

Abstract
Fanconi Anemia (FA) is a rare recessive disorder with chromosomal instability, congenital abnormalities and a high cancer risk. The breast cancer susceptibility gene BRCA2 (FANCD1) is one of the 16 genes involved in this recessive disease. We have identified a novel mutation of the splice donor site of intron 1 in the non-coding region of BRCA2 in a Japanese FA family. This mutation may account for the FA phenotype in a patient originally reported to have biallelic mutations in BRCA2. Subsequent functional studies revealed that one of the mutations, K2729N, was a neutral change. As reported here, a more careful analysis resulted in the identification of a novel splice site mutation. Functional analysis using a mouse embryonic stem cell-based assay revealed that it causes aberrant splicing, reduced transcript levels and hypersensitivity to DNA damaging agents, suggesting that it is likely to be pathogenic. Although similar pathogenic variants in the non-coding region of BRCA1 and 2 were not identified in a cohort of 752 familial breast cancer cases, we still think this finding is relevant for mutation analysis in HBOC families in a diagnostic setting.
Keywords: Fanconi Anemia, BRCA2, Familial breast cancer, aberrant splicing
Linkage studies in the 90's identified BRCA2 (MIM# 600185) as a high risk breast cancer gene [Wooster et al., 1995]. Patients with a mutation in BRCA2 have lifetime risks of getting breast and ovarian cancer of 50-60% and a 5-20%, respectively [Mavaddat et al., 2013]. In addition to the association with ‘Hereditary Breast and Ovarian Cancer Syndrome’ (HBOC), BRCA2 is also associated with Fanconi anemia (FA) in biallelic mutation carriers [Howlett et al., 2002]. FA is a rare recessive disorder characterized by chromosomal instability, bone marrow failure, congenital abnormalities and a high cancer risk, in particular acute myeloid leukemia (AML) and head and neck squamous cell carcinomas at young age. Until now, 16 different genes have been shown to cause FA [Kottemann and Smogorzewska, 2013].
Compared to other FA subtypes, patients with biallelic BRCA2 or PALB2 (FA-D and FA-N) mutations have a more severe phenotype with high risk of solid childhood tumors particularly Wilms tumor and medulloblastoma [Howlett et al., 2002; Reid et al., 2007; Xia et al., 2007]. Cells of FA patients are in general characterized by hypersensitivity to cross-linking agents such as mitomycin C (MMC) and enhanced levels of spontaneous as well as MMC-induced chromosomal aberrations [Auerbach, 2009]. An additional hallmark of the BRCA2 and PALB2-deficient cells is their inability to recruit Rad51 to double strand breaks, essential for homologous repair [Roy et al., 2012]. One of our reported FA-D1 patients carried a nonsense mutation c.8732C>A (p.S2835X) in BRCA2 on one allele and a missense mutation c.8415G>T (p.K2729N) on the other allele [Ikeda et al., 2003]. The patient was diagnosed with AML at the age of 2 years. Recently, Biswas et al. used a mouse embryonic stem cell-based assay to characterize BRCA2 variants associated with FA. In this study, the missense variant K2729N was found to be neutral. Embryonic stem cells with this variant were indistinguishable from wild type BRCA2 and exhibited no effect on cell survival, sensitivity to DNA-damaging agents and were proficient in RAD51 focus formation and homologous recombination [Biswas et al., 2011]. No other possible pathogenic variants were identified in the initial screening of BRCA2 and resequencing of the coding exons did not reveal any new variants. To determine the cause of FA in the patient carrying the K2729N allele, we decided to analyze cDNA from the patient cells. However, as reported previously, a MMC-sensitive lymphoblastoid cell line (LCL) was not available from the patient [Ikeda et al., 2003]. The patient presented with acute leukemia and after a chemotherapy protocol, he received a bone marrow transplantation from his HLA-identical sister. In addition, the established AML cell lines from the patient appeared to be reverted. Reversion of the cellular FA phenotype to wild type often occurs. In these cases, a spontaneous correction of a mutant allele by mitotic recombination, gene conversion, or secondary mutations can restore the function of the affected FA gene [Ikeda et al., 2003; Waisfisz et al., 1999]. However, early passages of the AML cells appeared sensitive in a chromosomal breakage test. These cells, with minimal passages, were used for sequence analysis of BRCA2 cDNA. We observed that both alleles were detected at equal levels, based on heterozygous SNPs in the BRCA2 coding region. We expected the allele carrying the premature stop codon (p.S2835X) to be present at a reduced level due to nonsense mediated mRNA decay. Indeed, inhibition of nonsense mediated mRNA decay by cyclohexamide treatment of the cells stabilized the transcript with the premature stop codon while the transcript of the other allele remained relatively low (data not shown). These results suggested that the transcripts of both alleles were present at reduced levels. To identify a mutation that could cause this low expression which was unaffected by the inhibition of nonsense mediated mRNA decay we subsequently analyzed the region encoding the 5′UTR- and 3′UTR-region of BRCA2 DNA by sequencing. A splice site mutation, c.-40+1G>A, disrupting the donor site of intron 1 was identified. Analysis of this variant in the family showed that one of the parents was heterozygous for this splice site mutation and that the healthy sister carried the premature stop mutation and not the splice site mutation (Supp. Table S1). No material was available from the other parent and information about the cancer history in the heterozygous carriers was lacking.
