

Abstract
The performance of differential IMS (FAIMS) analyzers is much enhanced by gases comprising He, especially He/N2 mixtures. However, electrical breakdown has limited the He fraction to ~50 %–75 %, depending on the field strength. By the Paschen law, the threshold field for breakdown increases at shorter distances. This allows FAIMS using chips with microscopic channels to utilize much stronger field intensities (E) than “full-size” analyzers with wider gaps. Here we show that those chips can employ higher He fractions up to 100 %. Use of He-rich gases improves the resolution and resolution/sensitivity balance substantially, although less than for full-size analyzers. The optimum He fraction is ~80 %, in line with first-principles theory. Hence, one can now measure the dependences of ion mobility on E in pure He, where ion-molecule cross section calculations are much more tractable than in other gases that form deeper and more complex interaction potentials. This capability may facilitate quantitative modeling of high-field ion mobility behavior and, thus, FAIMS separation properties, which would enable a priori extraction of structural information about the ions.
Keywords: Ion mobility spectrometry, Differential ion mobility spectrometry, FAIMS, Peptides, Lipids
Introduction
Ion mobility spectrometry (IMS) methods separate and characterize ions based on their gas-phase transport driven by electric fields [1–3]. The drift-tube (DT) IMS [1, 2], traveling-wave (TW) IMS [4], and other “conventional” IMS techniques sort ions by the absolute mobility (K) mostly in the low-field regime, where K is independent of the field intensity (E) [1]:
| (1) |
Here k is the Boltzmann constant, T and N are the gas temperature and number density, m and ze are the ion mass and charge, M is the gas molecule mass, and Ω is the orientationally-averaged ion–molecule collision cross section determined by the interaction potential, Φ.
Thus, ion mobilities depend on the gas via (1) mass and (2) properties influencing Φ, such as the molecular geometry, polarizability (a), and dipole moment [1, 3]. Lighter molecules are usually smaller and less polarizable, which means closer repulsive walls and shallower potentials. Therefore, both (1) and (2) lead to higher mobilities in lighter gases. Conventional IMS has mainly utilized He or N2. As He atoms are lighter than N2 molecules (4 versus 28 Da) and also smaller and less polarizable (a = 0.2 versus 1.8 Å3), the Ω values are greater in N2 than in He. For homologous ions, the difference diminishes with increasing size as the cross section becomes controlled by size rather than molecular properties [3]. For example, that difference drops from ~300 % for some atomic ions [5–7] to ~110 % for glycine, ~50 % for tetraglycine [8], and ~25 % and ~15 % for the kemptide and neurotensin peptides with 7 and 13 residues, respectively [9]. Hence, ion mobilities are higher in He than N2 by a factor that exceeds and typically varies from ~6 to 9 for atomic and small polyatomic ions to ~3 for macromolecules. Nonetheless, He and N2 buffers produce similar DTIMS separations [10, 11]. The maximum resolving power is independent of the ion mobility [2, 12]:
| (2) |
Here L is the drift length. Indeed, the measured R values do not depend on the gas [10]. While specific ions may be resolved only in some gases [13], the total peak capacity is unaffected [10].
However, as the He atom cannot rotate or vibrate, is not a dipole, and has the smallest size and polarizability of all molecules, it forms the simplest and least deep potentials with ions that can be approximated by pairwise Lennard-Jones interactions or even repulsive hard shells [14]. This allows more robust mobility calculations in He than other gases [14, 15], and He is preferred for structural elucidation by IMS.
At high enough E, the mobilities of all ions in gases deviate from the low-field limit [1]. Differential IMS exploits that to sort ions by the difference between mobility (ΔK) in strong and relatively weak fields [3, 16]. As that difference is elicited by a temporally periodic asymmetric field, the technique is also called field asymmetric waveform IMS (FAIMS). The field of some amplitude ED (“dispersion field”) is established in the gap between two electrodes by applying a high-voltage waveform. Ions, entrained in a gas flow, transit that gap and oscillate in the field but, as the mobilities during the alternating segments of high and low E differ, also drift across the gap and are lost on the electrodes. Selected ions can pass when the drift is offset by compensation field EC created by a compensation voltage (CV) applied to an electrode, and scanning EC reveals the spectrum of species entering the gap [3, 16].
