

Abstract
Despite ongoing advances in stroke imaging and treatment, ischemic and hemorrhagic stroke continue to debilitate patients with devastating outcomes at both the personal and societal levels. While the ultimate goal of therapy in ischemic stroke is geared towards restoration of blood flow, even when mitigation of initial tissue hypoxia is successful, exacerbation of tissue injury may occur in the form of cell death, or alternatively, hemorrhagic transformation of reperfused tissue. Animal models have extensively demonstrated the concept of reperfusion injury at the molecular and cellular levels, yet no study has quantified this effect in stroke patients. These preclinical models have also demonstrated the success of a wide array of neuroprotective strategies at lessening the deleterious effects of reperfusion injury. Serial multimodal imaging may provide a framework for developing therapies for reperfusion injury.
Key Words : Collaterals, Ischemia-reperfusion injury, MRI, Neuroprotection, Stroke
Introduction
Reperfusion is characterized by initial restriction of blood supply followed by subsequent vascular restoration and concomitant reoxygenation of downstream tissue. In spite of mitigating initial tissue hypoxia, exacerbation of tissue injury may occur. This transpires both in global hypoxic injury as well as in focal ischemic injury secondary to stroke. In this article, we review the extensive basic science research which has been conducted on the mechanism and treatment of ischemia-reperfusion injury. Given that no study to date has systematically detailed the dynamic course of reperfusion injury in humans and how this clinically manifests in acute ischemic stroke, we also consider how the recent development of multimodal imaging has allowed for studying the chronology of serial perfusion, allowing for the specific quantification of tissue-based changes. This may lend itself well to the development of protective treatments for ischemia-reperfusion injury in humans.
Cerebral Ischemia
Cerebral ischemia may be global, affecting numerous vascular territories as in the case of cardiac arrest and cerebral hypoperfusion, or alternatively target a focal region or arterial distribution as in the case of ischemic stroke. The end result in both of these cases is tissue hypoxia, cellular dysfunction and cell death. Circulation in the central nervous system is distinctive due to the fact that large vessels including those of the circle of Willis as well as the extra- and intracranial vessels contribute to approximately half of cerebrovascular resistance [1,2]. Experiencing an episode of prolonged ischemic insult leads to fatal consequences for neurons that extend beyond the failure of cellular energy resources. The cellular changes associated with resultant death include nuclear fragmentation, chromatin condensation and cell body shrinkage [3,4,5]. Changes also occur at the level of the membrane phospholipid structure, which is comprised in part of phosphatidylethanolamine, phosphatidylcholine, phosphatidylserine and sphingomyelin. Studies have proposed a regulatory role for the aforementioned phosphatidylethanolamine in blebbing and in the formation of apoptotic bodies [6]. This phospholipid also serves a role in mediation of cellular necrosis as its externalization may serve in part to contribute to cytoskeletal organization [6].
Stroke
Stroke, whether ischemic or hemorrhagic in nature, has the ability to culminate in devastating clinical outcomes. According to the American Heart Association update in 2012, in the USA someone experiences a stroke every 40 s and, on average, a person dies from stroke every 4 min [7]. The mechanisms of ischemic stroke are variable, complex and multifactorial in nature. These include thromboembolic stroke secondary to atherosclerosis in the elderly patient population and structural cardiac or vasculopathic/metabolic etiologies in younger patients. Atherosclerosis in itself is one of the most common causes of vascular thrombosis and occlusion and is mediated by atherogenesis [8]. Borderzone infarctions can lead to ischemia in the brain parenchyma, or tissue, by hypoxia to areas in between principal arterial territories. They may occur between the anterior and middle cerebral artery in the case of anterior cortical borderzone infarcts and between the anterior, middle and posterior cerebral territories in the case of posterior borderzone infarctions [9]. Lacunar infarcts, previously thought to result solely from lipohyalinosis, are now thought to ensue from platelet or fibrin clots within an environment of diffuse atherosclerotic small vessel disease [10]. While common causes of embolic stroke include arrhythmias (most prominently atrial fibrillation), structural cardiac pathology such as persistent patent foramen ovale or artery-to-artery thromboembolic phenomenon, another etiology for embolic infarction is septic emboli. These occur typically in the context of systemic infection with seeding of heart valves and more commonly in immunocompromised patients with atypical pathogens including fungal infections and in immunocompetent patients with more common bacterial infections [11]. In these cases, parenchymal damage is mediated by pathogen-mediated inflammation (further contributing to disturbance of the blood/brain barrier, BBB, with cerebral edema), resultant reduction cerebral blood flow and, subsequently, cellular hypoxia and death [12,13,14]. The diagnosis of cerebral venous infarctions, which are less common than arterial infarctions, has become more readily identified with the improvement of imaging techniques including multimodal CT and MRI with venography. Pathophysiologically, it is believed that pressure changes in the vascular beds largely contribute to the mechanism of infarct development and resultant parenchymal damage [15,16,17], and while it was once thought that little interplay existed between venous and arterial infarction, recently there has been more data to suggest not only an association but also an overlapping risk in developing arterial and venous conditions [18].
Ischemia-Reperfusion Injury
Restoration of vascular supply to an organ temporarily deprived of blood flow, while effective in providing oxygenation, often paradoxically results in injury of the affected tissue bed [19,20]. This concept has been demonstrated in a variety of organ systems including the brain, heart and kidneys as in the cases of stroke, myocardial infarction and acute kidney injury, respectively. It has also been seen in the setting of multiorgan ischemia-reperfusion during trauma, cardiac arrest and sleep apnea as well as during surgical procedures including solid organ transplant where acute graft failure and early rejection may be the resultant outcome. Eltzschig and Eckle [20], in 2011, provided a schematic overview of the cellular and molecular mechanisms believed to be involved in reperfusion injury involving both innate and adaptive immune systems as well as the complement system, platelets and coagulation factors. Subsequent to activation of these systems, cell death can occur through a number of mechanisms including both necrosis with a hallmark of cellular swelling and apoptosis involving intricate cell signaling mechanisms for cellular demise [21]. Both processes have been linked to further stimulation of the inflammatory system to include the release of nucleotides acting as signals to promote phagocytosis in the apoptotic tissue bed, leading to more extensive reperfusion injury [19,22]. Additional injury to reperfused tissue has been extensively shown to be mediated by reactive radical oxide species in the brain as well as in other tissues [23,24,25,26,27,28]. The mechanism of tissue damage occurs at both the cellular and molecular levels. The activity of inducible nitric oxide synthase is incurred by cerebral ischemia. Subsequently, this leads to the production of nitric oxide which, after forming peroxynitrite, acts as one of the main reactive oxide species leading to both necrosis and apoptosis. It does so by mediating lipid peroxidation, DNA damage and disruption of ATP production in the case of necrosis and by the upregulation of p53 protein and downregulation of Bcl-2 protein in turn, leading to apoptotic cell death [29,30]. Additionally, studies have shown a role of NADPH oxidases as some of the most well-studied molecules involved in the generation of reactive oxygen species [31]. The vascular consequences of injury extend beyond the outcome of cell death to also include a change in the nature of the vascular system as a whole. As a result of the general dysfunction in the cellular metabolic milieu, proinflammatory cytokines lead to endothelial cell inflammation and increased permeability of the vascular system. This injury lasts beyond the period of ischemia, as animal studies have shown the sustained effect of oxidative stress on pericytes in the microvasculature of a middle cerebral artery occlusion murine model, despite arterial recanalization [32].
Additional Consequences of Reperfusion Injury in the Brain
Brain parenchymal damage in the setting of ischemia followed by reperfusion injury (as in other organ systems) occurs as a result of a complex series of events. These events at initiation include an interruption in blood flow to the affected tissue followed by depletion of cellular energy resources and glycolysis at an anaerobic substrate level, subsequently followed by lactic acidosis, failure of sodium potassium pump, release of glutamate, cytotoxic edema, free radical formation and activation of both innate and adaptive immune responses [33,34]. The excessive generation of free radicals overwhelms the system, which then is thought to become inefficient in scavenging these molecules, leading to cellular demise. While ischemic stroke resulting from vascular occlusion by nature limits tissue bed oxygenation, it is believed that cerebral ischemia and hypoxia occur even in the setting of primary hemorrhagic stroke. The mechanism by which this is proposed to occur happens through a ‘multihit theory’. In essence, it is believed that in patients with predisposing factors such as microangiopathy secondary to atherosclerotic disease or amyloid angiopathy, the threshold for tolerance of ischemia is lower and, in the presence of a prothrombotic environment subsequent to hemorrhage, the failure of autoregulation, the increase in intracranial pressure or iatrogenic hypotensive therapy may all act as factors contributing to tissue hypoxia and ischemia [35].
