

Abstract
Because phages use their host translation machinery, their codon usage should evolve toward that of highly expressed host genes. We used two indices to measure codon adaptation of phages to their host, rRSCU (the correlation in relative synonymous codon usage [RSCU] between phages and their host) and Codon Adaptation Index (CAI) computed with highly expressed host genes as the reference set (because phage translation depends on host translation machinery). These indices used for this purpose are appropriate only when hosts exhibit little mutation bias, so only phages parasitizing Escherichia coli were included in the analysis. For double-stranded DNA (dsDNA) phages, both rRSCU and CAI decrease with increasing number of transfer RNA genes encoded by the phage genome. rRSCU is greater for dsDNA phages than for single-stranded DNA (ssDNA) phages, and the low rRSCU values are mainly due to poor concordance in RSCU values for Y-ending codons between ssDNA phages and the E. coli host, consistent with the predicted effect of C→T mutation bias in the ssDNA phages. Strong C→T mutation bias would improve codon adaptation in codon families (e.g., Gly) where U-ending codons are favored over C-ending codons (“U-friendly” codon families) by highly expressed host genes but decrease codon adaptation in other codon families where highly expressed host genes favor C-ending codons against U-ending codons (“U-hostile” codon families). It is remarkable that ssDNA phages with increasing C→T mutation bias also increased the usage of codons in the “U-friendly” codon families, thereby achieving CAI values almost as large as those of dsDNA phages. This represents a new type of codon adaptation.
Keywords: bacteriophage, codon adaptation, phage-host coevolution, mutation bias, deamination, Escherichia coli
Introduction
Efficient production of proteins is essential for survival and reproduction and strongly affects the fitness of a genotype, especially in unicellular organisms and viruses where rapid replication is essential for propagating the genotype into future generations. Efficient translation depends on the efficiency of the three subprocesses of translation, that is, initiation, elongation, and termination. Codon–anticodon adaptation directly impacts elongation efficiency. Ever since the empirical documentation of the correlation between codon usage and transfer RNA (tRNA) abundance (Ikemura 1981), codon–anticodon adaptation has been well documented in bacterial and fungal genomes (Ikemura 1981, 1992; Gouy and Gautier 1982; Xia 1998) as well as in mitochondrial genomes in vertebrates (Xia 2005; Xia et al. 2007) and fungi (Carullo and Xia 2008; Xia 2008). In short, differential tRNA availability almost invariably leads to biased codon usage, with most frequently used codons corresponding to the most abundant tRNA species. Optimizing codon usage according to host codon usage has been shown to increase the production of viral proteins (Haas et al. 1996; Ngumbela et al. 2008) or transgenic genes (Hernan et al. 1992; Kleber-Janke and Becker 2000; Koresawa et al. 2000). Studies on codon–anticodon adaptation have progressed in theoretical elaboration (Bulmer 1987, 1991; Xia 1998, 2008; Higgs and Ran 2008; Jia and Higgs 2008; Palidwor et al. 2010), in critical tests of alternative theoretical predictions (Xia 1996, 2005; Carullo and Xia 2008; van Weringh et al. 2011), and in formulation and implementation of codon bias indices such as relative synonymous codon usage (RSCU, Sharp et al. 1986), effective number of codons (Nc, Wright 1990; Sun et al. 2013), and Codon Adaptation Index (CAI, Sharp and Li 1987; Xia 2007). Although a recent study has questioned the relationship between codon usage and protein production (Kudla et al. 2009), its conclusion has been found to be unwarranted (Tuller et al. 2010).
Bacteriophage needs to have efficient translation to survive among alternative phage genotypes. Because phages depend mainly on the translation machinery of their host for protein translation, their codon adaptation is shaped by mutation and selection of the host tRNA pool (Grosjean et al. 1978; Gouy 1987; Kunisawa et al. 1998; Sahu et al. 2005; Carbone 2008; Lucks et al. 2008). Although some studies have suggested that extrinsic factors such as temperature (Sau and Deb 2009) and host diversity (Sau et al. 2007) may also affect phage codon usage, such factors should act indirectly through mutation and selection.
To study factors contributing to phage codon adaptation, we first use two codon usage indices, rRSCU (correlation of RSCU values between the host and the phage) and CAI, to measure phage codon adaptation. As explained in the next section, these indices are appropriate measures of phage codon adaptation when the host exhibits little nucleotide bias indicating little mutation bias. We then derive testable predictions on factors that contribute to phage codon adaptation.
Two Codon Usage Indices to Measure Phage Codon Adaptation
Assuming that the codon usage of highly expressed host genes are well adapted to their own translation machinery, we expect the phage genes to evolve a codon usage pattern similar to that of highly expressed host genes (Sharp et al. 1984). This suggests that concordance in codon usage between the host and the phage may be used as a proxy of phage codon adaptation. A simple measure of such concordance could be the correlation between host RSCU and phage RSCU, referred to hereafter as rRSCU.
rRSCU as a measure of phage codon adaptation has two problems. First, it can be increased not only by selection for codon adaptation but also by biased mutation. For example, strongly AT-biased mutations shared by both the host and the phage will lead to a high rRSCU. Such a high rRSCU cannot be equated to a high degree of codon adaptation because adaptation, by definition, arises in response to selection. There is, however, one special case where rRSCU can be reasonably used as a proxy of phage codon adaptation and that is when we study phages parasitizing the same host and when the host has roughly equal nucleotide frequencies indicating unbiased mutations.
Escherichia coli is approximately such a host species. Its genomic nucleotide frequencies are roughly equal, being 0.2462, 0.2541, 0.2537, and 0.2460 for nucleotides A, C, G and T, respectively. This indicates that mutations in E. coli do not lead to strong codon usage bias, in contrast to AT-biased or GC-biased mutations in many other bacterial species that can cause strong codon usage bias without any selection (Muto and Osawa 1987). Increasing the rate of unbiased mutations will lead to more randomized RSCU values and smaller rRSCU values.
The benefit of using a host with equal genomic nucleotide frequencies (presumably resulting from unbiased mutation) is that the effect of tRNA-mediated selection is often unequivocally detectable. Table 1 illustrates E. coli codon usage of four codon families in which tRNA-mediated selection favors A-, G-, C-, and U-ending codons, respectively. The most frequently used codon in each codon family matches the tRNA species with the highest gene copy numbers (table 1). For example, there are four tRNAGlu/UUC genes forming Watson–Crick base pair with Glu codon GAA but no tRNAGlu/CUC. As tRNA gene copy number is well correlated with experimentally measured tRNA abundance (Percudani et al. 1997), tRNA-mediated selection therefore should favor GAA, which is true (table 1). What is remarkable is that this association between major codon and tRNA abundance is visible when tRNA-mediated selection favors A-, G-, C-, and U-ending codons, respectively (table 1). If the E. coli genome had experienced strong AT-biased mutation, then tRNA-mediated selection for C-ending or G-ending codons may be invisible (i.e., A-ending and T-ending codons may still be the most frequently observed in spite of tRNA-mediated selection favoring C-ending and G-ending codons when AT-biased mutation dominates over the tRNA-mediated selection). For this reason, phages studied here are all E. coli phages.
Table 1.
The Effect of tRNA-Mediated Selection in Escherichia coli, Whose Genomic Sequence Has Equal Nucleotide Frequencies, Presumably Resulting from Little Mutation Bias.
| AA | Codon | Na | tRNAb | CF |
|---|---|---|---|---|
| Glu | GAA | 4,683 | 4 | A-ending |
| GAG | 1,459 | 0 | ||
| Phe | UUC | 2,229 | 2 | C-ending |
| UUU | 872 | 0 | ||
| Leu4c | CUA | 54 | 1 | |
| CUG | 5,698 | 4 | G-ending | |
| CUC | 541 | 1 | ||
| CUU | 357 | 0 | ||
| Arg4c | CGA | 34 | 0 | |
| CGG | 33 | 1 | ||
| CGC | 1,530 | 0 | ||
| CGU | 2,995 | 3 | U-ending |
Note.—CF, codon favored by tRNA.
aNumber of codons in highly expressed E. coli genes compiled in the EMBOSS package (Rice et al. 2000).
bNumber of E. coli tRNA genes with anticodon forming Watson–Crick pairing with the associated codon. Nucleotide A at the first anticodon position is mostly modified to inosine.
cLeu and Arg are coded by a four-codon subfamily and a two-codon subfamily. Leu4 and Arg4 refer to their respective four-codon subfamily.