The differential mobility underlying FAIMS is more sensitive than the mobility itself to the form of Φ and thus gas molecule characteristics. Early studies were conducted with N2 or air; other gases, including CO2, SF6, and their mixtures with He, were tested later [3, 17, 18]. Even the sign of ΔK can change [17] depending on the gas, with ions switching between the types A (ΔK > 0) and C (ΔK < 0). The peak inversions upon gas substitution are more prevalent in FAIMS [19] than DTIMS. With any FAIMS gap geometry [3, 20, 21], lighter gases generally increase R by raising absolute EC. For planar devices (where the field is homogeneous), the effect is augmented by peak widths (w) decreasing in proportion to ~ K−1/2 (at moderate ED) [22]. In particular, He/N2 mixtures tend to provide better resolution than N2 or heavier gases, and higher He content improves separations [21–28]. The problem is that the electrical breakdown threshold in He/N2 precipitously drops with growing He content (e.g., for a 2 mm gap at ambient conditions, from ~150 Td in N2 to ~15 Td in He) [29], which limited the He fraction (vol/vol) to ≤75 % (at ED/N = 87 Td) and ≤50 % at the maximum 117 Td feasible with the existing power supply [23–25]. Hydrogen resists breakdown better than He, permitting H2/N2 compositions with up to ~85 % H2 (at 117 Td) that provide better resolution than He/N2 mixtures [26, 27, 30]. However, H2 is flammable, and its higher polarizability and diatomic nature (that breaks the spherical symmetry of Φ and allows vibrational and rotational degrees of freedom) complicate ion mobility modeling. With theory and experiment pointing to continued resolution gains at higher He concentrations, moving to >75 % He could be advantageous. Also, with the relative simplicity of ion-He interactions, measuring K(E) curves in He could facilitate building a priori capability to predict separation parameters and thus deduce the ion structures from FAIMS data.
As the FAIMS technology has advanced, the minimum gap width (g) has been reduced from ~2 mm to 0.5 mm [31–33] and, recently, to 35 or 100 μm in microchips with multiple channels for concurrent ion processing [34–38]. By the Paschen law [29], the breakdown field for any gas goes up at lower gN. Hence, microscopic gaps permit extreme fields, up to ~170 kV/cm or ~700 Td with g = 35 μm (for N2 at ambient conditions). The resolving power is approximately proportional to ED3 and t1/2 (where t is the ion filtering time), therefore elevating ED permits compressing t as ~ ED−6 at equal separation quality [34]. This steep dependence enables chips to reduce t by over a 1000-fold (to ~20–200 μs from ~100–500 ms for the “full-size” (fs) units with g ~ 2 mm), which tremendously facilitates FAIMS after chromatography or electrophoresis. Unfortunately, the utility of chips has been constrained by lower resolution, as stronger fields only partially compensate for shorter separations [34].
While chips were previously operated with N2 or air only, their narrow gaps must elevate the breakdown point for He as well, permitting buffers with >75 % He. Here we use these chips to perform FAIMS in He/N2 with up to 100 % He. As anticipated, He-rich gases broadly benefit separations, improving the resolving power and resolution/sensitivity (r/s) balance.
Experimental
We employed FAIMS chips from Owlstone (Cambridge, UK): the early version (I) with g = 35 μm, installed on the Thermo Scientific LTQ ion trap mass spectrometer [35], and current ultraFAIMS (II) with g = 100 μm, coupled to the Agilent 6538 time-of-flight (ToF) MS platform [38]. One difficulty of integrating FAIMS using large He or H2 fractions with commercial MS instruments is the aspiration of hard-to-evacuate light gases into MS vacuum, where they challenge pumping designed for air. With I, we kept the pressure in the trap below the interlock trigger point by heating the MS inlet to 260 °C (the mass conductance through a capillary scales with the gas density that decreases at higher temperatures.) With II, we joined the FAIMS unit to the ToF instrument via a continuous-mode drift tube (filled with 4 Torr of N2 [11]) that isolated the MS vacuum from He inflow. Ions entered the tube via a 63-mm long capillary with 0.5 mm i.d., maintained at 120 °C. The inlet of Agilent ToF instrument is wider (i.d. = 0.6 mm) but longer (180 mm) and has nearly the same conductance, hence the t values in our platform are close to those in the FAIMS/ToF system [38]. The He/N2 mixture was prepared using flow meters (MKS Instruments), filtered, and delivered at the rates of Qin = 2–5 L/min. With FAIMS devices not vacuum-sealed to the MS inlet [23–28, 32], Qin governs the gas flow through the gap (based on the ratio of aperture areas in the curtain plate region) and thus t scales as 1/Qin. Here, the chip is sealed to the inlet, thus the flow through it and t are given by the flow Qout sucked from the chamber behind the chip (0–1 L/min with I and 0 with II) plus capillary conductance Qms (estimated at 0.8 L/min with I and 1.2 L/min with II) [35]. This allows optimizing Qin for best ion signal and signal/noise ratio without impacting the FAIMS resolution. Overall, higher He fractions require larger Qin values for proper ion desolvation.
Chips have open surface areas of 4 mm2 (I) and 7.8 mm2 (II), channel lengths of 0.3 mm (I) and 0.7 mm (II), and thus internal volumes of 1.2 mm3 (I) and 5.5 mm3 (II). Hence, the gas flow is much slower and t longer for II, improving resolution for same ED and temperature. We operated I at 40–60 °C (with heating from the adjacent capillary) and the maximum enabled [34] ED = 61 kV/cm or ED/N ~260–280 Td, which avoided breakdown even for pure He. The highest ED in II is slightly lower (55 kV/cm), but this chip can be directly heated up to 150 °C, where that field corresponds to ED/N = 320 Td. At a lower 120 °C here (equal to the drift tube inlet temperature), same ED translates into ED/N = 297 Td. However, a wider gap in II facilitates breakdown, which occurs at ~270 Td for 70 % He and ~190 Td for 95 % He. We evaluated II at up to 95 % He, while keeping ~5 %–7 % below those ED/N thresholds. Approaching them may cause arcing that would damage the chip and/or the control unit.
