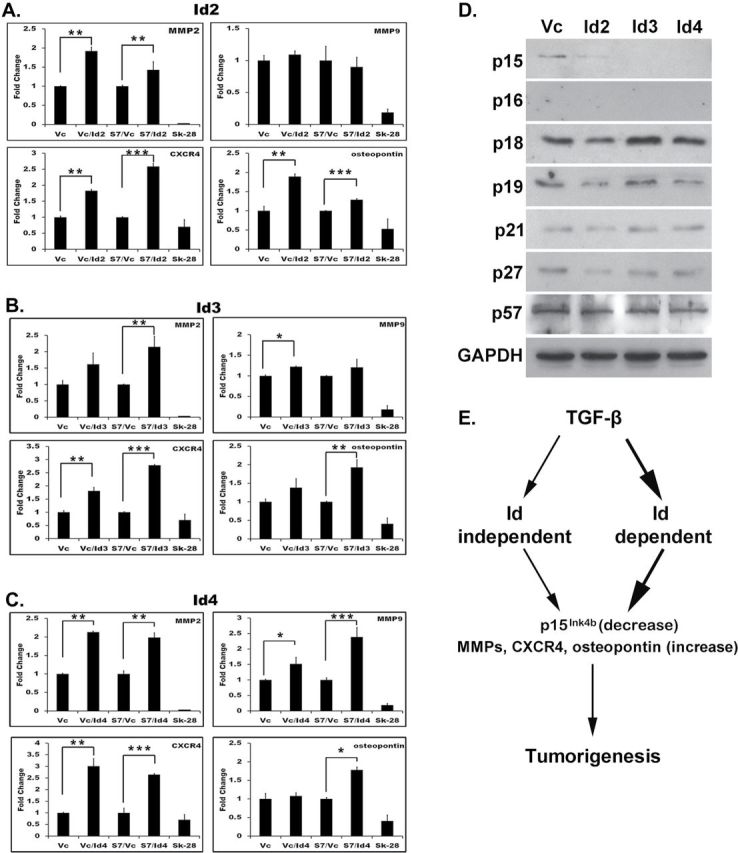
Id2–4 increases expression of tumor-promoting genes and reduces expression of p15Ink4b. Id2 (A), Id3 (B) and Id4 (C) were transiently expressed in 1205Lu/Vc 1205Lu/S7 cells and subjected to quantitative reverse transcription–PCR using primers specific to MMP2, MMP9, osteopontin and CXCR4. Fold-change in expression was calculated after normalization against β-actin. Sk-28 cells with low metastatic potential were used a negative control. Cells were collected after 48h. Results show the mean ± SE of three independent experiments, where each experiment was performed in triplicate. *P < 0.05, **P < 0.01, and ***P < 0.001. (D) Cyclin-dependent kinase inhibitors expression profile in response to Id2, Id3 or Id4 in 1205Lu cells expressing Smad7. (E) Model of alternate pathways for the role of Ids in TGF-β-mediated melanomagenesis.
Discussion
Id2, Id3 and Id4 are each able to overcome the potent tumorigenic block provided by Smad7 in 1205Lu metastatic melanoma cells, thus generating TGF-β-independent tumors in the previously TGF-β-dependent 1205Lu melanoma cells. Previous work has shown that both in the presence of Smad7, the endogenous TGF-β antagonist, as well as synthetic TGF-β inhibitors, melanomagenesis is blocked in 1205Lu cells, establishing the requirement for TGF-β in transformation (22,23). Observations shown here suggest an Id-related tumorigenic mechanism acting downstream or independently of TGF-β. We believe that Ids mediate tumorigenesis primarily downstream of TGF-β (Figure 5E, thick arrows) because (i) Smad7 blocks Id expression and (ii) re-expression of Ids overcome Smad7-mediated repression of tumorigenesis. We further hypothesize that the mechanism responsible was due, in part, to elevated MMP2, MMP9, CXCR4 and osteopontin expression shown previously to be downregulated in response to Smad7 (23). In our model, these four genes are re-upregulated, to varying degrees, in response to Id2, Id3 and Id4 expression, with CXCR4 demonstrating the most robust response. MMPs have a strong link to invasion and tumorigenic progression and are Id-dependent, as Id2 and Id3 silencing results in a potent loss of MMP gene expression (34); however, the association between MMPs and Id4 was unknown prior to this work. Ectopic Id4 expression has also been shown to elevate upstream stimulatory factor-1 expression levels in cervical cancer cells (35). Increased CXCR4 expression in response to Id2–4 in this study may, therefore, be due to upregulation of upstream stimulatory factor-1, which can associate with an E-box at −260 bp in the CXCR4 promoter, stimulating its expression (36). The induction of osteopontin that we observed may be due to the interaction of Ids with their known partner E47. This would disrupt E47/Twist basic helix-loop-helix heterodimers, relieving repression of osteopontin. This was shown to be the mechanism by which ectopic Id expression in mesenchymal cells induces osteopontin (37).
