

Abstract
Macaques are the most widely distributed nonhuman primates and used as animal models in biomedical research. The availability of full-genome sequences from them would be essential to both biomedical and primate evolutionary studies. Previous studies have reported whole-genome sequences from rhesus macaque (Macaca mulatta) and cynomolgus macaque (M. fascicularis, CE), both of which belong to the fascicularis group. Here, we present a 37-fold coverage genome sequence of the Tibetan macaque (M. thibetana; TM). TM is an endemic species to China belonging to the sinica group. On the basis of mapping to the rhesus macaque genome, we identified approximately 11.9 million single-nucleotide variants), of which 3.9 million were TM specific, as assessed by comparison two Chinese rhesus macaques (CR) and two CE genomes. Some genes carried TM-specific homozygous nonsynonymous variants (TSHNVs), which were scored as deleterious in human by both PolyPhen-2 and SIFT (Sorting Tolerant From Intolerant) and were enriched in the eye disease genes. In total, 273 immune response and disease-related genes carried at least one TSHNV. The heterozygosity rates of two CRs (0.002617 and 0.002612) and two CEs (0.003004 and 0.003179) were approximately three times higher than that of TM (0.000898). Polymerase chain reaction resequencing of 18 TM individuals showed that 29 TSHNVs exhibited high allele frequencies, thus confirming their low heterozygosity. Genome-wide genetic divergence analysis demonstrated that TM was more closely related to CR than to CE. We further detected unusual low divergence regions between TM and CR. In addition, after applying statistical criteria to detect putative introgression regions (PIRs) in the TM genome, up to 239,620 kb PIRs (8.84% of the genome) were identified. Given that TM and CR have overlapping geographical distributions, had the same refuge during the Middle Pleistocene, and show similar mating behaviors, it is highly likely that there was an ancient introgression event between them. Moreover, demographic inferences revealed that TM exhibited a similar demographic history as other macaques until 0.5 Ma, but then it maintained a lower effective population size until present time. Our study has provided new insight into the macaque evolutionary history, confirming hybridization events between macaque species groups based on genome-wide data.
Keywords: Tibetan macaque, whole-genome sequencing, SNVs, genetic divergence, introgression, demographic trajectories
Introduction
The Old World monkey genus Macaca is the most widespread nonhuman primates, having diversified 5–5.5 Ma, and subsequently spread throughout Asia (Delson et al. 2000). Macaques have long been used as important animal models in biomedical research because they are closely related to humans, sharing a last common ancestor about 25 Ma (Kumar and Hedges 1998). The genus contains about 19–22 extant species belonging to four well-defined Asian macaque groups (sylvanus, silenus, sinica, and fascicularis), including a single African species (Macaca sylvanus; Fooden 1976; Tosi et al. 2003; Ziegler et al. 2007; Li et al. 2009; Perelman et al. 2011). The most widely used macaque in biomedical research is the rhesus macaque (M. mulatta). However, a ban on the export of rhesus macaques by the Indian government has increased the requirement of other macaque species (subspecies) for research, such as the Chinese rhesus macaque (CR, M. mulatta lasiota), cynomolgus macaque (CE, M. fascicularis), Japanese macaque (M. fuscata), the bonnet macaque (M. radiata), and the Tibetan macaque (TM, M. thibetana). Because of their great significance as animal models in human evolutionary and biomedical research, whole-genome sequences of rhesus macaque and CE were released in 2007, 2011, and 2012 (Rhesus Macaque Genome Sequencing and Analysis Consortium 2007; Fang et al. 2011; Yan et al. 2011; Higashino et al. 2012). However, the sequenced genomes are within the fascicularis group, genome information of macaques from the sinica group is still unavailable to this date.
The TM (M. thibetana), a sinica group species endemic to China, differs substantially from fascicularis species in their geographical range, body size, and a variety of morphological, physiological, and behavioral characteristics. Compared with rhesus macaque, TM has bigger body size, longer life span, calmer temperament, and is easier to train (Groves et al. 2005). TM has already been used in biomedical researches, such as in intraocular pressure (IOP) research (Liu et al. 2011) and liver transplantation studies. Although all macaques share a common ancestor up to 5 Ma, the sinica and fascicularis groups diverged from each other around 3 Ma (Tosi et al. 2003; Perelman et al. 2011). It is already known that different species (subspecies) of macaque react differently and show different levels of pathogenesis to human infectious diseases such as AIDS (Ling et al. 2002) and malaria (Schmidt et al. 1977). Therefore, it is highly desirable to fully assess the genetic backgrounds of the different macaque species.
In addition, the relationships between different macaque species are still controversial due to the existence of historic and ongoing episodes of hybridization (Perelman et al. 2011), which occurred particularly intensively among macaques of Asian origin. For example, hybridization has been detected in the pig-tailed macaques (Tosi et al. 2003), various Sulawesi macaques (Evans et al. 2003), and between rhesus macaque and CE (Yan et al. 2011). Furthermore, some studies suggested that there were ancient hybridizations between two lineages (sinica and fascicularis) (Tosi et al. 2002, 2003; Li et al. 2009). However, there is no report inferring hybridization in TM. The whole-genome sequencing of TM would let us examine whether there were any admixture events between TM and other macaques.
The released whole-genome sequences of the Indian rhesus macaque (IR) provided a great reference for the resequencing strategy of other macaque species (Fang et al. 2011; Higashino et al. 2012). In this study, we generated a TM genome sequence with approximately 37-fold coverage using the Illumina Hiseq 2000 system based on resequencing strategy. The sequences were then mapped to the IR genome (rheMac2) to generate genotype calls. Combined with two previously sequenced CRs (CR1 from Yunnan Province, CR2 from Sichuan Province) and two previously sequenced CEs (CE1 from Vietnam and CE2 from Malaysia) genomes, we further identified TM-specific variants. Notably, we identified specific variants within important human immune response and diseases genes. In addition, we explored the genome-wide genetic divergence and the evolutionary history of these macaque species.
Results and Discussion
Genome Sequencing and Variant Discovery
A wild female TM (12–15 years old) captured at Mabian County in Sichuan province, China, was used for whole-genome sequencing. DNA was extracted from its blood, and whole-genome sequencing was performed with the Illumina HiSeq 2000 platform. More than 1,275 million clean reads were generated, which covered approximately 114.75 Gbp. The clean reads have been deposited in the National Center for Biotechnology Information (NCBI) Short Read Archive under accession number SRP032525. In total, about 1,209 million reads could be mapped to the IR reference genome (rheMac2), and more than 900 million reads (72.8%) were unique concordantly mapped (supplementary file S1, table S1, Supplementary Material online). The effective genome-wide mean coverage for TM was 36.92-fold. The alignment files of TM and other macaques were processed with our customized genotyping pipeline to get the genotype calls of five macaques (fig. 1A). Then, we applied conservative data quality filters to further control the quality of genotype calls (see supplementary note, Supplementary Material online, for details). After imposing genome and sample filters (SFs), we could genotype 91.09% of the genome (20 autosomes). A total of 11,937,445 single-nucleotide variants (SNVs) were identified in TM compared with the reference. The transitions/transversions (Ti/Tv) ratio in TM (Ti/Tv = 2.17; supplementary file S1, table S2, Supplementary Material online) was in accordance to the criteria of approximately 2–2.2 in the 1000 Genomes and other genome-wide sequencing studies (DePristo et al. 2011; Lachance et al. 2012). Among all SNVs, 82.84% of them (9,889,106) were homozygous in TM (table 1). To further quantify genome-wide heterozygosity, we calculated the ratio of heterozygous SNVs against all passed filters sites, which yielded an autosomal heterozygosity of only 0.000898 in TM (table 1).
Fig. 1.

Genotyping pipeline and mean genome wide coverage. (A) Overview of the sequence alignment and customized genotyping pipeline in our study. Details of the major steps and postgenotype filters can be found in supplementary file S4, Supplementary Material online. (B) Proportion of the covered genome per sample as a function of minimum depth of coverage. The numbers in legend are the mean genome wide coverage.
Table 1.