The performance was gauged for standards analyzed by fs-FAIMS units: peptides and reserpine (~1–4 μM solutions in 50:49:1 water/methanol/acetic acid [24]), reduced and alkylated tryptic digest of bovine serum albumin (BSA) in 50:49.8:0.2 water/methanol/acetic acid [23], and the lipid triacylglycerol (TG) 18:1/18:1/16:0 9Z (~10 μM solution in 30:66.5:3.5 chloroform/methanol/water with 10 mM ammonium acetate) [39]. Samples were infused to the electrospray ionization (ESI) emitter at ~0.5 μL/min.
Results and Discussion
Increased Resolving Power
We started the investigation from Chip I. Peptide Syntide 2 (S2, PLARTLSVAGLPGKK, 1508 Da), adopted as a benchmark in FAIMS [23–25, 27], yields protonated ions with z = 2–4. Their FAIMS spectra at selected He fractions are presented in Figure 1, with the separation metrics summarized and compared to those for a fs unit [24] in Figure 2. With N2 gas, the EC values increase from 2+ to 4+ to 3+ species as in fs units [23–25], but all three peaks are much broader and not fully resolved (Figure 1). Adding He up to ~85 % transposes these features to higher EC, then 2+ begins shifting back while 3+ and 4+ continue further up to 100 % He. The move is faster for 4+ than 3+, and the apexes coincide at ~60 % He, where the peak order changes to 2+, 3+, 4+ (Figure 2). The peak widths remain about constant for 2+ and 3+, and narrow for 4+. With these trends, the separation of 2+ and 4+ substantially improves at higher He fractions, reaching baseline (customarily, at 10 % peak height) at ~70 % He, but that of 2+ and 3+ increases only slightly (Figure 1). The order inversion of 3+ and 4+ may be due to unfolding of 3+ (normally manifested by transition to lower EC) [40, 41], induced by stronger field heating of ions upon He addition [23–25]. In fs devices, going from 0 % to 50 %–75 % He consistently raises EC for z = 2–4 with no order inversions [23–25]. This is likely because the magnitude of field heating above the gas temperature (ΔT) scales3 as (ED/N)2 and, thus, is ~5–10 times higher in FAIMS chips (with ED/N ~260–320 Td) than in fs analyzers (ED/N ~90–120 Td). At ~98 % He, the peaks for 2+ and 3+ abruptly broaden (Figure 1), possibly resulting from incomplete desolvation of ions with He in the curtain plate region. (This behavior, highly dependent on the species but not significantly shifting EC for any, is unlikely to reflect solvent vapors from the source penetrating into the FAIMS gap. In any event, such peak broadening was not observed for other species, as seen below.) Nevertheless, the 3+ and 4+ ions are well-resolved from 2+ even in He.
Figure 1.

FAIMS spectra for 2+, 3+, and 4+ ions of Syntide 2, measured using He/N2 with 0–100 % He (as labeled). The peak widths [full width at half maximum (FWHM)] are given on each panel. The data at 82 %, 88 %, and 97 % He are not shown, but their metrics are included into Figure 2
Figure 2.

Metrics of the separations in Figure 1 depending on the He fraction: normalized compensation field (a), (b), peak width (c), (d), resolving power (e), (f), and resolution of the features for 3+ and 4+ from 2+ (g), (h). Absolute quantities are in the left column, values relative to those with N2 gas are in the right column. Circles stand for the measurements, short-dash lines are cubic regressions through each dependence. The peak width and resolution data in pure He are marked by empty circles and not entered into the regressions. Solid lines (right column) are the trends (at 0–50 % He) with a planar FAIMS unit at ED = 29 kV/cm (reference [24])
The overall resolving power and peak resolution maximize at ~90 %–95 % He (Figure 2). At that point, the EC/N values are ~75 %–110 % above those with N2, whereas the resolving power is greater by ~80 %–130 % for 2+ and 3+ and ~210 %–250 % for 4+. While significant, these increases are somewhat less than ~95 %–130 % for EC/N and ~140 %–180 % (for 2+ and 3+) and ~250 % (for 4+) for R provided [24] by just 50 % He with fs-FAIMS units (Figure 2). Here, use of He improves the resolution of 3+ and 4+ peaks from 2+ by about two and four times, respectively (Figure 2). Though slightly lower than the three- to 5-fold obtained with fs devices [24], these are large gains. The benefit of He is smaller here because (1) EC increase more gradually, and (2) peaks narrow less (if at all). Still, per Figure 1, species poorly separated in N2 can be cleanly resolved using He-rich buffers. A broad proportionality of resolution to the resolving power for single species confirms R as a proxy for the separation quality as a function of He percentage.