The effect of the splice site mutation on mRNA splicing was further analyzed. Primer pairs were designed for amplifying a fragment ranging from non-coding exon 1 to coding exon 2 and cDNA was analyzed from the parent carrying the splice site mutation, the FA patient and a wild type control. After PCR amplification, a smaller fragment was detected in the cDNA from the AML cells of the patient, which was absent in the control and sibling LCL sample (Figure 1A). In cDNA from the parent this smaller transcript was expressed at almost undetectable levels as compared to the wild type transcript. The expression of this aberrant transcript was again not influenced by the treatment with cyclohexamide (data not shown). The fragment was subsequently cloned into a vector (Zero Blunt® PCR Cloning Kit, Invitrogen) to generate clones with the aberrant transcript. Sequence analysis of these clones showed that the splice site mutation c.-40+1G>A leads to disruption of this donor site and the use of an alternative donor site in the middle of exon 1 (Figure 1B). To test whether this was a naturally occurring transcript we designed a primer set specific for this transcript (Forward primer: 5′-TGTCTTTTGCGGCGACT-3′ and Reverse primer: 5′-ACGATATTCCTCCAATGCA-3′) of which the forward primer spans the aberrant exon 1 to exon 2 transcript. PCR analysis confirmed that the aberrant transcript was clearly present in cDNA of samples with the splice site mutation c.-40+1G>A while control samples showed only very little or no product (Figure 1C). As described above, the aberrant transcript was present at reduced levels in the AML cells. This reduction might be due to a lower stability of this transcript or due to lower efficiency of splicing itself. It is known that regulatory proteins interact with specific sequences (exonic splicing enhancers or silencers) in the pre-mRNAs and subsequently stimulate or repress exon recognition [Maniatis and Tasic, 2002]. Possibly, the aberrant pre-mRNA transcript is missing such an exonic splicing enhancer.
Figure 1. Analysis of human BRCA2 splice site mutation c.-40+1G>A.

A Primers to exon 1 and to exon 2 (Forward primer: 5′-CGAGCTTCTGAAACTAGGC-3′ and Reverse primer: 5′- CTTTGTTGCAGCGTGTCTTA-3′) were used to PCR amplify cDNA fragments from the patient AML cells, LCL of parent, sibling and wild type control. The PCR fragments of the patient AML cells were cloned into a blunt vector and subsequently sequenced. The wild type and aberrant fragment is shown on electrophoresis and in a schematic picture. B Genomic sequence of exon 1 and 2 of BRCA2 (Reference sequence NM_000059.3, indicated in grey). Sequencing of cDNA revealed that the splice site mutation led to the disruption of the original splice donor site in intron 1 and use of an alternative splice donor site within exon1, resulting in a 99 nt shorter transcript. The original and alternative splice donor sites and the start codon are indicated in black. The nucleotide found mutated in the FA patient is underlined. C Primers specific for this aberrant transcript (Forward primer: 5′-TGTCTTTTGCGGCGACT-3′ and Reverse primer: 5′- TTCACAGGCCAAAGACGGTA -3′) confirmed that the aberrant transcript (1235 bp) was clearly present in cDNA of the parent and patient with the splice site mutation c.-40+1G>A and not or hardly present in the sibling and control sample without this mutation.
To provide direct evidence that the splice site mutation c.-40+1G>A affects the function and expression of BRCA2 we examined c.-40+1G>A using a mouse embryonic stem (ES) cell-based in vitro functional assay [Kuznetsov et al., 2008]. We generated the c.-40+1G>A mutation in a BAC clone containing full-length human BRCA2 and expressed it in PL2F7 cells that have a mutant and a conditional allele of mouse Brca2. We confirmed the presence of the 5′ and the 3′ end of the human BRCA2 transgene in the ES cells by PCR and also confirmed the expression of the transgene by RT-PCR using primers to exons 11 and 18 (data not shown).