Numbers of Useable Sites, Autosomal Heterozygosity, and SNV Rate in Five Macaques.
| Sample | Nonvariant Sites | SNVs |
Total Useable Sites | Heterozygosity | SNV Rate | ||
|---|---|---|---|---|---|---|---|
| Heterozygous | Homozygous | Total | |||||
| CR1 | 2,254,758,652 | 5,925,877 | 3,458,482 | 9,384,359 | 2,264,143,011 | 0.002617 | 0.004145 |
| CR2 | 1,631,118,438 | 4,277,233 | 1,974,865 | 6,252,098 | 1,637,370,536 | 0.002612 | 0.003818 |
| CE1 | 2,233,731,233 | 6,746,357 | 5,004,945 | 11,751,302 | 2,245,482,535 | 0.003004 | 0.005233 |
| CE2 | 2,249,104,923 | 7,188,355 | 4,812,493 | 12,000,848 | 2,261,105,771 | 0.003179 | 0.005308 |
| TM | 2,269,701,317 | 2,048,339 | 9,889,106 | 11,937,445 | 2,281,638,762 | 0.000898 | 0.005232 |
Small Indels in TM
Using the mapping information within the alignments, we also genotyped small indels in TM with the Genome Analysis Toolkit (GATK). The threshold for the minimum base quality was 20. In total, we detected 1,125,876 deletions and 1,032,913 insertions on the 20 autosomes (table 2). We further identified indels within genes and their respective locations. There were 367,317 genic deletions (32.62%) and 336,218 genic insertions (32.55%), as well as 5,268 untranslated region (UTR) deletions and 4,894 UTR insertions (table 2). Nine hundred and sixty-two deletions and 875 insertions were found within exons. Among these exon indels, 623 deletions and 613 insertions could cause frame shifting (non-3x-bp length). In addition, more than 60% of them were homozygous (deletions: 424; 68% and insertions: 481; 78%). Future studies will be needed to investigate the effect of these potential frame shifts. The proportion of the 3x-bp-length indels within the coding regions (32.7%) was much higher than that within the noncoding regions (13.4%), indicating that there probably is purifying selection on frame-shifting indels within the coding regions. A similar pattern has been observed in the genome of CE2 (Higashino et al. 2012). The distribution of the small indel lengths is shown in figure 2. More than 50% of them correspond to single base pairs (deletions: 50.3%; insertions: 54.4%).
Table 2.
Numbers of Small Indels (Deletions and Insertions) in TM.
| Type | Numbers |
Cause Frame Shifting | |||
|---|---|---|---|---|---|
| Total | Within Gene | Within Exon | Within UTR | ||
| Deletions | 1,125,876 | 367,317 | 962 | 5,268 | 623 |
| Insertions | 1,032,913 | 336,218 | 875 | 4,894 | 613 |
Note.—Numbers of total indels and numbers of indels within genes/exon/UTR of genes are shown. “Cause frame shifting” indels mean the indels within the exon of genes could cause frame shifting because they are not 3x-bp length.
Fig. 2.

Distribution of small indel lengths detected in the TM genome. (A) Total numbers of indels and the numbers of genic indels. (B) Numbers of exon indels.
SNVs Distribution in TM
Based on gene annotations from the reference genome, a total of 3,833,049 SNVs of TM were located in TM genic regions, including 63,626 SNVs within exons (table 3). We examined the exons of each gene with variants by translating the respective codons using the standard genetic code and found 52,987 synonymous variants and 33,612 nonsynonymous variants (supplementary file S1, table S3, Supplementary Material online). We next searched for immune- and drug-response genes in humans that carried nonsynonymous SNVs in TM because these genes are of particular interest in biomedical research (Fang et al. 2011; Yan et al. 2011; Higashino et al. 2012). In addition, considering the big body size of TM and that TM is more prone to develop obesity and high blood sugar levels than other macaques, we examined variants in glucose metabolic process genes and insulin-related genes. In total, we searched six gene ontology (GO) terms. We found that there were 1,224 nonsynonymous variants in immune response genes and 270 in drug response genes. There were 136 nonsynonymous variants located in glucose metabolic process genes, as well as more than 200 in three insulin-related GO terms (supplementary file S1, table S3, Supplementary Material online). The number of genes exhibiting at least one homozygous nonsynonymous variant are listed in supplementary table S3, Supplementary Material online, including 347 genes related to immune response, 78 genes involved in drug response, and 41 genes related to glucose metabolic process, as well as 37 genes related to insulin secretion, 35 genes associated with the insulin receptor signaling pathway, and 5 genes related to insulin receptor binding.
Table 3.
SNV Distributions in TM.
| Total | Homozygous | Heterozygous | |
|---|---|---|---|
| Total SNVs | 11,937,445 (3,936,546) | 9,889,106 (2,861,190) | 2,048,339 (1,075,356) |
| Gene region | 3,833,049 (1,332,527) | 3,228,837 (999,602) | 604,212 (332,925) |
| Exon | 63,626 (26,205) | 52,992 (19,784) | 10,634 (6,421) |
| Intron | 3,262,692 (1,130,354) | 2,750,155 (847,520) | 512,537 (282,834) |
| UTR | 50,118 (18,892) | 41,776 (14,454) | 8,342 (4,438) |
| Noncoding | 456,613 (157,076) | 383,914 (117,844) | 72,699 (39,232) |
Note.—In parentheses are the numbers of SNVs that are specific to the TM when compared with other macaque individuals.
Genetic Diversity among Macaques
To determine the relationship between TM and other macaque species, we applied the same pipeline to process other four available macaque genomes (CR1, CR2, CE1, and CE2). We obtained high coverage (>20-fold) for three of the genomes (CR1, CE1, and CE2) and lower coverage (10.52-fold) for the CR2 genome (fig. 1B), and all exhibited approximately 90% useable sites of the genome (supplementary file S1, table S4, Supplementary Material online). Notably, more than half of the SNVs in CRs and CEs were heterozygous (CR1: 63.15%, CR2: 68.41%; CE1: 57.41%, CE2: 59.90%), and their autosomal heterozygosity rates were approximately three times higher compared with that of TM (table 1). Hence, these large differences in SNV types and heterozygosity rates between TM and the fascicularis macaques revealed important differences in their genetic background.
By combining with previously reported macaque genomes from one reference genome (IR), two CRs, and two CEs, we explored the genetic divergence between TM and other macaques at the genome-wide level. First, we used a 50 kb nonoverlapping windows to scan each chromosome, suing only homozygous sites within each individual to estimate the divergence between macaque species (Yan et al. 2011). As expected, the divergences between TM and the fascicularis macaques were higher than those within the fascicularis group. The divergence between TM and IR was the highest (whole-genome average: 0.384%), whereas the divergences between TM and CRs (TM/CR1: 0.300%; TM/CR2: 0.301%) were very similar to those between TM and CEs (TM/CE1: 0.289%; TM/CE2: 0.280%), but the TM was slightly closer to the CEs (fig. 3A). With respect to the divergence within the fascicularis group (supplementary file S1, table S5, Supplementary Material online), the divergences between two CRs and two CEs were the smallest (CR1/CR2: 0.042%; CE1/CE2: 0.086%), whereas the divergences between IR and CEs were much higher (IR/CE1: 0.179%; IR/CE2: 0.191%). We found lower divergences between IR and CRs (IR/CR1: 0.115%; IR/CR2: 0.105%), which is expected because IR and CR correspond to two subspecies of rhesus macaque, and these results are also consistent with a previous study (Yan et al. 2011). In addition, we detected a similar pattern confirming these results by repeating the calculation based on a 100 kb window size (supplementary file S2, fig. S1, Supplementary Material online).
Fig. 3.

Single-nucleotide divergence and pairwise differences. (A) Single-nucleotide divergence between TM and other macaques in 50 kb nonoverlapping windows across the 20 autosomes. The heterozygous variants were not used in this test. (B) Pairwise differences between and within macaques estimated in 50 kb nonoverlapping windows across the genome. We calculated the genetic distance by using the genetic distance metric from Gronau et al. (2011). (C) The divergence between TM and CRs (TM/CRs) and compared it with different control sets in 50 kb nonoverlapping windows. Divergences within two CRs and between CRs and CEs were used as control sets. Numbers in parentheses are the percentages of windows, which TM/CRs minus control sets is smaller than zero. The divergence ratio is smaller than zero means the divergence between TM and CRs is lower than that of control sets.
However, more than half of the variants observed in two CRs and two CEs were heterozygous (table 1), thus only using homozygous variants could potentially introduce bias, because all the information from heterozygous variants was ignored. Therefore, all the variants were employed to calculate the pairwise genetic distances by using the genetic distance metric from Gronau et al. (2011). We performed pairwise comparisons between and within macaques across the genome with 50 kb (fig. 3B) and 100 kb (supplementary file S2, fig. S2, Supplementary Material online) nonoverlapping window sizes. Of note, the two different window sizes yielded similar results (supplementary file S2, fig. S2, Supplementary Material online). Consistent with the results in supplementary file S1, table S5, Supplementary Material online, the level of diversity within CR was the lowest, whereas the genetic diversity within CE was higher. With respect to pairwise differences between TM and other macaques, such as the pattern shown in figure 3A, TM exhibited similar pairwise genomic divergence from CRs and CEs. Interestingly, in contrast to the relationship detected with only homozygous variants (fig. 3A), most genomic regions exhibited lower divergence between TM and CRs than between TM and CEs, indicating that TM was more closely related to CRs than to CEs (fig. 3B).