Field heating in FAIMS can also fragment fragile ions, which is of potential concern for peptides with labile post-translational modifications (PTM). While no PTM abstraction from modified peptides in FAIMS has been reported, those experiments employed fs devices with ~5–10 times lower maximum ΔT values (as explained above). With ΔT increasing upon He addition as we stated, the risk of PTM severance would maximize at the highest He content. However, no PTM elimination at any He fraction was found for either 2+ or 3+ ions of the phosphopeptide APLpSFRGSLPKSYVK (1729 Da) previously analyzed with fs units [42]. The evolution of separation properties (Figure 3) tracks that for S2 (Figure 2). On the way from 0 % to 100 % He, the EC values for both charge states increase more gradually than with fs devices. (Again, a slight maximum appears at ~85 % He for z = 2). The peaks narrow by ~20 %–30 % and the resolving power improves by ~120 %–140 %, with the resolution of 2+ and 3+ peaks increasing 3-fold (Figure 3). These results prove chips suitable for modified peptides, even at the highest He fractions.
Figure 3.

Same as Figure 2 for 2+ and 3+ ions of phosphopeptide APLpSFRGSLPKSYVK (1729 Da). Here, the values for resolving power and resolution of two peaks (the latter plotted in black) are combined in (e), (f), and data with pure He are included in the regressions. The values with a planar FAIMS unit used in (b) are from reference [42]
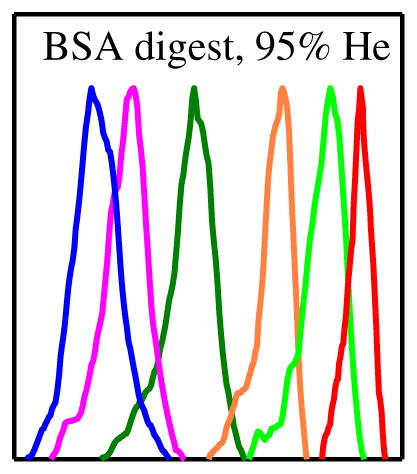
The advantage of using He for complex mixture analyses is obvious in the FAIMS spectra for BSA tryptic digest. Again, the EC/N values for all peptide ions increase upon He addition, with those for larger m/z (here above ~800) maximizing at ~80 %–90 % He (Figure 4). As seen for selected species (representative of the charge states and m/z range for multiply-charged ions), the peak resolution is drastically better with ~90 % He than with N2. The peak capacity (the EC range between bookend peak apexes divided by mean peak width) increases from (barely useful) 1.3 to 5.3 at 88 % He and 6.8 at 95 % He. The gain results from both the expansion of separation space (from 0.9 Td with N2 to 2.2 Td with 88 % He and 2.7 Td with 95 % He) and peak narrowing (from 0.7 to 0.4 Td with either He fraction).
Figure 4.

FAIMS spectra for selected peptide ions from the BSA tryptic digest, obtained using N2 and 95:5 He/N2 buffers. Features are color-coordinated with labels showing the measured m/z and (in parentheses) charge state
Both fs and chip FAIMS units are employed to analyze singly-charged ions typical for metabolites [38, 39]. To understand the utility of He in this context, we have looked at a customary MS standard reserpine (609 Da) observed as the protonated ion and TG lipid (859 Da) that produces the ammoniated ion (Figure 5). The EC values for these species also increase upon He addition, but rather disparately. The trend for reserpine resembles that for 4+ ion of S2 (Figure 2): the rise is uniform and rapid, with EC/N in He exceeding that in N2 by ~130 %. The situation for TG is closer to that for the phosphopeptide (Figure 3) with z = 2: the rise maximizes at ~80 % He and is slow (~40 % in the maximum and ~30 % for pure He). Thus, the EC curves for reserpine and TG cross at ~65 % He. The peak widths also behave differently, slightly broadening for reserpine but narrowing for TG. As the consequence, the resolution of reserpine and TG peaks drops from 0.5 in N2 to zero at ~65 % He, then improves to ~1 in pure He. Hence, using nearly pure He may also help resolve 1+ ions (notably, here almost no separation is seen at the previously maximum He fraction of 75 %). However, as shown for reserpine (Figure 5), the effects of He on EC and peak width are, again, less pronounced here than with fs analyzers.
Figure 5.

Same as Figure 3 for 1+ ions of triacylclycerol and reserpine. No resolving power is shown, and the resolution between two peaks (plotted in black) is added to (c). Long-dash lines (b), (d) indicate the trends at 0–75 % He with a planar FAIMS unit at ED = 21 kV/cm (reference [24])
Improved Resolution/Sensitivity Balance
One can also increase FAIMS resolving power at constant gas composition by raising ED or extending separation. As is common for filtering systems, improving instrumental resolution diminishes ion transmission and thus sensitivity. Hence, the key criterion when comparing different analyzers or experimental regimes is not R per se, but the r/s balance: the question is which approach provides the maximum R at a given sensitivity or maximum sensitivity for given R. We have used chip II to measure this balance for two peptide ion pairs: melittin (GIGAVLKVLTTGLPALISWIKRKRQQ, 2847 Da) with z = 3 and 4, and melittin 4+ and neurotensin (pyroELYENKPRRPYIL, 1673 Da) with z = 3 (Figure 6). In both cases, the r/s balance is optimized (the curve furthest out of the r = 0, s = 0 corner) near 80 % He, with notably worse performance at 70 % or 90 % He (Figure 6a, b). The gains in terms of either resolution or sensitivity are large. The resolution at equal sensitivity exceeds that with N2 by up to 4-fold [r = 1.6 versus 0.4 (Figure 6a) and r=0.9 versus 0.2 (Figure 6b) at ~20 %–25 % ion transmission], allowing substantial separation of previously “co-eluting” species (Figure 6c, d). These peaks are sparsely defined because of discrete ToF detection, but the improved resolution was reproduced with differing origin locations and thus samplings, as well as using different gas flow rates (Figure 6a, b). The relative sensitivity for same resolution increases by up to ~10–30 times: from s = 0.06 to 0.7 for r~1 (Figure 6a) and from 0.03 to 0.9 for r~0.5 (Figure 6b). These results demonstrate the practical utility of He-rich gases in FAIMS microchips.