We next examined the effect of the c.-40+1G>A mutation on splicing in two PL2F7 clones containing the BRCA2 mutant BAC (A2D7 and H2B1) using primers to exon 1 and exon 9, followed by a nested PCR using primers to exon 1 and exon 2. The correctly spliced form of 465 bp, was observed in the ES cells carrying the wild type BRCA2 (WT) while in both A2D7 and H2B1 mutant clones two fragments, one 306 bp and another 1159 bp in size were observed (Supp. Figure S1A). Sequence analysis confirmed that the 306 bp band in the mutant samples was the aberrantly spliced form with loss of 99 nucleotides due to the use of an alternative donor site in the middle of exon 1 (Supp. Figure S1B and S1C), similar to the patient samples. The 1159 bp band, however, retained the entire intron 1. This splice form was also detected with specific primer directed to intron 1 in comparable levels between patient and control lymphoblast samples (Supp. Figure S2). In human lymphoblasts this transcript seems to be a lowly expressed naturally occurring splice form.
To examine the effect of aberrant splicing on transcript stability, we quantified the levels of the BRCA2 transcripts in mutant clones by q-PCR. The transcript levels were normalized to the BAC copy number. As shown in Figure 2A, we observed a significant reduction in transcript levels (A2D7, p=0.013 and H2B1, p=0.035) in the two mutants compared to WT, consistent with the results obtained in the FA patient. At present we do not fully understand why we did detect the transcript retaining intron 1 especially in the mouse samples with c.-40+1G>A mutation. It is possible that splicing or degradation of mRNA differs between cell types or between human and mice. We next examined the consequences at the cellular level of the c.-40+1G>A mutation. We first tested its ability to rescue the viability of ES cells following deletion of endogenous Brca2. We have previously shown that Brca2-null ES cells are not viable. The mutants exhibited a 13-16 fold reduction (A2D7; 0.63% and H2B1; 0.52%) in the number of viable Brca2-null cells compared to cells carrying the WT BRCA2 (8.5%), indicating that the c.-40+1G>A mutation severely affects BRCA2 function. Next, we assessed the DNA repair proficiency of the mutants by testing their sensitivity to DNA damaging agents and gamma-irradiation (IR). Both mutant clones, A2D7 and H2B1, were hypersensitive to the DNA damaging agents cisplatin, methyl methanesulfonate (MMS), MMC and IR compared to WT, suggesting a compromised DNA repair function (Figure 2B).
Figure 2. Analysis of the BRCA2 c.-40+1G>A splice site mutation in the mouse ES cell model.
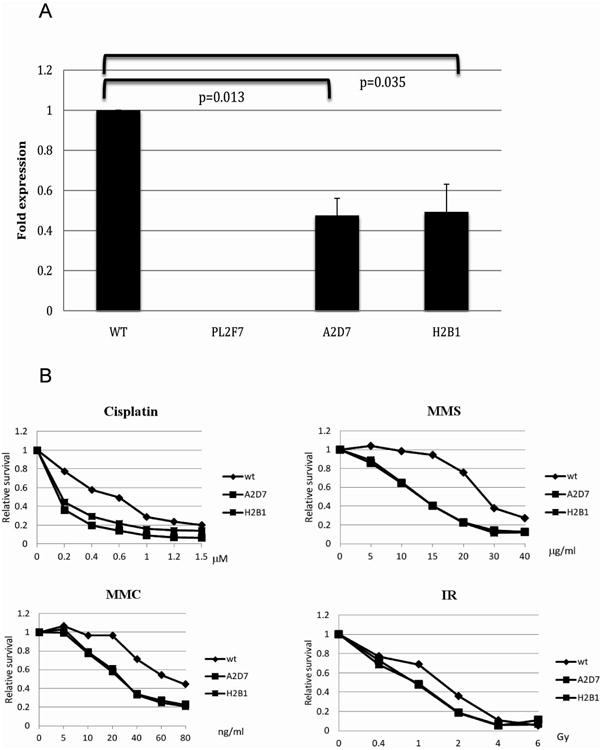
A BRCA2 transcripts are present at lower levels in mutant clones (A2D7 and H2B1) compared to WT (Negative control - PL2F7 cells do not carry human BRCA2) B Assessment of survival following exposure to DNA damaging agents. A metabolic cell proliferation assays (XTT) of cells 48h after treatment with cisplatin, MMS, MMC and gamma irradiation (IR) showed that both c.-40+1G>A mutant clones were hypersensitive compared to wt cells.
In most sequencing facilities, the non-coding exons of BRCA1 and BRCA2 are not sequenced. Therefore, we tested the contribution of BRCA1/2 non-coding exon mutations to familial breast cancer. A cohort of 752 unrelated familial non-BRCA1/2 breast cancer cases were selected from the department of Clinical Genetics at VU University Medical Center Amsterdam. Cases were selected by clinical geneticists based on current clinical guidelines. Genomic DNA was isolated from peripheral blood samples by standard methods. PCR primers were designed to cover exon and exon/intron boundaries of the non-coding exon 1 from BRCA1 and BRCA2. This mutation screening did not reveal any mutations in the splice donor sites. This is in agreement with a previously published study, where the BRCA1 promotor/5′UTR region was screened in 150 Polish familial breast and/or ovarian cancer cases and no disease causing mutations in or close to non-coding exon 1 were reported [Pamula et al., 2006]. Supp. Table S2 shows the observed variants and their prediction in splice site programs. The variants are also added to the LOVD database (www.lovd.nl/BRCA2).