Increased necrosis (Figure 4D) was observed in tumors expressing S7/Id4 when compared with 1205Lu, yet 1205Lu formed even larger tumors than mice xenografted with S7/Id4 (Figure 3B and C). Further analysis of S7/Id4 tumor histology revealed an innate immune cell infiltration composed of tissue histiocytes, which are present in athymic nude mice (data not shown). We believe this phagocytic component found in S7/Id4 tumors may have also contributed to the observed necrosis and therefore reduced proliferation detected by Ki-67 staining. The mechanism for histiocyte infiltration and necrosis is currently under investigation.
In conclusion, we find that Id2, Id3 and Id4 are each able to contribute significantly to melanomagenesis independent of TGF-β in 1205Lu melanoma cells. Further work is required to address whether TGF-β inhibitors might inadvertently select for Id-positive tumors and could be used as adjuvants with Id-antagonists, such as Id aptamers, which are currently under study (38,39). At present, only anti-Id4 appears to be a reliable measure of Id protein levels for use on paraffin sections in our hands. However, in order to fully address any relationship between Id expression and prognosis in melanoma, future studies will need to examine Id status in patient-derived tumors and determine correlation with overall survival.
Funding
National Cancer Institute; National Institutes of Health (1RO1 CA100443-01A1 to D.S.R); Georgetown University School of Medicine, Office of the Dean for Research.
Acknowledgements
The authors would like to sincerely thank Drs A.Mauviel and D. Javelaud for providing 1205Lu/Smad7 cells and the PIRL (Georgetown University) for aid with live-animal in vivo imaging.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations:
- HFM
human foreskin melanocytes
- Ids
inhibitors of differentiation
- MMP
matrix metalloproteinase
- TGF-β
transforming growth factor-β
- SE
standard error
- siRNA
small interfering RNA.
References
- 1. Imoto S., et al. (2003). Regulation of transforming growth factor-beta signaling by protein inhibitor of activated STAT, PIASy through Smad3. J. Biol. Chem., 278, 34253–34258 [DOI] [PubMed] [Google Scholar]
- 2. Kang Y., et al. (2003). A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol. Cell, 11, 915–926 [DOI] [PubMed] [Google Scholar]
- 3. Ruzinova M.B., et al. (2003). Id proteins in development, cell cycle and cancer. Trends Cell Biol., 13, 410–418 [DOI] [PubMed] [Google Scholar]
- 4. Krasagakis K., et al. (1995). Growth control of melanoma cells and melanocytes by cytokines. Recent Results Cancer Res., 139, 169–182 [DOI] [PubMed] [Google Scholar]
- 5. Rodeck U., et al. (1999). Independent regulation of growth and SMAD-mediated transcription by transforming growth factor beta in human melanoma cells. Cancer Res., 59, 547–550 [PubMed] [Google Scholar]
- 6. Reed J.A., et al. (1994). Expression of transforming growth factor-beta 2 in malignant melanoma correlates with the depth of tumor invasion. Implications for tumor progression. Am. J. Pathol., 145, 97–104 [PMC free article] [PubMed] [Google Scholar]
- 7. Javelaud D., et al. (2008). Transforming growth factor-beta in cutaneous melanoma. Pigment Cell Melanoma Res., 21, 123–132 [DOI] [PubMed] [Google Scholar]
- 8. Benezra R., et al. (2001). The Id proteins and angiogenesis. Oncogene, 20, 8334–8341 [DOI] [PubMed] [Google Scholar]
- 9. Fong S., et al. (2004). Id genes and proteins as promising targets in cancer therapy. Trends Mol. Med., 10, 387–392 [DOI] [PubMed] [Google Scholar]
- 10. Schlegel N.C., et al. (2009). Id2 suppression of p15 counters TGF-beta-mediated growth inhibition of melanoma cells. Pigment Cell Melanoma Res., 22, 445–453 [DOI] [PubMed] [Google Scholar]
- 11. Lasorella A., et al. (2000). Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature, 407, 592–598 [DOI] [PubMed] [Google Scholar]
- 12. Iavarone A., et al. (1994). The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev., 8, 1270–1284 [DOI] [PubMed] [Google Scholar]
- 13. Sharma P., et al. (2012). Id1 and Id3 expression is associated with increasing grade of prostate cancer: Id3 preferentially regulates CDKN1B. Cancer Med., 1, 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shepherd T.G., et al. (2008). Autocrine BMP4 signalling regulates ID3 proto-oncogene expression in human ovarian cancer cells. Gene, 414, 95–105 [DOI] [PubMed] [Google Scholar]