The second problem with rRSCU is that it does not capture all aspects of codon adaptation. This is illustrated in table 2, which shows fictitious codon count and RSCU of highly expressed host genes and two phage genes (PG1 and PG2). RSCU values for codons in PG1 and PG2 are exactly the same, so rRSCU for PG1 and PG2 will also be the same. However, PG2 is expected to be translated more efficiently than PG1 for the following reason. We notice that highly expressed host genes strongly avoid UUU in the Phe codon family (table 2), suggesting that UUU cannot be translated efficiently by the host translation machinery. Given this, PG2 as a whole should be translated faster than PG1 because PG2 has only 90 “bad” UUU codons, whereas PG1 has 180 “bad” UUU codons. In this case, the Gly codon family is “U-friendly” because an increased number of U-ending codons will in fact improve translation. In contrast, the Phe codon family is “U-hostile” because increasing the number of U-ending codons will reduce translation efficiency. A single-stranded DNA (ssDNA) phage that cannot avoid high C→T mutations can nonetheless evolve codon adaptation by reducing the usage of codons in U-hostile codon families and increase the usage of codons in U-friendly codon families as PG2 does (table 2). This kind of adaptation is invisible to rRSCU but can be detected by CAI. We use the mean CAI value, computed from all genes in a phage genome with highly expressed host genes as a reference set, as an alternative measure of phage codon adaptation. The reason for using highly expressed host genes is that phage translation depends on host translation machinery, that is, efficient translation elongation of phage mRNA depends on whether the phage mRNA would overuse codons preferred by highly expressed host genes.
Table 2.
Fictitious Codon Usage for Highly Expressed Host Genes (HOST) and Two Phage Genes (PG1 and PG2).
| AA | Codon | Count |
RSCU |
||||
|---|---|---|---|---|---|---|---|
| HOST | PG1 | PG2 | HOST | PG1 | PG2 | ||
| Gly | GGA | 400 | 50 | 75 | 0.8889 | 1 | 1 |
| GGG | 300 | 30 | 45 | 0.6667 | 0.6 | 0.6 | |
| GGC | 100 | 20 | 30 | 0.2222 | 0.4 | 0.4 | |
| GGU | 1,000 | 100 | 150 | 2.2222 | 2 | 2 | |
| Phe | UUC | 2,000 | 20 | 10 | 1.8182 | 0.2 | 0.2 |
| UUU | 200 | 180 | 90 | 0.1818 | 1.8 | 1.8 | |
Note.—rRSCU between HOST and PG1 is identical to that between HOST and PG2, but PG2 will have higher CAI than PG1 when CAI is computed with HOST as the reference set of genes.
Phages are essentially a mosaic of genes sampled from a pool of frolicking phage genomes. For example, although many related tailed phages have nearly identical genome organization such as “DNA packaging-head-tail-tail fiber-lysis-lysogeny-DNA replication-transcription regulation” (Desiere et al. 2001), essentially any function in a phage can be fulfilled by one of many distinct genes with homologous function but little sequence similarity (Brussow and Kutter 2005). In other words, horizontal gene transfer is rampant in phage, so that individual genes in each phage could differ dramatically in evolutionary history and different codon usage. Consequently, a mean/median CAI may not be representative of all genes in a phage genome. For this reason, we have added standard deviation of CAI values in the supplementary files S1-S3, Supplementary Material online, to show that the among-gene difference in CAI is actually quite small.
Effect of Phage-Encoded tRNA Genes on Phage Codon Usage
Some phage genomes are long known to encode tRNA genes (Chattopadhyay and Ghosh 1988; Mandal and Ghosh 1988), for example, Enterobacteria phage WV8 carries 20 tRNA genes on its genome. Phage-encoded tRNAs tend to have anticodons decoding codons overused in the phage genes but rarely used in host genes (Kunisawa 1992, 2000; Bailly-Bechet et al. 2007; Enav et al. 2012). Such phage-encoded tRNAs would alter host tRNA pool, render the phage less dependent on the host tRNAs, and reduce the need (selection pressure) for the phage genes to evolve toward a codon usage pattern similar to that of the host genes. In other words, such tRNA genes would tend to reduce rRSCU and CAI and need to be taken into consideration in studying phage codon adaptation, especially in characterizing the difference between double-stranded DNA (dsDNA) and ssDNA phages because the latter do not encode tRNA genes in their genomes.
Effect of C→T Mutation Bias on Codon Usage of ssDNA Phages
Mutation rate differs much between ssDNA and dsDNA phages. Although dsDNA is well protected against mutation agents, ssDNA is subject to a high rate of DNA decay, especially spontaneous deamination leading to C→T mutations, the rate of which is about 100 times higher in ssDNA than in dsDNA (Frederico et al. 1990). Oxidative deamination leading to high C→U/T transitional mutation rates has been reported in ssDNA phage M13 (Kreutzer and Essigmann 1998). The high mutation rate of ssDNA phages relative to dsDNA phages impact strongly on genomic GC content (Xia and Yuen 2005) and codon usage bias (Cardinale and Duffy 2011). For this reason, one would predict that, given the same tRNA-mediated selection for codon usage bias, dsDNA phages would achieve better codon adaptation than ssDNA phages.
Coevolution Time and Maximum rRSCU
We have predicted that tRNA-mediated selection will increase rRSCU and that increased mutation rate will decrease rRSCU in E. coli phage. However, testing these predictions is confounded by coevolution time between phages and their host. Suppose a group of phages, given sufficient coevolution time with E. coli, would reach a maximum rRSCU. When we sample these phage lineages, some may have coevolved sufficiently long to have reached the maximum rRSCU, whereas others may be far from reaching the maximum because they may have invaded E. coli only recently. Thus, both dsDNA and ssDNA phages may have some of their members with low rRSCU values, but we predict that the maximum rRSCU value should be much greater for dsDNA phages than for ssDNA phages.
In short, we predict that 1) for dsDNA phages, rRSCU should decrease with the number of tRNA genes encoded by the phage genome, with phage-encoded tRNAs likely decoding codons overused by phage mRNAs but rarely used by host mRNAs, 2) rRSCU should be greater for dsDNA phages than ssDNA phages when the effect of phage-encoded tRNA genes has been taken into consideration, and maximum rRSCU should in particular be much greater for dsDNA phages than for ssDNA phages, and 3) ssDNA phages with a strong C→T mutation bias may evolve to increase the usage of codons in U-friendly codon families and reduce the usage of codons in U-hostile codon families. We report results confirming these predictions.
Results
Twenty-two dsDNA phage species encode tRNA genes in their genomes (13 from Myoviridae, 4 from Podoviridae, and 5 from Siphoviridae; supplementary file S1, Supplementary Material online), whereas none of the ssDNA phage genomes carry tRNA genes. Before making comparisons in codon usage between dsDNA and ssDNA phages, it is important to test if phage-encoded tRNA genes can affect codon usage. The presence of an effect implies that the fair comparison should only be carried out between ssDNA phages and those dsDNA phages that do not carry tRNA genes.
Effect of Phage-Encoded tRNA on Codon Adaptation in dsDNA Phage
We have reasoned before that phage-encoded tRNA genes may reduce rRSCU, especially if these tRNAs tend to decode codons overused in the phage genes but underused in host genes. There is indeed a highly significant (P < 0.0001) negative relationship between rRSCU and the number of tRNA genes encoded in the phage genome (fig. 1). The use of an exponential decay to fit the negative relationship is based on the rationale that, if the number of tRNA genes in the phage approaches infinity, then the codon usage of the phage would approach complete independence of the host tRNA pool, with rRSCU approaching zero. A significant (P = 0.0260) negative relationship is also observed between CAI and the number of tRNA genes encoded in the phage genome.
Fig. 1.
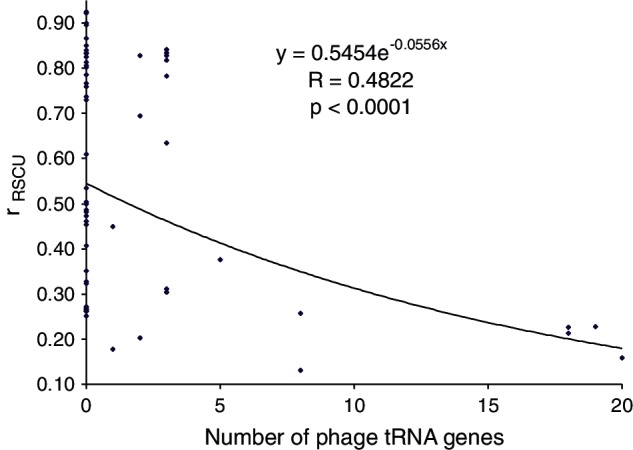
Codon adaptation of the phage genes, measured by rRSCU, decreases with increasing number of tRNA genes encoded in phage genomes.