We further examined how many genomic regions (50 kb and 100 kb) support that TM was more closely related to CR than to CE. We calculated the divergence ratio between TM and CRs and compared it with the divergence ratio between TM and CEs (TM/CR – TM/CE). In the 50 kb window size analyses, up to 60.75% (56.59–60.75%) of the genomic windows displayed a lower divergence between TM and CRs, in comparison to that between TM and CEs (supplementary file S2, fig. S3, Supplementary Material online), thus suggesting a closer relationship between TM and CRs than between TM and CEs. Furthermore, when the window size was increased to 100 kb, slightly more regions (59.64–65.23%) supported these findings (supplementary file S2, fig. S3, Supplementary Material online).
Introgression between TM and CR
Given that the widely occurring introgression among different macaque species (Tosi et al. 2002; Kanthaswamy et al. 2008), we further tested whether there was admixture between TM and CR, even though no previous studies have reported admixture between TM and other macaques. We calculated the divergence between TM and CRs and compared it with different control sets by using pairwise differences metrics, as previously described. Given that TM and CR correspond to different species group, both the value of TM/CRs minus CR1/CR2 (within species) and the value of TM/CRs minus CRs/CEs (within species group) was expected to be positive at all or at most windows. Surprisingly, some proportion of windows supported that the divergence level of TM/CRs was lower than the control sets (fig. 3C). Indeed, comparison of the divergence within species group (the divergence of CRs/CEs) using a 50 kb window size analysis revealed that 5.26% (TM/CR1 – CR2/CE1) to 9.95% (TM/CR1 – CR1/CE2) of windows were negative (fig. 3C). These results suggest a lower divergence between TM and CRs than that within fascicularis group. Even when compared with the divergence within species (the divergence of CR1/CR2), up to 0.48% of windows supported a lower divergence for TM/CRs (fig. 3C). Of note, the results were subject to some variation that was dependent to whether CE1/CR or CE2/CR was used as a control set. This is likely because CE1 and CE2 were obtained from different populations (see Inference of demography) and possibly due to admixture between CE1 and CRs (Yan et al. 2011). Similarly, the 100 kb window sizes analyses yielded similar results, although with a less proportion of windows showing lower divergence between TM and CRs than that of control sets (0.19–5.47%; supplementary file S2, fig. S4, Supplementary Material online).
The unusual low divergence regions detected in our results could be the result of either hybridization between TM and CR or persistence of ancestral polymorphisms. Therefore, we applied statistical criteria to detect putative introgression regions (PIRs) in the TM genome, as described previously (Yan et al. 2011). Six comparisons with different control sets were performed with a series of window sizes ranging from 10 to 1,000 kb. Additionally, because there were two CRs in our data set, we further checked whether the same possible PIRs could be detected in both CRs. Before applying statistical criteria, the results showed that more possible PIRs could be found in smaller window sizes, with a peak value at 10 kb window size. The maximum number of possible PIRs calculated was 557,890 kb, and 437,000 kb of these could be found in both CRs (supplementary file S1, tables S6 and S7, Supplementary Material online).
To determine cut-off values of Rdiff for PIRs described earlier, we performed coalescent simulations adopting the demographic parameters of three macaque populations (CE1, CR1, and IR) estimated by Yan et al. (2011) using ∂a∂i software (Gutenkunst et al. 2009). Then, we calculated Rdiff for each window size in the simulated data. Because it was clear that fewer possible PIRs could be found in large window sizes, we only calculated the Rdiff from 10 to 100 kb window sizes in the simulated data. The 1% and 5% quantiles of Rdiff in the simulated data were used as cutoffs (Rcutoff). These two different cutoffs from the simulated data were applied to the real data. An Rdiff smaller than Rcutoff and 0 indicates this region is a PIR in TM genome. Using a stringent P value (0.01), we identified roughly 35,000 kb PIRs in the TM genome, which comprised approximately 1.3% of the genome (table 4). These results were consistent between different window sizes and showed a peak at 20 kb window size. However, more PIRs could be detected with a less stringent P value of 0.05 (table 4). Indeed, up to 239,620 kb PIRs (8.84% of the genome) were found in the 20 kb window size (fig. 4). The 20 kb window could find more PIRs than that within the 10 kb window (215,640 kb; 7.95% of the genome) was likely due to smaller power within the 10 kb window. The sizes of PIRs decreased markedly in larger window sizes (40–100 kb), suggesting that the introgression between TM and CR was an ancient event, because the length of the PIRs could reflect the time at which the gene flow occurred (Pool and Nielsen 2009). Previous studies based on different markers (mtDNA, cDNA, and X chromosome) suggested that the ranges of divergence time between cynomolgus and rhesus macaques were from 0.5 to 2.0 Ma (Hayasaka et al. 1996; Blancher et al. 2008; Osada et al. 2008, 2013). The introgression events should have occurred after the divergence of cynomolgus and rhesus macaques because this criterion was used to identify PIRs. However, we noticed that there were more mutations between TM and the other macaques than that between CE1 and CR1, which may also increase the statistical power for small window sizes.
Table 4.
Size and Proportion of the PIRs in TM Genome with Different P Values under Different Window Sizes.
| P | Terms | Different Window Sizes (kb) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 10 | 20 | 30 | 40 | 50 | 60 | 80 | 100 | ||
| 0.01 | Cutoff Rdiff | −0.5 | −0.31 | −0.233 | −0.172 | −0.135 | −0.115 | −0.069 | −0.043 |
| Sizes (kb) | 35,880 | 36,660 | 34,020 | 35,400 | 36,450 | 33,360 | 34,320 | 36,600 | |
| Proportion of the genome (%) | 1.324 | 1.353 | 1.255 | 1.306 | 1.345 | 1.231 | 1.267 | 1.351 | |
| 0.05 | Cutoff Rdiff | −0.131 | −0.032 | 0.0125 | 0.045 | 0.062 | 0.076 | 0.104 | 0.123 |
| Sizes (kb) | 215,640 | 239,620 | 218,100 | 171,000 | 141,200 | 99,300 | 82,720 | 63,300 | |
| Proportion of the genome (%) | 7.958 | 8.843 | 8.049 | 6.311 | 5.211 | 3.665 | 3.053 | 2.336 | |
Note.—The 1% and 5% quantiles of Rdiff in the simulated data are used as cutoff Rdiff, which are equal to P value 0.01 and 0.05 when applied them to the real data. The control set in the real data and the simulated data is the divergence between CR1 and CE1.
Fig. 4.

Distribution of PIRs in the TM genome. The red bar represents the total sizes of PIRs in each chromosome, and the blue plot represents the proportion of the PIRs in each chromosome (based on the full size of the reference genome). The last column “all” means the sum of the 20 autosomes. (A) With the P value = 0.01 as cutoff. (B) With the P value = 0.05 as cutoff.
Historic and ongoing episodes of hybridization in macaques have been reported to occur mostly within one species group (Perelman et al. 2011), such as between various Sulawesi macaques (Evans et al. 2003), the recently described Arunachal macaque (Chakraborty et al. 2007), and rhesus and CEs (Bonhomme et al. 2009; Yan et al. 2011). Furthermore, hybridization has been inferred among macaques from different groups. A previous study suggested that extensive hybridization between early sinica and fascicularis group members in a Pleistocene forest refugium gave rise to the species of M. arctoides (Tosi et al. 2003). This hypothesis was further supported by a study based on Alu elements data in which the results suggested that M. arctoides was the result of male-mediated introgression from TM to M. fascicularis (Li et al. 2009). Although current hybridization is rare among macaque groups, the isolated forest refugium associated with the Pleistocene glacial periods may have provided rich opportunities for interspecific mating. In the case of TM and CR, their geographical distribution areas overlap in southwestern China (supplementary file S2, fig. S5, Supplementary Material online) providing an opportunity for hybridization. Additionally, during the Middle Pleistocene, TM and CR shared the same refuge in southwestern China (Sichuan and Yunnan Provinces) (supplementary file S2, fig. S5, Supplementary Material online; Eudey 1979; Delson 1980; Fa 1989). Moreover, there are many similarities regarding the mating behavior. For example, both of them are multiple mount-to-ejaculation species, and their reproductive seasons overlap (Zhao 1994; Berard 1999). Therefore, ancient introgression between TM and CR is highly likely and could explain the observed extraordinarily low divergence regions and PIRs found in our study.
