

Abstract
Inhibition of mixed lineage kinase 3 (MLK3) is a potential strategy for treatment of Parkinson’s Disease and HIV-1 Associated Neurocognitive Disorders (HAND), requiring an inhibitor that can achieve significant brain concentration levels. We report here URMC-099 (1) an orally bioavailable (F = 41%), potent (IC50 = 14 nM) MLK3 inhibitor with excellent brain exposure in mouse PK models and minimal interference with key human CYP450 enzymes or hERG channels. The compound inhibits LPS-induced TNFα release in microglial cells, HIV-1 Tat-induced release of cytokines in human monocytes, and up-regulation of phospho-JNK in Tat-injected brains of mice. Compound 1 likely functions in HAND preclinical models by inhibiting multiple kinase pathways, including MLK3 and LRRK2 (IC50 = 11 nM). We compare the kinase specificity and BBB penetration of 1 with CEP-1347 (2). Compound 1 is well tolerated, with excellent in vivo activity in HAND models, and is under investigation for further development.
Keywords: Mixed Linage Kinase, MLK3, LRRK2, HIV-Associated Neurocognitive Disorder, HAND, Blood Brain Barrier, Kinase Inhibitor, Parkinson’s Disease, CNS Penetrant
INTRODUCTION
We report here the discovery, synthesis and characterization of URMC-099 (1), a new inhibitor of mixed lineage kinase type 3 (MLK3) with excellent blood-brain barrier penetration properties, which has shown neuroprotective and anti-neuroinflammatory properties in in vitro and in vivo models of HIV-1 Associated Neurocognitive Disorders (HAND)1. Combination antiretroviral therapy (cART) has greatly increased both the life expectancy and quality of life for HIV-1 seropositive individuals and is one of the greatest success stories of modern drug development. However, as the population of AIDS patients has aged it has become apparent that neurological impairments resulting from HIV infection are not controlled by cART and may indeed be exacerbated by some CNS penetrating antiretroviral agents used in HIV therapy.2 HAND encompasses a broad range of neurologic deficits that range from mild cognitive impairment to frank dementia and is the result of damage to normal synaptic architecture that is likely mediated by dysregulation of immune cells in the CNS. In the U.S., greater than 50% of AIDS patients experience some symptoms of HAND with a significant percentage (15%) exhibiting neurologic morbidity severe enough to preclude normal activities of daily living with substantial economic impact for their healthcare.2
The hallmarks of HAND include: 1) a dysregulation of inflammatory cytokines and chemokines, 2) the recruitment of monocytes to the CNS, 3) viral infection of microglia leading to interruption of their normal function, and 4) extensive synaptodendritic damage, which ultimately impacts polysynaptic pathways that are the substrate for HAND in affected regions of the brain. A host of inflammatory mediators have been implicated in cellular models of HAND, where TNF-α release and signaling likely play a major central role. A more limited subset of mediators has been identified as being up-regulated in the cerebrospinal fluid (CSF) and post-mortem brain tissues of HAND patients. These mediators/effectors include TNFα, the chemokine monocyte chemoattractant protein (MCP-1), and from preclinical models, mixed-lineage kinase 3 (MLK3), an important control point in MAPK kinase regulated inflammation pathways.3
Mixed lineage kinases are mitogen activated protein kinase kinase kinases (MAPKKKs) with features of both serine-threonine and tyrosine kinases that regulate the c-Jun N-terminal kinase (JNK) mitogen activated protein kinase (MAPK) signaling cascade, and also regulate p38 and extracellular signal-regulated kinase (ERK).4,5,6 MLK3 (MAP3K11) is the most widely expressed MLK family member,4,5,6 and is expressed in neurons7 (as well as other cell types).8 At the cellular level, MLK3 is activated by stress, including reactive oxygen species, ceramide, and TNFα.10,11 At the molecular level, it is activated by Cdc42 and Rac, which interact with MLK3, and can cause it to dimerize via a leucine zipper interface, resulting in autophosphorylation at Thr277 and Ser281 within the protein activation loop, and enzyme activation.12,13 HIV-1 Tat also leads to phosphorylation at these same residues in primary rat neurons14 and to activation of glycogen synthase kinase (GSK-3β) in neurons.15,16 This is important because MLK3 can be activated as a result of direct phosphorylation by GSK-3β.17
Previously published MLK3 inhibitors: CEP-134718 (2), K252a6 (3), CEP-70119 (4), CEP-1100420 (5), and compound 621 (Fig. 1) have been based largely on the protein kinase-promiscuous staurosporine scaffold. Compound 2 has been used as a tool compound to explore the effects of MLK3 inhibition for HAND and Parkinson’s disease,22,23,24 in cellular and animal models, although the compound is in no way specific for MLK3. Compound 2 has also been a central player in the target validation of mixed lineage kinases for HAND. Compound 2 protected primary rat hippocampal neurons as well as dorsal root ganglion neurons from the otherwise lethal effects of exposure to HIV-1 coat protein gp120.25, 26 Tat and gp120 induce autophosphorylation of MLK3 in primary rat neurons, which was abolished by the addition of 2. Compound 2 also enhanced survival of both rat and human neurons and inhibited the activation of human monocytes after exposure to Tat and gp120.14 Compound 2 is neuroprotective in an in vivo model of HIV-1 infection, reversing microglial activation and restoring normal synaptic architecture, as well as restoring macrophage secretory profiles to a trophic vs. toxic phenotype in response to HIV-1 infection.27
Figure 1.
