

Abstract
Protein acetylation, which is central to transcriptional control as well as other cellular processes, is disrupted in Huntington's disease (HD). Treatments that restore global acetylation levels, such as inhibiting histone deacetylases (HDACs), are effective in suppressing HD pathology in model organisms. However, agents that selectively target the disease-relevant HDACs have not been available. SirT1 (Sir2 in Drosophila melanogaster) deacetylates histones and other proteins including transcription factors. Genetically reducing, but not eliminating, Sir2 has been shown to suppress HD pathology in model organisms. To date, small molecule inhibitors of sirtuins have exhibited low potency and unattractive pharmacological and biopharmaceutical properties. Here, we show that highly selective pharmacological inhibition of Drosophila Sir2 and mammalian SirT1 using the novel inhibitor selisistat (selisistat; 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide) can suppress HD pathology caused by mutant huntingtin exon 1 fragments in Drosophila, mammalian cells and mice. We have validated Sir2 as the in vivo target of selisistat by showing that genetic elimination of Sir2 eradicates the effect of this inhibitor in Drosophila. The specificity of selisistat is shown by its effect on recombinant sirtuins in mammalian cells. Reduction of HD pathology by selisistat in Drosophila, mammalian cells and mouse models of HD suggests that this inhibitor has potential as an effective therapeutic treatment for human disease and may also serve as a tool to better understand the downstream pathways of SirT1/Sir2 that may be critical for HD.
INTRODUCTION
Huntington's disease (HD) is a progressive neurodegenerative disease, caused by an expanded CAG repeat in the HD gene on chromosome 4. This mutation produces an extended N-terminal polyglutamine stretch in the huntingtin (Htt) protein and results in progressive clinical symptoms and neuronal cell death. This process is accompanied by significant transcriptional dysregulation and reduced chromatin acetylation (1–3). Genetic manipulations that influence histone acetylation such as reduction of histone deacetylases (HDACs) can restore mutant Htt (mHtt)-challenged cells, flies and mice toward their predisease state (4–6).
Three classes of HDACs can be defined based on their homology to yeast proteins and co-factor requirements (7) with homologs of the yeast silent information regulator 2 (Sir2) being unique among HDACs in that they employ nicotinamide adenine dinucleotide (NAD+) as a cofactor and comprise the Class III HDACs (8). Genetic and pharmacologic studies with a Drosophila model of HD suggest that partial reduction (but not complete deletion) of Sir2 is protective in animals challenged with mHtt exon 1 fragments (9). In addition, genetic manipulations found reduced SirT1/Sir2 to be beneficial in other neurodegenerative models, such as oxidatively challenged neurons (10) and a Drosophila model expressing mutant Ataxin-3 (11). Further, in transgenic mice, Alzheimer's disease pathology was improved by treating mice with the non-selective sirtuin inhibitor nicotinamide (12). These observations suggest that pharmacological inhibition of Sir2 may be effective for the treatment of HD and possibly other diseases. Accordingly, we sought to test the efficacy of pharmacologic inhibition of SirT1 in multiple preclinical models of HD. Unfortunately, drug-like and highly selective inhibitors of SirT1 with good biopharmaceutical properties have been few. Nicotinamide acts as a competitive inhibitor by binding SirT1 to regenerate NAD+ and reduce SirT1 deacetylase activity (13–15), but presumably has a similar effect on all other NAD+ requiring sirtuins. Sirtinol inhibits SirT1 with an IC50 of 131 μm and SirT2 with an IC50 of 40 μm (16,17). Other inhibitors, including guttiferone G, hyperforin and aristoforin, inhibit both SirT1 and SirT2 in the low-micromolar range (18). A recently described set of indole-based molecules exhibits a 500-fold improvement over previously described SirT1 inhibitors and exhibits promising biopharmaceutical properties with a high degree of specificity and selectivity towards SirT1 as opposed to other sirtuins (14,19,20). Here we report that the highly specific SirT1/Sir2 inhibitor selisistat (selisistat; SEN0014196, EX-527, 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide; CAS RN 49843-98-3) is effective in suppressing HD pathology in Drosophila, mammalian cells and mouse models. We report target validation studies demonstrating that the in vivo target responsible for these effects is indeed SirT1/Sir2. We also show that activity of both Drosophila Sir2 and human SirT1 is decreased by treatment of transfected cells with selisistat. Selisistat is currently in clinical trials in HD patients and has proven to be safe and well tolerated in healthy human volunteers, potentially making this drug an exciting option for SirT1-lowering regimens in humans that may be effective in treating HD and possibly other diseases.
RESULTS
Genetic or pharmacological inhibition of Sir2/SirT1 is protective in a Drosophila HD model
To determine if deacetylation activity driven by Sir2 mitigates neurodegeneration, we tested whether the pathology observed in transgenic Drosophila expressing an expanded mutant human Htt exon 1 fragment in all neurons is affected when the animals are homozygous or heterozygous for a null mutation of Sir2. We measured both morphological loss of photoreceptor neurons (Fig. 1A) and physiological loss of motor function (Fig. 1B). The compound eye of Drosophila contains ∼1000 ommatidia each containing eight retinal neurons (photoreceptor cells). Seven of these can be detected by visualizing rhabdomeres (the light gathering organ of each photoreceptor neuron) with the pseudopupil technique (6,21). Animals expressing mHtt (Httex1pQ93) pan neuronally exhibit loss of retinal neurons. When Htt-expressing animals are also heterozygous for a mutation of the Sir2 (+/−), the extent of neuronal loss is reduced (Fig. 1A). However, animals with no functioning Sir2 (−/−) show more neuronal loss than siblings with two functioning copies (Fig. 1A). Similarly, Htt-challenged animals heterozygous for Sir2 (+/−) show improved motor function compared to siblings with normal Sir2 (+/+) levels. In contrast, complete loss of Sir 2 (−/−) severely compromises climbing ability (Fig. 1B). Thus, genetic loss of a single copy of Sir2 alleviates pathology by both measures while loss of both copies of Sir2 exhibits reduced rescue compared with Sir2 heterozygotes (Fig. 1AB).
Figure 1.

Genetic and pharmacologic modulation of Sir2 affects mHTT phenotypes in Drosophila. (A) Reducing Sir2 dose by half (Sir2 +/−) protects photoreceptor neurons from degeneration in flies challenged with mHttex1p Q93, but complete loss of Sir2 (Sir2 −/−) is deleterious (*P < 0.05, ***P < 0.005). (B) Animals heterozygous for Sir2 (+/−, black) show better climbing behavior than Htt-challenged animals with two doses of Sir2 (+/+, gray solid), while Sir2 −/− animals (dashed) have severely reduced climbing ability. (C) Selisistat rescues retinal neuron degeneration of mHttex1p Q93-challenged flies in a dose-dependent manner (*P < 0.05, ***P < 0.005). (D) Treatment with 10 µm selisistat (black squares) tends to improve survival of mHtt-challenged animals with the greatest effect seen in the 10–20 day survival rates (**P < 0.01) compared with no drug controls (gray diamonds). Control males not expressing mHtt (gray circles) survive much longer than mHtt-challenged females and are not affected by selisistat (not shown).
Treatment with selisistat rescues neuronal degeneration and extends lifespan in Htt-challenged Drosophila
Genetic reduction of Sir2 levels points to Sir2 as an attractive target for pharmacologic intervention. Accordingly, we tested whether pharmacologic suppression of Sir2 by selisistat would be effective in suppressing mHtt-mediated pathology in vivo using our Drosophila model of HD. Adult flies expressing mHtt exon 1 were placed on food containing 0.1, 1 or 10 μm selisistat on the first day after eclosion (i.e. emergence as an adult). In untreated animals, neurodegeneration as measured by photoreceptor neuron survival (22) gets progressively worse until death on or about Day 7 posteclosion. Sibling animals treated with selisistat show a concentration-dependent rescue of photoreceptor neurons (Fig. 1C). At 10 μm, selisistat is able to rescue degeneration to at least the same level as genetic loss of one copy of the Sir2 gene (Supplementary Material, Fig. S1). These results show that treatment with selisistat rescues neurodegeneration in a Drosophila model of HD and that the rescue is at least as robust as the maximum rescue achieved genetically.