Variants Specific in TM
To identify the variants specific at sampled macaques in this study, we created a merged SNV data set by combining the SNVs of all the macaques. In total, there were 31,868,376 informative SNVs among all the macaques. After dispelling any missing data in the samples, there were still 20,035,210 SNVs in the merged SNV data set. By comparison with other five macaques (including the reference genome), 3.9 million TM-specific SNVs were identified, including more than 1.3 million located within genic regions and 26,205 in exons (table 3). The amount of TM-specific SNVs accounted for about one-third of the total SNVs found in TM, which were far more than the CRs- and CEs-specific SNVs (CR1: 2,038,702; CR2: 2,005,637; CE1: 2,876,161; CE2: 3,452,073; supplementary file S1, table S8, Supplementary Material online). These results are consistent with the phylogenetic evolutionary position of TM, which exhibits a deeper divergence from the reference genome when compared with CR and CE.
We identified 27,348 homozygous nonsynonymous SNVs in the TM genome, compared with the reference genome. Among these SNVs, 10,106 were TM-specific when compared with other four macaques (supplementary file S1, table S3, Supplementary Material online). We then investigated how many TM-specific SNVs were within the six GO terms. We found 397 TM-specific homozygous nonsynonymous variants (TSHNV) related to immune response and 97 involved in drug response genes. Dozens of TSHNVs were also identified in the other four GO terms. Importantly, 203 immune response genes, 48 drug response genes, 17 glucose metabolic process genes, 21 insulin secretion genes, two insulin receptor binding genes, and 21 insulin receptor signaling pathway genes carried at least one TSHNV (supplementary file S1, table S3, Supplementary Material online). In total, 86 genes within the six GO terms carried more than one TSHNV (supplementary file S3, table S9, Supplementary Material online). Some specific genes of particular importance for human disease conditions, such as autoimmune regulator (AIRE) and Mediterranean fever gene, are discussed later.
We examined the effect of 10,106 TSHNVs on protein structure and functionality by PolyPhen-2 prediction tests. PolyPhen-2 predicts the effects of missense mutations by comparing the properties of the ancestral allele with those of the derived allele according to sequence and structural features of the protein. The results showed that 2,235 sites scored as probably or possibly damaging for humans. In total, 1,496 different genes carried at least one of the probably or possibly damaging TSHNV. Thus, we performed functional enrichment analyses to determine which pathways or functions were enriched by the genes containing predicted deleterious TSHNVs. Within enriched categories, we observed numerous human disease classes, including diabetes mellitus, eye problems, and skin problems (supplementary file S1, table S10, Supplementary Material online). Indeed, we found a significant enrichment in genes related to diabetes mellitus (HP:0000819), with 14 genes in this category: AIRE, ALMS1, ATM, BLM, CNGB1, LEMD3, LOC703862, LOC718993, LOC719052, NOP10, PPP1R3A, PROM1, RP1, and ZMPSTE24. In addition, child levels of diabetes mellitus were also significantly enriched with 9 of the 14 genes above. Moreover, a group in China recently reported a significant positive correlation between TM weight/age and blood sugar level and also found significantly higher blood sugar levels in TM than that in rhesus macaques (Yang et al. 2010). Eye problems were also prevalently observed, such as glaucoma (HP:0000501), pigmentary retinopathy (HP:0000580), abnormality of the choroid (HP:0000610), inflammatory abnormality of the eye (HP:0100533), chorioretinal abnormality (HP:0000532), and abnormality of the vasculature of the eye (HP:0008047).
Additionally, we used Sorting Tolerant From Intolerant (SIFT) to further estimate the functional changes of the 2,235 TSHNVs that were estimated as deleterious by PolyPhen-2. SIFT calculates normalized probabilities for all possible substitutions in the alignment and predicts that positions with normalized probabilities smaller than 0.05 are “deleterious." Additionally, the program outputs the median conservation of the proteins used in the alignment, which is recommended to be between 2.75 and 3.5. When the value is more than 3.25, the prediction is made based on highly similar sequences and is therefore not as reliable (Kumar et al. 2009). We found 707 TSHNVs with deleterious scores, which were located in 556 different genes. Enrichment analyses found less significant categories than data set above, but some overlaps were found, such as sensory perception (supplementary file S1, table S11, Supplementary Material online). Although the eye-related categories in supplementary table S10, Supplementary Material online, were not observed, other similar categories were detected (visual perception, GO:0007601; supplementary file S1, table S11, Supplementary Material online). Liu et al. (2011) reported that TM had been trained in captivity to allow health checking (such as IOP), and the authors further suggested that TM appeared to be a suitable model for glaucoma research. Genes carrying damaging TSHNVs showed significant enrichment at multiple categories in eye problems and diabetes mellitus (supplementary file S1, tables S10 and S11, Supplementary Material online), thus indicating that TM could be a potentially good biomedical model for studying these diseases.
We also examined loss-of-function variants (lose start codon, lose/gain stop codon, and frame shifting) in TM (supplementary file S1, table S12, Supplementary Material online). We found 158 transcripts in TM containing homozygous specific loss-of-function variants (supplementary file S1, table S13, Supplementary Material online) and 68 transcripts containing heterozygous specific loss-of-function variants (supplementary file S1, table S14, Supplementary Material online). Most of the corresponding genes are uncharacterized or predicted genes. We further examined whether these genes are within the six GO terms analyzed previously. We found four and one immune response genes containing homozygous- and heterozygous-specific loss-of-function variants, respectively.
SNVs are valuable genetic markers for assessment of interspecies or interindividual variability. Previously, a relatively small panel of SNVs was used to distinguish rhesus macaque populations from Chinese and Indian origin (Ferguson et al. 2007). Higashino et al. (2012) detected 1,368,528 completely differentiated SNVs between CE and rhesus macaque genomes through four genomes comparison (IR, CR1, CE1, and CE2). In this study, we increase the number of specific SNVs in each sampled macaque by genome-wide comparison with more macaque species or individuals and discovered 10,106 TSHNVs, which provide a plethora of markers that will greatly improve the study of regional populations, animal ancestry and origins, and even hybrids between macaque species. The newly discovered SNVs constitute an important resource of candidate variants for determining species-specific responses to drugs and pathogens and significantly extend the power and resolution of genotyping approaches in population genetics or genotype–phenotype association studies.
Allele Frequency of TSHNVs in Disease Genes
As shown earlier, we identified a substantial number of TSHNVs by comparing the TM genome with the reference genome and four other macaques’ genomes. To determine whether these TSHNVs are found at high frequency or even entirely fixed in the TM population, we estimated their frequency in this population through polymerase chain reaction (PCR) sequencing. A group of 29 TSHNVs in 14 well-studied immune and disease-related genes were selected to perform the PCR resequencing in 18 unrelated TM individuals from Sichuan province, China (supplementary file S3, table S15, Supplementary Material online). The resequencing experiment generated more than 8,000 bp Sanger sequences in each individual and allowed us to calculate allele frequencies for each TSHNV in the TM population. The individual TM03 used for genome sequencing was also included to validate our genotype calls. Resequencing results from all 29 TSHNVs showed identical genotypes to the whole-genome sequencing results, thus confirming the confidence of our genotype calls (supplementary file S3, table S16, Supplementary Material online). Moreover, the allele frequency distribution of the 29 TSHNVs showed that the alternative alleles of 23 of these were fixed in the sequenced TM individuals. The other six TSHNVs had higher than 50% allele frequencies as well (fig. 5; supplementary file S3, table S16, Supplementary Material online).
Fig. 5.
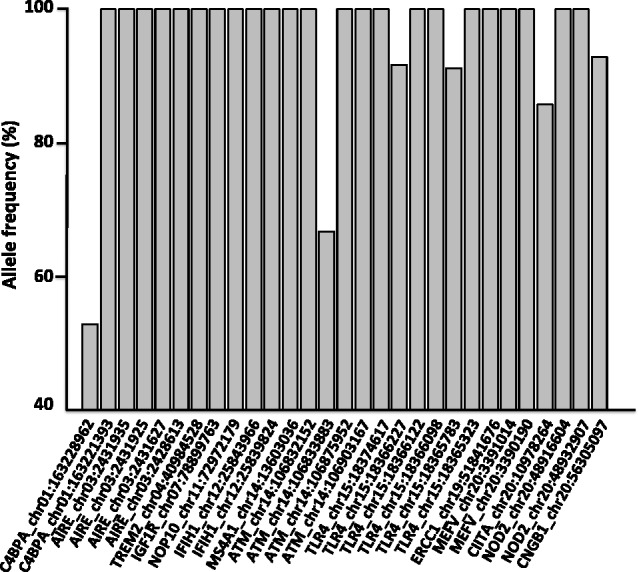
The allele frequency of 29 TSHNVs. The allele frequency of 29 TSHNVs at 14 important immune or disease genes was generated by PCR resequencing in 14–18 unrelated TM individuals from Sichuan province, China. The gene symbol, chromosome location, and genomic position of the variants were shown, and the details for these variants and genes could be found in supplementary file S3, table S15, Supplementary Material online. The detail sequencing results could be found in supplementary file S3, table S16, Supplementary Material online.