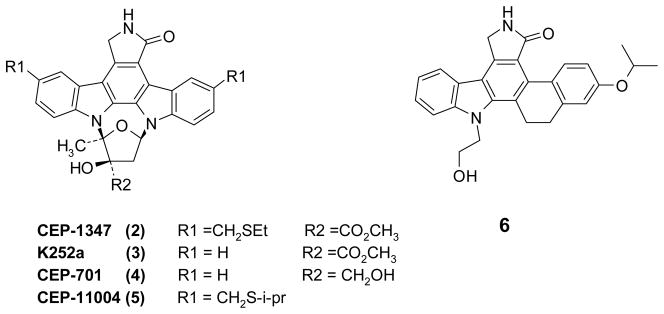
Examples of Known Potent MLK3 Inhibitors
Unfortunately 2 failed to show efficacy in a large Phase II clinical trial for early stage Parkinson’s Disease.28 Compound 2 has a high molecular weight (MW = 615) with a large polar surface area (PSA = 95 Å2), properties which are not conducive to blood brain barrier (BBB) penetration. There are additional reasons to suspect that 2 likely did not maintain therapeutic levels in the brains of significant numbers of patients. No published data for CNS penetration is available for this compound, however 2 is known to interact with and inhibit CYP450 enzymes.29 Plasma PK concentration data for human subjects receiving 2 and antiviral therapy showed very large patient to patient variation in blood levels of 2, which may be indicative of induction of metabolic liver enzymes. In silico assessment using Optibrium30 software suggested poor CNS penetration of 2, confirmed by blood brain barrier studies reported here, as well as the likelihood of interaction with liver enzymes, which could lead to further degradation of CNS exposure over time. Therefore our goal was to identify a new potent MLK3 inhibitor with improved pharmacokinetics and significant CNS penetration, which might serve as a first-in-class treatment, adjunctive to cART, for HAND.
RESULTS AND DISCUSSION
We have identified a 7-azaindole based MLK3 inhibitor 1, with potent in vitro activity against HIV-1 Tat mediated release of cytokines in human and mouse immune cells, including cultured mouse microglial BV-2 cells; it is also neuroprotective in mice exposed to HIV-1 Tat.1 In mouse brain imaging experiments, compound 1 has demonstrated its in vivo efficacy for preservation of normal synaptic architecture and reversal of neuroinflammation following CNS exposure to HIV-1 Tat.1 Because of a compelling profile of central anti-inflammatory effects and neuroprotection, compound 1 is being evaluated for adjunctive therapy with or without cART in humanized mice engrafted with human CD34 cells that can sustain peripheral and central infection with HIV-1.31
A proprietary focused library of kinase inhibitors from BioFocus Ltd. containing approximately 15,000 unique compounds was screened against MLK3 in a radiometric filter plate assay format. A large number of active compounds with consistent structure activity themes were identified from structurally distinct chemical libraries. We focused our initial hit-to-lead development on the imidazopyrazinone series based around screening hit (7) (see Fig. 3). This compound gave variable potency in enzyme inhibition assays (MLK3 IC50 ranged between 40 – 120 nM); we believed this variability was due to solubility issues. Cellular assays also suggested poor ability to penetrate cells (LPS-stimulated TNF-α release in BV-2 mouse microglial cells, IC50 = 23.6 μM). However the simple structure, well-developed SAR from screening hits, ease of synthesis and potential for scaffold hopping from this series of compounds more than compensated for its initial drawbacks. During the modification of this scaffold, the design of compounds for optimization focused on low molecular weight compounds (<450 MW) with small numbers of hydrogen bond donors (<3), and low polar surface area to maximize chances of CNS penetration. Analysis of virtual libraries of potential synthetic targets with Stardrop™ (Optibrium Ltd.) guided us towards the 7-azaindole type scaffold and compounds related to the indole containing (8), a direct analog of 7. Compound 8 (see figures 2 and 3) was much more potent against MLK3 and showed significant inhibition of the release of TNFα in BV-2 cells stimulated with LPS. We rapidly synthesized a number of potent analogs around this structure type with the goal of simultaneously optimizing (1) high potency in a biochemical MLK3 inhibition assay, (2) inhibitory activity in macrophages/microglia to prevent HIV-1 Tat-induced stimulation of a relevant panel of cytokines of validated importance in HAND, (3) metabolic stability, (4) CNS penetration, and (5) high brain and plasma concentrations upon oral dosing. Ultimately compound 1 emerged as an attractive candidate in a small focused library of very active compounds and was designed through a strategy to introduce solubilizing groups known to be effective in marketed kinase inhibitors. Our goal was to replace the trimethoxy aryl residue, with a group that could exhibit very low polar surface area in order to increase the potential for CNS penetration. The benzyl-linked N-methyl piperazine moiety met these criteria and yielded compound 1, both highly potent against MLK3 and with very low polar surface area.