To determine whether selisistat also affects the early death phenotype caused by expressing mHtt (Httex1pQ93) with a pan-neuronal driver (9), we monitored the lifespan of mHtt-challenged flies treated with 0 and 10 μm selisistat. Treatment with 10 μm selisistat tends to extend the lifespan of mHtt-challenged animals compared to untreated flies with the greatest effect seen in the 10–20 day survival (P < 0.01) (Fig. 1D). These effects are not due to modulation of mHtt transgene expression in the animals, as transgene mRNA levels are not altered in treated flies (Supplementary Material, Fig. S2).
Sir2 is the in vivo target of selisistat activity in Drosophila
To further assess whether Sir2 is the in vivo target of selisistat in Drosophila, we compared the ability of selisistat to rescue neurodegeneration in HD animals with a normal Sir2 gene dosage compared to its ability to rescue siblings homozygous for a null allele of Sir2. If Sir2 is really the in vivo target of selisistat, absence of any functioning Sir2 gene should abolish the ability of selisistat to improve pathology. Results show that mHtt-challenged flies with two normal copies of Sir2 show an ∼40% recovery of retinal neurons when treated with 10 μm selisistat (Fig 2A). However, when sibling HD flies homozygous for a null allele of Sir2 are treated with different concentrations of selisistat, no significant change in photoreceptor degeneration is observed when compared with untreated flies even with 10 μm selisistat (Fig. 2A). Similarly, mHtt-challenged animals with a normal (double) dose of Sir2 show improved motor function when fed up to 10 μm selisistat (Fig. 2B). However, the response to selisistat is completely abolished in mHtt-challenged animals that are also homozygous null for Sir2 (−/−). These results suggest that selisistat not only inhibits Sir2 in vitro, but that the beneficial effects of selisistat in vivo are completely dependent on the presence of functioning Sir2.
Figure 2.

Target validation: dSir2 is the in vivo target of selisistat in mHtt-challenged flies. (A) In mHTT flies challenged with Httex1p Q93 and bearing the normal two doses of Sir2 (+/+), selisistat rescues photoreceptors, while in the absence of Sir2 (−/−), the beneficial effects of selisistat on photoreceptor neurodegeneration are eliminated. Retinal neuron survival that is typically improved by treatment with selisistat is abolished if the presumed target gene is removed genetically (***P < 0.005). (B) Elimination of the Sir2 target also eliminates the effect of selisistat treatment on motor phenotype amelioration. Treatment of mHtt-expressing animals harboring two doses of Sir2 (+/+) with 10 µm selisistat (gray solid line) improved motor function compared with untreated animals (gray dashed line). Genetic reduction of Sir2 (−/−) in mHtt-challenged animals (black dashed line) decreased climbing ability and was not rescued by treatment with 10 µm selisistat (black solid line). (**P < 0.01 at 5 s).
Selisistat specificity in mammalian cells
To determine the specificity and activity of selisistat on sirtuins, HEK293 cells were transfected with GCN5 (a histone acetyltransferase), and the nuclear factor kappa B (NFκB) p65 subunit (a characterized SirT1 substrate) (23). GCN5 actively acetylates p65 as indicated by the ratio of acetylated p65 to total p65 protein in transfected cells (Fig. 3A, B, D and E). When human SirT1 is also co-transfected along with GCN5 and p65, the level of p65 acetylation is reduced by ∼80% (Fig. 3A and B). When Drosophila Sir2 is co-transfected into cells, the GCN5 acetylation of p65 is reduced by ∼70% (Fig. 3D and E). The addition of selisistat to these cells suppresses the SirT1 deacetylation restoring ∼50% of the p65 acetylation at 10 μm (Fig. 3A and C). Similarly, selisistat blocks the ability of Drosophila Sir2 to deacetylate p65 as indicated by the 60% recovery of the inhibited acetylation activity (Fig. 3D and F). These data show that selisistat inhibits the deacetylation activity of both Drosophila Sir2 as well as human SirT1.
Figure 3.

Selisistat inhibits the deacetylation activity of both human SirT1 and Drosophila Sir2. (A and D) WB analysis of total cell lysates from HEK293 cells transiently transfected with GCN5, p65 subunit and SirT1 (A) or Sir2 (D) (ratio 1: 0.5: 1). p65 acetylation was measured using a specific antibody against acetylated K310 and normalized to the total p65 content. The acetylation level in cells transfected with p65 and GCN5 only served as the control to evaluate the deacetylation activity of SirT1 or Sir2. The acetylation level in cells transfected with p65, GCN5 and SirT1 or Sir2 and treated with DMSO served as the control to evaluate the effect of selisistat treatment. The WBs shown are representative of three independent experiments. (B and E) Band intensity ratios normalized within each independent experiment (n = 3) show the extent of deacetylation by transfected SirT1 (B) or Sir2 (E). p65 deacetylation by SirT1 and Sir2, tested by applying the one-sample t-test versus the theoretical mean of 100, found the samples significantly different from respective controls (**P < 0.01). (C and F) Band intensity ratios normalized within each experiment (n = 3) show the extent to which p65 acetylation is restored by the drug treatment in cells transfected with SirT1 (C) or Sir2 (F). One-way ANOVA analysis followed by the Tukey–Kramer test for multiple comparisons found all concentrations of selisistat tested significantly different from DMSO control in SirT1-transfected cells (***P < 0.005) (C) and selisistat tested at 10 µm was significantly different from DMSO controls in Sir2-transfected cells (**P < 0.01) (F).
Selisistat is protective in cultured mammalian cell models of HD
Given the robust positive effects of genetically reducing Sir2 on HD pathology in Drosophila, we sought to determine whether selisistat exhibited positive effects in mammalian models of HD. Rat pheochromocytoma cells (PC-12) expressing mHtt exon 1 fragments have been widely employed to study mHtt toxicity and aggregation (24). PC-12 cells inducibly expressing an exon 1 fragment of human Htt with an expanded polyglutamine repeat present with aggregates, transcriptional changes and cytotoxicity upon transgene expression (24). In this model, induction of mHtt expression results in a robust increase in toxicity (measured as lactate dehydrogenase [LDH] release), which was significantly reduced by treatment with selisistat at the concentrations of 1 and 10 μm (Fig. 4A).
Figure 4.