IFIH1 (interferon-induced with helicase C domain 1) gene is a putative RNA helicase. Genetic variation in IFIH1 gene has been shown to be associated with diabetes mellitus insulin-dependent type 19 (Smyth et al. 2006; Liu et al. 2009; Nejentsev et al. 2009). We found two nonsynonymous IFIH1 gene variants in TM. Furthermore, these two variants were found to be TSHNVs. The Sanger sequencing confirmed that both were entirely fixed in the TM population (supplementary file 3, table S16, Supplementary Material online).
Both AIRE and ataxia telangiectasia mutated (ATM) genes were within the diabetes mellitus (HP:0000819) and the eye problem categories obtained from GO analysis (supplementary file S1, table S10, Supplementary Material online). Both genes were enriched with nonsynonymous variants in the TM genome. Six nonsynonymous variants, including four TSHNVs, were found in each gene. The AIRE gene encodes a transcriptional regulator that plays an important role in immunity by regulating the expression of autoantigens and negative selection of autoreactive T-cells in the thymus. Mutations in this gene can cause rare autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome (APECED) in humans (Finnish-German APECED Consortium 1997). In addition, homozygous disruption of AIRE expression in a mouse model revealed that inactivating this gene results in immune system dysfunction (supplementary file S3, table S15, Supplementary Material online). The ATM gene encodes a protein belonging to the phosphatidylinositol 3-kinase family that responds to DNA damage. Mutations in this gene are associated with Ataxia-telangiectasia (Gilad et al. 1996) and susceptibility to breast cancer (Stredrick et al. 2006). PCR resequencing found that all the four TSHNVs at AIRE gene were fixed, whereas three TSHNVs at ATM were fixed and one TSHNV (chr14: 106833883) displayed an allele frequency of 66.67% (supplementary file S3, table S16, Supplementary Material online). Analysis of the allele frequency of TSHNVs at 11 other important immune response genes (supplementary file S4, Supplementary Material online) also revealed markedly high allele frequencies in TM individuals (fig. 5; supplementary file S3, table S16, Supplementary Material online).
Next, we examined the amino acid residue replacement from these variants in the human–macaque orthologs. The results showed that 22 of these exhibited different amino acid residues in human (supplementary file S3, table S15, Supplementary Material online). Although the phenotypes described earlier have been reported in human studies, the TSHNVs we found in TM could still provide useful information about the genetic diversity between TM and other macaques or humans. Despite the high conservation of the primate immune system, clinical trials have shown that responses in macaques are not good predictors of human defense reactions, likely due to mutations in immune-related genes (Suntharalingam et al. 2006). The identification and characterization of distinct genotypes in TM population may further reveal associations between macaque genetic background and pharmacology, drug response, or metabolism. However, our resequencing results showed that most of the variants from selected SNVs were almost fixed in the TM population from Sichuan province. Our results suggested that the TM exhibited remarkably very low heterozygosity, which was consistent with the genome wide results (table 1).
Inference of Demography
In TM, we observed that the genome wide heterozygosity rate was only 0.0898% per bp (table 1). This heterozygosity level was approximately three times lower than those observed in other macaque individuals (CR1: 0.2617% per bp; CR2: 0.2612% per bp; CE1: 0.3004% per bp; CE2: 0.3179% per bp) and might indicate serious bottlenecks in TM evolutionary history. Therefore, we used the pairwise sequentially Markovian coalescent (PSMC) model to infer ancestral demographic trajectories. The results revealed at least two population bottlenecks in TM and the two CEs, whereas only one bottleneck in two CRs (fig. 6). Thus, approximately 1.0–1.2 Ma, all macaques began experiencing a first population bottleneck, which lasted until roughly 0.5 Ma. Subsequently, the two CRs showed evidence of population growth although the increase between 0.1 and 0.5 Ma was slight. Both CE1 and CE2 showed a remarkable population increase between 0.2 and 0.5 Ma, but subsequently experienced a second decline. The effective population size (Ne) of CE2 was smaller than that of CE1 between 0.06 and 0.2 Ma. However, CE2 (Malaysian CE) showed evidence of growth around 0.08 Ma, whereas the decrease in CE1 (Vietnamese CE) lasted until more recent times. TM exhibited a similar demographic trajectory to the other four macaques until around 0.5 Ma, but the population growth and the Ne during 0.2–0.5 Ma were lower than that of the two CEs. When the two CRs began the second increase, TM started the second decline, and the resulting decline in Ne has continued until present days. Notably, the TM Ne at recent periods was low compared with other macaques. To validate the confidence in the PSMC results described earlier, we ran 100 bootstrap replicates for each genome. The results from all five genomes confirmed the confidence of our findings (supplementary file S2, fig. S6, Supplementary Material online). However, it should be noted that the genome coverage might have influenced the PSMC results, especially for the CR2 because it was only approximately 10-fold.
Fig. 6.
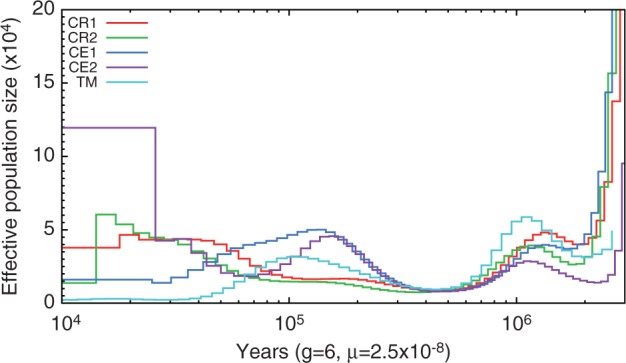
Historical changes in effective population size. Reconstruction of historical patterns of effective population size for five macaque genomes based on the genomic distribution of heterozygous sites by using the pairwise sequential Markovian coalescent (PSMC) method.
The population bottlenecks in macaques may correspond to the different glacial periods in history (Higashino et al. 2012). The TM population declines started around 1.0 and 0.1 Ma, around the same time as the two large Pleistocene glaciations in China, the Naynayxungla Glaciation (0.78–0.50 Ma) and the last glacial period (0.08–0.01 Ma) (Zheng et al. 2002). A second possible reason may involve distribution and habitat loss. Rhesus macaques and CEs display a wider distribution than TM. CEs inhabit Southeast Asia, including the Indonesian Islands, Philippine Islands, and Indochina, whereas rhesus macaques are found in an even more extensive region of Asia (Kanthaswamy et al. 2008; Osada et al. 2010). However, TM is endemic to China. Although it has a wide historical distribution ranging from southern to eastern China, its habitats have declined with farmland development. Currently, the most serious threat to TM comes from human impacts, which include habitat destruction, illegal poaching, and human-transmitted diseases. As a result, the TM population has been reduced dramatically (Wang 1998). TM subspecies are also isolated from each other, thus leading to limited gene flows between them. Previous studies have shown significant genetic differences between Sichuan, Anhui, Yunnan-Guizhou, and Fujiang TM populations (Liu et al. 2006; Li et al. 2008; Yao et al. 2013; Zhong et al. 2013). Specifically for the Sichuan populations, no haplotypes have been shared with Anhui and Yunnan-Guizhou populations, as assessed by mitochondrial DNA analysis (Liu et al. 2006; Li et al. 2008; Zhong et al. 2013). The 8,000 bp Sanger sequence data of 18 TM from Sichuan province in this study demonstrated that TM exhibited markedly low heterozygosity (fig. 5). As a consequence, it is not surprise to observe low genetic diversity and Ne in TM.
The different demographic history between CE1 and CE2 after 0.2 Ma was possibly associated with their origins. CEs were genetically grouped into Indonesian-Malaysian, Philippine, Indochinese, and Mauritian macaques (Kanthaswamy et al. 2008; Osada et al. 2010). Indochinese CEs (e.g., CE1) displayed admixture with rhesus macaques (Stevison and Kohn 2008; Bonhomme et al. 2009; Yan et al. 2011), whereas Indonesian-Malaysian CEs (e.g., CE2) originated from a putative ancestral population according to the fossil evidence showed the highest genetic diversity (Delson 1980; Higashino et al. 2012). Therefore, the genetic backgrounds of CE1 and CE2 were very different. The different demographic trajectories and larger Ne at recent times we observed in PSMC of them were consistent with fossil records and previous evidences.