What tRNA genes would benefit dsDNA phages that carry them? Translation of codons that are overused in phage genes but decoded by few host tRNAs would benefit from having extra cognate tRNAs from the phage genomes. Take R-ending codon, for example (where R stands for purine). If the host tRNA pool favors G-ending codon, but A-ending codon is overused by phage genes, then it is beneficial for the phage to carry tRNA genes with a wobble U to decode the overused A-ending codons. Similarly, if the host has few tRNAs decoding G-ending codons and uses few G-ending codons, but the phage uses many more G-ending codons, then it would be beneficial for phage tRNAs to have a wobble C to decode its relatively more frequently used G-ending codons.
Three general rules can be derived from the results in table 3, which shows the R-ending codon usage of highly expressed E. coli genes and two dsDNA phages each carrying a set of tRNA genes. First, if phage codon usage bias is the same as that of E. coli (e.g., GAR, AAR, and AGR codons for amino acids E, K, and R, respectively), then the phage-encoded tRNAs will decode the most frequently used codon. Second, if phage codon usage bias is opposite to that of the host (e.g., GGR, UUR, CCR, and UCR codons for amino acids G, L, P, and S, respectively), then the phage-encoded tRNAs will decode the codon overused in the phage but underused in the host. Third, if phage genes use the two R-ending codons roughly equally (e.g., CAR codons for amino acid Q), then the phage may carry tRNAs for both codons. Although only two phage species are included in table 3, the three rules are shared among other phage species with phage-encoded tRNAs.
Table 3.
Number of A- or G-Ending Codons (Ncod), RSCU, and Number of tRNA Genes (NtRNA) for Escherichia coli and Two Phage Species (WV8 and bV_EcoS_AKFV33).
| AA | Codon |
E. colia |
WV8 |
bV_EcoS_AKFV33 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ncod | RSCU | NtRNA | Ncod | RSCU | NtRNA | Ncod | RSCU | NtRNA | ||
| E | GAA | 4,683 | 1.525 | 4 | 1,125 | 1.259 | 1 | 1,489 | 1.365 | 1 |
| E | GAG | 1,459 | 0.475 | 662 | 0.741 | 692 | 0.635 | |||
| G | GGA | 118 | 0.068 | 1 | 245 | 0.584 | 1 | |||
| G | GGG | 267 | 0.154 | 1 | 150 | 0.357 | ||||
| K | AAA | 4,129 | 1.595 | 5 | 1,262 | 1.195 | 1 | 1,551 | 1.364 | 1 |
| K | AAG | 1,050 | 0.406 | 851 | 0.805 | 1 | 723 | 0.636 | 1 | |
| L | CUA | 54 | 0.033 | 1 | 233 | 0.745 | 1 | 544 | 1.335 | 1 |
| L | CUG | 5,698 | 3.427 | 3 | 318 | 1.017 | 433 | 1.063 | ||
| L | UUA | 210 | 0.774 | 1 | 718 | 1.453 | 1 | |||
| L | UUG | 333 | 1.227 | 1 | 270 | 0.547 | ||||
| P | CCA | 474 | 0.564 | 1 | 408 | 2.032 | 1 | 428 | 1.558 | 1 |
| P | CCG | 2,509 | 2.983 | 1 | 62 | 0.309 | 154 | 0.561 | ||
| Q | CAA | 550 | 0.355 | 2 | 481 | 1.058 | 1 | 593 | 1.06 | 1 |
| Q | CAG | 2,548 | 1.645 | 2 | 428 | 0.942 | 1 | 526 | 0.94 | 1 |
| R | AGA | 21 | 1.235 | 8 | 438 | 1.581 | 1 | 317 | 1.461 | 1 |
| R | AGG | 13 | 0.765 | 1 | 116 | 0.419 | 117 | 0.539 | ||
| S | UCA | 189 | 0.261 | 1 | 498 | 1.64 | 1 | |||
| S | UCG | 275 | 0.380 | 1 | 38 | 0.125 | ||||
| T | ACA | 181 | 0.160 | 1 | 447 | 1.002 | 1 | |||
| T | ACG | 526 | 0.465 | 1 | 164 | 0.368 | ||||
| V | GUA | 1,329 | 0.805 | 5 | 765 | 1.508 | 1 | |||
| V | GUG | 1,784 | 1.080 | 231 | 0.455 | |||||
Note.—See text for reasons of including only R-ending codons.
aFrom highly expressed E. coli genes, as compiled in the EMBOSS distribution (Rice et al. 2000).
The three rules are generally consistent with the interpretation that phage-encoded tRNAs facilitate translation of phage mRNAs. Similar findings, but less complete, have also been reported in previous studies on T4-like phages (Kunisawa 1992; Bailly-Bechet et al. 2007; Enav et al. 2012). They are also consistent with previous experiments in which alteration of E. coli tRNA pool is associated with changed translation efficiency of transgenes (Kleber-Janke and Becker 2000).
One may note that table 3 includes only R-ending codons. Can we extend the pattern to Y-ending codons (where Y stands for pyrimidine)? Suppose that the host overuses C-ending codons, with many tRNAs with a wobble G, but the phage overuses U-ending codons. Should we not predict that phage genomes should encode tRNAs with a wobble A to decode its overused U-ending codons? However, this prediction cannot be tested because a tRNA with wobble A would interfere with translation. That is, once such a tRNA is in the P-site, it interferes with the tRNA at the A-site (Lim 1994). Thus, Y-ending codons are decoded by either tRNAs with a wobble G or tRNA with a wobble A-derived inosine. This was overlooked in a previous study on tRNAs encoded in bacteriophage T4 (Kunisawa 1992).
Difference in rRSCU between dsDNA and ssDNA Phages
Given the significant effect of phage-encoded tRNA on rRSCU (fig. 1 and table 3), all phage genomes with encoded tRNA genes were excluded in all comparisons between dsDNA phages and ssDNA phages because none of the ssDNA phage genomes encode tRNA genes. This leaves 38 dsDNA phages and 11 ssDNA phages for further comparisons in rRSCU.
rRSCU is significantly greater for dsDNA phages than for ssDNA phages (0.5917 for the former and 0.3273 for the latter, t = 3.6533, DF = 47, P = 0.0008, table 4). To test if it is the C→T-biased mutation that is chiefly responsible for the reduced rRSCU values for the ssDNA phages, we computed the rRSCU values separately for the R-ending codons and Y-ending codons (table 5). The rRSCU values for the R-ending codons (rRSCU.R) are significantly greater than those for the Y-ending codons (rRSCU.Y), with the mean being 0.5217 for rRSCU.R and 0.1074 for rRSCU.Y (table 5). The difference is highly significant (paired-sample t-test: t = 17.2872, DF = 10, P < 0.0001), assuming data independence.
Table 4.
Mean and Distribution of rRSCU Values for Various dsDNA and ssDNA Phage Families.
| Type | Phage Family | n | Minimum | Maximum | Average | SD |
|---|---|---|---|---|---|---|
| dsDNA | Myoviridae | 9 | 0.3437 | 0.9207 | 0.6953 | 0.2359 |
| Podoviridae | 12 | 0.2553 | 0.8034 | 0.4216 | 0.1859 | |
| Siphoviridae | 16 | 0.2412 | 0.8955 | 0.6600 | 0.2355 | |
| Tectiviridae | 1 | 0.6084 | 0.6084 | 0.6084 | NA | |
| ssDNA | Inoviridae | 4 | 0.2700 | 0.3922 | 0.3449 | 0.0524 |
| Microviridae | 7 | 0.2757 | 0.3709 | 0.3173 | 0.0409 |
Note.—NA, not applicable.
Table 5.
Contrasting rRSCU Values for R-Ending Codons and for Y-Ending Codons (designated by rRSCU.R and rRSCU.Y, respectively).
| Family | ACCN | rRSCU.R | rRSCU.Y |
|---|---|---|---|
| Microviridae | NC_001330 | 0.6504 | 0.0854 |
| Microviridae | NC_001420 | 0.4530 | 0.0332 |
| Microviridae | NC_007856 | 0.4652 | 0.0447 |
| Microviridae | NC_007817 | 0.4168 | 0.0200 |
| Microviridae | NC_001422 | 0.4497 | 0.0843 |
| Microviridae | NC_012868 | 0.6009 | 0.1118 |
| Microviridae | NC_007821 | 0.6030 | 0.1158 |
| Inoviridae | NC_001332 | 0.5475 | 0.1709 |
| Inoviridae | NC_001954 | 0.4753 | 0.2154 |
| Inoviridae | NC_002014 | 0.5892 | 0.2105 |
| Inoviridae | NC_003287 | 0.4876 | 0.0894 |
| Mean | 0.5217 | 0.1074 |
Because some phages may not have enough time coevolving with their host, their rRSCU may not have reached the maximum possible. For example, if a dsDNA phage has recently switched to a host with a different codon usage pattern, then we would not expect it to have a high rRSCU value because codon adaptation takes time to evolve. However, given enough time, we expect dsDNA phages to reach a higher rRSCU than ssDNA phages whose mutation rate is higher than that of dsDNA phages. The mean and distribution of rRSCU values for the dsDNA and ssDNA phage (table 4) is consistent with this interpretation. The maximum rRSCU observed is only 0.3922 for ssDNA phages but 0.9207 for dsDNA phages (Enterobacteria phage Mu in Myoviridae). The mean and standard variation of rRSCU values for ssDNA phage is 0.3273 and 0.0450, respectively, so that the probability of having an rRSCU value as large as 0.5 is less than 0.0001 for ssDNA phages.