The BBB, due to its unique nature, provides the ability to achieve and maintain cerebral homeostasis. The BBB is constituted by endothelial cells, basement membrane and pericytes, as well as the end feet of astrocytes. Unlike elsewhere in the body, endothelial cells which contribute to the BBB connect via both adherent and tight junctions and lack fenestration. It does, however, afford low permeability due to pinocytic activity [36]. Vascular perfusion pressure and capillary blood flow surrounding the barrier is afforded by contractile elements of pericytes [37]. Finally, extracellular matrix proteins contribute to the formation of the basal lamina that connects to endothelial cells by virtue of integrin molecules. Astrocytic end feet help regulate cerebral capillary blood flow, contributing to both the creation and maintenance of the BBB environment [38]. As in the case of reactive oxygen species mediating their long-lasting effects on endothelium and altering the permeability of the vascular system, they are also thought to play a role in altering the permeability of the BBB. Kahles and Brandes [39] review the targets of reperfusion injury on the neurovascular unit, outlining the effects of matrix metalloprotease activation on basal lamina leading to the following: degradation (expression of cell adhesion molecules and stress fiber formation on endothelial cells), followed by contraction (altered expression/distribution/phosphorylation on tight junctions), leading to disassembly and the effect of lipid oxidation (changes in DNA and protein expression in all of the cells of the neurovascular unit) and, finally, oxidative cell damage [39]. Not only can damage related to ischemia-reperfusion occur at the molecular and cellular levels, but a disruption in the BBB contributes to further damage as an increase in vascular permeability causes edema which contributes to impedance in adequate perfusion to the affected tissue bed [40]. In addition to the effects of reactive oxygen species on the neurovascular unit and in altering BBB endothelial cells, excitotoxicity also deleteriously influences astrocyte function, leading in part to inhibition of astrocytic repair as well as end feet detachment, which together disable the BBB [13]. The functional outcome of these events is noted clinically, as studies have shown that the degree of disruption in BBB also correlates with severity of patient outcomes. Using CT perfusion as a reflection of BBB permeability, increased infarct permeability area was proposed as a predictor of poor outcome associated with increased likelihood for undergoing hemicraniectomy in patients with malignant middle cerebral artery infarction [40]. The permeability of the BBB can also be readily quantified using dynamic contrast-enhanced MRI protocols [41,42].
The unique cerebral autoregulatory mechanisms which the brain possesses may also be deleteriously affected as a result of reperfusion injury and result in a loss of adequate control of cerebral blood flow, contributing to further damage [43,44,45,46,47]. From a purely hemodynamic perspective, cerebral perfusion pressure in an adult with normal blood pressure ranges between 60 and 150 mm Hg [48]. Beyond the lower and upper limits of these ranges cerebral autoregulation is disrupted, resulting in the dependence of cerebral blood flow on mean arterial pressure [49]. Given that vasodilation occurs in the context of cerebral ischemia, reperfusion may lead to hyperemia, which exacerbates neuronal damage by virtue of reperfusion as well as in response to increased perfusion pressures [50,51,52,53,54].
Hemorrhagic transformation of ischemic infarction is perhaps the most well-recognized consequence of ischemia, followed by reperfusion. The disruption of BBB and subsequent edema, neuroinflammation and continued damage mediated by free radical oxides contribute to the hemorrhagic transformation of ischemic tissue beds following the return of blood flow to the area which once experienced restriction [55]. The concern for this complication, which can lead to worsening of already devastating outcomes of ischemic stroke, influences clinician treatment decisions for aggressive intervention at initial patient presentation. As with the pathophysiology of primary hemorrhagic stroke, subsequent to hemorrhagic transformation, parenchymal injury occurs at multiple levels. It is thought that one of the ways by which this occurs is due to a mechanism akin to traumatic brain injury, given increased pressure and tissue compression following hemorrhage [56]. Furthermore, activated blood may contain factors which are toxic to the parenchymal milieu and have been proposed to include excess glutamate, proapoptotic mediators (TNFα and Fas ligand), thrombin, hemoglobin and iron sulfate [57,58,59,60,61,62,63]. The question becomes whether, in addition to the tissue directly impacted by sheer pressure associated with hemorrhage, the cytotoxic nature of these blood products also contributes to damage. Additionally, ischemia of borders surrounding the zone of hemorrhage has been reported with mixed evidence [64,65]. Figure 1 provides an overall summary demonstrating the possible outcomes of vessel recanalization following acute ischemic stroke, whether that becomes successful and salvages tissue, leads to no-reflow or results in the deleterious outcome of reperfusion injury.
Fig. 1.
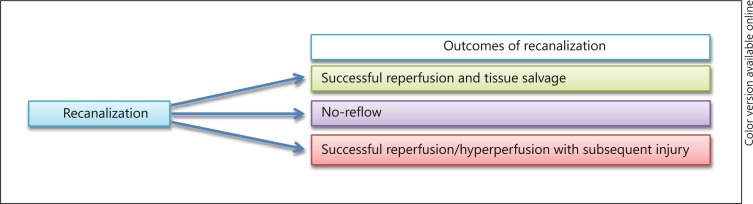
The outcomes of recanalization following vessel occlusion. In a percentage of patients, both recanalization and reperfusion are successful and lead to tissue salvage and recovery. The no-reflow phenomenon occurs when, in spite of successful mechanical recanalization, no flow is restored to the ischemic tissue bed and is thought in part to be a result of downstream vascular resistance. The third scenario, which has been demonstrated widely in animal models of stroke, is the instance where recanalization and reperfusion are both successful. However, the outcome is cell death or hemorrhagic transformation rather than tissue salvage.
Imaging of Injury in Stroke
Brain imaging techniques have undergone substantial advancements in the past 2 decades, providing more efficient, precise and reproducible assessments of brain parenchyma and vessels [66]. Neuroimaging in stroke extends beyond its role in diagnosis to reveal underlying pathophysiological mechanisms as well as to guiding clinical treatment decisions. Permutations of CT technology using multimodal CT which includes CT angiography and perfusion is more sensitive and informative than noncontrast CT in early stroke. In the acute stroke setting, multimodal CT typically includes noncontrast head CT and CT angiogram of the head and neck, as well as CT perfusion which offers crucial information regarding cerebral blood flow, cerebral blood volume and mean transient time.
Despite its higher cost and relatively longer acquisition time, MRI has shown itself to be extremely valuable as a first-line modality in the assessment of stroke [67], even in the hyperacute setting. The choice between multimodal CT and MRI is typically made on a patient-by-patient basis, preferring CT in patients for whom MRI is contraindicated. MRI sequences typically acquired in the acute stroke setting include diffusion-weighted imaging, apparent diffusion coefficient, perfusion-weighted imaging, fluid-attenuated inversion recovery (FLAIR) and gradient-recalled echocardiogram (GRE), as well as MR angiography of the head and neck.