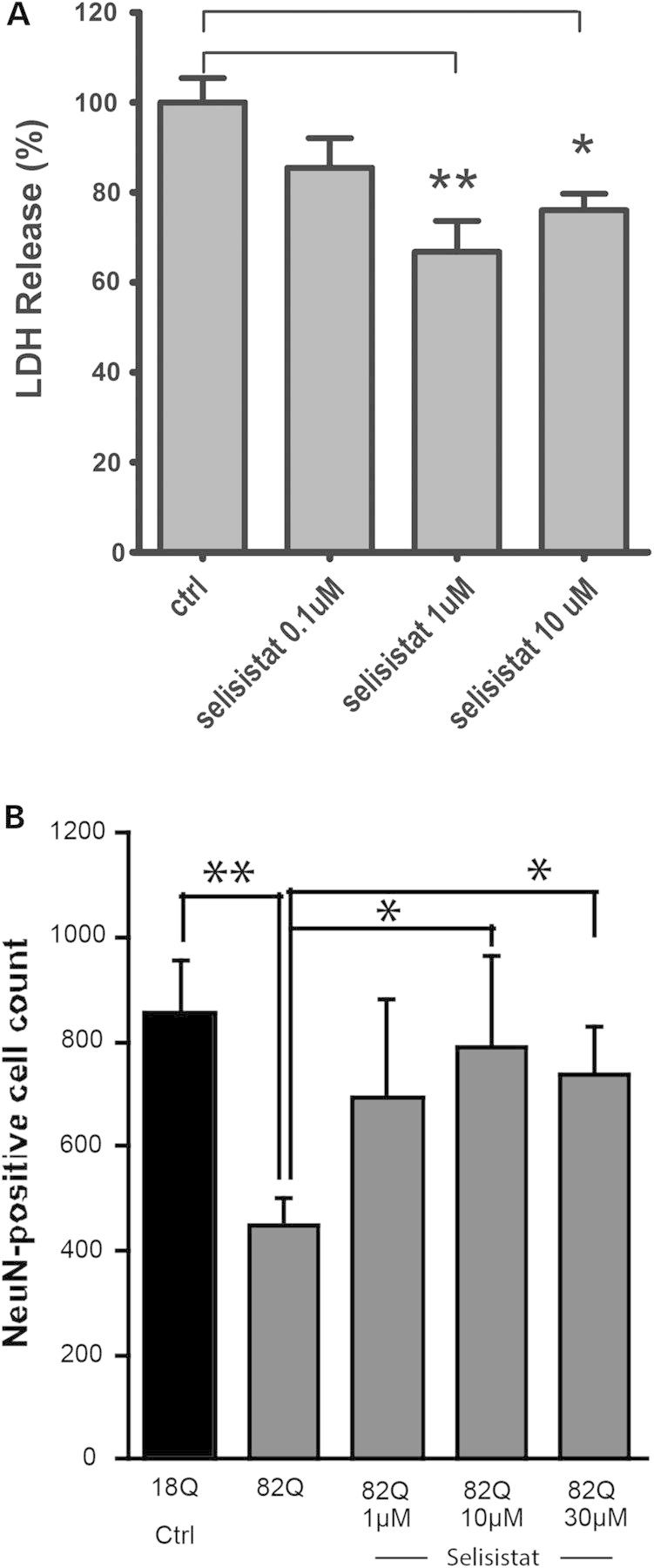
Pharmacological inhibition of SirT1 rescues mHTT-mediated toxicity in mammalian cells. (A) In PC12 cells inducibly expressing a human HTT exon 1 fragment bearing 72 glutamine repeats, selisistat suppresses toxicity induced by expression of the mHtt transgene. Data are represented as mean and SEM of normalized data (induced control only—cited in the figure as ctrl = 100%; uninduced ctrl = 0% not shown) (controls are DMSO-treated only). (*P < 0.05, **P < 0.01 post-ANOVA Tukey–Kramer test for multiple comparisons). (B) In primary rat striatal cultures infected with lentiviral vectors expressing mutant (82Q) or wild type (18Q), selisistat treatment abolished 82Q-HTT-induced neuronal loss in a concentration-dependent manner. Data are displayed as mean and SEM (n = 4). (*P < 0.05, **P < 0.01 by two-sample Student's t-test for indicated comparisons).
The neuroprotective activity of selisistat was also investigated in a postmitotic, primary neuronal model. Infecting primary cultures of rat striatal neurons with viral vectors expressing mHtt fragments (25,26) leads to decreased culture viability and aggregates of Htt when the Htt transgene bears an expanded (mutant) polyglutamine repeat (27). Therefore, striatal cultures infected with lentiviruses expressing an N171 fragment of human Htt encoding either 18 or 82 glutamines were treated with selisistat to investigate the neuroprotective activity of the compound. selisistat was able to significantly protect cultured primary neurons against cell loss (measured as NeuN counts) resulting from infection with a mHtt-expressing lentivirus (Fig. 4B).
Selisistat is protective in the R6/2 mouse model of HD
Finally, we sought evidence that chronic administration of selisistat is protective in an in vivo mammalian model of HD. The compound, which displays low clearance and complete oral bioavailability as well as a 2:1 brain:plasma ratio in mice and absence of overt toxicity in the mouse at dose levels up to 100 mg/kg (Table 1), was therefore tested in a transgenic mouse model of HD. Among transgenic mouse models, the R6/2 mouse (28) represents one of the most widely employed and characterized in vivo models for preclinical studies and has been the model of choice for a variety of pharmacological preclinical trials (10,29,30). The R6/2 mouse expresses a human exon 1 fragment encoding an expanded polyglutamine repeat under control of the human Htt promoter and displays rapid symptomatic onset and robust disease progression (31). This model is characterized, among other phenotypes, by decreased survival, loss of body weight, impaired motor activity, striatal degeneration and the presence of Htt-positive aggregates in the brain and other tissues.
Table 1.
Pharmacokinetic parameters of selisistat in R6/2 mice (mean ± SD, n = 3)
| Dose (mg/kg) | 5 mg/kg | 10 mg/kg |
20 mg/kg | |
|---|---|---|---|---|
| Plasma | Brain | Plasma | Plasma | |
| Cmax (μm) | 6.9 ± 6.9 | 21.5 ± 3.3 | 10.5 ± 3.6 | 29.3 ± 6.4 |
| tmax (h) | 0.3 ± 0.1 | 1.0 ± 0.0 | 0.8 ± 0.4 | 0.5 ± 0.0 |
| AUC0–τ(last) (μm h) | 9.9 ± 4.5 | 71.1 ± 9.1 | 35.4 ± 10.4 | 77.5 ± 9.7 |
| t1/2,z (h) | 2.7 ± 2.3 | 2.8 ± 0.4 | 1.4 ± 0.5 | 0.9 ± 0.2 |
| Css,avg (μm) | 0.4 ± 0.2 | 3.0 ± 0.4 | 1.5 ± 0.4 | 3.2 ± 0.4 |
Maximal plasma concentrations (Cmax) achieved at time tmax. AUC0–τ is the area under the time versus plasma concentration curve at steady state and the Css,avg indicates the average plasma concentration, i.e. the AUC0–τ divided by the dosing interval, τ (24 h). t1/2,z is the terminal plasma half-life. Brain versus plasma was determined for 10 mg/kg only.
To investigate the effects of selisistat, the compound was administered to transgenic R6/2 mice beginning at 4.5 weeks of age to death at doses of 5 and 20 mg/kg. The animals were subjected to a battery of standardized, disease-relevant phenotypic tests in a specialized contract research organization (PsychoGenics, Inc.). Treatment with selisistat resulted in a significant increase of survival of mice receiving the 20 mg/kg dose with a notable median lifespan increase of 3 weeks in comparison with vehicle-treated animals (Fig. 5A). Significant improvements were also observed when examining voluntary locomotor activity (Open Field parameters, Fig. 5C and D). Namely, administration of selisistat ameliorated the deficits of R6/2 mice detected both in distance travelled in the center (Fig. 5C) and the average velocity in the Open Field (Fig. 5D) for the 5 and 20 mg/kg regimens, respectively. Body weight (Fig. 5B), rotarod performance and grip strength (data not shown) were not significantly affected by treatment with selisistat.
Figure 5.

Effect of chronic administration of selisistat (5 and 20 mg/kg, PO, QD) on survival, body weight and behavioral parameters in mice. (A) R6/2 mice show premature death. Improved survival was associated with selisistat administration (one-sided log-rank Mantel–Cox statistic: *P < 0.05). Pairwise comparison of individual dose groups with vehicle-treated mice indicated no effect in the 5 mg/kg group (P > 0.5) but significant increase in survival in the 20 mg/kg group (P < 0.05) increasing median survival of 3 weeks. (B) The effect of chronic administration of selisistat (5 and 20 mg/kg, PO, QD) on body weights of the R6/2 mice from 4 to 25 weeks of age is depicted. R6/2 mice display lowered body weight compared with wild-type mice (data not shown). There was no statistically demonstrable effect of treatment on body weight although statistical analysis of body weight could only be carried out to 19 weeks due to the need for multiple subjects in all groups to satisfy conditions for ANOVA. (C and D) The effect of chronic administration of selisistat (5 and 20 mg/kg, PO, QD) in R6/2 mice on two different parameters of Open Field performance, distance traveled in center (C) and average velocity (D). (C) A significant effect of treatment on distance traveled in the center of the open field was observed with compound-treated mice. Individual comparison of drug-treated groups with the vehicle group across all ages revealed significant increases in center locomotion in the 5 mg/kg group (**P < 0.01). (D) A strong trend for treatment to increase velocity in the open field was detected in the 5 mg/kg group (P = 0.08), and individual group comparisons with vehicle revealed significant effects for the 20 mg/kg group (*P < 0.05).