When a phage species has a small rRSCU value, it could be due to weakened selection (e.g., the phage carries a large number of its own tRNA genes), strong mutation pressure disrupting codon adaptation, or insufficient coevolution time. Given that the three dsDNA phage families and the two ssDNA phage families all have multiple phage lineages parasitizing E. coli, we may assume that the phages should have coevolved with E. coli for sufficiently long time for codon adaptation to reach a mutation-selection equilibrium. Also, the comparison above between the dsDNA and ssDNA phages excluded phages with phage-encoded tRNA genes, so all these phages should have experienced roughly the same host tRNA-mediated selection. The most plausible explanation for the difference in rRSCU between the dsDNA and ssDNA phages is the higher mutation pressure in ssDNA phages that disrupt codon adaptation.
Effect of Life Cycle (Temperate vs. Virulent) on rRSCU in dsDNA Phages
dsDNA phages differ in their life cycles, some being temperate with a lysogenic phage and some are virulent with only lytic phase, although lysogenic phages can become lytic through mutations at lysogenic conversion genes (van Vliet et al. 1978; Brussow and Kutter 2005). Temperate phages are expected to have better concordance in codon usage with the host (i.e., higher rRSCU values) than lytic phages for two reasons. First, a prophage and its lysogen share the same mutation spectrum as the host DNA. Second, they have increased chance of recombining with or acquiring host genes or gene segments. For example, phage λ and phage µ carry a piece of host genome when they switch from the lysogenic phase to the lytic phase.
The expectation is borne out by empirical data (table 6), with rRSCU significantly greater in temperate phages than in virulent phages with two-sample t-tests (DF = 7, t = 11.5914, P < 0.0001 for Myoviridae; DF = 9, t = 5.7328, P = 0.0003 for Podoviridae; DF = 12, t = 10.4545, P < 0.0001 for Siphoviridae). A two-way analysis of variance accounts for 91.24% of total variance in rRSCU, with rRSCU differing highly significantly between temperate and virulent phages (F = 280.9918, DFmodel = 1, DFerror = 28, P < 0.0001), significantly among the three dsDNA phage families (F = 5.095, DF = 2, P = 0.0130), but with no significant interaction (F = 0.2101, DF = 2, P = 0.81175).
Table 6.
Effect of Life Cycle of dsDNA Phages on Codon Usage Concordance between Phage and Host, Measured by rRSCU.
| PhageFam | PhageName | Accession | LifeCycle | rRSCU |
|---|---|---|---|---|
| Myoviridae | Enterobacteria phage Mu | NC_000929 | Temperate | 0.9207 |
| Myoviridae | Enterobacteria phage P2 | NC_001895 | Temperate | 0.9011 |
| Myoviridae | Enterobacteria phage P4 | NC_001609 | Temperate | 0.8287 |
| Myoviridae | Enterobacteria phage SfV | NC_003444 | Temperate | 0.8750 |
| Myoviridae | Escherichia phage D108 | NC_013594 | Temperate | 0.9207 |
| Myoviridae | Enterobacteria phage JSE | NC_012740 | Virulent | 0.4789 |
| Myoviridae | Enterobacteria phage Phi1 | NC_009821 | Virulent | 0.4971 |
| Myoviridae | Enterobacteria phage phiEcoM-GJ1 | NC_010106 | Virulent | 0.3437 |
| Myoviridae | Enterobacteria phage RB49 | NC_005066 | Virulent | 0.4917 |
| Podoviridae | Escherichia phage phiV10 | NC_007804 | Temperate | 0.7308 |
| Podoviridae | Stx2 converting phage I | NC_003525 | Temperate | 0.8034 |
| Podoviridae | Enterobacteria phage 13a | NC_011045 | Virulent | 0.3181 |
| Podoviridae | Enterobacteria phage EcoDS1 | NC_011042 | Virulent | 0.4021 |
| Podoviridae | Enterobacteria phage K1-5 | NC_008152 | Virulent | 0.2629 |
| Podoviridae | Enterobacteria phage K1E | NC_007637 | Virulent | 0.2553 |
| Podoviridae | Enterobacteria phage K1F | NC_007456 | Virulent | 0.2553 |
| Podoviridae | Enterobacteria phage N4 | NC_008720 | Virulent | 0.2661 |
| Podoviridae | Enterobacteria phage T3 | NC_003298 | Virulent | 0.5306 |
| Podoviridae | Enterobacteria phage T7 | NC_001604 | Virulent | 0.3274 |
| Podoviridae | Enterobacteria phage BA14 | NC_011040 | Virulent | 0.4504 |
| Siphoviridae | Enterobacteria phage BP-4795 | NC_004813 | Temperate | 0.8049 |
| Siphoviridae | Enterobacteria phage cdtI | NC_009514 | Temperate | 0.8307 |
| Siphoviridae | Enterobacteria phage HK022 | NC_002166 | Temperate | 0.7416 |
| Siphoviridae | Enterobacteria phage HK97 | NC_002167 | Temperate | 0.7303 |
| Siphoviridae | Enterobacteria phage lambda | NC_001416 | Temperate | 0.8520 |
| Siphoviridae | Enterobacteria phage N15 | NC_001901 | Temperate | 0.8955 |
| Siphoviridae | Escherichia Stx1 converting bacteriophage | NC_004913 | Temperate | 0.8108 |
| Siphoviridae | Stx2-converting phage 1717 | NC_011357 | Temperate | 0.8335 |
| Siphoviridae | Enterobacteria phage SSL-2009a | NC_012223 | Temperate | 0.7853 |
| Siphoviridae | Enterobacteria phage EPS7 | NC_010583 | Virulent | 0.2583 |
| Siphoviridae | Enterobacteria phage JK06 | NC_007291 | Virulent | 0.2565 |
| Siphoviridae | Enterobacteria phage RTP | NC_007603 | Virulent | 0.2412 |
| Siphoviridae | Enterobacteria phage T1 | NC_005833 | Virulent | 0.4637 |
| Siphoviridae | Enterobacteria phage TLS | NC_009540 | Virulent | 0.4734 |
Note.—The phages are organized by phage families (PhageFam) and then by life cycle (LifeCycle: temperate or virulent) within each phage family.
A New Type of Codon Adaptation Mediated by C→T-Biased Mutation
Some ssDNA phages have strong C→T mutations as measured by SKEWTC defined as
| (1) |
where NT and NC are the count of nucleotides T and C, respectively. SKEWTC is expected to increase with increased C→T mutation rate and result in overuse of U-ending codons. For example, Enterobacteria phage Ike (NC_002014, Inoviridae) has a SKEWTC value of 0.2893, with U-ending codons being the most frequent in all Y-ending or N-ending codon families. The effect of biased mutation on codon usage has also been shown for several other ssDNA phages (Cardinale and Duffy 2011). This bias in favor of U-ending codons interferes with codon adaptation because E. coli translation machinery does not favor U-ending codons in most codon families. Highly expressed E. coli genes, as compiled in the EMBOSS distribution (Rice et al. 2000) or in Ran and Higgs (2012), have U-ending codons being the most frequent in four codon families, that is, Gly, Arg4 (the CGN codon subfamily for Arg), Ser4 (the UCN codon subfamily for Ser), and Val. Take the Val (GUN) codon family, for example. The RSCU values for GUA, GUC, GUG, and GUU are 0.8047, 0.4989, 1.0802, and 1.6161, respectively, based on the EMBOSS distribution (Rice et al. 2000). Such a codon family is “U-friendly” because U-ending codons are preferred and C→T-biased mutation will consequently improve translation elongation. In contrast, the other codon families containing U-ending codons have C-ending codons more frequent than U-ending codons based on the highly expressed E. coli protein-coding genes. These codon families will be designated as U-hostile. T-biased mutation in ssDNA phages would enhance codon adaptation in the four U-friendly codon families but would go against codon adaptation in the U-hostile codon families.