Figure 6.

Resolution/sensitivity diagrams for pairwise separations of protonated peptide ions depending on the He content (as marked): (a) melittin 3+ and 4+; (b) melittin 4+ versus neurotensin 3+. Here, “sensitivity” is signal (for the ion of lower m/z) relative to all-pass FAIMS operation (ED = 0). Quadratic regressions through the points are drawn for the optimum case of 80 % He. Bottom panels show the spectra for melittin 3+ and 4+ at 25 % ion transmission, obtained using N2 (c) and 4:1 He/N2 (d)
In the present platforms, the gas entering the FAIMS unit gap also flows through the curtain plate region and around the ESI emitter, and high He fractions degrade ion desolvation and ESI stability (as exemplified above). As this suppresses the ion signal, the benefit of He would be even greater if the gases in ionization region and FAIMS unit were decoupled. This would also raise the He content for best r/s balance, but only slightly as the resolving power itself often peaks at ~90 %–95 % He.
Theoretical Treatment of the Effect of Helium and Non-Blanc Phenomena
A shortcoming of FAIMS is the unpredictability of EC values for polyatomic ions a priori (unlike with conventional IMS, where the drift times are reliably computed by first-principles molecular dynamics [14]). The major reasons are: (1) most derivations of ion-molecule cross sections assume the low-field limit and are far less tractable at high fields [1] relevant to FAIMS and (2) the ΔK quantity underlying FAIMS is much smaller than the absolute K values and, thus, ΔK obtained by subtracting one calculated K from the other is inaccurate. Such calculations require sophisticated quantum chemistry and molecular dynamics to set the realistic potentials Φ and propagate trajectories in them [14]. However, at extreme fields and, thus, ion-molecule velocities (v) in chips, the attractive well of Φ becomes less important (especially for larger ions such as studied here) and EC is largely controlled by the repulsive wall profile [3, 17]. Hence, virtually all species with m > 100 Da are [35] “type C” (where mobilities diminish at higher E and, thus, EC is positive) with N2, and should shift to even greater EC in He that forms much harder potentials with all ions [17]. Indeed, in DTIMS of atomic ions, the K(E) function decreases steeper with He than with N2 or other gases [3, 5–7]. Same should apply to larger ions, in line with our observation of uniformly higher EC in He compared with N2.
To quantify the EC shift upon transition from N2 to He, we note that the FAIMS effect (except for large proteins that arguably exhibit dipole alignment [43]) mainly reflects that v deviates from a Maxwell-Boltzmann distribution when the drift velocity at peak field (vD = KED) is not negligible relative to the mean ion-molecule velocity in Brownian motion (vBr). Thus, the scaling of EC as ED3 (for type C ions) must fundamentally be due to the proportionality of ΔK to (vD/vBr)3. Combining
| (3) |
with Equation (1), we exclude the ion and gas masses to derive:
| (4) |
and the magnitudes of EC in gases A and B relate as the inverse cubes of cross-sections:
| (5) |
Then EC values are greater in the lighter of two gases, with the relative difference decreasing for larger ions. Based on the measured cross sections for kemptide and neurotensin given above, the EC values in He should exceed those in N2 by factors of roughly (1.25)3 ~2 for small peptides and (1.15)3 ~1.5 for large ones. Although clearly crude, these estimates are in agreement with present results (Figures 2 and 3). A lesser effect for the larger lipid than smaller reserpine ion (Figure 5) also follows this model.
Unlike for conventional IMS, the FAIMS separation properties in gas mixtures are not averages of those in components since the Blanc’s law (by which the Ω value for ion in a mixture is the weighted mean of those in the components) is valid at low fields only [18, 19]. At high E pertinent to FAIMS, the law fails and EC for ions in mixtures often grossly deviate from the interpolations between those in constituents and even lie outside their range [18, 19]. The effect grows with greater disparity between the shapes of Φ for two gases, and mixtures of light gases (e.g., He or N2) with heavy gases (e.g., SF6) support spectacular non-Blanc phenomena [18, 19]. By theory [18], those elevate ΔK, raising absolute EC and, thus, the resolving power for type A species (where mobilities increase at higher E and, thus, EC is negative [18]). This was confirmed in experiments using He/N2, He/SF6, He/CO2, and N2/CO2 compositions [18, 19]. Such phenomena have not heretofore been reported for type C ions, but are now evident from EC maximizing at ~85 % He for species of higher m/z (Figures 2, 3, and 5). The ~80 % He fraction for maximum effect is close to that for type A ions in He/N2 and well-explained by first-principles physics [18]. However, the magnitude of effect (the elevation of EC at maximum over the interpolation between values in He and N2) is modest, much smaller than that frequent for type A ions [18, 19]. This is likely due to extreme fields in present chips, which render the sampled (here, repulsive) parts of ion potentials with N2 and He more similar than at lower fields in fs units.