Conclusions
In this study, we reported a 37-fold coverage genome of a sinica species of TM using a resequencing strategy. Using the IR as a reference genome, these data enabled genome-wide discovery of 11.9 million high-quality SNVs in TM, including 3.9 million TM-specific SNVs through comparison with four available genomes of rhesus and CEs. The observed genome wide genetic differences between TM and fascicularis group macaques, as well as the genetic variations at particular immune- and disease-related genes confirmed by PCR resequencing, may lead to functional differences in clinical studies. The autosomal heterozygosity rate of TM was approximately one-third of that of CRs and CEs indicating a very low heterozygosity in TM. PCR resequencing data also demonstrated low heterozygosity in the TM Sichuan population. Moreover, genes carrying deleterious TSHNVs were significantly enriched in diabetes mellitus and eye disease ontology categories, which suggest that TM could be a potentially good biomedical model for studying these diseases. The genome-wide comparison revealed that TM was more closely related to CR than to CE, and some unusual low divergence regions between TM and CR were detected. After applying statistical criteria, up to 239,620 kb PIRs (8.84% of the genome) were identified in the TM genome. In addition, TM and CR display a highly overlapping geographical distribution, and they shared the same refuge during the Middle Pleistocene. They also exhibit similar mating behavior. Therefore, all these results indicate that probably there was an ancient introgression event between TM and CR. Furthermore, demographic inference analysis revealed that TM exhibited a similar demographic history as other macaques until 0.5 Ma, but then it maintained a lower effective population size until present days. In conclusion, our study has provided new insight about the macaque evolutionary history, confirming hybridization events between different macaque species groups based on genome-wide data.
Materials and Methods
Sample Information
A wild 12- to 15-year-old female TM captured from Mabian County, Sichuan province, China, was used for whole-genome sequencing. Genomic DNA was extracted from whole blood using the standard phenol–chloroform method. Two TM-specific and one Stump-tailed macaque-specific (M. arctoides) Alu locus (Li et al. 2009) and mitochondrial DNA fragment were amplified to confirm the species identification, because TM and Stump-tailed macaque are morphologically very similar. Further details can be found in supplementary file S4, Supplementary Material online. The short read sequences of CR1, CR2, and CE1 used in this study were downloaded from NCBI (accession numbers SRA023856, SRA037810, and SRA023855) (Fang et al. 2011; Yan et al. 2011). The alignment files (format as bam files) of the Malaysian CE (CE2) were kindly provided by Dr Osada (Department of Population Genetics, National Institute of Genetics, Japan) and described in Higashino et al. (2012).
Genome Sequencing and Mapping Short Reads
TM whole-genome sequencing was performed using an Illumina Hiseq 2000 at Beijing Genomics Institute (BGI). Two paired-end libraries with insert sizes of approximately 500 bp were generated. Library preparation and all sequencing runs were performed according to manufacturer’s protocols. The 100-bp pair-end (PE) short reads of TM were aligned to IR genome (rheMac2) using Bowtie2 (Langmead and Salzberg 2012) under local alignment algorithm with very sensitive model and proper insert sizes. Default options were used for other parameters. The short reads of CR1, CR2, and CE1 were also mapped to rheMac2 using the same pipeline. The SOLiD data of CE2 were originally mapped to rheMac2 using BioScope (Higashino et al. 2012), and the alignment files were used directly in the downstream genotype pipeline.
Genotyping Pipeline
After mapping the short reads to the reference genome, we applied Picard (http://picard.sourceforge.net, last accessed March 26, 2014) and GATK toolsets (DePristo et al. 2011) to process the alignments to SNV and indel calls for the five macaques. The whole pipeline converted the short reads to bam format alignment files and then generated genotype calls in Variant Call Format (http://www.1000genomes.org/node/101, last accessed March 26, 2014) (fig. 1A). Further details of our pipeline can be found in supplementary file S4, Supplementary Material online.
Postgenotype Filters
After obtaining the genotype calls from GATK, we applied several data quality filters to control the data quality. Previously, different genomic studies used different filters (Higashino et al. 2012; Lachance et al. 2012; Prüfer et al. 2012). Here, we sought to minimize the effects of sequencing and alignment errors (Nielsen et al. 2011) and also sought to exclude the regions that show accelerated evolutionary rates, which are not caused by positive selection. Therefore, we used both genome filters, which are based on the reference genome’s features and polymorphism across samples, and SF, which are based on the genotype calls of each sample. We described the details of filters in supplementary file S4, Supplementary Material online.
Gene Annotation
We obtained SeqGene files from the FTP section of the NCBI database as the gene annotation source for the reference genome (rheMac2; ftp://ftp.ncbi.nih.gov/genomes/Macaca_mulatta/mapview/, last accessed March 26, 2014). The retrieved files included annotated gene names and symbols, genomic coordinate of coding exons, and UTRs of both confirmed and predicted genes. After removing duplicated records, PSEUDO genes, and genes without transcript ID, the final annotation contained 25,045 transcripts from 19,544 genes. Using this gene annotation, we classified the location of all detected SNVs as nongenic and genic. The genic SNVs were further classified into noncoding, UTR, intron, and exon SNVs.
SNVs in Important Pathways and Genes
We checked how many variants in TM fell into immune response genes and drug response genes. In addition, because TM has the largest body size in genus Macaca and might be a good medical model for obesity and diabetes, we searched TM variants in glucose metabolic process genes and insulin-related genes.
We obtained GO lists from the GO project (http://www.geneontology.org, last accessed March 26, 2014) for the following categories in human: Immune response (GO0006955); Response to drug (GO0042493); Glucose metabolic process (GO0006006); Insulin secretion (GO:0030073); Insulin receptor binding (GO:0005158); Insulin receptor signaling pathway (GO:0008286).
Variant Functional Impact and Enrichment Analyses
We predicted the effects of nonsynonymous mutations on protein structure and functionality using PolyPhen-2 (Adzhubei et al. 2010). All protein sequences used as input for the program were obtained by translating the corresponding transcript sequences, and all TSHNVs were submitted as a batch file for HumDiv model. Genome assembly was preformed choosing hg19. Sites were ranked according to the predicted qualitative effects of the amino acid substitution (“benign,” “possibly damaging," or “probably damaging"). Complementary analyses were performed with SIFT for the TSHNVs estimated as “possibly damaging” or “probably damaging” in PolyPhen-2 (Kumar et al. 2009; http://sift.jcvi.org/, last accessed March 26, 2014). Then, genes containing TSHNVs that were classified as damaging (probably or possibly) in PolyPhen-2 were tested for significant enrichment in GO categories, Kegg/Reactome pathways (KGR), and Human Phenotype Ontologies (HPO) using the online tool g:Profiler (Reimand et al. 2011). The sources/databases used were GO terms, which aim to summarize relevant information about gene function with respect to their molecular function, the biological process, and cellular compartments (http://www.geneontology.org, last accessed March 26, 2014); the KGR, which are curated reference databases for biological pathways (http://www.genome.jp/kegg/pathway.html, last accessed March 26, 2014), and the HPO, which establishes standardized terms for phenotypic abnormalities encountered in human disease (http://www.human-phenotype-ontology.org/, last accessed March 26, 2014). We also did the same test for the TSHNVs that were estimated as deleterious in both PolyPhen-2 and SIFT. All genes of rhesus macaque annotated in Ensembl were used as background set, and the Benjamini–Hochberg false discovery rate (Benjamini and Hochberg 1995) was applied to correct for multiple testing. We only reported significantly enriched categories that included ≥5 genes and with multiple testing corrected P value ≤ 0.05.
PCR Resequencing of Selected Variants
To validate the allele frequency of the TM-specific homozygous SNVs, we checked 29 TSHNVs in 14 genes in 14–18 unrelated TM individuals from Sichuan province through PCR resequencing method (supplementary files S3 and S4, Supplementary Material online). These individuals were collected from Mabian, Emei, Ganluo, and Pengzhou counties, and none of them shared grandparents (supplementary file S4, fig. S9, Supplementary Material online). Three criteria were used to select these genes and SNVs: 1) well-studied important immune-related or human disease genes; 2) carried at least one TM-specific damaging homozygous SNV estimated by PolyPhen-2; and 3) the selected SNVs should cause nonsynonymous changes. Primer sets for PCR amplification are listed in supplementary file S3, table S17, Supplementary Material online. PCR products were subsequently sequenced using an ABI 3730XL sequencer (Applied Biosystems).
Genetic Divergence
To estimate the divergence between TM and other macaques, we followed the method described in a previous macaque genome study (Yan et al. 2011). Only homozygous variants were used (Yan et al. 2011), and two different nonoverlapping window sizes were applied (50 kb and 100 kb). Genetic distance was also calculated using the genetic distance metric described by Gronau et al. (2011). The genome was scanned with two different window sizes (50 kb and 100 kb nonoverlapping windows), and then we made pairwise comparison between and within different species across the genome.