What can ssDNA phages do to increase their translation elongation efficiency in face of the C→T mutation? One obvious solution to the problem is illustrated in table 2 with codon frequencies of two codon families (Gly and Phe) from two fictitious phage genes (designated as PG1 and PG2, respectively) and from the host. We can infer U-friendliness of the host translation machinery based on codon usage of host genes. The Gly codon family is U-friendly, with the host machinery strongly preferring U-ending codons. The Phe codon family is U-hostile with host translation machinery strongly favoring C-ending codons (table 2). The total number of codons for the two genes is the same and equal to 400, and the RSCU for each codon is also identical for two genes (table 2). Thus, rRSCU between PG1 and host would be exactly the same as that between PG2 and host. However, we note that the PG2 could be translated more efficiently than PG1 because the former has only 90 “bad” UUU codons, whereas the latter has 180. This differential translation elongation efficiency is not reflected by RSCU but is by CAI. For example, with the data in table 2 and assuming no other codons except for those listed in table 2, we have CAI being 0.2577 for PG1 but 0.3686 for PG2 when host codon frequencies are used as the reference set.
The example illustrated above suggested that E. coli ssDNA phages with strong C→T mutation bias can improve their translation elongation efficiency by overusing the codons in the four U-friendly codon families and decreasing the codons in the U-hostile codon families. This leads to the prediction that the summed frequencies of codons in the four U-friendly codon families, designated as F4, should increase with SKEWTC. That is, when U-ending codons are increased by U-biased mutations, these U-ending codons should be more concentrated in the four U-friendly codon families. This prediction is strongly supported by data from the 11 ssDNA E. coli phages (fig. 2), with the correlation between F4 and SKEWTC3 = 0.707 (P = 0.0151). Furthermore, F4 is significantly and positively correlated with mean CAI from the 11 ssDNA phages (r = 0.6595, P = 0.0273). The result in figure 2 is consistent with the interpretation that increased C→T mutation drives the increased use of codons in the four U-friendly codon families. Thus, although the ssDNA phages cannot fight against the C→T mutation, they have evolved to minimize the disruptive effect of this biased mutation on codon adaptation by coding more amino acids in the four U-friendly codon families.
Fig. 2.
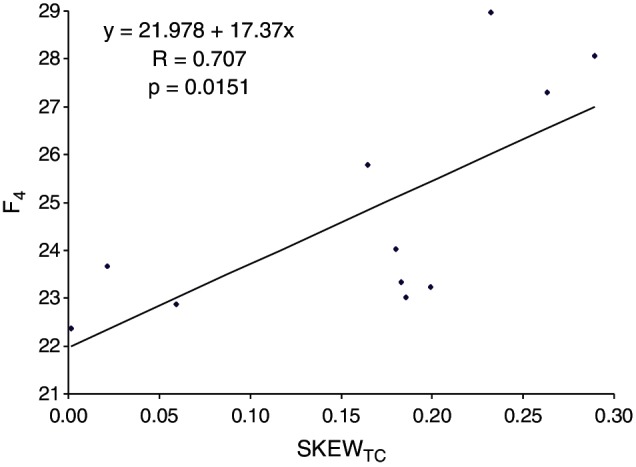
Positive association between SKEWTC, defined as (NT – NC)/(NT + NC) where Ni is the number of nucleotide i in a phage genome, and F4, the percentage of codons in four codon families (Gly, Arg4, Ser4, and Val) in which highly expressed E. coli genes prefer U-ending codons against C-ending codons. Results are from 11 ssDNA E. coli phages. We noted that, because U-rich codons will increase, and C-rich codons decrease, with increasing C→T mutation bias, only Gly codon family should be used for testing the predicted positive correlation, which would lead to r = 0.6837 and P = 0.02036.
The usage of Ser codons for Enterobacteria phage Ike (NC_002014, Inoviridae) illustrates this special codon adaptation well. Ser is coded by the four-codon UCN and the two-codon AGY codon subfamilies. In the AGY codon subfamily, highly expressed E. coli genes prefer AGC against AGU, suggesting that AGU is a “bad” codon. C→T mutations will lead to many “bad” AGU codons if Ser is largely encoded by the AGY subfamily. In contrast, in the UCN subfamily, highly expressed E. coli genes strongly prefer UCU against other synonymous codons, suggesting that UCU is a “good” codon. C→T mutations will lead to many “good” UCU codons if Ser is largely encoded by the UCN subfamily. In this conceptual framework, it is easy to understand that 88.4% of Ser codons in Enterobacteria phage Ike belong to the UCN subfamily. Because of this adaptive trick, the mean CAI value for ssDNA phages is almost as large as that for dsDNA phages (0.4768 for dsDNA phages and 0.4743 for ssDNA phages, excluding the 22 phages with phage-encoded tRNA genes), with no statistically significant difference.
The type of codon adaptation outlined earlier, that is, by switching codon usage from U-hostile codon families to U-friendly codon families, implies increased nonsynonymous substitution with increased C→T mutation. A simple way to check this is to test the change of UUC and CCN frequencies with increased C→T mutation rate. We used TC skew at the third codon position (SKEWTC3) to measure C→T mutation and checked how the frequencies of UUN and CCN codons would change SKEWTC3. The frequency of UUN codons increases (P = 0.0008, fig. 3) and that of CCN codons decreases (P = 0.0320, fig. 3), with increasing SKEWTC3, consistent with the expectation. However, the sharp increase in UUN codons and the relatively slow decrease in CCN codons (fig. 3) suggest that the increase in UUN codon is not entirely due to the decrease of CCN codons. Similar response of nonsynonymous mutation rate to directional mutation pressure has also been documented in several other studies (Sueoka 1961; Lobry 2004; Urbina et al. 2006).
Fig. 3.
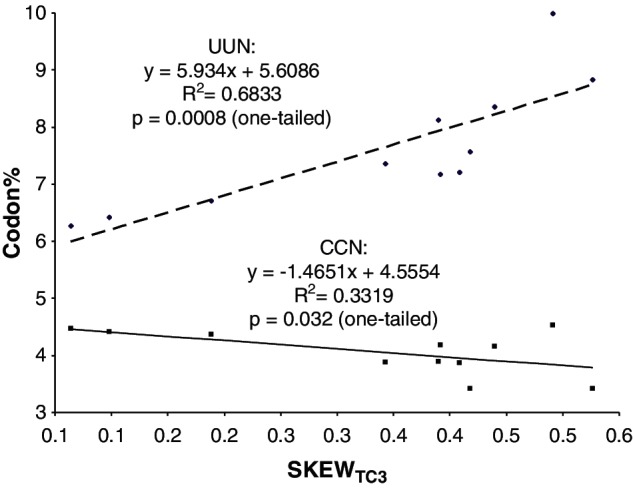
UUN codons increases, and CCN codons decreases, with C→T mutation measured by TC skew at the third codon position (SKEWTC3), but at different extent.
The results above suggest to us that our empirical test of the new type of codon adaptation in figure 2 is incorrect. For example, the Val codon family (coded by GUN) is U-friendly and its usage increases with C→T mutation bias, thus supporting the prediction from the hypothesized new type of codon adaptation. However, the increase may have nothing to do with codon adaptation but may be simply due to the increase of all U-containing codons and the decrease of C-containing codons with increasing C→T mutation bias. Thus, only codon families that do not contain C or U at the first and second codon positions are relevant to test the prediction of a positive association between the usage of U-friendly codon families and SKEWTC3. Among the U-friendly codon families, only the Gly codon family (coded by GGN) fulfills this criterion. The hypothesis is still supported as the percentage of Gly codons increased with SKEWTC3 (r = 0.6837, P = 0.0204).
Discussion
Studying codon adaptation in bacteriophage is important not only in understanding the biology of translation but also in practical applications. Several phages have been used to remove infectious biofilms (Azeredo and Sutherland 2008; Gladstone et al. 2012), to deliver vaccines (Clark and March 2004), or to treat human infections (Sau et al. 2005; Ranjan et al. 2007; Sau 2007; Skurnik et al. 2007; Goodridge 2010; Timms et al. 2010; Abedon et al. 2011), especially those caused by bacterial pathogens that have developed resistance to antibiotics. However, many of these phages do not have optimal codon usage for efficient replication. Studying codon adaptation in phages contributes to the theoretical foundation for re-engineering more efficient phages for therapeutic or industrial purposes (Skiena 2001). A database has been created to facilitate the study of phage codon adaptation to their hosts (Hilterbrand et al. 2012).