Figure 3.
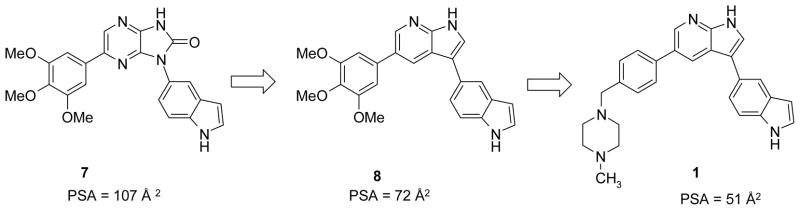
Optimization Design Path to Compound 1
Figure 2.

Potent and Drug-like 7-Azaindole MLK3 Inhibitors Selected for Initial Profiling
Inhibition of LPS-induced TNFα release in murine BV-2 microglial cells
After obtaining potent MLK3 inhibition data, we used an ELISA-based assay to gauge if our potent MLK3 inhibitor compounds were affecting the MAPK kinase pathway, which regulates TNFα release in microglial cells, one of the key cytokines responsible for inflammatory effects in HAND.32–35 This secondary assay allowed us to identify chemotypes that readily penetrated relevant immune cells and elicited functional activity in reduction of this key pro-inflamatory cytokine. We profiled 132 potent compounds for activity in MLK3 inhibition, TNFα release inhibition assays, as well as drug-like properties (see Figure 2) and selected six compounds (1, 8, 9, 10, 11, 12) for preliminary pharmacokinetic screening in mice (see Figure 2).
Screening Phamacokinetics Studies and Blood Brain Barrier Penetration
It is a widely held belief that few potent protein kinase inhibitors penetrate the CNS, although published data are scant regarding CNS exposure for these mostly anti-cancer drugs. Recent strategies in kinase drug development have skewed the propensity for these drugs to have properties unfavorable for BBB penetration.36
Our strategy for finding CNS penetrant compounds for proof-of-concept studies was to keep structures as simple as possible, with non-linear shapes, few hydrogen bond donors, LogD between 2 and 5, and low polar surface area. We tested the most promising compounds in screening PK studies for BBB penetration in mice. Test compounds were dosed i.v. in C57BL/6 male mice, through the tail vein, for a total dose of 10 mg/kg per animal, and standard pharmacokinetic parameters were determined (Table 1). Both 1 and 8 exhibited significant brain concentrations (brain AUC (μg/kg*hr) = 5130 and 1535, respectively) over the time course of the experiment with excellent brain/plasma ratios. The brain concentrations of 1 remained well above the in vitro MLK3 inhibition IC50 for over 6 hours after injection. The volume of distribution for 1 is within acceptable bounds and does not indicate that the compound will accumulate in tissues. The brain and plasma concentrations for 8 were very closely matched, giving B/P ratios of ~1 throughout the entire experiment. A B/P ratio of 1 suggests that the compound partitions fairly freely across the BBB, indicating few issues with PGP efflux pumps or accumulation of compound in brain tissue. However, compound 8 was rapidly metabolized in mice. In our experience with the 7-azaindole based inhibitors, polar surface area, molecular weight and number of hydrogen bond donors were most important in determining whether a compound would exhibit good CNS exposure. Thus compounds such as 12 with high polar surface area or 10 with large number of hydrogen bond donors, exhibited poor BBB penetration in this preliminary study (See Table 1.)
Table 1.
Screening PK Study (iv, 10 mg/kg, C57 BL/6 Mice)
| Parameter | 1 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|
| AUC (μg/L*hr) | 3410 | 1560 | 1400 | 1630 | 3150 | 1840 |
| T1/2 (hr) | 2.72 | 0.69 | 1.13 | 1.21 | 1.95 | 0.29 |
| Vzb (L/Kg) | 11.5 | 6.43 | 11.7 | 10.7 | 8.92 | 2.2 |
| CLz (L/hr/Kg) | 2.9 | 6.42 | 7.1 | 6.2 | 3.2 | 5.4 |
| Cmax (μg/L) | 1848 | 2744 | 4742 | 4823 | 9788 | 4250 |
| Brain AUC (μg/kg*hr) | 5130 | 1535 | 213 | 83 | 693 | 136 |
| Brain Cmax (μg/kg) | 4685 | 3736 | 530 | 314.5 | 834 | 283 |
| Brain/Plasma Ratio@1hr | 1.6 | 0.99 | 1.2 | BQLa | 1.02 | 0.1 |
. Below Quantification Level.