Detection of PIRs
PIRs were detected in the TM genome by defining a statistic value, Rdiff, to quantify the difference between the divergences between two different comparisons as described previously (Yan et al. 2011). Genetic distance was calculated according to the method by Gronau et al. (2011). Analyses were performed with different window sizes ranging from 10 to 1,000 kb. To filter out false-positive PIRs, we applied the following approach to determine the cut-off values for the Rdiff. We performed coalescent simulations adopting the demographic parameters of three macaque populations (CE1, CR1, and IR) estimated by Yan et al. (2011) using ∂a∂i software (Gutenkunst et al. 2009). For simplicity, we assumed a model without gene flow between different macaques, as described previously (Yan et al. 2011). We first estimated the divergence time between TM and the other three populations using the data presented in table 1. Because the reference genome (IR) is a haploid genome, thus to estimate the average genetic divergence, we simply summed up homozygous SNV counts and a half of heterozygous SNV counts and then divided the counts by the total number of analyzed sites. The average genetic divergence between TM and IR chromosomes was subtracted from the average genetic divergence between CE1 and IR chromosomes. If we assume the ancestral population sizes of TM/IR and CE1/IR are equal, which is consistent with the estimation by Yan et al. (2011), the difference corresponds to the difference of divergence time between TM/IR and CE1/IR. The time was scaled to the unit of 4Nm (4Nm; ancestral population size) in the model of Yan et al. and incorporated into a new model with TM. In coalescent simulations, we also incorporated the effect of recombination, because the sizes of window are not negligibly small for large window sizes. For each locus, we randomly sampled recombination rate per base pair from the exponential distribution with the average of 7.34 × 10−9, which was estimated from rhesus macaques (Rogers et al. 2006; Osada et al. 2013), and then generated a genealogy using ms software (Hudson 2002). We next calculated Rdiff using the same equation as we used in the real data. The procedure was repeated 100,000 times for each window size.
Inference of Demography
We used the PSMC method (Li and Durbin 2011) to infer the demographic history of TM, two CRs, and two CEs. Briefly, the method uses the distribution of heterozygote sites across the genome and a PSMC model that defines a hidden Markov model. The following parameters were used: numbers of iterations = 25, time interval = 1 × 6 + 58 × 1, mutation rate per generation = 2.5 × 10−8, and generation time = 6. The mutation rate and generation time were used in a previous macaque genome study (Higashino et al. 2012). We note that different mutation rate and generation time may have a big impact on the time estimation of PSMC result. To validate the confidence in PSMC findings, we ran 100 bootstrap replicates for each genome. To sample a bootstrap replicate, we divided the genome into segments of 5 Mb, sampled with replacement from those segments until we obtained a sequence with approximately the same length as the original genome as defined by using the “-b” option in the PSMC software, and reran the EM-based Ne estimation procedure.
Supplementary Material
Supplementary files S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
Z.F., B.Y., and J.L. contributed to the design of this research. Y.Y. and H.W. collected the samples. G.Z., P.L., and R.S. performed the experiments. Z.F., L.D., J.X., N.O., and P.S. contributed to data analysis. Z.F., G.Z., J.X., X.Z., R.S., B.Y., and J.L. wrote the manuscript. All authors read and approved the final manuscript. This work was supported by the National Natural Science Foundation of China (No. 31270431), the Sichuan Youth Science and Technology Foundation (2011JQ0022), the Project Sponsored by the Scientific Research Foundation for the Returned Overseas Scholars, State Education Ministry (20111568-8-3), the National Key Technology R&D Program (2012BAC01B06), the National Institutes of Health to J.X. (USA, R00 HG005846), and a PhD grant from Fundação para a Ciência e a Tecnologia to P.S. (Portugal) (SFRH/BD/60549/2009). The authors thank Diego Ortega Del Vecchyo at University of California, Los Angeles (UCLA) for help with the genetic diversity tests. They are grateful to Prof. Robert Wayne (UCLA), Rena Schweizer (UCLA), and Johann Bergholz at Sichuan University for reading the whole manuscript and helping to modify the language.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Berard J. A four-year study of the association between male dominance rank, residency status, and reproductive activity in rhesus macaques (Macaca mulatta) Primates. 1999;40:159–175. doi: 10.1007/BF02557708. [DOI] [PubMed] [Google Scholar]
- Blancher A, Bonhomme M, Crouau-Roy B, Terao K, Kitano T, Saitou N. Mitochondrial DNA sequence phylogeny of 4 populations of the widely distributed cynomolgus macaque (Macaca fascicularis fascicularis) J Hered. 2008;99:254–264. doi: 10.1093/jhered/esn003. [DOI] [PubMed] [Google Scholar]
- Bonhomme M, Cuartero S, Blancher A, Crouau-Roy B. Assessing natural introgression in 2 biomedical model species, the rhesus macaque (Macaca mulatta) and the long-tailed macaque (Macaca fascicularis) J Hered. 2009;100:158–169. doi: 10.1093/jhered/esn093. [DOI] [PubMed] [Google Scholar]
- Chakraborty D, Ramakrishnan U, Panor J, Mishra C, Sinha A. Phylogenetic relationships and morphometric affinities of the Arunachal macaque Macaca munzala, a newly described primate from Arunachal Pradesh, northeastern India. Mol Phylogenet Evol. 2007;44:838–849. doi: 10.1016/j.ympev.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Delson E. Fossil macaques, phyletic relationships and a scenario of deployment. In: Lindburg DG, editor. The macaques: studies in ecology, behavior, and evolution. New York: Van Nostrand Reinhold Co; 1980. pp. 10–30. [Google Scholar]
- Delson E, Tattersall I, Van Couvering JA, Brooks AS. Cercopithecidae. In: Delson E, Tattersall I, Van Couvering JA, Brooks AS, editors. Encyclopedia of human evolution and prehistory. 2nd ed. New York: Garland; 2000. pp. 166–171. [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eudey AA. 1979. Differentiation and dispersal of macaques (Macaca spp.) in Aria [Ph.D. thesis] [Davis (CA)]: University of California, Davis. [Google Scholar]
- Evans BJ, Supriatna J, Andayani N, Setiadi MI, Cannatella DC, Melnick DJ. Monkeys and toads define areas of endemism on Sulawesi. Evolution. 2003;57:1436–1443. doi: 10.1554/02-443. [DOI] [PubMed] [Google Scholar]
- Fa JE. The genus Macaca: a review of taxonomy and evolution. Mammal Rev. 1989;19:45–81. [Google Scholar]
- Fang X, Zhang Y, Zhang R, Yang L, Li M, Ye K, Guo X, Wang J, Su B. Genome sequence and global sequence variation map with 5.5 million SNPs in Chinese rhesus macaque. Genome Biol. 2011;12:R63. doi: 10.1186/gb-2011-12-7-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson B, Street SL, Wright H, Pearson C, Jia Y, Thompson SL, Allibone P, Dubay CJ, Spindel E, Norgren RB., Jr Single nucleotide polymorphisms (SNPs) distinguish Indian-origin and Chinese-origin rhesus macaques (Macaca mulatta) BMC Genomics. 2007;8:43. doi: 10.1186/1471-2164-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet. 1997;17:399–403. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- Fooden J. Provisional classifications and key to living species of macaques (primates: Macaca) Folia Primatol. 1976;25:225–236. doi: 10.1159/000155715. [DOI] [PubMed] [Google Scholar]
- Gilad S, Bar-Shira A, Harnik R, Shkedy D, Ziv Y, Khosravi R, Brown K, Vanagaite L, Xu G, Frydman M, et al. Ataxia-telangiectasia: founder effect among North African Jews. Hum Mol Genet. 1996;5:2033–2037. doi: 10.1093/hmg/5.12.2033. [DOI] [PubMed] [Google Scholar]
- Gronau I, Hubisz MJ, Gulko B, Danko CG, Siepel A. Bayesian inference of ancient human demography from individual genome sequences. Nat Genet. 2011;43:1031–1034. doi: 10.1038/ng.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves CP, Wilson DE, Reeder DM. Order primates mammal species of the world: a taxonomic and geographic reference. 3rd ed. Vol. 1. Baltimore (MD): Johns Hopkins University Press; 2005. [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 2009;5:e1000695. doi: 10.1371/journal.pgen.1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayasaka K, Fujii K, Horai S. Molecular phylogeny of macaques: implications of nucleotide sequences from an 896-base pair region of mitochondrial DNA. Mol Biol Evol. 1996;13:1044–1053. doi: 10.1093/oxfordjournals.molbev.a025655. [DOI] [PubMed] [Google Scholar]