Phage-Encoded tRNA Affects Phage Codon Usage
We found that the number of tRNA genes carried by dsDNA phage genomes reduced the need for the phages to evolve a codon usage pattern similar to that of their hosts and that these phage-encoded tRNA facilitate the translation of overused phage codons, especially when the host provides few tRNAs for these phage codons (fig. 1 and table 3). Several viral species have been found to alter host tRNA pool to favor the translation of the viral genes. HIV-1 viruses selectively enrich rare host tRNAs to decode A-ending codons overused in HIV-1 genes but rarely used by host genes (van Weringh et al. 2011), and such selective enrichment has also been found in vaccinia and influenza A viruses (Pavon-Eternod et al. 2013).
Translation efficiency is sensitive to the change of tRNA pool (Kleber-Janke and Becker 2000). A gain/loss of a tRNAMet/UAU gene has resulted significant change in AUA codon frequencies, in both bivalve mitochondria and tunicate mitochondria (Xia et al. 2007; Xia 2012). All these findings on the association of tRNA pool and codon usage suggest that translation efficiency of a target gene can not only be improved by optimizing the codon usage of the target gene but also by modifying the tRNA pool where the target gene is translated. This latter approach has the advantage over the former because the former sometimes will alter the structure of the mRNA leading to reduced translation initiation efficiency (Kudla et al. 2009).
Phage-encoded tRNA genes provide phages with the opportunity to parasitize hosts with different codon usage and may therefore increase their host diversity (Sau et al. 2007). However, existing data do not allow the characterization of phage-encoded tRNA and host diversity because few phage species have their host diversity characterized. One way to characterize host diversity is by subjecting phages to a diverse array of hosts and checking for lytic activities (Villegas et al. 2009). Unfortunately, few such studies have been carried out.
Mutation Plays a Significant Role in Phage Codon Adaptation
The rate of spontaneous deamination leading to C→T mutation is about 100 times higher in ssDNA than in dsDNA (Frederico et al. 1990), and such high mutation rate mediated by oxidative deamination has been reported in a ssDNA phage M13 (Kreutzer and Essigmann 1998). These high C→T mutations prevent ssDNA phages from evolving a codon usage pattern as close to that of the host as dsDNA phages. This is substantiated by the observation that rRSCU for R-ending codons are significantly greater than rRSCU for Y-ending codons in ssDNA phages (table 5).
Although our result is consistent with the mutation hypothesis, the lack of selection for Y-ending codons may also play a role in the poor concordance in RSCU for Y-ending codons between ssDNA phages and E. coli. A previous study (Xia 2008) strongly suggests that tRNAs with a wobble G are equally efficient in decoding C-ending and U-ending codons. This implies that C→T mutations will not be counterchecked by selection, leaving the ratio of U-ending to C-ending codons entirely to the mercy of mutation bias.
A New Type of Codon Adaptation in ssDNA Phage in Response to the C→T Mutation Pressure
The C→T mutation pressure has driven ssDNA phages to evolve a previously unknown type of codon adaptation by biased usage of codon families. That is, they overuse U-friendly codon families in which C→T-biased mutations improve codon adaptation and avoid U-hostile codon families in which the biased mutation hampers codon adaptation (fig. 2). We have illustrated this adaption strategy with the codon usage in the Ser codon family for Enterobacteria phage Ike (NC_002014, Inoviridae) with a strong SKEWTC indicating a strong C→T mutation bias. This simple strategy allows the protein-coding genes in ssDNA phages to have CAI values comparable to those of dsDNA phages.
We have noticed an analogous codon adaptation in the six-codon Leu, Arg, and Ser compound codon families in the yeast, Saccharomyces cerevisiae, in which the number of tRNA genes differ much between the four-codon subfamily and the two-codon subfamily. The yeast genome has 17 tRNALeu genes for the two-codon UUR subfamily but only four tRNALeu genes for the four-codon CUN codon family. The UUR codons account for 84% of Leu codons in highly expressed yeast genes compiled in the EMBOSS distribution (Rice et al. 2000). A similar pattern is observed for the Arg codon family. There are 16 tRNASer genes for the four-codon UCN subfamily and only two for the two-codon AGY codon subfamily. As expected, the UCN codons account for 89% of all Ser codons in highly expressed yeast genes. In short, whenever possible, selection for increased translation efficiency would drive protein-coding genes to maximize the use of codons that have many tRNAs to decode them.
Our study can be advanced in two ways. First, it should take into consideration the role of translation initiation in addition to translation elongation. Genes with poor translation initiation are not expected to increase their protein production with optimized codon usage. It is only genes with efficient translation initiation that are expected to increase protein production with improved codon–anticodon adaptation (Tuller et al. 2010).
Second, the existing phage genomic sequences still do not allow the construction of a sufficiently large phylogeny for phylogeny-based comparisons (Felsenstein 1985; Xia 2013), mainly due to 1) the rapid evolution of phage genomes, especially ssDNA phage genomes, and 2) few homologous genes identifiable among phage species parasitizing E. coli. However, one could argue that, given the rapid evolutionary erosion of coancestry among these phage lineages, the data from different phage lineages may indeed be considered nearly independent. Phages are essentially a mosaic of genes sampled from a pool of frolicking phage genomes. For example, although a number of “related” tailed phages have nearly identical genome organization at function level such as “DNA packaging-head-tail-tail fiber-lysis-lysogeny-DNA replication-transcription regulation” (Desiere et al. 2001), essentially any function in a phage can be fulfilled by one of many distinct genes with “homologous” function but little sequence homology (Brussow and Kutter 2005). In other words, horizontal gene transfer is so rampant that, coupled with rapid evolution, phylogenetic reconstruction based on sequence homology is nearly impossible. For example, a large number of phages have DNA polymerase, but these DNA polymerases apparently belong to a number of nonhomologous classes. Supplementary files S1-S3, Supplementary Material online, list all E. coli phage genes that share functional similarity but not necessarily sequence similarity, so that future researchers can add to it with newly sequenced phage genomes.
The difficulty in building a reliable phage tree also prevents an interesting question to be addressed. The loss/gain of tRNA genes may be related to host tRNA pool. Take AAR (Lys) codon family, for example. If a phage species overusing AAA codons originally parasitizes a host overusing AAG codons and having abundant tRNALys/CUU but rare tRNALys/UUU, then the phage would benefit from retaining a tRNALys/UUU gene decoding its overused AAA codons. If the phage subsequently switched to a host overusing AAA codons and having abundant tRNALys/UUU, then the phage-encoded tRNALys/UUU gene would be of little value and would be prone to gene loss. Addressing such a question would be straightforward if one can build a reliable phage tree, so that the gain/loss of tRNA genes can be mapped onto the tree.
Materials and Methods
Genomic Data and Processing
The genome sequences of 469 dsDNA phages, 41 ssDNA phages, and their corresponding bacterial hosts were downloaded from GenBank, of which 71 have E. coli specified as their host in the “/HOST” tag in “FEATURES” table, including 60 dsDNA phages and 11 ssDNA phages. All phage genomes were searched for encoded tRNAs by using tRNAscan-SE Search Server (Schattner et al. 2005). The complete compilation with phage name, phage family, phage accession, phage genome length, genomic GC%, number of coding sequences (CDSs) in each phage genome, genomic TC skew defined as (NT − NC)/(NT + NC) where NC and NT are the genomic counts of nucleotides C and T, number of tRNA genes encoded in each phage genome, rRSCU, and CAI were included in a supplementary file S1, Supplementary Material online.
Escherichia coli has many strains sequenced, but the “/Host” tag in most annotated viral genomes gives only species name (i.e., E. coli), with no strain-specific information. For this reason, the host GC% and RSCU are computed from the average of all E. coli genomes (The difference among E. coli strains is minimal.). The mean E. coli genome length is 5,024,514 nt, mean number of CDSs is 4,692.2, and mean genomic GC% is 50.68. The genomic accession numbers of all E. coli strains used to compute the average statistics are also included in the supplementary file S1, Supplementary Material online. The classification of phages into temperate and virulent categories is based on three publications (Lima-Mendez et al. 2007; Deschavanne et al. 2010; McNair et al. 2012).
Indices of Codon Adaptation
CDSs and tRNA genes in each phage and host genomes were extracted and RSCU computed by using DAMBE (Xia 2013). rRSCU (correlation between host and phage RSCU values) is taken as a measure of phage codon adaptation to the host translation machinery, with justifications outlined in the Introduction. Single-codon families such as the Met (coded by AUG) and Trp (coded by UGG) were excluded from computing rRSCU because the RSCU value is 1 for the two codons regardless of codon usage. CAI was computed with the improved implementation (Xia 2007) and highly expressed E. coli genes as the reference gene set. Throughout the text, the codon usage of highly expressed E. coli genes refers to the codon usage table compiled and distributed with the EMBOSS package (Rice et al. 2000). The median CAI for protein-coding genes for each phage is used as an alternative measure of phage codon adaptation.