. Apparent volume of distribution during terminal phase.
Oral PK Study with Compound 1
To gauge the potential of compound 1 to function as a drug and to establish that oral dosing would be allowed in animal models, we initiated an oral PK study where compound 1 was dosed at 10 mg/kg in C57BL/6 mice (see tables 2 and 3). In order to determine %F; iv dosing was repeated at a lower concentration (2.5 mg/kg) to insure that metabolic mechanisms would not be saturated by a high dose of test article dissolved in DMSO/PEG400. Using iv (2.5 mg/kg) and PO (10 mg/kg) dosing in C57Bl/6 mice, analysis of plasma concentrations of compound 1 yielded standard pharmacokinetic parameters confirming proportionally similar exposures as measured by AUC levels at lower doses and similar half-life (terminal half-life iv = 2.14 hr, oral = 1.92 hr) and good oral bioavailability (%F = 41) using a simple standard oral dosing vehicle containing 0.4% Tween-80, and 0.5 % hydroxypropylmethyl cellulose in pH 7.4 buffered saline.
Table 2.
Low Dose iv Administration of Compound 1 in C57 BL/6 Mice (2.5 mg/kg)
| Parameter | 1 |
|---|---|
| AUC (ug/L*hr) | 1400 |
| T1/2 (hr) | 2.14 |
| Vz (L/Kg) | 5.6 |
| CLz (L/hr/Kg) | 1.8 |
| Cmax (ug/L) | 570 |
Table 3.
Oral Dosing of Compound 1 in C57 BL/6 Mice (10 mg/kg)
| Parameter | 1 |
|---|---|
| AUC (μg/L*hr) | 2300 |
| T1/2 (hr) | 1.92 |
| Cmax (μg/L) | 2670 |
| Tmax (hr) | 4.0 |
| %F | 41 |
Blood Brain Barrier Penetration of 2
Our stated goal was to identify a compound with significantly improved CNS penetration over 2. To rapidly assess and compare the ability of a large number of compounds to penetrate the CNS, we adopted a simple screening model utilizing C57 BL/6 mice which were also employed in our imaging efficacy assays.1 In the screening BBB penetration assay, mice were dosed iv at 10 mg/kg and brain and plasma levels are measured at three time points utilizing averages from three mice per time point. (See Table 4.) In this assay format 1 again achieved significant brain levels (e.g., at 3 hours the whole brain concentration was 950 ng/g, with a B/P ratio of 0.81). We utilized this rapid assessment of BBB penetration to compare 2 with our compounds. In this iv dosing assay, 2 showed significantly lower plasma and brain levels at all time points, with poor B/P ratios. For example, by three hours after iv dosing at the same starting dose (10 mg/kg), the brain concentration of compound 1 (ng/g) was measured as fourteen times higher than that observed for 2. While these numbers may not necessarily translate into high free fraction concentrations in human brain, taken together with human microsomal and hepatocyte stability data, they suggest that compound 1 is much more likely to achieve significant CNS levels. Our method does not determine free brain/free plasma fraction concentration ratios. This determination may be important when drugs are very tightly protein bound in the brain (>99%.) With very high protein binding the drug may be present in the brain but may not engage the therapeutic target. Compounds showing weaker protein binding (< 99%) obviously are in equilibrium with an apparent binding constant that is usually much weaker than the highly potent Kd or Km of drugs with significantly increased activity for the target kinase MLK3. It is well known that for such drugs, the protein bound fraction represents a reservoir of displaceable drug in equilibrium with free drug. We have provided data (see Figure 2) to rank order compounds for protein binding in human plasma. This data shows that protein binding for compound 1 is approximately 98%, and we expect the reservoir of drug in the brain to be in free equilibrium and available to bind to the MLK 3 target enzyme with high affinity (Ki ~ 10 nM). This conclusion is supported by the fact that the terminal half-life for brain concentrations for compound 1 has been measured in mice at between 1.5 and 2 hours; very close to the plasma half-life, suggesting that compound 1 in mice is not highly protein bound in the brain, and is available to be metabolized by systemic CYP450 enzymes with modest Km affinity and is also available for target engagement with high affinity for MLK3.
Table 4.
Blood Brain Barrier Penetration Comparison (iv dosing, 10 mg/Kg).
| Compound | Sample | B/P Ratio | B/P Ratio | B/P Ratio | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| 30 min | 60 min | 180 min | 30 min | 60 min | 180 min | ||
|
|
|||||||
| 1 | Plasmaa | 4163 | 2920 | 1167 | |||
| Brainb | 2833 | 2249 | 950 | ||||
| 0.68 | 0.77 | 0.81 | |||||
| 13 | Plasmaa | 3613 | 2566 | 936 | |||
| Brainb | 164 | 179 | 130 | ||||
| 0.04 | 0.07 | 0.138 | |||||
| 2 | Plasmaa | 2403 | 1716 | 326 | |||
| Brainb | 594 | 463 | 67 | ||||
| 0.25 | 0.27 | 0.21 | |||||
. (ng/mL).