Conclusions
Differential ion mobility (FAIMS) spectrometers can utilize diverse buffers, and mixtures containing the lightest gases (He or H2) are particularly conducive to higher resolution [20–28, 30]. Such compositions were not previously explored with FAIMS chips capable of ultrafast separations in microscopic gaps [34–38]. As expected from the Paschen law, the narrow gap widths dramatically raise the electrical breakdown thresholds [29]. This permits employing He/N2 with 75 % He up to ~200–250 Td (versus ~90 Td in “full-size” FAIMS units [23]), and, for the first time, exceeding ~75 % He and even use of pure He.
As with full-size units, adding He improves the resolving power and resolution of chips. The EC and R values for some species (apparently with larger m/z) maximize at ~80 %–90 % He, which matches the theory of non-Blanc effects [18] and represents their first observation for type C ions. However, the gains at equal He fraction are smaller than those with full-size analyzers, presumably as the ion-molecule potentials for different gases (here, He and N2) are more similar at higher energies. The resolution/sensitivity balance also improves, with an optimum at ~80 % He. For the exemplary peptide ion pairs, the gain is up to ~4× for resolution at equal sensitivity and ~30× for sensitivity at equal resolution: the benefit of using He with FAIMS chips is quite significant, even if less than that with full-size devices. For example, the separation peak capacity for a tryptic digest increased 5-fold.
Present capabilities provide K(E) curves for ions in He over a wide E range. As ion mobilities can be computed much more easily and accurately in He than other gases [14, 15], this information may open the path to a quantitative description of FAIMS separations and, thus, ability to extract the ion structural information from the data. This expectation is buttressed by a simple formula derived here, fairly predicting the relative EC values with N2 and He.
Full-size devices perform similarly in He/N2 and H2/N2 with equal N2 fractions [30]; hence, H2 should also improve the resolving power and resolution/sensitivity balance with FAIMS chips. A major motivation for replacing He by H2 has been the much higher breakdown threshold of H2 that permits greater light gas fractions (up to ~85 % H2 versus ~50 % He at the maximum ED/N = 117 Td) and, thus, higher resolution [30]. That is largely immaterial here, as even pure He works at or near maximum enabled field without breakdown. However, practical advantages of H2 (low cost, broad availability, and possible in situ production) may still favor it over He with microchips as well.
Acknowledgments
The authors thank Owlstone for providing their FAIMS stages, Dr. Keqi Tang, Ronald Moore, Karl Weitz, and Dr. Danielle Toutoungi for major experimental help, Dr. Giorgis Mezengie for the lipid sample, Professor Helen Cooper (University of Birmingham, UK) for the phosphopeptide sample, and Bruce Harrer for useful discussions. This work was supported in part by the PNNL Technology Assistance Program, PNNL Technology Commercialization Office, NIGMS (8 P41 GM103493-10), and the USDOE OBER, and carried out in the Environmental Molecular Sciences Laboratory, a DOE national scientific user facility at PNNL.