Other novel techniques have been described in the literature, which include 99mTc-duramycin with SPECT imaging. As a modality, this was shown to be useful for both the detection and quantification of apoptotic neuronal cell death induced by ischemia-reperfusion injury in rats [6]. In nonhuman primates, diffusion tensor imaging was used in animal models of middle cerebral artery occlusion and provided temporal insight into the evolution of cerebral reperfusion injury and infarction growth [68]. Despite the valuable information provided by animal studies, the analysis of reperfusion injury in humans is still at its early stages, yet understanding it is instrumental to improving patient outcomes [69]. Although there has been an interesting report of 4 cases with early reperfusion and observed reperfusion injury with corresponding long-term effects [70], identifying ischemic reperfusion injury from a large cohort of images can be challenging. We recently introduced a framework [71] using serial imaging which allows for the identification and quantification of reperfusion injury using a voxel-based analysis of tissue fate. Serial acquisition of MR perfusion images were obtained at patient presentation and 3-6 h following recanalization therapy and were then processed to extract Tmax parameters. These images were coregistered serially at the voxel level with tissue fate outcomes evaluated by both FLAIR and GRE sequences 4-5 days following presentation (fig. 2). Reperfusion was defined by serial changes observed in Tmax voxels after which time volumes of reperfusion-injury as well as tissue fate relative to initial perfusion profile were calculated. Our preliminary results in 58 middle cerebral artery stroke patients demonstrated that nonhemorrhagic forms of reperfusion injury are common in acute stroke patients treated with standard thrombolytic therapies. Figure 3a and b demonstrates the use of this imaging methodology for capturing the extent and heterogeneity of reperfusion injury following ischemic stroke in 2 affected patients.
Fig. 2.

Schematic drawing of method for assessment of reperfusion injury in stroke patients. PWI = Perfusion-weighted images. Perfusion-weighted MRI images are acquired during the acute phase and after revascularization attempts. Reperfusion injury assessed by voxel-based comparison of the reperfused area and then subsequently undergoing irreversible cell death as measured by FLAIR sequence and/or hemorrhage as demonstrated by GRE sequence.
Fig. 3.

Demonstration of ischemia-reperfusion injury using serial multimodal MRI. a A 68-year-old woman with untreated atrial fibrillation presenting with aphasia, hemianopsia, right-sided weakness and sensory loss. The patient was treated with intravenous tPA followed by mechanical recanalization with MERCI device (AOL 3, TICI 2a). Serial change in Tmax shows improved perfusion; the perfusion deficit (Tmax >6 s) changed from 17.9 to 7.1 ml with resultant cell death (FLAIR) in spite of improved perfusion at follow-up day 5. b A 78-year-old woman with untreated paroxysmal atrial fibrillation presenting with left-sided hemiparesis, neglect and dysarthria. The patient was treated with intravenous tPA followed by mechanical recanalization with Penumbra device (AOL 2, TICI 2b). Serial change in Tmax shows improved perfusion; the perfusion deficit (Tmax >6 s) changed from 16.5 to 3.4 ml with resultant hemorrhagic transformation (GRE) in spite of improved perfusion at follow-up day 5.
Reperfusion Strategies
The role of intravenous tPA in the treatment of acute stroke and the current extended guidelines for treatment stem from the Third European Cooperative Acute Stroke Study (ECASS-3) [72]. This study was able to demonstrate the clinical benefit of tPA for a time period of up to 4.5 h, given that patients do not meet the exclusion criteria of age above 80, stroke with NIH stroke scale greater than 25 and history of diabetes and stroke [72]. Other thrombolytic therapies that have been under investigation for extended treatment (3-9 h from symptom onset) include desmoteplase. Although the trial assessing this therapy, Desmoteplase in Acute Ischemic Stroke (DIAS-2) [73], did not show favorable outcomes, the lack of evidence for successful treatment effect was confounded by a number of variables limiting study design. The same applied in the case of other thrombolytic therapies including tenecteplase, which was piloted in a limited number of patients (n = 15) and demonstrated high rates of successful recanalization and reperfusion, although the data remained largely inconclusive when taken together as a result of incongruities in imaging-based selection [74]. Aside from pharmacological means of clot lysis, ultrasound therapy aimed at mechanical clot disruption has been investigated as a potential treatment. In these studies, although patients receiving sonothrombolysis with microbubbles showed early clinical improvement and rates of recanalization, this was found to be associated with higher rates of hemorrhage, whether petechial or frank infarction/transformation [75,76]. This certainly invokes the delicate balance that is recanalization with beneficial versus deleterious reperfusion. Other pharmacological and mechanical, direct treatments of large artery recanalization include endovascular methods of targeting the occlusive clot. Aside from concomitant use of intravenous and intra-arterial tPA for clot thrombolysis, many devices exist for mechanical recanalization of occlusion. The combination of intravenous and potentially subsequent intra-arterial or clot retrieval treatments may prove to be very effective in the continued improvement of recanalization rates [77]. Current device options which have been closely studied for mechanical retrieval include the MERCI retrieval system, Penumbra aspiration system, SOLITAIRE flow restoration device and TREVO stent-like retriever [78,79,80,81], which allow for treatment in an extended time window typically under 8 h following symptom onset. When taken together, these treatments collectively aim at recanalization and subsequent reperfusion by treatment of the occlusive lesion responsible for initial parenchymal ischemia.
Although mechanical and pharmacological revascularization therapies generally aim at complete recanalization, downstream reperfusion is not always achieved. This concept has been described at length in the cardiac literature and referred to as the no-reflow phenomenon. It is believed that, in spite of clot removal, it is likely that the inability to then reperfuse tissue parenchyma is related to microvascular obstruction [82].
Role of Collateral Circulation in Reperfusion Therapy
While reperfusion is typically thought solely to be the result of recanalization of an arterial blockage in the setting of ischemic stroke, in recent years attention has been increasingly focusing on the role of collateral circulation in the reperfusion of ischemic tissue. Imaging plays a crucial role in defining the mechanism by which stroke debilitates patients and provides informative guidance as to the predicted evolution of damage as well as lending itself to guiding treatment options. Whether treatment consists of arterial recanalization due to direct clot lysis/retrieval or whether reperfusion is stimulated by augmentation of downstream collaterals, both strategies, alone or in combination, should theoretically contribute to the reversal of the effects of tissue hypoxia. Many methods have been used in an effort to promote collaterals in the setting of cerebral ischemia. These methods include induced hypertension, the results of which have been efficacious, mostly on a patient-by-patient situation, but with no clear supportive clinical evidence when systematically studied [83]. A number of other methods have been studied as mediators of supporting collaterals, including external counterpulsation. This is a technique which titrates pressure in the extremities in a synchronized manner and has been shown to support collaterals in the coronary circulation [84]. In the cerebral circulation, this technique was demonstrated to elevate arterial blood flow velocities as measured by transcranial Doppler ultrasonography [85]. In a study systematically evaluating the effect of this therapy in a randomized, crossover study, it was found that in spite of no significant differences in velocity seen between early (weeks 1-7) and late (weeks 8-14) treatment groups, clinical and functional outcome improvement correlated with earlier external counterpulsation treatment [86]. Modulation of blood flow and cerebral vascular collaterals has also been investigated in the context of partial aortic occlusion, showing that at least in animal studies and early human reports this treatment may be beneficial in increasing blood flow downstream from the occlusion and enhance cerebral perfusion [87,88,89,90,91].
Neuroprotective Strategies
Therapeutic strategies aimed at protecting from the deleterious effects of ischemia-reperfusion after vascular recanalization have been varied. Treatments in clinical trials range from those aimed at inhibition of apoptosis to promotion of angiogenesis, targeting the t cells of the immune system, inhibiting reactive oxygen species and modulating the metabolic processing systems [20]. Additionally, in an animal model of cerebral ischemia, the inhalation of hydrogen gas proved to serve as a neuroprotectant through its role in reducing oxidative stress under conditions where reperfusion injury was shown to lead to apoptotic cell death [92,93]. Other gases used therapeutically across tissue models of ischemia-reperfusion include nitric oxide, hydrogen sulfide and carbon monoxide [29,94,95,96].
Aside from gases, other attempts at providing neuroprotective effects include a number of pharmacologic agents. Sphingosine-1 phosphate receptor agonist, fingolimod, while currently well known for its therapeutic benefit in multiple sclerosis patients, has been previously described to act as a protecting agent from ischemia-reperfusion in the liver [97,98] and kidney [99,100] and, most recently, in murine models of ischemic stroke, demonstrating a reduction in infarct size, better functional neurological outcomes, decreased edema and a decreased number of activated immune cells [101]. Its mechanism of protection is thought to be mediated through its effects on the regulation of cytoskeletal organization, adhesion and migration, proliferation, inflammation and, finally, apoptosis, doing so through its ability to act as an agonist of 5 G-protein-coupled receptors termed S1P1-5[102] · These receptors, when activated, have been shown to reduce both hypoxic damage as well as ischemia-reperfusion injury in the cardiac system [103,104].