. (ng/g).
A direct control analog of compound 1 with poor CNS penetration
We have also identified a very closely related analog (13) of compound 1 with very limited CNS penetration. While compound 13 had similar MLK3 potency and activity compared to compound 1 in simple kinase specificity screens, it had good systemic exposure but much lower brain levels when administered iv to C57Bl/6 mice, with B/P ratios of 0.04, 0.07, and 0.13 at 0.5, 1, and 3 hrs. This result was not fully anticipated based on in silico predictions or results with similar analogs. The formal replacement of carbon by nitrogen provides both a new basic center, and a site for hydrogen bonding. It may be that the side chain azaindole pyridine nitrogen alters the preferred solution conformation compared to compound 1, allowing better exposure to hydrogen bond interactions with that side chain with proteins or water. Such increased H-bonding could impede BBB penetration.
Metabolic stability, CYP450 inhibition, hERG inhibition
When 1 was tested in vivo in mice, the terminal half-life after oral and iv administration was measured as 2.1 and 1.9 hrs, respectively. Data were obtained for the stability of 1 in the presence of human microsomes (45%), as well as human (55%), monkey (55%) and mouse hepatocytes (73%), where the data is expressed as “% remaining after 1h”. The human microsome and hepatocyte stability data are similar to several marketed drugs used as standards in the assays, suggesting that while the half-life is not ideal for once a day dosing, it likely would be acceptable for a first-in-class drug for HAND where no known treatment currently exists. There were no significant CYP450 inhibition issues for 1 identified from the three most common isoforms of interest for drug/drug interactions (see Table 5). Kinase inhibitors possessing amine-solubilizing groups such as the piperazine present potential risk for inhibition of hERG channels, with associated risks of QT-prolongation. The automated Cerep whole cell patch-clamp technique37 (Qpatch 16, Sophion Biosciences) was used to record outward potassium currents from single cells transfected with the hERG channel. The IC50 for 1 was determined as 21 μM, and was rated as low cardiac toxicity risk by the CEREP compound evaluation criteria.
Table 5.
CYP450 Inhibition (IC50, μM)
| CYP450 | 1 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|
| 2D6 | > 30 | > 30 | 3.7 | > 30 | 13.2 | > 30 |
| 3A4 | 16.2 | > 30 | 4.2 | > 30 | 21.8 | > 30 |
| 2C9 | > 30 | > 30 | 13.7 | > 30 | > 30 | > 30 |
Inhibition of Tat-induced pro-inflammatory cytokine release by primary human monocytes
We tested whether 1 could regulate the HIV-1 Tat-induced release of a set of pro-inflammatory cytokines from primary human monocytes. We focused on TNF-α, MCP-1, IL-6, and IL-8, since these are believed to be of importance of the pathogenesis of HAND. 32–40
Compound 1 potently inhibited all of the mediators in a dose dependent fashion. Statistically significant inhibition, compared to Tat-only treatment, was achieved at the lowest drug concentration tested (100 nM) for TNFα and IL-6, and at 300 nM for MCP1; in the case of IL-8, more than 50% inhibition of chemokine release was attained by 300 nM of drug, although this did not achieve statistical significance due to high variation in the Tat-only control samples. Similar findings were observed in human monocyte-derived macrophages (MDM, data not shown) with the most potent inhibition of our inhibitors against MCP-1 and TNF-α release from MDM.
In Vivo Inhibition of HIV-1 Tat induced JNK Signaling with Compound 1
HIV-1 Tat activates JNK in neurons and other cell types, via an MLK-dependent pathway.14, 41–44 JNKs are major regulators of mammalian apoptotic cell death pathways, and have also been proposed to have important role in neurodegeneration.45 However, direct inhibition of JNK may not be an acceptable strategy for disease modification, since the role of JNK in cell death is so ubiquitous in different cell types, tissues, and organs. Further, there is significant JNK phosphorylation and activity in neurons from healthy brain tissue, indicating that JNK plays multiple roles in the healthy CNS. A selective strategy of interfering with HIV-induced signaling upstream of JNK, such as MLK signaling, may provide an opportunity to develop a more selective treatment for HAND with less risk of side effects and undesired toxicity as well as a potential treatment for other neurodegenerative diseases.