The effects of selisistat on brain pathology were analyzed in a satellite group of animals that was sacrificed at 12 weeks of age (8 weeks of treatment). Ventricular volume and EM48 (Htt)-positive staining of aggregates were investigated as measures of striatal degeneration and Htt aggregate load, respectively. HD-like pathological indicators in R6/2 mice mimicked those seen in HD patients in the loss of striatal tissue with an associated increase in ventricular volume and accumulation of EM48-positive inclusions in the brain as seen in the top vehicle-treated panels of Figure 6A and B. In mice treated with 5 mg/kg selisistat, a statistically significant reduction of ventricular volume is evident in comparison with vehicle-treated animals (Fig. 6A and C). This improvement did not reach statistical significance versus vehicle controls in the 20 mg/kg-treated animals due to a somewhat larger variance, although a tendency towards reduced volume was observed (Fig. 6A and C).
Figure 6.
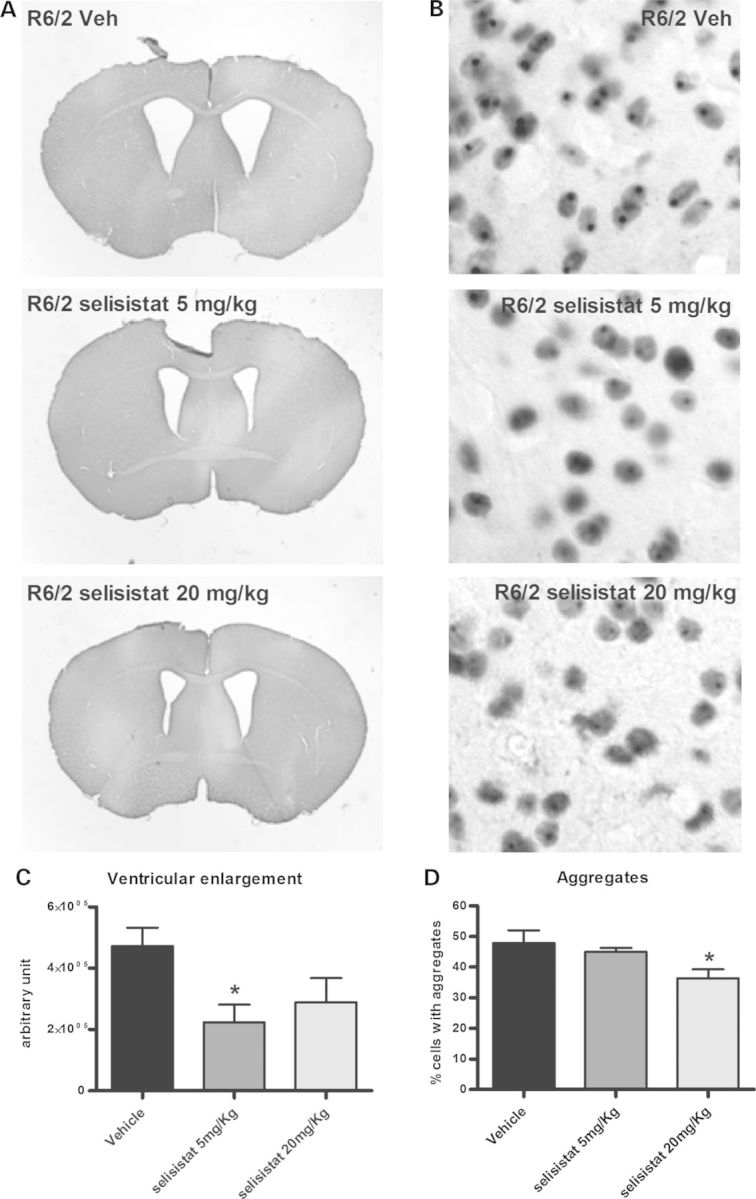
Pharmacological inhibition of SirT1 prevents the enlargement of ventricular volume and lowers Htt inclusion load in R6/2 mice. Brain slices from 12-week-old mice were analyzed for ventricular volume and inclusions using image analysis software. (A) Representative photomicrographs of brains from R6/2 mice treated with vehicle or 5 or 20 mg/kg selisistat. Note the reduction of ventricular enlargement in treated animals. (C) Ventricular volume is reduced in R6/2 animals treated with selisistat compared with untreated R6/2 animals. Data are displayed as mean and SEM (*P < 0.05 post-ANOVA Tukey–Kramer test for multiple comparisons). (B and D) Pharmacological inhibition of SirT1 lowers aggregation load in R6/2 mice striata. Brain slices from 12-week-old mice were stained to detect EM48-positive striatal inclusions. (B) Representative photomicrographs of striatal neurons from R6/2 mice treated with vehicle or 5 or 20 mg/kg selisistat. (D) Analysis of aggregate load in R6/2 mice striata in the different groups of animals expressed as percent of cells containing aggregates. Data are displayed as mean and SEM (*P < 0.05 post-ANOVA Tukey–Kramer test for multiple comparisons).
Brain slices were examined for the presence of inclusions of EM48-positive aggregates in vehicle- and selisistat-treated animals using automated image analysis. As evident in Figure 6B and D, a significant decrease in the number of inclusions was observed in selisistat-treated animals at the dose of 20 mg/kg and a trend toward reduction was also observed at 5 mg/kg.
Pharmacokinetic data (Table 1) indicate that the average steady-state plasma levels in R6/2 mice at 5, 10 and 20 mg/kg/day were 0.4, 1.5 and 3.2 μm, respectively, and that the brain exposure was about 2-fold that in the systemic circulation, effectively encompassing the drug concentrations used across the in vitro experiments. Collectively, these data suggest that selisistat can revert a number of mHtt-associated phenotypes when applied to in vitro and in vivo Drosophila and mammalian models of HD by targeting Sir2/SirT1 and inhibiting its activity.
DISCUSSION
Sir2 is a nuclear protein that primarily targets proteins involved in transcription (32). The activity of Sir2, first described in yeast as Silent Information Regulator, is central to the regulation of chromatin packaging. The deacetylase activity of Sir2 tends to silence transcription at target loci, many of which are involved in metabolic functions. The sirtuins are unique among the HDACs in their use of NAD+ as a cofactor. The requirement for NAD+ provides a tight link between cellular metabolism and transcriptional output. Sir2 removes acetyl groups from lysine residues in histones (33,34) and from other proteins including several transcription factors such as members of the FOXO family (35,36), NFκB (37), PGC1α (38) and p53 (34). Sir2 also represses the activity of the key chromatin-modifying protein p300 (39), a histone acetyl transferase related to CBP, which is also impaired in HD (6). Thus, the targets of Sir2/SirT1 are primarily transcription-modifying proteins such as histones, chromatin-modifying enzymes and transcription factors, thus making it a potentially attractive target for affecting the transcriptional dysregulation caused by pathogenic fragments of mHtt. However, the genetic manipulation of HDACs and of Sir2/SirT1 in particular in different preclinical models of HD, either through overexpression, RNA inhibition or knockout of Sir2/SirT1 alleles (9,40–43) has led to differing views regarding therapeutic strategies based on manipulation of Sir2/SirT1 activity. Our study and others support the notion that inhibition of Sir2 is protective in various neurodegenerative environments such as AD mice (12), Ataxin-3-challenged Drosophila (11), oxidatively damaged cells (10), Drosophila models of HD (9) and mHtt-challenged Drosophila, mammalian cells and mice challenged with mHtt exon 1 fragments (this study). Indeed, we have previously shown that lowering (but, importantly, not eliminating) Sir2 gene dosage (through the generation of heterozygous null alleles) or sirtuin inhibition provides substantial amelioration in a Drosophila model of HD (9). Consistent with this, complete lack of Sir2/SirT1 led to cell death and increased toxicity in PC12 cells inducibly expressing mHtt exon 1. Other genetic studies in mice suggest that increasing Sir2/SirT1 levels would provide relief through activation of the FOXO3A and CREB pathways (41,42), although overexpression of Sir2 in Drosophila neurons either enhances the phenotype when using an enhancer trap line that has a complex response (9,40,44) or is lethal when using the elav-Gal4/UAS system (44) (data not shown), making this an unlikely therapeutic option. On the other hand, SirT1 directly deacetylates and represses the activity of p300 (39), a histone acetyl transferase related to CREB-binding protein which is also impaired in HD (6), as well as Tip60, which promotes apoptosis (45), suggesting that decreasing the levels of Sirt1/Sir2 might be relevant to pathology by increasing the activities of key histone-acetylating enzymes, while overexpression may reduce the activity of key enzymes such as CBP leading to reduced viability. Until recently, the paucity of potent and selective pharmacological modulators (activators or inhibitors) of Sir2/SirT1 has prevented a careful assessment of the therapeutic potential of pharmacological SirT1 modulation in preclinical disease models.