In summary, we report a Fanconi anemia family with a novel mutation in the splice donor site of intron 1 of BRCA2. This mutation leads to reduced expression of a transcript that lacks part of the 5′-UTR in human cells. A mouse ES cell based in vitro functional assay confirmed that the c.-40+1G>A mutation affects splicing, and the resulting splice forms are unstable as seen by reduced transcript levels. Moreover, these experiments showed that BRCA2 containing c.-40+1G>A rescues cell survival at a very reduced frequency and results in hypersensitivity to DNA damaging agents and gamma irradiation. Screening for BRCA1/2 non-coding exon mutations in a Dutch cohort of familial breast cancer cases did not reveal similar pathogenic variants. Since the mutation was found in a Japanese FA patient it would have been interesting to screen a Japanese cohort of familial breast cancer cases for this specific splice site mutation to test for a possible founder effect. Unfortunately, we did not have access to such a cohort of patients. Although we did not observe breast cancer cases with intron 1 splice site mutations, an increased breast/ovarian cancer risk as a consequence of such mutations can not be excluded. We therefore recommend analyzing the non-coding exon of BRCA1 and 2 in the screening of familial breast and ovarian cancer. This study also highlights the importance of functional assays, where the mouse embryonic stem cell-based assay showed that the original missense variant (K2729N) in the FA patient was not pathogenic and consequently a novel pathogenic mutation in splice donor site of intron 1 of BRCA2 was identified.
Supplementary Material
Acknowledgments
We are very thankful to all participating families. This work is supported by the Center for Cancer Research Intramural Program, National Cancer Institute, National Institutes of Health of the U.S. Department of Health and Human Services (E.T. and S.K.S.). In addition we thank Roelien van der Eijk for collecting data in Amsterdam. The work in Amsterdam was funded by The Netherlands Organisation for Scientific Research (NWO) as part of a ZonMw/VIDI grant number 91756341.
Footnotes
Competing interests statement: The authors declare that they have no competing financial interests.
References
- Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res. 2009;668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas K, Das R, Alter BP, Kuznetsov SG, Stauffer S, North SL, Burkett S, Brody LC, Meyer S, Byrd RA, Sharan SK. A comprehensive functional characterization of BRCA2 variants associated with Fanconi anemia using mouse ES cell-based assay. Blood. 2011;118:2430–2442. doi: 10.1182/blood-2010-12-324541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Matsushita M, Waisfisz Q, Kinoshita A, Oostra AB, Nieuwint AW, De Winter JP, Hoatlin ME, Kawai Y, Sasaki MS, D'Andrea AD, Kawakami Y, et al. Genetic reversion in an acute myelogenous leukemia cell line from a Fanconi anemia patient with biallelic mutations in BRCA2. Cancer Res. 2003;63:2688–2694. [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov SG, Liu P, Sharan SK. Mouse embryonic stem cell-based functional assay to evaluate mutations in BRCA2. Nat Med. 2008;14:875–881. doi: 10.1038/nm.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Tasic B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature. 2002;418:236–243. doi: 10.1038/418236a. [DOI] [PubMed] [Google Scholar]
- Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, Evans DG, Izatt L, Eeles RA, Adlard J, Davidson R, Eccles D, et al. Cancer Risks for BRCA1 and BRCA2 Mutation Carriers: Results From Prospective Analysis of EMBRACE. J Natl Cancer Inst. 2013 doi: 10.1093/jnci/djt095. [DOI] [PubMed] [Google Scholar]
- Pamula J, Krzesniak M, Zientek H, Pekala W, Rusin M, Grzybowska E. Functional Impact of Sequence Alterations Found in BRCA1 Promoter/5'UTR Region in Breast/Ovarian Cancer Families from Upper Silesia, Poland. Hered Cancer Clin Pract. 2006;4:20–24. doi: 10.1186/1897-4287-4-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waisfisz Q, Morgan NV, Savino M, De Winter JP, van Berkel CG, Hoatlin ME, Ianzano L, Gibson RA, Arwert F, Savoia A, Mathew CG, Pronk JC, et al. Spontaneous functional correction of homozygous fanconi anaemia alleles reveals novel mechanistic basis for reverse mosaicism. Nat Genet. 1999;22:379–383. doi: 10.1038/11956. [DOI] [PubMed] [Google Scholar]
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- Xia B, Dorsman JC, Ameziane N, de VY, Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, Wang W, Livingston DM, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007;39:159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.