To verify in vivo inhibition of the HIV-1 Tat-induced JNK pathway activation by compound 1, C57BL/6 mice were injected intracerebrally with saline or HIV Tat1–72, 700μm deep in somatosensory cortex by stereotactic injection. This paradigm reproducibly results in sustained in vivo neuroinflammation after Tat injection.1 Half of the mice received i.p. pretreatment/treatment with 10mg/kg of compound 1, every twelve hours, which continued until sacrifice, with a total of 3 doses before Tat injection, and two doses post injection, while the other half were untreated (n = 3 for each of the four groups). Mice received the Tat injection at 4 to 6 hours after the third ip injection of compound 1. In separate single dose PK experiments plasma levels measured at 4 hours post injection of drug were 500 nM, and brain levels were also at 500 nM. At 6 hours post drug injection plasma levels were at 275 nM and brain levels were at 233 nM. Mice were anesthetized and sacrificed 24 hours after Tat injection. Levels of phosphorylated JNK (pJNK) were measured by Western blotting from whole brain lysates collected directly adjacent to the Tat injection site (the caudal medial quadrant of the left hemisphere). To control for variations in protein loading between experimental replicates, levels of pJNK were normalized to the expression of total α-Tubulin (see Fig. 5A). The untreated Tat exposed animals had a 75% increase in the levels of phosphorylated JNK compared to the saline control animals (Fig. 5B). Compound 1 treatment in the Tat exposed animals normalized phosphorylated JNK levels to those observed in saline controls (Fig. 5B) (*p<0.05. We analyzed data depicted in A-B by one-way ANOVA with Newman-Keuls post hoc test. Error bars indicate SEM), but the compound had no effect on pJNK levels in saline-treated animals.
Fig 5.

Intraperitoneal (i.p.) Compound 1 Treatment Prevents HIV-1 Tat Mediated Increase in JNK Phosphorylation In Vivo in Brain Tissue Ipsilateral to Stereotactically Injected Tat. Panel A depicts immunoblots of the expression of pJNK and α Tubulin in brain tissue of mice stereotactically injected with saline (Sa) vehicle and no treatment or Sa + systemic administration of 1 or stereotaxic injection of HIV-1 Tat and no treatment or stereotaxic injection of Tat with systemic administration of 1. Panel B shows densitometric analyses of the optical density of bands in Panel A, normalized to the expression of α Tubulin in each experimental replicate. * = p<0.05.
Molecular modeling studies with compound 1
Analysis of the potential binding modes of compound 1 were based on known SAR, as well as crystal structures of similar 7-azaindole based inhibitors co-crystallized in kinase active sites. Docking studies of compound 1 in the MLK1 ATP binding site were performed using the automated docking utilities contained within the molecular modeling suite MOE,46 as well as manual dockings and minimizations allowing complete flexibility of the protein and ligand. (Refer to Fig. 6.) MLK1 is an excellent model for MLK3 binding and 1 potently inhibits MLK1 (IC50 = 19 nM). In aggregate, these analyses suggested that the compound is likely a type 1 kinase inhibitor forming two hydrogen bonds to the hinge. The indole is likely positioned so that it may make potential key interactions with neighboring lysine, aspartic acid, asparagine, or phenylalanine residues. In these models the piperazine moiety interacts with a critical arginine residue and is also partially solvent exposed.
Fig. 6.

Potential Binding Site Interactions Analysis for MLK1 and Compound 1 based on manual docking and minimization studies.
To the best of our knowledge no X-ray crystal structure for MLK3 has yet been published. Cephalon has released an X-ray crystal structure (3DTC) of one of their inhibitors 6 co-crystallized in MLK1.22 All but one of the significant residues in the MLK1 ATP binding site are identical to MLK3 (Fig. 6). Phe222, present in the hinge region of MLK1, is a tyrosine in MLK3, however the side chain is outwardly facing and its impact is minimal on ligand binding. In our SAR studies we observed that the pyrrolo NH and the pyridine nitrogen of the scaffold were required for potency. In addition, for the side-chains, having an aromatic group such as an indole, aniline or phenol was associated with high in vitro enzyme inhibition potency as they provided a hydrogen bond donor from the para position of an aromatic ring.
In studying the sequences of related kinases, only MLK1, MLK3 and ZAP70 contained the Gly225-Gly226-Pro227 motif, positioned at the top of the hinge, however ten kinases contained a proline residue at the position equivalent to Pro227. One such kinase was spleen tyrosine kinase, SYK, which had been crystallized with the Rigel compound, R40647. In this structure the 3,4,5 trimethoxyphenyl group extended into the solvent exposed region where it was observed to buttress the proline and also interact with a lysine residue in the equivalent position to Arg203. (MLK3 numbering) This group was recognized within our SAR in early compounds such as 7 and 8 and it was proposed that a similar interaction could take place with this group extending into the solvent exposed region.
The 7-azaindole scaffold, in recent years, has been demonstrated to be a privileged structure for forming interactions with the hinge region of ATP binding sites of several kinases and these compounds are the subject of several patents and studies of binding fragments for several protein kinases including AKT1, IKKB, JAK3, SGK1 and ABL1 mutants.48 A binding mode analysis performed against kinase X-ray structures containing ligands with an azaindole scaffold revealed consistency with our binding mode hypothesis identified through docking studies with homology models and PDB 3DTC.