Taking advantage of the knowledge that reactive oxygen species play a big role in the cellular damage associated with ischemia-reperfusion, many studies have aimed at tailoring neuroprotective therapies to target these players. These most notably include therapy using superoxide dismutase and NADPH oxidase as agents, which have been shown in animal models of ischemia-reperfusion to lessen injury and improve function in multiple organ systems [39,105,106,107,108]. Multiple isoforms of NADPH oxidase have been identified with a variable pattern of expression across the central nervous system. Of these Nox genes, Nox 4 is highlighted as one which is highly expressed in endothelial cells of the brain, as seen in rat basilar artery endothelial cells, but also ubiquitously expressed elsewhere in the body [109,110].
HMG-CoA reductase, atorvastatin, one of the commonly utilized antihyperlipidemics used in patients with vascular risk factors, has also been proposed to play a role in protection against ischemia-reperfusion injury. Its protective effects were shown to be mediated by protein gp19phox in a rat model of ischemia-reperfusion and were also shown to diminish BBB disruption [111,112]. In patients, preliminary studies using MR perfusion in a group of 31 patients, 12 of whom had prior use of statins, was found to be associated with greater early reperfusion and clinical improvements both in patients who received tPA as well as those who did not [113]. This study did not, however, specifically address the effects of reperfusion injury in these patients.
Other strategies of neuroprotection have focused on modulating vascular mitogens. Of the classes of proteins thought to be very important in both vascular and neural development, netrins, specifically netrin-1, have been implicated in their role in promoting bidirectional axonal guidance as well as stimulation of vasculature through its actions on endothelial cells [114,115,116]. Animal studies of middle cerebral artery occlusion demonstrated the potential for this therapeutic target as adeno-associated viral overexpression of netrin-1-enhanced post-stroke locomotor activity by postulated improvement in periinfarct vascular density, even though it did not significantly reduce infarct size per se [117].
Despite a wealth of basic science and animal studies showing the benefit of molecules acting as neuroprotective agents, in clinical practice the concept of oxidative stress mediating neuronal injury in the setting of ischemia-reperfusion and efficacy in targeting these molecules with clinically relevant benefit has been difficult to show or interpret at best. In heterogeneous patient populations and clinically varied ischemic tissue beds, Kahles and Brandes [39], in 2012, proposed a number of possible reasons to explain the disparity in beneficial outcomes seen in preclinical versus clinical studies of treatment with molecular antioxidants and free radical scavengers. These reasons include nontherapeutic compound dosing, an imbalance in the detoxification system (giving supplementation with only one type of antioxidant), the unequal competition for affinity of the reactive oxygen species with the therapeutic scavenger versus damaged tissue bed, site targeting of therapy and, finally, adverse effects of therapeutic agent [39]. In 1999, the initial Stroke Therapy Academic Industry Roundtable (STAIR) made one of its goals to improve the quality of preclinical studies for acute stroke treatments. However, reflecting back 10 years later, in 2009, an update to these recommendations found that adherence to the recommendations still lags behind and it was encouraged that more be done in order to truly achieve success with an agent from benchside to bedside [118]. Therefore, a strong need exists for careful, detailed, nonbiased and clinically relevant design of these therapeutic trials, testing them in appropriate animal models and, when shown to be initially safe and effective, in their human counterparts. In addition to the aforementioned explanations for disagreement in therapeutic success rates in preclinical versus clinical studies, evaluation of outcome measures is also essential. Monitoring success rates in patients may be done using clinical behavioral examination outcomes such as Rankin scores measured at intervals during recovery from stroke, although these outcome measures are clouded by many factors and do not serve well in providing a direct measure of experimental treatment success. Neuroimaging on the other hand, when used in a tailored fashion, may be very effective in providing clear, noninvasive, easily reproducible measures of success outcome monitored over time.
Ischemic Conditioning
Another valuable strategy which offers promise in mitigating the harmful effects of reperfusion injury is ischemic conditioning. The concept of ischemic conditioning, whether applied as preconditioning or postconditioning (or applied remotely from the event), is one where exposure to ischemia in short durations (which are nonpathological in themselves) offers a downstream protective effect against prolonged ischemia. Studies proposing the beneficial role for ischemic conditioning in stroke have attributed the benefit to a number of possible downstream consequences including the regulation of endothelial cells, histamine, inflammatory modulators and reactive oxygen species and the promotion of angiogenesis, partly through promoting VEGF and erythropoietin [119,120,121,122,123,124,125]. Hypoxia in oxygen-dependent signaling pathways has been shown to increase hypoxia-inducible factor, HIF-1α, which when dimerized acts to stimulate hypoxia response genes [126]. As a pharmacological target, modulation of HIF-α has also been shown to increase both VEGF and endothelial nitric oxide synthase and to be associated with improvement of cerebral blood flow in animal models of middle cerebral artery occlusion [127]. Again, while this concept has had very positive results in animal and tissue-based studies, the clinical benefits have not been shown to be as robust, again perhaps limited by therapeutic end points for the detection of these benefits. In the heart, however, a randomized, controlled study demonstrated the concept of postconditioning in a group of 30 patients who underwent short episodes of ischemia during the therapeutic reperfusion procedure. This was done by the application of four episodes, 1 min each, of limitation in flow by the inflation and subsequent deflation of the balloon catheter. In doing so, they found clinical benefit in the patients who underwent postconditioning versus their counterparts who did not [128]. Thibault et al. [129] also showed the benefit of postconditioning to last for up to 1 year following the treatment in cardiac disease. While this concept has been breached clinically in the cardiovascular setting, it has been investigated clinically to a lesser extent in the realm of neurological disease.
Summary and Future Considerations
Although an expansive body of literature has detailed the numerous molecular and cellular pathophysiological mechanisms associated with reperfusion injury in animal models, the impact of reperfusion injury in routinely treated clinical stroke cases remains unquantified. As current revascularization approaches have become highly successful and continue to improve, the concept of reperfusion injury will become more prominent with respect to patient treatment and supportive care. The future of acute stroke therapy will lay in the advancement of our understanding of revascularization beyond the concept of recanalization alone. The systematic use of imaging as a tool in stroke management and treatment will allow for a better understanding of the no-reflow phenomenon in the brain, the contribution of collaterals, the incidence of hyperperfusion and, most importantly, the heterogeneity of tissue fate. The need, therefore, exists for the use of combined approaches for stroke treatment that employ concomitant use of neuroprotective and vasoprotective agents with revascularization treatments. The use of serial multimodal imaging will serve well in providing a framework for the development and testing of therapies targeted at the prevention of ischemia-reperfusion injury in patients.