Access to a selective and effective Sir2/SirT1 inhibitor has allowed a reassessment of the therapeutic potential of Sir2 inhibition for HD using both new genetic and pharmacological tools. We find that lowering Sir2 gene dosage using genetically targeted knockouts improves neuronal survival, motor function and lifespan in a Drosophila model of HD. Importantly, these effects can be phenocopied by a potent and selective SirT1 inhibitor (selisistat) which possesses a profile suitable for clinical testing. Through the use of combined genetic and pharmacological approaches, we have established that the endogenous HD-relevant target of selisistat in the fly is indeed Sir2, thus confirming that the beneficial effects of reduced Sir2 in this model are not due to alterations in non-HDAC SirT1 functions and providing proof of target engagement for the compound. The demonstrated efficacy of selisistat to inhibit mammalian SirT1 as well as rescue neurons is promising, although the possibility of off-target effects in mammalian systems cannot be strictly ruled out. As Sir2 in turn can engage multiple downstream protein targets, many of which have been identified as relevant to HD pathology (e.g. histones, PGC1a, FOXO), inhibition of Sir2 can be thought of as an intervention that is likely to engage multiple disease-relevant pathways that are modulated by Sir2 activity.
In this study, we have tested the effects of genetic and pharmacologic manipulation of Sir2/SirT1 activity in a robust, naturally occurring disease-relevant model of HD (i.e. mHtt exon 1 expression). We find that selisistat displays cyto- and neuroprotective activity in both mitotic and postmitotic mammalian cellular models of HD and that it displays positive effects on disease-relevant phenotypes in both transgenic Drosophila and R6/2 mice. Selisistat has completed clinical studies in healthy volunteers to assess safety, tolerability and pharmacokinetics as well as short-term studies (up to 12 weeks) in HD patients, aimed at assessing safety, tolerability and pharmacodynamic parameters, and satisfies all criteria to qualify as a candidate for clinical efficacy studies in patients with HD.
MATERIALS AND METHODS
Drosophila crosses
To compare phenotypes of Htt-expressing animals in normal versus a Sir2-altered background, flies that were elav-Gal4[C155]; Sir2[17]/CyO were crossed to UAS-Httex1p Q93 homozygotes (line p463) (6). To produce the homozygous deletion of Sir2 in an HD background, flies that were elav-Gal4[C155]; Sir2[17]/CyO were crossed to UAS-Httex1p Q93 that contained Sir2[17] and a second chromosome marker. Crosses were performed at 22.5°C. After eclosion, adult flies were reared at 25°C on standard cornmeal molasses medium for genetic studies or medium containing either 0.1% DMSO or the indicated concentration of selisistat (0.1–10 µm) for pharmacological studies. Fresh food was provided daily. Pseudopupil analysis was performed at 7 days as described (6,21). For longevity experiments, freshly enclosed virgins were aged in groups of 25–30 animals. Longevity was determined by counting the number of surviving animals to calculate percent survival, and flies were passed every 2–3 days. Climbing of aged 7-day-old flies was performed in polystyrene shell vials 9.5 cm in height with a diameter of 2.4 cm (Fisher Scientific, Pittsburgh, PA, USA). Vials were placed in a holding box with the front and back open. A light box was placed behind the vials to improve visibility of flies. The flies were video recorded using an Exilim EX-FH20 camera with 40 f.p.s. The percentage of flies that climbed past the midpoint of the vial was calculated as a function of time after shakedown. Approximately 10–15 flies per vial were used for the climbing assay. For overexpression of Sir2, the UAS-Sir2 line was generously provided by Dr. Karen Chang at USC.
PC-12 exon 1-expressing mutant/wild-type Htt cell lines
PC-12 7210 (exon 1 mutQ74) cells (PC12-Q74) stably expressing a GFP-tagged-exon 1 fragment of the human HD gene were obtained from Prof. Rubinsztein's laboratory (24). The tetracycline (Tet-on)-inducible mHTT construct comprises nucleotides 1–297 of the human Htt sequence (NM_00211) and includes a 74 CAG repeat expansion that, once expressed, is toxic to the cells. Cells were seeded in a 96-well poly-d-lysine (MW 70–150 kDa) (Sigma, MO, USA) precoated plate at a density of 45K cells/100 μl medium/well in DMEM-containing 2% HS, 1% FBS, 100 mU/ml penicillin/streptomycin and 1% glutamax, then grown for 24 h prior to the experiment in an incubator at 37°C, with 90% of relative humidity and 10% CO2 atmosphere. The day of the experiment, the same medium but devoid of serum was added to the wells in order to obtain a final dilution of the previous serum concentration to 1:3. For transgene induction, the serum-free medium was complemented with doxycycline (final concentration 1 μg/ml). Selisistat was added (from a DMSO 10 mm stock solution) to obtain the final concentrations described in the results, omitting its addition in the controls that received DMSO only. The final concentration of DMSO in all treatments and controls was 0.1%. At the 72 h time point, cell death was assessed by measuring levels of LDH released from cells in the medium using an LDH-Mix Cytotoxicity Test Kit (Roche, Switzerland), absorbance was measured at 490 nm (reading) and 720 nm (blank) with a spectrophotometer (Benchmark Plus, Bio-Rad, Hercules, CA, USA).
Preparation and culture of primary striatal neurons
Cultures were obtained after dissection of the brain ganglionic eminence obtained from rat embryos at E15, after their removal from a pregnant Sprague-Dawley rat (Janvier Labs), humanely euthanized in accordance with applicable laws and Animal Ethics approvals. Tissue was minced with forceps and cells were mechanically dissociated in a solution of BSA (1 mg/ml) in DMX medium by gently flushing the cell suspension up and down with a tip-flamed Pasteur pipette. The solution was allowed to settle until the remaining cell aggregates could sediment to the bottom. The supernatant containing dissociated cells was removed with the Pasteur pipette and transferred to a new tube. A DMX/BSA solution was added to the remaining cell aggregates, again dissociated mechanically by gently flushing the cell suspension in the Pasteur pipette. The supernatant was removed with the Pasteur pipette and transferred to the new tube. A 5% BSA-PBS solution was added slowly to the bottom of the tube containing the cell suspension. This step allowed the formation of a BSA gradient, which removed any debris from the cell suspension before plating. The cell suspension was centrifuged at 300g for 8 min at RT. Then, cells were resuspended in Neurobasal medium containing B27, 100 mU/ml penicillin/streptomycin, l-glutamine (0.5 mm), KCl (15 mm), plated in poly-l-lysine-coated multi-well plates at a density of 150K cells/cm2 and placed in cell incubator set at 37°C with a 5% CO2 atmosphere.