We did not use Nc (Wright 1990; Sun et al. 2013) as a measure of codon adaptation for the following reason. For an E. coli phage, selection by the host tRNA pool is expected to increase rRSCU and CAI. In contrast, mutation, biased or not, will decrease rRSCU and CAI. The effect of mutation and tRNA-mediated selection on Nc is more difficult to distinguish. In general, tRNA-mediated selection will decrease Nc, but biased mutation will also decrease Nc. For this reason, Nc is not good for measuring codon adaptation in E. coli phages.
Supplementary Material
Supplementary files S1–S3 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
This study was supported by the Discovery Grant of Natural Science and Engineering Research Council (NSERC) of Canada to X.X. The authors thank S. Aris-Brosou, N. Corradi, and A. Golshani for comments. Two anonymous reviewers provided excellent comments leading to significantly improvement of the manuscript.
References
- Abedon ST, Kuhl SJ, Blasdel BG, Kutter EM. Phage treatment of human infections. Bacteriophage. 2011;1:66–85. doi: 10.4161/bact.1.2.15845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azeredo J, Sutherland IW. The use of phages for the removal of infectious biofilms. Curr Pharm Biotechnol. 2008;9:261–266. doi: 10.2174/138920108785161604. [DOI] [PubMed] [Google Scholar]
- Bailly-Bechet M, Vergassola M, Rocha E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007;17:1486–1495. doi: 10.1101/gr.6649807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brussow H, Kutter E. Genomics and evolution of tailed phages. In: Kutter E, Sulakvelidze A, editors. Bacteriophages: biology and applications. Boca Raton (FL): CRC Press; 2005. pp. 91–128. [Google Scholar]
- Bulmer M. Coevolution of codon usage and transfer RNA abundance. Nature. 1987;325:728–730. doi: 10.1038/325728a0. [DOI] [PubMed] [Google Scholar]
- Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone A. Codon bias is a major factor explaining phage evolution in translationally biased hosts. J Mol Evol. 2008;66:210–223. doi: 10.1007/s00239-008-9068-6. [DOI] [PubMed] [Google Scholar]
- Cardinale DJ, Duffy S. Single-stranded genomic architecture constrains optimal codon usage. Bacteriophage. 2011;1:219–224. doi: 10.4161/bact.1.4.18496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carullo M, Xia X. An extensive study of mutation and selection on the wobble nucleotide in tRNA anticodons in fungal mitochondrial genomes. J Mol Evol. 2008;66:484–493. doi: 10.1007/s00239-008-9102-8. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay S, Ghosh RK. Characterization of phage-specific transfer RNA molecules coded by Vibrio eltor phage e4. Virology. 1988;165:606–608. doi: 10.1016/0042-6822(88)90606-x. [DOI] [PubMed] [Google Scholar]
- Clark JR, March JB. Bacterial viruses as human vaccines? Exp Rev Vaccines. 2004;3:463–476. doi: 10.1586/14760584.3.4.463. [DOI] [PubMed] [Google Scholar]
- Deschavanne P, DuBow MS, Regeard C. The use of genomic signature distance between bacteriophages and their hosts displays evolutionary relationships and phage growth cycle determination. Virol J. 2010;7:163. doi: 10.1186/1743-422X-7-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desiere F, Mahanivong C, Hillier AJ, Chandry PS, Davidson BE, Brussow H. Comparative genomics of lactococcal phages: insight from the complete genome sequence of Lactococcus lactis phage BK5-T. Virology. 2001;283:240–252. doi: 10.1006/viro.2001.0857. [DOI] [PubMed] [Google Scholar]
- Enav H, Beja O, Mandel-Gutfreund Y. Cyanophage tRNAs may have a role in cross-infectivity of oceanic Prochlorococcus and Synechococcus hosts. ISME J. 2012;6:619–628. doi: 10.1038/ismej.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Phylogenies and the comparative method. Am Nat. 1985;125: 1–15. [Google Scholar]
- Frederico LA, Kunkel TA, Shaw BR. A sensitive genetic assay for the detection of cytosine deamination: determination of rate constants and the activation energy. Biochemistry (Mosc) 1990;29:2532–2537. doi: 10.1021/bi00462a015. [DOI] [PubMed] [Google Scholar]
- Gladstone EG, Molineux IJ, Bull JJ. Evolutionary principles and synthetic biology: avoiding a molecular tragedy of the commons with an engineered phage. J Biol Eng. 2012;6:13. doi: 10.1186/1754-1611-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodridge LD. Designing phage therapeutics. Curr Pharm Biotechnol. 2010;11:15–27. doi: 10.2174/138920110790725348. [DOI] [PubMed] [Google Scholar]
- Gouy M. Codon contexts in enterobacterial and coliphage genes. Mol Biol Evol. 1987;4:426–444. doi: 10.1093/oxfordjournals.molbev.a040450. [DOI] [PubMed] [Google Scholar]
- Gouy M, Gautier C. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982;10: 7055–7064. doi: 10.1093/nar/10.22.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H, Sankoff D, Jou WM, Fiers W, Cedergren RJ. Bacteriophage MS2 RNA: a correlation between the stability of the codon: anticodon interaction and the choice of code words. J Mol Evol. 1978;12:113–119. doi: 10.1007/BF01733262. [DOI] [PubMed] [Google Scholar]
- Haas J, Park E-C, Seed B. Codon usage limitation in the expression of HIV-1 envelope glycoprotein. Curr Biol. 1996;6:315–324. doi: 10.1016/s0960-9822(02)00482-7. [DOI] [PubMed] [Google Scholar]
- Hernan RA, Hui HL, Andracki ME, Noble RW, Sligar SG, Walder JA, Walder RY. Human hemoglobin expression in Escherichia coli: importance of optimal codon usage. Biochemistry (Mosc) 1992;31:8619–8628. doi: 10.1021/bi00151a032. [DOI] [PubMed] [Google Scholar]
- Higgs PG, Ran W. Coevolution of codon usage and tRNA genes leads to alternative stable states of biased codon usage. Mol Biol Evol. 2008;25:2279–2291. doi: 10.1093/molbev/msn173. [DOI] [PubMed] [Google Scholar]
- Hilterbrand A, Saelens J, Putonti C. CBDB: the codon bias database. BMC Bioinformatics. 2012;13:62. doi: 10.1186/1471-2105-13-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J Mol Biol. 1981;146:1–21. doi: 10.1016/0022-2836(81)90363-6. [DOI] [PubMed] [Google Scholar]
- Ikemura T. Correlation between codon usage and tRNA content in microorganisms. In: Hatfield DL, Lee BJ, Pirtle RM, editors. Transfer RNA in protein synthesis. Boca Raton (FL): CRC Press; 1992. pp. 87–111. [Google Scholar]
- Jia W, Higgs PG. Codon usage in mitochondrial genomes: distinguishing context-dependent mutation from translational selection. Mol Biol Evol. 2008;25:339–351. doi: 10.1093/molbev/msm259. [DOI] [PubMed] [Google Scholar]
- Kleber-Janke T, Becker WM. Use of modified BL21(DE3) Escherichia coli cells for high-level expression of recombinant peanut allergens affected by poor codon usage. Protein Expr Purif. 2000;19:419–424. doi: 10.1006/prep.2000.1265. [DOI] [PubMed] [Google Scholar]
- Koresawa Y, Miyagawa S, Ikawa M, Matsunami K, Yamada M, Shirakura R, Okabe M. Synthesis of a new Cre recombinase gene based on optimal codon usage for mammalian systems. J Biochem. 2000;127:367–372. doi: 10.1093/oxfordjournals.jbchem.a022617. [DOI] [PubMed] [Google Scholar]
- Kreutzer DA, Essigmann JM. Oxidized, deaminated cytosines are a source of C → T transitions in vivo. Proc Natl Acad Sci U S A. 1998;95:3578–3582. doi: 10.1073/pnas.95.7.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisawa T. Synonymous codon preferences in bacteriophage T4: a distinctive use of transfer RNAs from T4 and from its host Escherichia coli. J Theor Biol. 1992;159:287–298. doi: 10.1016/s0022-5193(05)80725-8. [DOI] [PubMed] [Google Scholar]
- Kunisawa T. Functional role of mycobacteriophage transfer RNAs. J Theor Biol. 2000;205:167–170. doi: 10.1006/jtbi.2000.2057. [DOI] [PubMed] [Google Scholar]
- Kunisawa T, Kanaya S, Kutter E. Comparison of synonymous codon distribution patterns of bacteriophage and host genomes. DNA Res. 1998;5:319–326. doi: 10.1093/dnares/5.6.319. [DOI] [PubMed] [Google Scholar]
- Lim VI. Analysis of action of wobble nucleoside modifications on codon-anticodon pairing within the ribosome. J Mol Biol. 1994;240:8–19. doi: 10.1006/jmbi.1994.1413. [DOI] [PubMed] [Google Scholar]
- Lima-Mendez G, Toussaint A, Leplae R. Analysis of the phage sequence space: the benefit of structured information. Virology. 2007;365:241–249. doi: 10.1016/j.virol.2007.03.047. [DOI] [PubMed] [Google Scholar]