References
- 1.Paulson OB, Strandgaard S, Edvinsson L. Cerebral autoregulation. Cerebrovasc Brain Metab Rev. 1990;2:161–192. [PubMed] [Google Scholar]
- 2.Shapiro HM, Stromberg DD, Lee DR, Wiederhielm CA. Dynamic pressures in the pial arterial microcirculation. Am J Physiol. 1971;221:279–283. doi: 10.1152/ajplegacy.1971.221.1.279. [DOI] [PubMed] [Google Scholar]
- 3.Broughton BRS, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–e339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- 4.Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- 5.Balasubramanian K, Schroit AJ. Aminophospholipid asymmetry: a matter of life and death. Annu Rev Physiol. 2003;65:701–734. doi: 10.1146/annurev.physiol.65.092101.142459. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Stevenson GD, Barber C, Furenlid LR, Barrett HH, Woolfenden JM, Zhao M, Liu Z. Imaging of rat cerebral ischemia-reperfusion injury using 99mTc-labeled duramycin. Nucl Med Biol. 2013;40:80–88. doi: 10.1016/j.nucmedbio.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, et al. Heart disease and stroke statistics – 2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berliner JA, Navab M, Fogelman AM, Frank JS, Demer LL, Edwards PA, Watson AD, Lusis AJ. Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation. 1995;91:2488–2496. doi: 10.1161/01.cir.91.9.2488. [DOI] [PubMed] [Google Scholar]
- 9.Momjian-Mayor I, Baron J-C. The pathophysiology of watershed infarction in internal carotid artery disease: review of cerebral perfusion studies. Stroke. 2005;36:567–577. doi: 10.1161/01.STR.0000155727.82242.e1. [DOI] [PubMed] [Google Scholar]
- 10.Ay H, Oliveira-Filho J, Buonanno FS, Ezzeddine M, Schaefer PW, Rordorf G, Schwamm LH, Gonzalez RG, Koroshetz WJ. Diffusion-weighted imaging identifies a subset of lacunar infarction associated with embolic source. Stroke. 1999;30:2644–2650. doi: 10.1161/01.str.30.12.2644. [DOI] [PubMed] [Google Scholar]
- 11.Syrjänen J. Infection as a risk factor for cerebral infarction. Eur Heart J. 1993;14(suppl K):17–19. [PubMed] [Google Scholar]
- 12.Ruttmann E, Willeit J, Ulmer H, Chevtchik O, Höfer D, Poewe W, Laufer G, Müller LC. Neurological outcome of septic cardioembolic stroke after infective endocarditis. Stroke. 2006;37:2094–2099. doi: 10.1161/01.STR.0000229894.28591.3f. [DOI] [PubMed] [Google Scholar]
- 13.Chen ZL, Indyk JA, Bugge TH, Kombrinck KW, Degen JL, Strickland S. Neuronal death and blood-brain barrier breakdown after excitotoxic injury are independent processes. J Neurosci. 1999;19:9813–9820. doi: 10.1523/JNEUROSCI.19-22-09813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finelli PF, Uphoff DF. Magnetic resonance imaging abnormalities with septic encephalopathy. J Neurol Neurosurg Psychiatry. 2004;75:1189–1191. doi: 10.1136/jnnp.2003.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van den Bergh WM, Van der Schaaf I, Van Gijn J. The spectrum of presentations of venous infarction caused by deep cerebral vein thrombosis. Neurology. 2005;65:192–196. doi: 10.1212/01.wnl.0000179677.84785.63. [DOI] [PubMed] [Google Scholar]
- 16.Bousser M-G, Ferro JM. Cerebral venous thrombosis: an update. Lancet Neurol. 2007;6:162–170. doi: 10.1016/S1474-4422(07)70029-7. [DOI] [PubMed] [Google Scholar]
- 17.Stam J. Thrombosis of the cerebral veins and sinuses. New Engl J Med. 2005;352:1791–1798. doi: 10.1056/NEJMra042354. [DOI] [PubMed] [Google Scholar]
- 18.Franchini M, Mannucci PM. Association between venous and arterial thrombosis: clinical implications. Eur J Intern Med. 2012;23:333–337. doi: 10.1016/j.ejim.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eltzschig HK, Eckle T. Ischemia and reperfusion – from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. New Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–286. doi: 10.1038/nature08296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters O, Back T, Lindauer U, Busch C, Megow D, Dreier J, Dirnagl U. Increased formation of reactive oxygen species after permanent and reversible middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1998;18:196–205. doi: 10.1097/00004647-199802000-00011. [DOI] [PubMed] [Google Scholar]
- 24.Olmez I, Ozyurt H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochem Int. 2012;60:208–212. doi: 10.1016/j.neuint.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 25.Vitturi DA, Patel RP. Current perspectives and challenges in understanding the role of nitrite as an integral player in nitric oxide biology and therapy. Free Radic Biol Med. 2011;51:805–812. doi: 10.1016/j.freeradbiomed.2011.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Zhang H, Zhang C. Role of inflammation in the regulation of coronary blood flow in ischemia and reperfusion: mechanisms and therapeutic implications. J Mol Cell Cardiol. 2012;52:865–872. doi: 10.1016/j.yjmcc.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kietadisorn R, Juni RP, Moens AL. Tackling endothelial dysfunction by modulating NOS uncoupling: new insights into its pathogenesis and therapeutic possibilities. Am J Physiol Endocrinol Metab. 2012;302:E481–E495. doi: 10.1152/ajpendo.00540.2011. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez F, Bonacasa B, Fenoy FJ, Salom MG. Reactive oxygen and nitrogen species in the renal ischemia/reperfusion injury. Curr Pharm Des. 2013;19:2776–2794. doi: 10.2174/1381612811319150014. [DOI] [PubMed] [Google Scholar]
- 29.Shen J, Qiu X, Jiang B, Zhang D, Xin W, Fung PCW, Zhao B. Nitric oxide and oxygen radicals induced apoptosis via bcl-2 and p53 pathway in hypoxia-reoxygenated cardiomyocytes. Sci China C Life Sci. 2003;46:28–39. doi: 10.1007/BF03182682. [DOI] [PubMed] [Google Scholar]
- 30.Bolaños JP, Almeida A. Roles of nitric oxide in brain hypoxia-ischemia. Biochim Biophys Acta. 1999;1411:415–436. doi: 10.1016/s0005-2728(99)00030-4. [DOI] [PubMed] [Google Scholar]
- 31.Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, Maier CM, Narasimhan P, Goeders CE, et al. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal. 2011;14:1505–1517. doi: 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- 33.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 34.Lee JM, Grabb MC, Zipfel GJ, Choi DW. Brain tissue responses to ischemia. J Clin Invest. 2000;106:723–731. doi: 10.1172/JCI11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prabhakaran S, Naidech AM. Ischemic brain injury after intracerebral hemorrhage: a critical review. Stroke. 2012;43:2258–2263. doi: 10.1161/STROKEAHA.112.655910. [DOI] [PubMed] [Google Scholar]
- 36.Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43:348–364. doi: 10.1080/10715760902751902. [DOI] [PubMed] [Google Scholar]
- 37.Fisher M. Pericyte signaling in the neurovascular unit. Stroke. 2009;40(3 suppl):S13–S5. doi: 10.1161/STROKEAHA.108.533117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009;335:75–96. doi: 10.1007/s00441-008-0658-9. [DOI] [PubMed] [Google Scholar]
- 39.Kahles T, Brandes RP. NADPH oxidases as therapeutic targets in ischemic stroke. Cell Mol Life Sci. 2012;69:2345–2363. doi: 10.1007/s00018-012-1011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bektas H, Wu T-C, Kasam M, Harun N, Sitton CW, Grotta JC, Savitz SI. Increased blood-brain barrier permeability on perfusion CT might predict malignant middle cerebral artery infarction. Stroke. 2010;41:2539–2544. doi: 10.1161/STROKEAHA.110.591362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kassner A, Thornhill R. Measuring the integrity of the human blood-brain barrier using magnetic resonance imaging. Methods Mol Biol. 2011;686:229–245. doi: 10.1007/978-1-60761-938-3_10. [DOI] [PubMed] [Google Scholar]