Of particular interest to us were 7-azaindole ligands bound in PDB 3ETA (insulin-like growth factor-1 receptor (IGF-1R) tyrosine kinase and 2Z60 (T315I mutant of ABL1) which exhibited a binding mode we had originally focused on from docking studies with homology models. In these structures the pyrrolo hydrogen of the inhibitor donates a hydrogen bond to the carbonyl of the Hinge +1 residue, and the pyridine nitrogen accepts a hydrogen bond from the hinge +3 N-H moiety. 3-position substituents would likely interact with Phe155 in MLK1 and Asn273. 5-Position substituents on the 7-azaindole scaffold may interact with Arg230 and may be largely solvent exposed allowing the incorporation of solubilizing groups. This model is consistent with all structure activity data.
Protein Kinase Specificity
Previously, little protein kinase specificity data has been published for MLK3 inhibitors. Compound 3 is a fairly promiscuous inhibitor.49 Compound 4 was originally identified as a potent inhibitor of FLT3 or JAK isoforms, but is also a fairly potent inhibitor of MLK kinases and a close analog of 2.49 Compound 6 was reported to be a potent inhibitor of MLK1 and a weak inhibitor of TrkA and Fyn.21 Compound 1 (MLK3 IC50 = 14 nM) inhibits both MLK1 (IC50 = 19 nM) as well as MLK2 (IC50 = 42 nM), and the related MLK family member DLK (IC50 = 150 nM) - as expected from the analysis of the active sites of these enzymes.
We initially utilized a screen of nine protein kinases to profile potential compounds (AMPK, CDK1, ERK2, GSK3B, LCK, P38a, SRK, SYK, JNK1, and ZAP70; see Table 6). In this panel of important diverse kinases from TK and AGC families only LCK, a fairly promiscuous kinase, yielded an IC50 potency below 500 nM. As interest in this compound progressed with uniformly superior activity in cellular and in vivo models of HAND, we began to explore reasons for the unique activity. Using a bioinformatics approach we identified six kinases which have known high affinity for closely related azaindole structures for further screening of the inhibition profile of 1. Two of these kinases had crystal structures of bound 7-azaindole inhibitors available to allow us to explore modeling of kinase specificity based on the observed sub-site interactions.
Table 6.
IC50s (nM) a Determined for Selected Protein Kinases for Compound 1
| Kinase | IC50 |
|---|---|
| AMPK | 1512 |
| AurA | 108 |
| AurB | 123 |
| AurC | 290 |
| CDK1 | 1125 |
| CDK2 | 1180 |
| c-MET | 177 |
| ERK2 | 6290 |
| FLT1 | 39 |
| GSK3B | >10,000 |
| IGF1R | 307 |
| IR | 200 |
| JNK1 | 3280 |
| LCK | 333 |
| MEKK2 | 661 |
| P38a | 12,050 |
| ROCK1 | 1030 |
| ROCK2 | 111 |
| SGK | 67 |
| SGK1 | 201 |
| SRC | 4330 |
| SYK | 731 |
| TRKA | 85 |
| TRKB | 217 |
| ZAP70 | 5050 |
| ABL1 | 6.8 |
. Average of two determinations (nanomolar). IC50s for various kinases in specificity screens were conducted by Reaction Biology, Inc. (Malvern, Pennsylvania) as described in ref. 51.
Although compound 1 had initially appeared fairly selective on the original screening panel, on these six selected kinases compound 1 exhibited significant potency for several of the kinases in these biochemical assays for inhibition of phosphorylation of substrates (Table 7). We expanded our investigation of specificity using the binding kinome scan ScanMax™ assay from DiscoverRx (originally conducted at Ambit Biosciences) so comparison could be made with compounds described in the literature.
Table 7.
IC50 Valuesa for Kinase Panel Selected by Bioinformatics Analysis of Kinase Ligands
| Compounds | ||
|---|---|---|
|
| ||
| Kinase /Assay | 1 | 13 |
| ABL1 (T315I) | 3 | 285 |
| CDK2 | 1180 | 720 |
| FLT3 | 4 | 9.5 |
| IKKa | 591 | 330 |
| IKKb | 257 | 288 |
| IR | 200 | 1290 |
| FLT3(MEF Cells) | 560 | 730 |
. Average of two determinations (nanomolar). IC50s for various kinases in specificity screens were conducted by Reaction Biology, Inc. (Malvern, Pennsylvania) as described in ref. 51. MEF cell experiments described in Experimental Section.
In this assay Compound 1 was screened against a panel of 442 kinases, and showed greater than 90% inhibition against 111 wild type human protein kinases at 1 μM. (Data in Supporting Information.) The ScanMax™ assay from DiscoverRx49,50 is widely used to compare kinase inhibitor specificity based on extrapolated kinase/inhibitor binding and to produce thermodynamic binding constants, Kd. The assay measures the ability of significantly modified kinases to bind to a solid support immobilized ligand and utilizes conditions not present in living cells, such as the absence of ATP. To confirm the results obtained with this assay, we therefore also conducted a scan of 342 human wild type kinases for inhibition of substrate phosphorylation using a high throughput ATP-P33 radiolabeled assay from Reaction Biology Corp, at a dose of 1 μM in the presence of 10 μM ATP (Data in Supporting Information).51 We followed up with individual radioligand binding data for ATP uptake to determine IC50 values for some of the more interesting kinases. (See tables 6 and 7) In the ScanMax binding format assay, at a concentration of 1 μM, compound 1 exhibited greater than 50% inhibition of binding for 265 kinases and showed greater than 99% inhibition of binding for 36 kinases. In the Reaction Biology format in a screen of 342 human wild type kinases compound 1 exhibited 50% or greater inhibition of 202 kinases, with 15 showing greater than 99% inhibition (see Supporting Information).