Lentiviral infection of cultured striatal neurons
In vitro models of HD were realized using lentiviral vectors as described (27). These models involve the lentiviral-mediated overexpression of N-terminal 171 amino acid fragments of wild-type Htt (with 18 glutamine repeats, 18Q) or mHtt (with 82 glutamine repeats, 82Q) in striatal neuronal cultures. For lentiviral-mediated protein expression, cultures were infected 24 h after seeding. On Day 4, half of the medium was replaced with the fresh medium supplemented with selisistat in 2× concentration. Treatments with compound were performed once a week thereafter by adding fresh medium with compound at 1× concentration. The strong promoter constructs (high expression, 5–10 times endogenous) resulted in polyQ-dependent cell death within 2–4 weeks in vitro, as assessed by reduced NeuN-positivity and NeuN-positive cell numbers. Htt-N171-82Q- but not Htt-N171-18Q-exposed cells also develop intracellular Htt inclusions at 1–2 weeks (high expression) or 2–4 weeks (moderate expression) in vitro. For the neurotoxicity assessment, primary cultured striatal neurons were washed with PBS and fixed with 4% paraformaldehyde in PBS (Fluka) for 15 min at RT. After fixation, cells were washed three times with PBS and blocked with 10% normal goat serum (NGS; Invitrogen) in PBS supplemented with 0.1% Triton X-100 (Sigma). An anti-NeuN/Fox-3 monoclonal antibody (Millipore) was diluted 1:500 in PBS with 5% NGS and 0.1% Triton X-100 and incubated with the cells overnight at 4°C. Cells were washed three times with PBS and incubated for 1 h with goat-anti-mouse Cy3-conjugated antibody (1:1000 dilution, Invitrogen) in PBS with 1% NGS, followed by three washes with PBS. Images were acquired employing an automated high-throughput imaging system BD Pathway 855 Bioimager (Becton Dickinson) using a 4× objective. Image processing and cell counting was performed using the public domain software ImageJ. Thresholds for fluorescence intensity, size and circularity of fluorescent objects were set to uniform parameters for each experiment.
HEK293 cell transfection and treatments
HEK 293-T cells were grown in DMEM (Gibco, Invitrogen) containing 10% FBS, 1% Penstrep (Gibco), 1% G-Max (Gibco) at 37°C and 10% CO2. 8 × 105 cells were seeded on MW6 plates (Corning) and after 24 h, cells were transfected with 2.5 µg of total plasmid DNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Plasmids expressing GCN5 (NM_021078.1), p65 (NM_021975.3), human_SirT1 (NM_012238.4) were purchased from OriGene Technologies, Rockville, MD, USA; plasmid expressing the Drosophila gene Sir2 cDNA (LD07439) was ordered from DGRC and cloned into a pcDNA vector. Four hours after transfection, the Opti-MEM medium was removed and selisistat was diluted to 0.1, 1 and 10 µm (DMSO 0.1%, v/v, as control) in the culture medium and added to the cells. At 24 h posttransfection, cells were collected and lysed in RIPA buffer (150 mm NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris, pH 8.0) with protease and phosphatase inhibitors (Complete EDTA-free protease inhibitor cocktail, Roche and PhosSTOP inhibitor cocktail, Roche). Total lysates were clarified by centrifugation at 3000g for 5 min and the protein amount quantified by BCA (Pierce) according to the manufacturer's instructions.
Western blotting
Protein lysates (20 µg) were loaded on AnyKD TGX gels (Bio-Rad) and transferred for 1 h at 100 V to PVDF membranes (Amersham). Membranes were blocked in 3% NFM for 1 h, washed with PBS containing 0.01% Tween-20 and incubated with appropriate antibodies in 3% NFM. Primary antibodies were used at the following concentrations: anti p65AcK310 at 1 µg/ml (Abcam), anti total p65 at 0.1 µg/ml (Abcam), anti β-actin AC-74 clone, at 0.05 µg/ml (Sigma-Aldrich). HRP-conjugated secondary antibodies (Bio-Rad) were diluted 1:20 000 in 3% NFM and incubated for 1 h at room temperature. ECL Prime substrate (GE Healthcare) was used to develop chemiluminescent signal that was acquired using Versadoc 4000 (Bio-Rad).
Mouse studies
Animals
R6/2 transgenic mice carrying the N-terminal region of a mutant human Htt gene (28) were bred by crossing ovarian transplanted females (from R6/2 CBAB6J female donors, Jackson Laboratories) with CBAxC57BL/6J F1 wild-type males. Genotype was determined by real-time PCR of tail snips collected ∼10 days of age. In mutant mice, the CAG repeat lengths were determined by Genemapper software by Laragen Inc (28). Only mice with at least 100 CAG repeats were included in the study. The average CAG repeat length in this study was 108.9 ± 0.5. Mice were handled on 2 consecutive days (1 min each day) between 19 and 21 days of age. Animals were tail tattooed at 20–21 days of age and weaned at 21–22 days of age. Mice from multiple litters were used for each treatment group (equally divided between genders). Mice were housed 4–5 mice/cage in Optimice cages (Animal Care Systems, CO, USA) with wood shavings, food and water; an enriched environment was created with the addition of play tunnels, shredded paper and plastic bones. Mice had free access to regular food and water and, in addition, all mice received from weaning wet powdered food placed inside a cup on the floor of the cage which was replaced fresh daily. All manipulations and handling were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals, NRC 2011. The protocol was approved by the Institutional Animal Care and Use Committee of PsychoGenics, Inc. (PHS OLAW animal welfare assurance number A4471-01), an AAALAC International accredited Institution (Unit #001213).
Body weight and survival
Mice were weighed twice a week and were examined daily for survival analysis. Death was determined by lack of heartbeat.
Behavioral tests
The following behavioral tests were performed during the light phase of the diurnal cycle.
Open field
Activity chambers (Med Associates Inc, St Albans, VT, USA; 27 × 27 × 20.3 cm) were equipped with infrared beams. Mice were placed in the center of the chamber and their behavior was recorded for 30 min. Quantitative analysis was performed on distant travelled (total and center), total rearing frequency and velocity. Mice were tested at 3.5–4, 6, 8, 10 and 12 weeks of age.
Rotarod
Mice were tested over 3 consecutive days at 3.5–4, 6, 8 and 10 weeks of age. Each daily session included a training trial of 6 min at 4 revolutions per minute (RPM) on the rotarod apparatus (AccuScan, OH, USA). One hour later, the animals were tested for three consecutive trials during which the speed accelerated from 0 to 40 RPM over 360 s, with an intertrial interval at least 30 min. The latency to fall from the rod was recorded. Mice remaining on the rod for >360 s were removed and their time scored as 360 s. Data for the training trial were not included.
Grip strength
Mice were scruffed by the lower back and tail and lowered towards the mesh grip piece on the gauge (Chatillion Force Gauge, San Diego Instruments, San Diego, CA, USA) until the animal grabbed on with both front paws. The animal was then lowered toward the platform and gently pulled straight back with consistent force until it released its grip. The forelimb grip force was recorded on the strain gauge and averaged over five trials. Mice were tested at 3.5–4, 6, 8, 10 and 12 weeks of age.
Drug treatments
Treatments were started at 4.5 weeks of age after mice had been tested at 3.5–4 weeks to establish baseline behavioral performance for all of the animals. Groups of 18 mice (9 per gender) were assigned to each R6/2 group. Mice were balanced across experimental groups by body weight, CAG repeats, date of birth and litter size before testing began. Mice were run in open field, rotarod and grip strength at 3.5–4 weeks and treatment designations were rebalanced before drug treatments were started to ensure that behavioral performance was initially similar between treatment groups. Additional data used for rebalancing the groups included rotarod fall time, total distance travelled and total rearing frequency in the open field and grip strength. Mice received daily (QD) oral gavage (PO; 10 ml/kg) of selisistat (5 and 20 mg/kg) or its vehicle (0.5% hydroxyl-propylmethylcellulose Methocel K4M Premium in sterile water; 0.5% HPMC). Suspensions were prepared weekly and aliquotted into amber vials (light sensitive) for daily dosing; powdered drug was stored in a desiccator at 4°C. Vehicle was prepared bimonthly and stored at 4°C. Each vial was vortexed prior to dosing and contained a small stir bar and remained on a stir plate during dosing. A satellite group of animals for pharmacokinetic assessments were dosed from 3 to 10 weeks of age with selisistat. Following the last dose, animals were terminated and trunk blood samples were collected from three mice per group at 0.25, 0.5, 1, 6 and 24 h postdose in heparin-coated tubes kept on wet ice until centrifugation at 2700 RPM at +4°C. The supernatant was removed and plasma stored at −80°C until analysis using an LC–MS/MS method with a lower limit of quantitation of 5 ng/ml. Pharmacokinetic parameter estimates were achieved using WinNonlin, v. 5.01.1, Pharsight CA, USA.