- 42.Scalzo F, Alger JR, Hu X, Saver JL, Dani KA, Muir KW, Demchuk AM, Coutts SB, Luby M, Warach S, Liebeskind DS. Multi-center prediction of hemorrhagic transformation in acute ischemic stroke using permeability imaging features. Magn Reson Imaging. 2013;31:961–969. doi: 10.1016/j.mri.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishigaya K, Yoshida Y, Sasuga M, Nukui H, Ooneda G. Effect of recirculation on exacerbation of ischemic vascular lesions in rat brain. Stroke. 1991;22:635–642. doi: 10.1161/01.str.22.5.635. [DOI] [PubMed] [Google Scholar]
- 44.Cipolla MJ, McCall AL, Lessov N, Porter JM. Reperfusion decreases myogenic reactivity and alters middle cerebral artery function after focal cerebral ischemia in rats. Stroke. 1997;28:176–180. doi: 10.1161/01.str.28.1.176. [DOI] [PubMed] [Google Scholar]
- 45.Kågström E, Smith ML, Siesjö BK. Local cerebral blood flow in the recovery period following complete cerebral ischemia in the rat. J Cereb Blood Flow Metab. 1983;3:170–182. doi: 10.1038/jcbfm.1983.24. [DOI] [PubMed] [Google Scholar]
- 46.Siesjö BK. Pathophysiology and treatment of focal cerebral ischemia. Part II. Mechanisms of damage and treatment. J Neurosurg. 1992;77:337–354. doi: 10.3171/jns.1992.77.3.0337. [DOI] [PubMed] [Google Scholar]
- 47.Siesjö BK. Pathophysiology and treatment of focal cerebral ischemia. Part I. Pathophysiology. J Neurosurg. 2008;108:616–631. doi: 10.3171/JNS/2008/108/3/0616. [DOI] [PubMed] [Google Scholar]
- 48.Phillips SJ, Whisnant JP. Hypertension and the brain. The National High Blood Pressure Education Program. Arch Intern Med. 1992;152:938–945. [PubMed] [Google Scholar]
- 49.Faraci FM, Baumbach GL, Heistad DD. Myogenic mechanisms in the cerebral circulation. J Hypertens Suppl. 1989;7:S61–S64. discussion S65. [PubMed] [Google Scholar]
- 50.Lassen NA, Agnoli A. The upper limit of autoregulation of cerebral blood flow – on the pathogenesis of hypertensive encephalopathy. Scand J Clin Lab Invest. 1972;30:113–116. doi: 10.3109/00365517209081099. [DOI] [PubMed] [Google Scholar]
- 51.Euser AG, Cipolla MJ. Cerebral blood flow autoregulation and edema formation during pregnancy in anesthetized rats. Hypertension. 2007;49:334–340. doi: 10.1161/01.HYP.0000255791.54655.29. [DOI] [PubMed] [Google Scholar]
- 52.Johansson B, Li CL, Olsson Y, Klatzo I. The effect of acute arterial hypertension on the blood-brain barrier to protein tracers. Acta Neuropathol. 1970;16:117–124. doi: 10.1007/BF00687666. [DOI] [PubMed] [Google Scholar]
- 53.Gourley JK, Heistad DD. Characteristics of reactive hyperemia in the cerebral circulation. Am J Physiol. 1984;246:H52–H58. doi: 10.1152/ajpheart.1984.246.1.H52. [DOI] [PubMed] [Google Scholar]
- 54.Sundt TM, Waltz AG. Cerebral ischemia and reactive hyperemia. Studies of cortical blood flow and microcirculation before, during, and after temporary occlusion of middle cerebral artery of squirrel monkeys. Circ Res. 1971;28:426–433. doi: 10.1161/01.res.28.4.426. [DOI] [PubMed] [Google Scholar]
- 55.Wang X, Lo EH. Triggers and mediators of hemorrhagic transformation in cerebral ischemia. Mol Neurobiol. 2003;28:229–244. doi: 10.1385/MN:28:3:229. [DOI] [PubMed] [Google Scholar]
- 56.Golding EM. Sequelae following traumatic brain injury. The cerebrovascular perspective. Brain Res Brain Res Rev. 2002;38:377–388. doi: 10.1016/s0165-0173(02)00141-8. [DOI] [PubMed] [Google Scholar]
- 57.Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab. 2003;23:137–149. doi: 10.1097/01.WCB.0000044631.80210.3C. [DOI] [PubMed] [Google Scholar]
- 58.Striggow F, Riek M, Breder J, Henrich-Noack P, Reymann KG, Reiser G. The protease thrombin is an endogenous mediator of hippocampal neuroprotection against ischemia at low concentrations but causes degeneration at high concentrations. Proc Nat Acad Sci USA. 2000;97:2264–2269. doi: 10.1073/pnas.040552897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J Neurochem. 2003;84:3–9. doi: 10.1046/j.1471-4159.2003.01268.x. [DOI] [PubMed] [Google Scholar]
- 60.Mayne M, Ni W, Yan HJ, Xue M, Johnston JB, Del Bigio MR, Peeling J, Power C. Antisense oligodeoxynucleotide inhibition of tumor necrosis factor-α expression is neuroprotective after intracerebral hemorrhage. Stroke. 2001;32:240–248. doi: 10.1161/01.str.32.1.240. [DOI] [PubMed] [Google Scholar]
- 61.Lenzlinger PM, Marx A, Trentz O, Kossmann T, Morganti-Kossmann MC. Prolonged intrathecal release of soluble Fas following severe traumatic brain injury in humans. J Neuroimmunol. 2002;122:167–174. doi: 10.1016/s0165-5728(01)00466-0. [DOI] [PubMed] [Google Scholar]
- 62.Regan RF, Panter SS. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci Lett. 1993;153:219–222. doi: 10.1016/0304-3940(93)90326-g. [DOI] [PubMed] [Google Scholar]
- 63.Meguro T, Chen B, Lancon J, Zhang JH. Oxyhemoglobin induces caspase-mediated cell death in cerebral endothelial cells. J Neurochem. 2001;77:1128–1135. doi: 10.1046/j.1471-4159.2001.00313.x. [DOI] [PubMed] [Google Scholar]
- 64.Yang GY, Betz AL, Chenevert TL, Brunberg JA, Hoff JT. Experimental intracerebral hemorrhage: relationship between brain edema, blood flow, and blood-brain barrier permeability in rats. J Neurosurg. 1994;81:93–102. doi: 10.3171/jns.1994.81.1.0093. [DOI] [PubMed] [Google Scholar]
- 65.Zazulia AR, Diringer MN, Videen TO, Adams RE, Yundt K, Aiyagari V, Grubb RL, Powers WJ. Hypoperfusion without ischemia surrounding acute intracerebral hemorrhage. J Cereb Blood Flow Metab. 2001;21:804–810. doi: 10.1097/00004647-200107000-00005. [DOI] [PubMed] [Google Scholar]
- 66.Scalzo F, Hao Q, Walczak A, Hu X, Hoi Y, Hoffmann K, Liebeskind D. Computational hemodynamics in intracranial vessels reconstructed from biplane angiograms. Advances in Visual Computing. 2010;6455:359–367. [Google Scholar]
- 67.Scalzo F, Hao Q, Alger JR, Hu X, Liebeskind DS. Regional Prediction of Tissue Fate in Acute Ischemic Stroke. Ann Biomed Eng. 2012;40:2177–2187. doi: 10.1007/s10439-012-0591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo J, Zheng H-B, Duan J-C, He L, Chen N, Gong Q-Y, Tang H-H, Li H-X, Wang L, et al. Diffusion tensor MRI for the assessment of cerebral ischemia/reperfusion injury in the penumbra of non-human primate stroke model. Neurol Res. 2011;33:108–112. doi: 10.1179/016164110X12761752770177. [DOI] [PubMed] [Google Scholar]
- 69.Domínguez C, Delgado P, Vilches A, Martín-Gallán P, Ribó M, Santamarina E, Molina C, Corbeto N, Rodríguez-Sureda V, et al. Oxidative stress after thrombolysis-induced reperfusion in human stroke. Stroke. 2010;41:653–660. doi: 10.1161/STROKEAHA.109.571935. [DOI] [PubMed] [Google Scholar]
- 70.Hwang Y-H, Kim Y-W, Kim J, Kim Y-S, Park S-P, Suh C-K. Subacute neurological deterioration with selective axonal injury in patients with acute ischemic stroke following reperfusion of middle cerebral artery occlusion. Case Rep Neurol. 2011;3:199–202. doi: 10.1159/000331450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nour M, Scalzo F, Liebeskind DS. Insight into human ischemia reperfusion injury in acute stroke: voxel-based MRI analysis of tissue fate. Int J Stroke, in press.