References
- 1.McDaniel EW, Mason EA. Transport properties of ions in gases. Wiley; NY: 1988. [Google Scholar]
- 2.Eiceman GA, Karpas Z. Ion mobility spectrometry. CRC Press; Boca Raton: 2004. [Google Scholar]
- 3.Shvartsburg AA. Differential ion mobility spectrometry: nonlinear ion transport and fundamentals of FAIMS. CRC Press; Boca Raton, FL: 2008. [Google Scholar]
- 4.Pringle SD, Giles K, Wildgoose JL, Williams JP, Slade SE, Thalassinos K, Bateman RH, Bowers MT, Scrivens JH. An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/oa-ToF instrument. Int J Mass Spectrom. 2007;261:1–12. [Google Scholar]
- 5.Thomson GM, Schummers JH, James DR, Graham E, Gatland IR, Flannery MR, McDaniel EW. Mobility, diffusion, and clustering of K+ ions in gases. J Chem Phys. 1973;58:2402–2411. [Google Scholar]
- 6.Viehland LA, Lozeille J, Soldán P, Lee EPF, Wright TG. Spectroscopy of K+·Rg and transport coefficients of K+ in Rg (Rg = He - Rn) J Chem Phys. 2004;121:341–351. doi: 10.1063/1.1735560. [DOI] [PubMed] [Google Scholar]
- 7.Thackston MG, Ellis HW, Pai RY, McDaniel EW. Experimental transverse diffusion and Monte Carlo simulation studies of Rb+ swarm drifting in He gas. J Chem Phys. 1976;65:2037–2038. [Google Scholar]
- 8.Beegle LW, Kanik I, Matz L, Hill HH. Effects of drift-gas polarizability on glycine peptides in ion mobility spectrometry. Int J Mass Spectrom. 2002;216:257–268. [Google Scholar]
- 9.Hill HH, Hill CH, Asbury GR, Wu C, Matz LM, Ichiye T. Charge location on gas phase peptides. Int J Mass Spectrom. 2002;219:23–37. [Google Scholar]
- 10.Ruotolo BT, McLean JA, Gillig KJ, Russell DH. Peak capacity of ion mobility separation. Utility of altering drift gas polarizability for tryptic peptide separation. J Mass Spectrom. 2004;39:361–367. doi: 10.1002/jms.592. [DOI] [PubMed] [Google Scholar]
- 11.Baker ES, Clowers BH, Li F, Tang K, Tolmachev AV, Prior DC, Belov ME, Smith RD. Ion mobility spectrometry—mass spectrometry performance using electrodynamic ion funnels and elevated drift gas pressures. J Am Soc Mass Spectrom. 2007;18:1176–1187. doi: 10.1016/j.jasms.2007.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Revercomb HE, Mason EA. Theory of plasma chromatography/gaseous electrophoresis. Review Anal Chem. 1975;47:970–983. [Google Scholar]
- 13.Asbury GR, Hill HH. Using different drift gases to change separation factors (alpha) in ion mobility spectrometry. Anal Chem. 2000;72:580–584. doi: 10.1021/ac9908952. [DOI] [PubMed] [Google Scholar]
- 14.Mesleh MF, Hunter JM, Shvartsburg AA, Schatz GC, Jarrold MF. Structural information from ion mobility measurements: effects of the long-term potential. J Phys Chem. 1996;100:16082–16086. [Google Scholar]
- 15.Shvartsburg AA, Schatz GC, Jarrold MF. Mobilities of carbon cluster ions: critical importance of the molecular attractive potential. J Chem Phys. 1998;108:2416–2423. [Google Scholar]
- 16.Guevremont R. High-field asymmetric waveform ion mobility spectrometry: a new tool for mass spectrometry. J Chromatogr A. 2004;1058:3–19. [PubMed] [Google Scholar]
- 17.Barnett DA, Ells B, Guevremont R, Purves RW, Viehland LA. Evaluation of carrier gases for use in high-field asymmetric waveform ion mobility spectrometry. J Am Soc Mass Spectrom. 2000;11:1125–1133. doi: 10.1016/S1044-0305(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 18.Shvartsburg AA, Tang K, Smith RD. Understanding and designing field asymmetric waveform ion mobility spectrometry separations in gas mixtures. Anal Chem. 2004;76:7366–7374. doi: 10.1021/ac049299k. [DOI] [PubMed] [Google Scholar]
- 19.Barnett DA, Purves RW, Ells B, Guevremont R. Separation of o-, m-, and p- phthalic acids by high-field asymmetric waveform ion mobility spectrometry (FAIMS) using mixed carrier gases. J Mass Spectrom. 2000;35:976–980. doi: 10.1002/1096-9888(200008)35:8<976::AID-JMS25>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 20.McCooeye MA, Ells B, Barnett DA, Purves RW, Guevremont R. Quantitation of morphine and codeine in human urine using high-field asymmetric waveform ion mobility spectrometry (FAIMS) with mass spectrometric detection. J Anal Toxicol. 2001;25:81–88. doi: 10.1093/jat/25.2.81. [DOI] [PubMed] [Google Scholar]
- 21.Cui M, Ding L, Mester Z. Separation of cisplatin and its hydrolysis products using electrospray ionization high-field asymmetric waveform ion mobility spectrometry coupled with ion trap mass spectrometry. Anal Chem. 2003;75:5847–5853. doi: 10.1021/ac0344182. [DOI] [PubMed] [Google Scholar]