Immunohistochemical analysis
A satellite group of animals (n = 3/4) for morphological studies was sacrificed at 12 weeks of age. The animals were perfused transcardially with ice-cold paraformaldehyde solution (4% in phosphate-buffer, pH 7.4). The brains were postfixed for 4 h and cryoprotected in 18% sucrose solution. Brains were sectioned in a cryostat into 30 µm-thick coronal sections and placed in anti-freezer solution (phosphate-buffered saline containing 30% ethylene glycol and 30% glycerol) and stored at −20°C until used for immunohistochemistry, according to the following schedule:
Day 1: For detection of cholinergic neurons, the monoclonal antibody EM48 raised against Htt protein (1:250 dilution) (Chemicon International, Temecula, CA, USA) was used. The sections were washed in PBS-Triton X-100 0.3% (PBS-TX), and then incubated free-floating overnight at RT with the primary antibody in PBS solutions containing albumin 5 mg/ml, Triton X-100 0.3% and sodium azide 0.1%.
Day 2: After washing in PBS, sections were incubated in biotinylated secondary antibody (1:1000 dilution, Vector Laboratories, Burlingame, CA, USA) and subsequently incubated in avidin–biotin–peroxidase complex (Vectastain, Vector Laboratories, Burlingame, CA, USA, final dilution 1: 500) and stained using diaminobenzidine (DAB) (Vector Laboratories, Burlingame, CA, USA) in the presence of NiCl. DAB-stained slices were examined using an Olympus BX40 (Milan, Italy) microscope and photographed using a digital camera (Olympus DP50).
Inclusions in EM48-positive cells and ventricular enlargements were quantified in DAB-stained slices by transforming digitized images to TIFF files and analyzing using Scion Image (Scion Corporation, Frederic, MD, USA) for Windows. The gray background (non-specific staining) was calculated as a threshold, automatically subtracted from each image and the area in pixels above the set threshold was measured automatically using SCION Image. Care was taken to maintain the same gray threshold in the images from control and treated slices from the same experiment and inclusions were scored by the software without operator intervention. The area above the set threshold was calculated in pixels.
Statistical analysis
Statistical analyses for the Drosophila pseudopupil assay and climbing assay and analysis of data from primary striatal neurons and p65 deacetylation by Sir2 and SirT1 were performed using Student's t-test. *P < 0.05, **P < 0.01; ***P < 0.005. Statistical analysis of data from in vitro experiments in PC-12 cells, band intensity ratios testing selisistat on Acp65K310 versus total p65 on SirT1/Sir2 and immunohistochemistry ex vivo experiments were performed using one-way ANOVA analysis.
In vivo studies in R6/2 mice: body weight, rotarod and open field data were analyzed using repeated-measures analyses, using mixed-effect models. All models were fitted using the PROC MIXED procedure in SAS 9.1.3 (SAS Institute, Cary, NC, USA). Treatment and age were the two independent factors assessed. The within-subject correlation structure was modeled through an autoregression: slight variations of those models were tested and the best model based on the Akaike and Bayesian Information Criteria was selected. Data from 4 weeks of age were used as baseline to compare against the behavioral measurements taken between 6 and 12 weeks.
Survival data were analyzed with Kaplan–Meier survival curves. Log-rank (Mantel–Cox) analysis was performed to test effects on survival. When appropriate, post hoc paired group analyses were conducted using the Tukey–Kramer test to correct for multiple comparisons.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by NS-45283 with ReEntrySupplement for M.R.S. and Siena Biotech SB-51857 to J.L.M.
Supplementary Material
ACKNOWLEDGEMENTS
This study represents the efforts of a consortium of groups that each brought unique expertise to the project. Authors are generally grouped by institution in order of appearance of their contribution with the bulk of the Drosophila work being done in Irvine, the cell work in Siena and Lausanne and the mouse work in Siena and in PsychoGenics Inc, Tarrytown, NY, USA. All groups have contributed intellectually to the course of the project. We thank Karen Chang (USC) and Linda Partridge (University College London) for UAS>Sir2 Drosophila stocks. We thank Evrim Arslan, Meredith Taylor, Renette Walker, Washington Arias, Judith Watson-Johnson, Luis Sanchez, Melinda Ruiz and Raynel Stevenson for their technical support.
Conflict of Interest statement. S.T. and R.L.C. have received consultancy fees from Siena Biotech SPA. J.L.M. and R.L.C. have received research grants from Siena Biotech SPA.
REFERENCES
- 1.Benn C.L., Sun T., Sadri-Vakili G., McFarland K.N., DiRocco D.P., Yohrling G.J., Clark T.W., Bouzou B., Cha J.H. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 2008;28:10720–10733. doi: 10.1523/JNEUROSCI.2126-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cha J.H. Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 2000;23:387–392. doi: 10.1016/s0166-2236(00)01609-x. [DOI] [PubMed] [Google Scholar]
- 3.McFarland K.N., Das S., Sun T.T., Leyfer D., Xia E., Sangrey G.R., Kuhn A., Luthi-Carter R., Clark T.W., Sadri-Vakili G., et al. Genome-wide histone acetylation is altered in a transgenic mouse model of Huntington's disease. PLoS ONE. 2012;7:e41423. doi: 10.1371/journal.pone.0041423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrante R.J., Kubilus J.K., Lee J., Ryu H., Beesen A., Zucker B., Smith K., Kowall N.W., Ratan R.R., Luthi-Carter R., et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J. Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sadri-Vakili G., Cha J.H. Mechanisms of disease: Histone modifications in Huntington's disease. Nat. Clin. Pract. Neurol. 2006;2: 330–338. doi: 10.1038/ncpneuro0199. [DOI] [PubMed] [Google Scholar]
- 6.Steffan J.S., Bodai L., Pallos J., Poelman M., McCampbell A., Apostol B.L., Kazantsev A., Schmidt E., Zhu Y.Z., Greenwald M., et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 7.McLaughlin F., La Thangue N.B. Histone deacetylase inhibitors open new doors in cancer therapy. Biochem. Pharmacol. 2004;68:1139–1144. doi: 10.1016/j.bcp.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 8.Dali-Youcef N., Lagouge M., Froelich S., Koehl C., Schoonjans K., Auwerx J. Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann. Med. 2007;39:335–345. doi: 10.1080/07853890701408194. [DOI] [PubMed] [Google Scholar]
- 9.Pallos J., Bodai L., Lukacsovich T., Purcell J.M., Steffan J.S., Thompson L.M., Marsh J.L. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease. Hum. Mol. Genet. 2008;17:3767–3775. doi: 10.1093/hmg/ddn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., Xu W., McBurney M.W., Longo V.D. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008;8:38–48. doi: 10.1016/j.cmet.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh S., Feany M.B. Comparison of pathways controlling toxicity in the eye and brain in Drosophila models of human neurodegenerative diseases. Hum. Mol. Genet. 2004;13:2011–2018. doi: 10.1093/hmg/ddh214. [DOI] [PubMed] [Google Scholar]
- 12.Green K.N., Steffan J.S., Martinez-Coria H., Sun X., Schreiber S.S., Thompson L.M., LaFerla F.M. Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 2008;28:11500–11510. doi: 10.1523/JNEUROSCI.3203-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grubisha O., Smith B.C., Denu J.M. Small molecule regulation of Sir2 protein deacetylases. FEBS J. 2005;272:4607–4616. doi: 10.1111/j.1742-4658.2005.04862.x. [DOI] [PubMed] [Google Scholar]