- 72.Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, et al. Thrombolysis with alteplase 3 to 4.5 h after acute ischemic stroke. New Engl J Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 73.Hacke W, Furlan AJ, Al-Rawi Y, Davalos A, Fiebach JB, Gruber F, Kaste M, Lipka LJ, Pedraza S, et al. Intravenous desmoteplase in patients with acute ischaemic stroke selected by MRI perfusion-diffusion weighted imaging or perfusion CT (DIAS-2): a prospective, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2009;8:141–150. doi: 10.1016/S1474-4422(08)70267-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parsons MW, Miteff F, Bateman GA, Spratt N, Loiselle A, Attia J, Levi CR. Acute ischemic stroke: imaging-guided tenecteplase treatment in an extended time window. Neurology. 2009;72:915–921. doi: 10.1212/01.wnl.0000344168.05315.9d. [DOI] [PubMed] [Google Scholar]
- 75.Molina CA, Barreto AD, Tsivgoulis G, Sierzenski P, Malkoff MD, Rubiera M, Gonzales N, Mikulik R, Pate G, et al. Transcranial ultrasound in clinical sonothrombolysis (TUCSON) trial. Ann Neurol. 2009;66:28–38. doi: 10.1002/ana.21723. [DOI] [PubMed] [Google Scholar]
- 76.Dinia L, Rubiera M, Ribo M, Maisterra O, Ortega G, Del Sette M, Alvarez-Sabin J, Molina CA. Reperfusion after stroke sonothrombolysis with microbubbles may predict intracranial bleeding. Neurology. 2009;73:775–780. doi: 10.1212/WNL.0b013e3181b6bb45. [DOI] [PubMed] [Google Scholar]
- 77.Mazighi M, Serfaty J-M, Labreuche J, Laissy J-P, Meseguer E, Lavallée PC, Cabrejo L, Slaoui T, Guidoux C, et al. Comparison of intravenous alteplase with a combined intravenous-endovascular approach in patients with stroke and confirmed arterial occlusion (RECANALISE study): a prospective cohort study. Lancet Neurol. 2009;8:802–809. doi: 10.1016/S1474-4422(09)70182-6. [DOI] [PubMed] [Google Scholar]
- 78.Saver JL, Jahan R, Levy EI, Jovin TG, Baxter B, Nogueira R, Clark W, Budzik R, Zaidat OO. SOLITAIRE™ with the Intention for Thrombectomy (SWIFT) trial: design of a randomized, controlled, multicenter study comparing the SOLITAIRE™ Flow Restoration device and the MERCI Retriever in acute ischaemic stroke. Int J Stroke, E-pub ahead of print. [DOI] [PubMed]
- 79.Alshekhlee A, Pandya DJ, English J, Zaidat OO, Mueller N, Gupta R, Nogueira RG. Merci mechanical thrombectomy retriever for acute ischemic stroke therapy: literature review. Neurology. 2012;79(suppl 1):S126–S134. doi: 10.1212/WNL.0b013e3182697e89. [DOI] [PubMed] [Google Scholar]
- 80.Nogueira RG, Lutsep HL, Gupta R, Jovin TG, Albers GW, Walker GA, Liebeskind DS, Smith WS. TREVO versus MERCI retrievers for thrombectomy revascularisation of large vessel occlusions in acute ischaemic stroke (TREVO 2): a randomised trial. Lancet. 2012;380:1231–1240. doi: 10.1016/S0140-6736(12)61299-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hussain SI, Zaidat OO, Fitzsimmons B-FM. The Penumbra system for mechanical thrombectomy in endovascular acute ischemic stroke therapy. Neurology. 2012;79(suppl 1):S135–S141. doi: 10.1212/WNL.0b013e31826958a8. [DOI] [PubMed] [Google Scholar]
- 82.Kloner RA. No-reflow phenomenon: maintaining vascular integrity. J Cardiovasc Pharmacol Ther. 2011;16:244–250. doi: 10.1177/1074248411405990. [DOI] [PubMed] [Google Scholar]
- 83.Geeganage CM, Bath PM. Relationship between therapeutic changes in blood pressure and outcomes in acute stroke: a metaregression. Hypertension. 2009;54:775–781. doi: 10.1161/HYPERTENSIONAHA.109.133538. [DOI] [PubMed] [Google Scholar]
- 84.Pagonas N, Utz W, Schulz-Menger J, Busjahn A, Monti J, Thierfelder L, Dietz R, Klauss V, Gross M, et al. Assessment of the effect of external counterpulsation on myocardial adaptive arteriogenesis by invasive functional measurements – design of the arteriogenesis network trial 2. Int J Cardiol. 2010;145:432–437. doi: 10.1016/j.ijcard.2009.05.050. [DOI] [PubMed] [Google Scholar]
- 85.Alexandrov AW, Ribo M, Wong KS, Sugg RM, Garami Z, Jesurum JT, Montgomery B, Alexandrov AV. Perfusion augmentation in acute stroke using mechanical counter-pulsation-phase IIa: effect of external counterpulsation on middle cerebral artery mean flow velocity in five healthy subjects. Stroke. 2008;39:2760–2764. doi: 10.1161/STROKEAHA.107.512418. [DOI] [PubMed] [Google Scholar]
- 86.Han JH, Leung TW, Lam WW, Soo YO, Alexandrov AW, Mok V, Leung Y-F V, Lo R, Wong KS. Preliminary findings of external counterpulsation for ischemic stroke patient with large artery occlusive disease. Stroke. 2008;39:1340–1343. doi: 10.1161/STROKEAHA.107.500132. [DOI] [PubMed] [Google Scholar]
- 87.Shuaib A, Bornstein NM, Diener H-C, Dillon W, Fisher M, Hammer MD, Molina CA, Rutledge JN, Saver JL, et al. Partial aortic occlusion for cerebral perfusion augmentation: safety and efficacy of NeuroFlo in Acute Ischemic Stroke trial. Stroke. 2011;42:1680–1690. doi: 10.1161/STROKEAHA.110.609933. [DOI] [PubMed] [Google Scholar]
- 88.Pranevicius O, Pranevicius M, Liebeskind DS. Partial aortic occlusion and cerebral venous steal: venous effects of arterial manipulation in acute stroke. Stroke. 2011;42:1478–1481. doi: 10.1161/STROKEAHA.110.603852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hammer MD, Schwamm L, Starkman S, Schellinger PD, Jovin T, Nogueira R, Burgin WS, Sen S, Diener HC, et al. Safety and feasibility of NeuroFlo use in eight- to 24-hour ischemic stroke patients. Int J Stroke. 2012;7:655–661. doi: 10.1111/j.1747-4949.2011.00719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hussain MS, Bhagat YA, Liu S, Scozzafava J, Khan KA, Dillon WP, Shuaib A. DWI lesion volume reduction following acute stroke treatment with transient partial aortic obstruction. J Neuroimaging. 2010;20:379–381. doi: 10.1111/j.1552-6569.2009.00407.x. [DOI] [PubMed] [Google Scholar]
- 91.Liebeskind DS. Aortic occlusion for cerebral ischemia: from theory to practice. Curr Cardiol Rep. 2008;10:31–36. doi: 10.1007/s11886-008-0007-3. [DOI] [PubMed] [Google Scholar]
- 92.Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K-I, Katayama Y, Asoh S, et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. 2007;13:688–694. doi: 10.1038/nm1577. [DOI] [PubMed] [Google Scholar]
- 93.Wood KC, Gladwin MT. The hydrogen highway to reperfusion therapy. Nat Med. 2007;13:673–674. doi: 10.1038/nm0607-673. [DOI] [PubMed] [Google Scholar]
- 94.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 95.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Nat Acad Sci USA. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Siriussawakul A, Chen LI, Lang JD. Medical gases: a novel strategy for attenuating ischemia-reperfusion injury in organ transplantation? J Transplant. 2012;2012:819382. doi: 10.1155/2012/819382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Man K, Ng KT, Lee TK, Lo CM, Sun CK, Li XL, Zhao Y, Ho JW, Fan ST. FTY720 attenuates hepatic ischemia-reperfusion injury in normal and cirrhotic livers. Am J Transplant. 2005;5:40–49. doi: 10.1111/j.1600-6143.2004.00642.x. [DOI] [PubMed] [Google Scholar]
- 98.Delbridge MS, Shrestha BM, Raftery AT, El Nahas AM, Haylor JL. Reduction of ischemia-reperfusion injury in the rat kidney by FTY720, a synthetic derivative of sphingosine. Transplantation. 2007;84:187–195. doi: 10.1097/01.tp.0000269794.74990.da. [DOI] [PubMed] [Google Scholar]