- 22.Shvartsburg AA, Smith RD. Scaling of the resolving power and sensitivity for planar FAIMS and mobility-based discrimination in flow-and field- driven analyzers. J Am Soc Mass Spectrom. 2007;18:1672–1681. doi: 10.1016/j.jasms.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 23.Shvartsburg AA, Danielson WF, Smith RD. High-resolution differential ion mobility separations using helium-rich gases. Anal Chem. 2010;82:2456–2463. doi: 10.1021/ac902852a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shvartsburg AA, Prior DC, Tang K, Smith RD. High-resolution differential ion mobility separations using planar analyzers at elevated dispersion fields. Anal Chem. 2010;82:7649–7655. doi: 10.1021/ac101413k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shvartsburg AA, Smith RD. Ultrahigh-resolution differential ion mobility spectrometry using extended separation times. Anal Chem. 2011;83:23–29. doi: 10.1021/ac102689p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shvartsburg AA, Zheng Y, Smith RD, Kelleher NL. Ion mobility separations of variant histone tails extending to the middle-down range. Anal Chem. 2012;84:4271–4276. doi: 10.1021/ac300612y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shvartsburg AA, Seim TA, Danielson WF, Norheim R, Moore RJ, Anderson GA, Smith RD. High-definition differential ion mobility spectrometry with resolving power up to 500. J Am Soc Mass Spectrom. 2013;24:109–114. doi: 10.1007/s13361-012-0517-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shvartsburg AA, Smith RD. High-resolution differential ion mobility spectrometry of a protein. Anal Chem. 2013;85:10–13. doi: 10.1021/ac3029129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meek JM, Craggs JD. Electrical breakdown of gases. Wiley; NY: 1978. [Google Scholar]
- 30.Shvartsburg AA, Smith RD. Accelerated high-resolution differential ion mobility separations using hydrogen. Anal Chem. 2011;83:9159–9166. doi: 10.1021/ac202386w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purves RW, Guevremont R. Electrospray ionization high-field asymmetric waveform ion mobility spectrometry-mass spectrometry. Anal Chem. 1999;71:2346–2357. doi: 10.1021/ac981380y. [DOI] [PubMed] [Google Scholar]
- 32.Shvartsburg AA, Li F, Tang K, Smith RD. High-resolution field asymmetric waveform ion mobility spectrometry using new planar geometry analyzers. Anal Chem. 2006;78:3706–3714. doi: 10.1021/ac052020v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller RA, Nazarov EG, Eiceman GA, King AT. A MEMS radio-frequency ion mobility spectrometer for chemical agent detection. Sensors Actuators A. 2001;91:301–312. [Google Scholar]
- 34.Shvartsburg AA, Smith RD, Wilks A, Koehl A, Ruiz-Alonso D, Boyle B. Ultrafast differential ion mobility spectrometry at extreme electric fields in multichannel microchips. Anal Chem. 2009;81:6489–6495. doi: 10.1021/ac900892u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shvartsburg AA, Tang K, Smith RD, Holden M, Rush M, Thompson A, Toutoungi D. Ultrafast differential ion mobility spectrometry at extreme electric fields coupled to mass spectrometry. Anal Chem. 2009;81:8048–8053. doi: 10.1021/ac901479e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown LJ, Toutoungi DE, Devenport NA, Reynolds JC, Kaur-Atwal G, Boyle P, Creaser CS. Miniaturized ultra-high field asymmetric waveform ion mobility spectrometry combined with mass spectrometry for peptide analysis. Anal Chem. 2010;82:9827–9834. doi: 10.1021/ac102125u. [DOI] [PubMed] [Google Scholar]
- 37.Brown LJ, Smith RW, Toutoungi DE, Reynolds JC, Bristow AWT, Ray A, Sage A, Wilson ID, Weston DJ, Boyle B, Creaser CS. Enhanced analyte detection using in-source fragmentation of field asymmetric waveform ion mobility spectrometry-selected ions in combination with time-of-flight mass spectrometry. Anal Chem. 2012;84:4095–4103. doi: 10.1021/ac300212r. [DOI] [PubMed] [Google Scholar]
- 38.Smith RW, Toutoungi DE, Reynolds JC, Bristow AWT, Ray A, Sage A, Wilson ID, Weston DJ, Boyle B, Creaser CS. Enhanced performance in the determination of ibuprofen 1-β-O-acyl glucuronide in urine by combining high field asymmetric waveform ion mobility spectrometry with liquid chromatography-time-of-flight mass spectrometry. J Chromatogr A. 2013;1278:76–81. doi: 10.1016/j.chroma.2012.12.065. [DOI] [PubMed] [Google Scholar]
- 39.Shvartsburg AA, Isaac G, Leveque N, Smith RD, Metz TO. Separation and characterization of lipids using differential ion mobility spectrometry. J Am Soc Mass Spectrom. 2011;22:1146–1155. doi: 10.1007/s13361-011-0114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Purves RW, Barnett DA, Ells B, Guevremont R. Investigation of bovine ubiquitin conformers separated by high-field asymmetric waveform ion mobility spectrometry: cross sections measurements using energy-loss experiments with a triple quadrupole mass spectrometer. J Am Soc Mass Spectrom. 2000;11:738–745. doi: 10.1016/S1044-0305(00)00136-7. [DOI] [PubMed] [Google Scholar]
- 41.Shvartsburg AA, Li F, Tang K, Smith RD. Characterizing the structures and folding of free proteins using 2-D gas-phase separations: observation of multiple unfolded conformers. Anal Chem. 2006;78:3304–3315. doi: 10.1021/ac060283z. [DOI] [PubMed] [Google Scholar]
- 42.Shvartsburg AA, Creese AJ, Smith RD, Cooper HJ. Separation of peptide isomers with variant modified sites by high-resolution differential ion mobility spectrometry. Anal Chem. 2010;82:8327–8334. doi: 10.1021/ac101878a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shvartsburg AA, Noskov SY, Purves R, Smith RD. Pendular proteins in gases and new avenues for characterization of macromolecules by ion mobility spectrometry. Proc Natl Acad Sci U S A. 2009;106:6495–6500. doi: 10.1073/pnas.0812318106. [DOI] [PMC free article] [PubMed] [Google Scholar]