- 14.Napper A.D., Hixon J., McDonagh T., Keavey K., Pons J.F., Barker J., Yau W.T., Amouzegh P., Flegg A., Hamelin E., et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 2005;48:8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- 15.Sauve A.A., Moir R.D., Schramm V.L., Willis I.M. Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Mol. Cell. 2005;17:595–601. doi: 10.1016/j.molcel.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 16.Grozinger C.M., Chao E.D., Blackwell H.E., Moazed D., Schreiber S.L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001;276:38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 17.Mai A., Massa S., Lavu S., Pezzi R., Simeoni S., Ragno R., Mariotti F.R., Chiani F., Camilloni G., Sinclair D.A. Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (Sirtuin) inhibitors. J. Med. Chem. 2005;48:7789–7795. doi: 10.1021/jm050100l. [DOI] [PubMed] [Google Scholar]
- 18.Gey C., Kyrylenko S., Hennig L., Nguyen L.H., Buttner A., Pham H.D., Giannis A. Phloroglucinol derivatives guttiferone G, aristoforin, and hyperforin: inhibitors of human sirtuins SIRT1 and SIRT2. Angew. Chem. 2007;46:5219–5222. doi: 10.1002/anie.200605207. [DOI] [PubMed] [Google Scholar]
- 19.Zhao X., Allison D., Condon B., Zhang F., Gheyi T., Zhang A., Ashok S., Russell M., MacEwan I., Qian Y., et al. The 2.5 A crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J. Med. Chem. 2013;56:963–969. doi: 10.1021/jm301431y. [DOI] [PubMed] [Google Scholar]
- 20.Gertz M., Fischer F., Nguyen G.T., Lakshminarasimhan M., Schutkowski M., Weyand M., Steegborn C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA. 2013;110:E2772–e42781. doi: 10.1073/pnas.1303628110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song W., Smith M.R., Syed A., Lukacsovich T., Barbaro B.A., Purcell J., Bornemann D.J., Burke J., Marsh J.L. Morphometric analysis of Huntington's disease neurodegeneration in Drosophila. Methods Mol. Biol. 2013;1017:41–57. doi: 10.1007/978-1-62703-438-8_3. [DOI] [PubMed] [Google Scholar]
- 22.Marsh J.L., Thompson L.M. Drosophila in the study of neurodegenerative disease. Neuron. 2006;52:169–178. doi: 10.1016/j.neuron.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 23.Rothgiesser K.M., Erener S., Waibel S., Luscher B., Hottiger M.O. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010;123:4251–4258. doi: 10.1242/jcs.073783. [DOI] [PubMed] [Google Scholar]
- 24.Wyttenbach A., Swartz J., Kita H., Thykjaer T., Carmichael J., Bradley J., Brown R., Maxwell M., Schapira A., Orntoft T.F., et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum. Mol. Genet. 2001;10:1829–1845. doi: 10.1093/hmg/10.17.1829. [DOI] [PubMed] [Google Scholar]
- 25.Perrin V., Regulier E., Abbas-Terki T., Hassig R., Brouillet E., Aebischer P., Luthi-Carter R., Deglon N. Neuroprotection by Hsp104 and Hsp27 in lentiviral-based rat models of Huntington's disease. Mol. Ther. 2007;15:903–911. doi: 10.1038/mt.sj.6300141. [DOI] [PubMed] [Google Scholar]
- 26.Runne H., Regulier E., Kuhn A., Zala D., Gokce O., Perrin V., Sick B., Aebischer P., Deglon N., Luthi-Carter R. Dysregulation of gene expression in primary neuron models of Huntington's disease shows that polyglutamine-related effects on the striatal transcriptome may not be dependent on brain circuitry. J. Neurosci. 2008;28:9723–9731. doi: 10.1523/JNEUROSCI.3044-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zala D., Benchoua A., Brouillet E., Perrin V., Gaillard M.C., Zurn A.D., Aebischer P., Deglon N. Progressive and selective striatal degeneration in primary neuronal cultures using lentiviral vector coding for a mutant huntingtin fragment. Neurobiol. Dis. 2005;20:785–798. doi: 10.1016/j.nbd.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 28.Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S.W., et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 29.Menalled L., El-Khodor B.F., Patry M., Suarez-Farinas M., Orenstein S.J., Zahasky B., Leahy C., Wheeler V., Yang X.W., MacDonald M., et al. Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol. Dis. 2009;35:319–336. doi: 10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menalled L.B., Patry M., Ragland N., Lowden P.A., Goodman J., Minnich J., Zahasky B., Park L., Leeds J., Howland D., et al. Comprehensive behavioral testing in the R6/2 mouse model of Huntington's disease shows no benefit from CoQ10 or minocycline. PLoS ONE. 2010;5:e9793. doi: 10.1371/journal.pone.0009793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sathasivam K., Neueder A., Gipson T.A., Landles C., Benjamin A.C., Bondulich M.K., Smith D.L., Faull R.L., Roos R.A., Howland D., et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA. 2013;110:2366–2370. doi: 10.1073/pnas.1221891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang T., Kraus W.L. SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim. Biophys. Acta. 2010;1804:1666–1675. doi: 10.1016/j.bbapap.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaquero A., Scher M., Lee D., Erdjument-Bromage H., Tempst P., Reinberg D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell. 2004;16:93–105. doi: 10.1016/j.molcel.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 34.Vaziri H., Dessain S.K., Ng Eaton E., Imai S.I., Frye R.A., Pandita T.K., Guarente L., Weinberg R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 35.Motta M.C., Divecha N., Lemieux M., Kamel C., Chen D., Gu W., Bultsma Y., McBurney M., Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 36.Yang Y., Hou H., Haller E.M., Nicosia S.V., Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005;24:1021–1032. doi: 10.1038/sj.emboj.7600570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeung F., Hoberg J.E., Ramsey C.S., Keller M.D., Jones D.R., Frye R.A., Mayo M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nemoto S., Fergusson M.M., Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J. Biol. Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 39.Bouras T., Fu M., Sauve A.A., Wang F., Quong A.A., Perkins N.D., Hay R.T., Gu W., Pestell R.G. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 2005;280:10264–10276. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- 40.Branco J., Al-Ramahi I., Ukani L., Perez A.M., Fernandez-Funez P., Rincon-Limas D., Botas J. Comparative analysis of genetic modifiers in Drosophila points to common and distinct mechanisms of pathogenesis among polyglutamine diseases. Hum. Mol. Genet. 2008;17:376–390. doi: 10.1093/hmg/ddm315. [DOI] [PubMed] [Google Scholar]
- 41.Jeong H., Cohen D.E., Cui L., Supinski A., Savas J.N., Mazzulli J.R., Yates J.R., 3rd, Bordone L., Guarente L., Krainc D. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 2012;18:159–165. doi: 10.1038/nm.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang M., Wang J., Fu J., Du L., Jeong H., West T., Xiang L., Peng Q., Hou Z., Cai H., et al. Neuroprotective role of Sirt1 in mammalian models of Huntington's disease through activation of multiple Sirt1 targets. Nat. Med. 2012;18:153–158. doi: 10.1038/nm.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kakefuda K., Fujita Y., Oyagi A., Hyakkoku K., Kojima T., Umemura K., Tsuruma K., Shimazawa M., Ito M., Nozawa Y., et al. Sirtuin 1 overexpression mice show a reference memory deficit, but not neuroprotection. Biochem. Biophys. Res. Commun. 2009;387:784–788. doi: 10.1016/j.bbrc.2009.07.119. [DOI] [PubMed] [Google Scholar]
- 44.Griswold A.J., Chang K.T., Runko A.P., Knight M.A., Min K.T. Sir2 mediates apoptosis through JNK-dependent pathways in Drosophila. Proc. Natl. Acad. Sci. USA. 2008;105:8673–8678. doi: 10.1073/pnas.0803837105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J., Chen J. SIRT1 regulates autoacetylation and histone acetyltransferase activity of TIP60. J. Biol. Chem. 2010;285:11458–11464. doi: 10.1074/jbc.M109.087585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.