- 99.Kaudel CP, Frink M, Schmiddem U, Probst C, Bergmann S, Krettek C, Klempnauer J, Van Griensven M, Winkler M. FTY720 for treatment of ischemia-reperfusion injury following complete renal ischemia; impact on long-term survival and T-lymphocyte tissue infiltration. Transplant Proc. 2007;39:499–502. doi: 10.1016/j.transproceed.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 100.Awad AS, Ye H, Huang L, Li L, Foss FW, Macdonald TL, Lynch KR, Okusa MD. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am J Physiol Renal Physiol. 2006;290:F1516–F1524. doi: 10.1152/ajprenal.00311.2005. [DOI] [PubMed] [Google Scholar]
- 101.Wei Y, Yemisci M, Kim H-H, Yung LM, Shin HK, Hwang S-K, Guo S, Qin T, Alsharif N, et al. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann Neurol. 2011;69:119–129. doi: 10.1002/ana.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 103.Karliner JS, Honbo N, Summers K, Gray MO, Goetzl EJ. The lysophospholipids sphingosine-1-phosphate and lysophosphatidic acid enhance survival during hypoxia in neonatal rat cardiac myocytes. J Mol Cell Cardiol. 2001;33:1713–1717. doi: 10.1006/jmcc.2001.1429. [DOI] [PubMed] [Google Scholar]
- 104.Jin Z-Q, Zhou H-Z, Zhu P, Honbo N, Mochly-Rosen D, Messing RO, Goetzl EJ, Karliner JS, Gray MO. Cardioprotection mediated by sphingosine-1-phosphate and ganglioside GM-1 in wild-type and PKC epsilon knockout mouse hearts. Am J Physiol Heart Circ Physiol. 2002;282:H1970–H1977. doi: 10.1152/ajpheart.01029.2001. [DOI] [PubMed] [Google Scholar]
- 105.Obal D, Dai S, Keith R, Dimova N, Kingery J, Zheng Y-T, Zweier J, Velayutham M, Prabhu SD, et al. Cardiomyocyte-restricted overexpression of extracellular superoxide dismutase increases nitric oxide bioavailability and reduces infarct size after ischemia/reperfusion. Basic Res Cardiol. 2012;107:305. doi: 10.1007/s00395-012-0305-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Reddy MK, Labhasetwar V. Nanoparticle-mediated delivery of superoxide dismutase to the brain: an effective strategy to reduce ischemia-reperfusion injury. FASEB J. 2009;23:1384–1395. doi: 10.1096/fj.08-116947. [DOI] [PubMed] [Google Scholar]
- 107.Kahles T, Brandes RP. Which NADPH oxidase isoform is relevant for ischemic stroke? The case for Nox 2. Antioxid Redox Signal. 2013;18:1400–1417. doi: 10.1089/ars.2012.4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Radermacher KA, Wingler K, Langhauser F, Altenhöfer S, Kleikers P, Hermans JJ, Hrabě de Angelis M, Kleinschnitz C, Schmidt HH. Neuroprotection after stroke by targeting NOX4 as a source of oxidative stress. Antioxid Redox Signal. 2013;18:1418–1427. doi: 10.1089/ars.2012.4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ago T, Kitazono T, Kuroda J, Kumai Y, Kamouchi M, Ooboshi H, Wakisaka M, Kawahara T, Rokutan K, et al. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke. 2005;36:1040–1046. doi: 10.1161/01.STR.0000163111.05825.0b. [DOI] [PubMed] [Google Scholar]
- 110.Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH, Takada J, Wakisaka M, Ibayashi S, Utsumi H, et al. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation. 2004;109:227–233. doi: 10.1161/01.CIR.0000105680.92873.70. [DOI] [PubMed] [Google Scholar]
- 111.Hong H, Zeng J-S, Kreulen DL, Kaufman DI, Chen AF. Atorvastatin protects against cerebral infarction via inhibition of NADPH oxidase-derived superoxide in ischemic stroke. Am J Physiol Heart Circ Physiol. 2006;291:H2210–H2215. doi: 10.1152/ajpheart.01270.2005. [DOI] [PubMed] [Google Scholar]
- 112.Kahles T, Luedike P, Endres M, Galla H-J, Steinmetz H, Busse R, Neumann-Haefelin T, Brandes RP. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 113.Ford AL, An H, D'Angelo G, Ponisio R, Bushard P, Vo KD, Powers WJ, Lin W, Lee J-M. Preexisting statin use is associated with greater reperfusion in hyperacute ischemic stroke. Stroke. 2011;42:1307–1313. doi: 10.1161/STROKEAHA.110.600957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park KW, Crouse D, Lee M, Karnik SK, Sorensen LK, Murphy KJ, Kuo CJ, Li DY. The axonal attractant netrin-1 is an angiogenic factor. Proc Nat Acad Sci USA. 2004;101:16210–16215. doi: 10.1073/pnas.0405984101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 1996;87:1001–1014. doi: 10.1016/s0092-8674(00)81795-x. [DOI] [PubMed] [Google Scholar]
- 116.Métin C, Deléglise D, Serafini T, Kennedy TE, Tessier-Lavigne M. A role for netrin-1 in the guidance of cortical efferents. Development. 1997;124:5063–5074. doi: 10.1242/dev.124.24.5063. [DOI] [PubMed] [Google Scholar]
- 117.Sun H, Le T, Chang TTJ, Habib A, Wu S, Shen F, Young WL, Su H, Liu J. AAV-mediated netrin-1 overexpression increases peri-infarct blood vessel density and improves motor function recovery after experimental stroke. Neurobiol Dis. 2011;44:73–83. doi: 10.1016/j.nbd.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, Lo EH. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dunn JF, Wu Y, Zhao Z, Srinivasan S, Natah SS. Training the brain to survive stroke. PloS One. 2012;7:e45108. doi: 10.1371/journal.pone.0045108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Iadecola C, Anrather J. Stroke research at a crossroad: asking the brain for directions. Nat Neurosci. 2011;14:1363–1368. doi: 10.1038/nn.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stowe AM, Altay T, Freie AB, Gidday JM. Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann Neurol. 2011;69:975–985. doi: 10.1002/ana.22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fan Y, Hu W, Dai H, Zhang J, Zhang L, He P, Shen Y, Ohtsu H, Wei E, et al. Activation of the central histaminergic system is involved in hypoxia-induced stroke tolerance in adult mice. J Cereb Blood Flow Metab. 2011;31:305–314. doi: 10.1038/jcbfm.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu J, Narasimhan P, Yu F, Chan PH. Neuroprotection by hypoxic preconditioning involves oxidative stress-mediated expression of hypoxia-inducible factor and erythropoietin. Stroke. 2005;36:1264–1269. doi: 10.1161/01.STR.0000166180.91042.02. [DOI] [PubMed] [Google Scholar]
- 124.Ara J, Fekete S, Frank M, Golden JA, Pleasure D, Valencia I. Hypoxic-preconditioning induces neuroprotection against hypoxia-ischemia in newborn piglet brain. Neurobiol Dis. 2011;43:473–485. doi: 10.1016/j.nbd.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 125.Tang Y, Pacary E, Fréret T, Divoux D, Petit E, Schumann-Bard P, Bernaudin M. Effect of hypoxic preconditioning on brain genomic response before and following ischemia in the adult mouse: identification of potential neuroprotective candidates for stroke. Neurobiol Dis. 2006;21:18–28. doi: 10.1016/j.nbd.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 126.Semenza GL. Regulation of physiological responses to continuous and intermittent hypoxia by hypoxia-inducible factor 1. Exp Physiol. 2006;91:803–806. doi: 10.1113/expphysiol.2006.033498. [DOI] [PubMed] [Google Scholar]
- 127.Nagel S, Papadakis M, Chen R, Hoyte LC, Brooks KJ, Gallichan D, Sibson NR, Pugh C, Buchan AM. Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2011;31:132–143. doi: 10.1038/jcbfm.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L'Huillier I, Aupetit J-F, Bonnefoy E, Finet G, et al. Postconditioning the human heart. Circulation. 2005;112:2143–2148. doi: 10.1161/CIRCULATIONAHA.105.558122. [DOI] [PubMed] [Google Scholar]
- 129.Thibault H, Piot C, Staat P, Bontemps L, Sportouch C, Rioufol G, Cung TT, Bonnefoy E, Angoulvant D, et al. Long-term benefit of postconditioning. Circulation. 2008;117:1037–1044. doi: 10.1161/CIRCULATIONAHA.107.729780. [DOI] [PubMed] [Google Scholar]