- 15. Castañon E., et al. (2013). Id1 and Id3 co-expression correlates with clinical outcome in stage III-N2 non-small cell lung cancer patieznts treated with definitive chemoradiotherapy. J. Transl. Med., 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richter J., et al. (2012). Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet., 44, 1316–1320 [DOI] [PubMed] [Google Scholar]
- 17. Carey J.P., et al. (2009). Inhibitor of differentiation 4 (Id4) is a potential tumor suppressor in prostate cancer. BMC Cancer, 9, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martini M., et al. (2013). Epigenetic silencing of Id4 identifies a glioblastoma subgroup with a better prognosis as a consequence of an inhibition of angiogenesis. Cancer, 5, 1004–1012 [DOI] [PubMed] [Google Scholar]
- 19. Yu L., et al. (2005). Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat. Genet., 37, 265–274 [DOI] [PubMed] [Google Scholar]
- 20. DiVito K.A., et al. (2010). Smad7 restricts melanoma invasion by restoring N-cadherin expression and establishing heterotypic cell-cell interactions in vivo. Pigment Cell Melanoma Res., 23, 795–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Javelaud D., et al. (2011). GLI2 and M-MITF transcription factors control exclusive gene expression programs and inversely regulate invasion in human melanoma cells. Pigment Cell Melanoma Res., 24, 932–943 [DOI] [PubMed] [Google Scholar]
- 22. Javelaud D., et al. (2005). Stable overexpression of Smad7 in human melanoma cells inhibits their tumorigenicity in vitro and in vivo. Oncogene, 24, 7624–7629 [DOI] [PubMed] [Google Scholar]
- 23. Javelaud D., et al. (2007). Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res., 67, 2317–2324 [DOI] [PubMed] [Google Scholar]
- 24. Norton J.D., et al. (1998). Coupling of cell growth control and apoptosis functions of Id proteins. Mol. Cell. Biol., 18, 2371–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilson J.W., et al. (2001). Expression of Id helix-loop-helix proteins in colorectal adenocarcinoma correlates with p53 expression and mitotic index. Cancer Res., 61, 8803–8810 [PubMed] [Google Scholar]
- 26. Kushimoto T., et al. (2001). A model for melanosome biogenesis based on the purification and analysis of early melanosomes. Proc. Natl Acad. Sci. USA, 98, 10698–10703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rosenthal D.S., et al. (1995). Engineered human skin model using poly(ADP-ribose) polymerase antisense expression shows a reduced response to DNA damage. J. Invest. Dermatol., 105, 38–43 [DOI] [PubMed] [Google Scholar]
- 28. Trabosh V.A., et al. (2009). Sequestration of E12/E47 and suppression of p27KIP1 play a role in Id2-induced proliferation and tumorigenesis. Carcinogenesis, 30, 1252–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Freshney R.I. (2010) Culture of Animal Cells. 6th edn Wiley-Blackwell, Hoboken, NJ. [Google Scholar]
- 30. Cumming G., et al. (2007). Error bars in experimental biology. J. Cell Biol., 177, 7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coppé J.P., et al. (2003). Id proteins in epithelial cells. Exp. Cell Res., 285, 131–145 [DOI] [PubMed] [Google Scholar]
- 32. Lyden D., et al. (1999). Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature, 401, 670–677 [DOI] [PubMed] [Google Scholar]
- 33. Samanta J., et al. (2004). Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development, 131, 4131–4142 [DOI] [PubMed] [Google Scholar]
- 34. Asirvatham A.J., et al. (2007). ID1-, ID2-, and ID3-regulated gene expression in E2A positive or negative prostate cancer cells. Prostate, 67, 1411–1420 [DOI] [PubMed] [Google Scholar]
- 35. Pagliuca A., et al. (1998). A role for Sp and helix-loop-helix transcription factors in the regulation of the human Id4 gene promoter activity. J. Biol. Chem., 273, 7668–7674 [DOI] [PubMed] [Google Scholar]
- 36. Moriuchi M., et al. (1999). USF/c-Myc enhances, while Yin-Yang 1 suppresses, the promoter activity of CXCR4, a coreceptor for HIV-1 entry. J. Immunol., 162, 5986–5992 [PubMed] [Google Scholar]
- 37. Hayashi M., et al. (2007). Comparative roles of Twist-1 and Id1 in transcriptional regulation by BMP signaling. J. Cell Sci., 120, 1350–1357 [DOI] [PubMed] [Google Scholar]
- 38. Lahn M., et al. (2005). TGF-beta inhibitors for the treatment of cancer. Expert Opin. Investig. Drugs, 14, 629–643 [DOI] [PubMed] [Google Scholar]
- 39. Mern D.S., et al. (2010). Targeting Id1 and Id3 by a specific peptide aptamer induces E-box promoter activity, cell cycle arrest, and apoptosis in breast cancer cells. Breast Cancer Res. Treat., 124, 623–633 [DOI] [PubMed] [Google Scholar]