- Lobry JR. Life history traits and genome structure: aerobiosis and G+C content in bacteria. Lect Notes Comput Sci. 2004;3039:679–686. [Google Scholar]
- Lucks JB, Nelson DR, Kudla GR, Plotkin JB. Genome landscapes and bacteriophage codon usage. PLoS Comput Biol. 2008;4:e1000001. doi: 10.1371/journal.pcbi.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal N, Ghosh RK. Characterization of the phage-specific transfer RNA molecules coded by cholera phage phi 149. Virology. 1988;166:583–585. doi: 10.1016/0042-6822(88)90529-6. [DOI] [PubMed] [Google Scholar]
- McNair K, Bailey BA, Edwards RA. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics. 2012;28:614–618. doi: 10.1093/bioinformatics/bts014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto A, Osawa S. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci U S A. 1987;84:166–169. doi: 10.1073/pnas.84.1.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngumbela KC, Ryan KP, Sivamurthy R, Brockman MA, Gandhi RT, Bhardwaj N, Kavanagh DG. Quantitative effect of suboptimal codon usage on translational efficiency of mRNA encoding HIV-1 gag in intact T cells. PLoS One. 2008;3:e2356. doi: 10.1371/journal.pone.0002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palidwor GA, Perkins TJ, Xia X. A general model of codon bias due to GC mutational bias. PLoS One. 2010;5:e13431. doi: 10.1371/journal.pone.0013431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon-Eternod M, David A, Dittmar K, Berglund P, Pan T, Bennink JR, Yewdell JW. Vaccinia and influenza A viruses select rather than adjust tRNAs to optimize translation. Nucleic Acids Res. 2013;41:1914–1921. doi: 10.1093/nar/gks986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percudani R, Pavesi A, Ottonello S. Transfer RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J Mol Biol. 1997;268:322–330. doi: 10.1006/jmbi.1997.0942. [DOI] [PubMed] [Google Scholar]
- Ran W, Higgs PG. Contributions of speed and accuracy to translational selection in bacteria. PLoS One. 2012;7:e51652. doi: 10.1371/journal.pone.0051652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan A, Vidyarthi AS, Poddar R. Evaluation of codon bias perspectives in phage therapy of Mycobacterium tuberculosis by multivariate analysis. In Silico Biol. 2007;7:423–431. [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Sahu K, Gupta SK, Sau S, Ghosh TC. Comparative analysis of the base composition and codon usages in fourteen mycobacteriophage genomes. J Biomol Struct Dyn. 2005;23:63–71. doi: 10.1080/07391102.2005.10507047. [DOI] [PubMed] [Google Scholar]
- Sau K. Studies on synonymous codon and amino acid usages in Aeromonas hydrophila phage Aeh1: architecture of protein-coding genes and therapeutic implications. J microbiol immunol infect. 2007;40:24–33. [PubMed] [Google Scholar]
- Sau K, Deb A. Temperature influences synonymous codon and amino acid usage biases in the phages infecting extremely thermophilic prokaryotes. In Silico Biol. 2009;9:1–9. [PubMed] [Google Scholar]
- Sau K, Gupta SK, Sau S, Ghosh TC. Synonymous codon usage bias in 16 Staphylococcus aureus phages: implication in phage therapy. Virus Res. 2005;113:123–131. doi: 10.1016/j.virusres.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Sau K, Gupta SK, Sau S, Mandal SC, Ghosh TC. Studies on synonymous codon and amino acid usage biases in the broad-host range bacteriophage KVP40. J Microbiol. 2007;45:58–63. [PubMed] [Google Scholar]
- Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33:W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH. The Codon Adaptation Index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Rogers MS, McConnell DJ. Selection pressures on codon usage in the complete genome of bacteriophage T7. J Mol Evol. 1984;21:150–160. doi: 10.1007/BF02100089. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Tuohy TM, Mosurski KR. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986;14:5125–5143. doi: 10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skiena SS. Designing better phages. Bioinformatics. 2001;17(Suppl 1):S253–S261. doi: 10.1093/bioinformatics/17.suppl_1.s253. [DOI] [PubMed] [Google Scholar]
- Skurnik M, Pajunen M, Kiljunen S. Biotechnological challenges of phage therapy. Biotechnol Lett. 2007;29:995–1003. doi: 10.1007/s10529-007-9346-1. [DOI] [PubMed] [Google Scholar]
- Sueoka N. Correlation bewteen base composition of deoxyribonucleic acid and amino acid composition of proteins. Proc Natl Acad Sci U S A. 1961;47:1141–1149. doi: 10.1073/pnas.47.8.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XY, Yang Q, Xia X. An improved implementation of effective number of codons (Nc) Mol Biol Evol. 2013;30:191–196. doi: 10.1093/molbev/mss201. [DOI] [PubMed] [Google Scholar]
- Timms AR, Cambray-Young J, Scott AE, Petty NK, Connerton PL, Clarke L, Seeger K, Quail M, Cummings N, Maskell DJ, et al. Evidence for a lineage of virulent bacteriophages that target Campylobacter. BMC Genomics. 2010;11:214. doi: 10.1186/1471-2164-11-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Waldman YY, Kupiec M, Ruppin E. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci U S A. 2010;107:3645–3650. doi: 10.1073/pnas.0909910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina D, Tang B, Higgs PG. The response of amino acid frequencies to directional mutation pressure in mitochondrial genome sequences is related to the physical properties of the amino acids and to the structure of the genetic code. J Mol Evol. 2006;62:340–361. doi: 10.1007/s00239-005-0051-1. [DOI] [PubMed] [Google Scholar]
- van Vliet F, Couturier M, Desmet L, Faelen M, Toussaint A. Virulent mutants of temperate phage Mu-1. Mol Gen Genet. 1978;160:195–202. [Google Scholar]
- van Weringh A, Ragonnet-Cronin M, Pranckeviciene E, Pavon-Eternod M, Kleiman L, Xia X. HIV-1 modulates the tRNA pool to improve translation efficiency. Mol Biol Evol. 2011;28:1827–1834. doi: 10.1093/molbev/msr005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas A, She YM, Kropinski AM, Lingohr EJ, Mazzocco A, Ojha S, Waddell TE, Ackermann H-W, Moyles DM, Ahmed R, et al. The genome and proteome of a virulent Escherichia coli O157:H7 bacteriophage closely resembling Salmonella phage felix O1. Virol J. 2009;6:41. doi: 10.1186/1743-422X-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. The “effective number of codons” used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Xia X. Maximizing transcription efficiency causes codon usage bias. Genetics. 1996;144:1309–1320. doi: 10.1093/genetics/144.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. How optimized is the translational machinery in Escherichia coli, Salmonella typhimurium and Saccharomyces cerevisiae? Genetics. 1998;149:37–44. doi: 10.1093/genetics/149.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. Mutation and selection on the anticodon of tRNA genes in vertebrate mitochondrial genomes. Gene. 2005;345:13–20. doi: 10.1016/j.gene.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Xia X. An improved implementation of Codon Adaptation Index. Evol Bioinformatics. 2007;3:53–58. [PMC free article] [PubMed] [Google Scholar]
- Xia X. The cost of wobble translation in fungal mitochondrial genomes: integration of two traditional hypotheses. BMC Evol Biol. 2008;8:211. doi: 10.1186/1471-2148-8-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. Rapid evolution of animal mitochondria. In: Singh RS, Xu J, Kulathinal RJ, editors. Evolution in the fast lane: rapidly evolving genes and genetic systems. Oxford: Oxford University Press; 2012. pp. 73–82. [Google Scholar]
- Xia X. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol. 2013;30:1720–1728. doi: 10.1093/molbev/mst064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Huang H, Carullo M, Betran E, Moriyama EN. Conflict between translation initiation and elongation in vertebrate mitochondrial genomes. PLoS One. 2007;2:e227. doi: 10.1371/journal.pone.0000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Yuen KY. Differential selection and mutation between dsDNA and ssDNA phages shape the evolution of their genomic AT percentage. BMC Genet. 2005;6:20. doi: 10.1186/1471-2156-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.