- Higashino A, Sakate R, Kameoka Y, Takahashi I, Hirata M, Tanuma R, Masui T, Yasutomi Y, Osada N. Whole-genome sequencing and analysis of the Malaysian cynomolgus macaque (Macaca fascicularis) genome. Genome Biol. 2012;13:R58. doi: 10.1186/gb-2012-13-7-r58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Kanthaswamy S, Satkoski J, George D, Kou A, Erickson BJ, Smith DG. Interspecies hybridization and the stratification of nuclear genetic variation of rhesus (Macaca mulatta) and long-tailed macaques (Macaca fascicularis) Int J Primatol. 2008;29:1295–1311. doi: 10.1007/s10764-008-9295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1082. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Kumar S, Hedges SB. A molecular timescale for vertebrate evolution. Nature. 1998;392:917–920. doi: 10.1038/31927. [DOI] [PubMed] [Google Scholar]
- Lachance J, Vernot B, Elbers CC, Ferwerda B, Froment A, Bodo JM, Lema G, Fu W, Nyambo TB, Rebbeck TR, et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell. 2012;150:457–469. doi: 10.1016/j.cell.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DM, Fan LQ, Ran JH, Yin HL, Wang HX, Wu SB, Yue BS. Genetic diversity analysis of Macaca thibetana based on mitochondrial DNA control region sequences. Mitochondr DNA. 2008;19:446–452. [PubMed] [Google Scholar]
- Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Han K, Xing J, Kim HS, Rogers J, Ryder OA, Disotell T, Yue BS, Batzera MA. Phylogeny of the macaques (Cercopithecidae: Macaca) based on Alu elements. Gene. 2009;448:242–249. doi: 10.1016/j.gene.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling B, Veazey RS, Luckay A, Penedo C, Xu K, Lifson JD, Marx PA. SIV(mac) pathogenesis in rhesus macaques of Chinese and Indian origin compared with primary HIV infections in humans. AIDS. 2002;16:1489–1496. doi: 10.1097/00002030-200207260-00005. [DOI] [PubMed] [Google Scholar]
- Liu G, Zeng T, Yu WH, Yan NH, Wang HX, Cai SP, Pang IH, Liu XY. Characterization of intraocular pressure responses of the Tibetan monkey (Macaca thibetana) Mol Vis. 2011;17:1405–1413. [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wang H, Jin Y, Podolsky R, Reddy MV, Pedersen J, Bode B, Reed J, Steed D, Anderson S, et al. IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum Mol Genet. 2009;18:358–365. doi: 10.1093/hmg/ddn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Li JH, Zhao JY. Sequence variation of mitochondrial DNA control region and population genetic diversity of Tibetan macaques Macaca thibetana in the Huangshan Mountain. Acta Zool Sin. 2006;52:724–730. [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada N, Hashimoto K, Kameoka Y, Hirata M, Tanuma R, Uno Y, Inoue I, Hida M, Suzuki Y, Sugano S, et al. Large-scale analysis of Macaca fascicularis transcripts and inference of genetic divergence between M. fascicularis and M. mulatta. BMC Genomics. 2008;9:90. doi: 10.1186/1471-2164-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada N, Nakagome S, Mano S, Kameoka Y, Takahashi I, Terao K. Finding the factors of reduced genetic diversity on X chromosomes of Macaca fascicularis: male-driven evolution, demography, and natural selection. Genetics. 2013;195:1027–1035. doi: 10.1534/genetics.113.156703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada N, Uno Y, Mineta K, Kameoka Y, Takahashi I, Terao K. Ancient genome wide admixture extends beyond the current hybrid zone between Macaca fascicularis and M. mulatta. Mol Ecol. 2010;19:2884–2895. doi: 10.1111/j.1365-294X.2010.04687.x. [DOI] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, Seuánez HN, Horvath JE, Moreira MA, Kessing B, Pontius J, Roelke M, Rumpler Y, et al. A molecular phylogeny of living primates. PLoS Genet. 2011;7:e1001342. doi: 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool JE, Nielsen R. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics. 2009;181:711–719. doi: 10.1534/genetics.108.098095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, Munch K, Hellmann I, Akagi K, Miller JR, Walenz B, Koren S, Sutton G, Kodira C, Winer R, et al. The bonobo genome compared with the chimpanzee and human genomes. Nature. 2012;486:527–531. doi: 10.1038/nature11128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Arak T, Vilo J. g:Profiler-a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39:W307–W315. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhesus Macaque Genome Sequencing and Analysis Consortium. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- Rogers J, Garcia R, Shelledy W, Kaplan J, Arya A, Johnson Z, Bergstrom M, Novakowski L, Nair P, Vinson A, et al. An initial genetic linkage map of the rhesus macaque (Macaca mulatta) genome using human microsatellite loci. Genomics. 2006;87:30–38. doi: 10.1016/j.ygeno.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Schmidt LH, Fradkin R, Harrison J, Rossan RN. Differences in the virulence of Plasmodium knowlesi for Macaca irus (fascicularis) of Philippine and Malayan origins. Am J Trop Med Hyg. 1977;26:612–622. doi: 10.4269/ajtmh.1977.26.612. [DOI] [PubMed] [Google Scholar]
- Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, Guja C, Ionescu-Tirgoviste C, Widmer B, Dunger DB, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38:617–619. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- Stevison LS, Kohn MH. Determining genetic background in captive stocks of cynomolgus macaques (Macaca fascicularis) J Med Primatol. 2008;37:311–317. doi: 10.1111/j.1600-0684.2008.00292.x. [DOI] [PubMed] [Google Scholar]
- Stredrick DL, Garcia-Closas M, Pineda MA, Bhatti P, Alexander BH, Doody MM, Lissowska J, Peplonska B, Brinton LA, Chanock SJ, et al. The ATM missense mutation p.ser49cys (c.146C-G) and the risk of breast cancer. Hum Mutat. 2006;27:538–544. doi: 10.1002/humu.20323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. New Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- Tosi AJ, Morales JC, Melnick DJ. Y-chromosome and mitochondrial markers in Macaca fascicularis indicate introgression with Indochinese M. mulatta and a biogeographic barrier in the Isthmus of Kra. Int J Primatol. 2002;23:161–178. [Google Scholar]
- Tosi AJ, Morales JC, Melnick DJ. Paternal, maternal, and biparentalmolecular markers provide unique windows onto the evolutionary history of macaque monkeys. Evolution. 2003;57:1419–1435. doi: 10.1111/j.0014-3820.2003.tb00349.x. [DOI] [PubMed] [Google Scholar]
- Wang S. China Red Data Book of Endangered Animal (Mammalia) Beijing: Science Press; 1998. [Google Scholar]
- Yan G, Zhang G, Fang X, Zhang Y, Li C, Ling F, Cooper DN, Li Q, Li Y, van Gool AJ, et al. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat Biotechnol. 2011;29:1019–1023. doi: 10.1038/nbt.1992. [DOI] [PubMed] [Google Scholar]
- Yang F, Wang HX, Zhou L, Ai YX, Zeng T. A primary analyze and measurement on partial biochemistry index of peripheral blood cells of Macaca thibetana. Sichuan J Zool. 2010;29:256–258. [Google Scholar]
- Yao YF, Zhong LJ, Liu BF, Li JY, Ni QY, Xu HL. Genetic variation between two Tibetan macaque (Macaca thibetana) populations in the eastern China based on mitochondrial DNA control region sequences. Mitochondr DNA. 2013;24:267–275. doi: 10.3109/19401736.2012.748040. [DOI] [PubMed] [Google Scholar]
- Zhao QK. Mating competition and intergroup transfer of males in Tibetan macaques (Macaca thibetana) at Mt Emei, China. Primates. 1994;35:57–68. [Google Scholar]
- Zheng BX, Xu QQ, Shen YP. The relationship between climate change and Quaternary glacial cycles on the Qinghai-Tibetan Plateau: review and speculation. Quaternary Int. 2002;97–98:93–101. [Google Scholar]
- Zhong LJ, Zhang MW, Yao YF, Ni QY, Mu J, Li CQ, Xu HL. Genetic diversity of two Tibetan macaque (Macaca thibetana) populations from Guizhou and Yunnan in China based on mitochondrial DNA D-loop sequences. Genes Genomics. 2013;35:205–214. [Google Scholar]
- Ziegler T, Abegg C, Meijaard E, Perwitasari-Farajallah D, Walter L, Hodges JK, Roos C. Molecular phylogeny and evolutionary history of Southeast Asian macaques forming the M. silenus group. Mol Phylogenet Evol. 2007;42:807–816. doi: 10.1016/j.ympev.2006.11.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.