A large number of kinases showed very significant affinity for Compound 1 in the ScanMax binding assay. These included the following important kinases that showed >90% inhibition at 1 μM: ABL1, CDK11, CDK4, CDKL2, CLK1, CLK2, CLK4, DYRK1B, FLT3, KIT, MELK, PDGFRB, SRPK2, ALK, ARK5, AXL, IKKalpha, IKKbeta, ROCK1, TYK2. Additionally, several kinases that are potential targets for anti-neurodegenerative kinase inhibitor programs showed substantial inhibition: DLK (88%, Kd = 70 nM, IC50 = 150 nM), LRRK2 (92%, IC50 =11 nM), LRRK2 (G2019S mutant, 96%), MARK1 (92%), MARK2 (49%), IGF1R (48%), SGK1 (70%). Compound 1 is essentially inactive against two kinases known to interact with MLK3 (GSK3β IC50 > 10 μM, as well as AKT1) and several other pathway-related or interesting kinases including AKT2, AKT3, ERK1, ERK2, p38 (all isoforms), BRAF and EGFR. It is also noteworthy that 1 inhibited binding of JNK kinases potently in the ScanMax format, but that high JNK1, 2 and 3 activity was not confirmed in the biochemical assay measuring inhibition of substrate phosphorylation. For example: JNK1 showed 99% binding inhibition at 1 μM in the ScanMax assay, but the IC50 for phosphorylation inhibition was 3.2 μM.
Comparison Kinome Scan of Compound 2
To define the specificity of 2 and to compare and contrast activity at potential control nodes for anti-inflammatory and neuroprotective pathways we synthesized 252 as a standard and conducted kinase inhibition scans using the ScanMax assay to examine activity on 456 kinases - including most known human kinases and important mutants thereof. Compound 2 was characterized by 1H-NMR, LC-MS and high-resolution mass spectrometry and the activity was confirmed in enzyme inhibition assays. As expected, 2 was a very potent inhibitor of the three major MLK enzymes with IC50s (average of two determinations) for MLK3 = 6 nM, MLK2 = 2 nM, MLK1 < 1 nM (Refer to Experimental Section: Method B).
In the ScanMax binding format assay, at a concentration of 1 μM, 2 exhibited greater than 50% inhibition of binding of 185 kinases and showed greater than 99% inhibition of binding of 28 kinases including LRRK2, FLT3, CAMK1D, KIT, MLK1 and Aurora Kinases A and C. (See Fig. 7 and Supporting Information Section.) Overall, both compound 1 and 2 showed only modest kinase specificity with notable differences in selectivity. Compound 1 showed potent activity on the ABL1 family and important mutants, whereas 2 lacked potent activity in this family. In contrast, 2 showed high activity against certain CAMK family kinases, unlike compound 1. Activity against CAMK2A (84% inhibition for 2) and CAMK2D (87% inhibition for 2) is highly correlated with positive activity in micronucleus screening models for kinase induced chromosome damage, as are BRSK2 (94% inhibition), and AMPK-alpha2 (98% inhibition).53 Unlike 1, compound 2 also strongly interacted with a subset of AGC family kinases - inhibiting RSK1 and RSK4 and related kinases.
Fig 7.

Percent Inhibition Profiles in the DiscoverRx ScanMax Kinome Scan for Compounds 1 and 2.
Both compounds 1 and 2, as well as other MLK3 inhibitors not reported here, strongly interact with members of the STE family kinases (MAP4K4, TNIK, and MINK) involved in MAPK pathways. Our working hypothesis is that the remarkable effects observed in in vivo efficacy studies1 of 1 may be the result of Compound 1 being a “selectively non-selective kinase inhibitor.” Indeed recent strategies in kinase drug discovery now recognize that the most efficacious drugs modulate multiple kinase pathways, but a key issue remains in finding safe compounds with multiple activities.54 Two kinases, FLT3 and LRRK2, are both very potently inhibited by both Compound 1 and 2, and seem potentially interesting with regard to off-target and neuroprotective effects.
FLT3 Inhibition
To date every compound showing potent activity in our human monocyte assay for inhibition of cytokine induction by HIV-1 Tat have been potent inhibitors of FLT3 and MLK3. Moreover, a literature survey revealed that most previously described MLK3 inhibitors also potently inhibit FLT3 (Table 8).
Table 8.
Kinase Inhibitors with High Affinity for MLK3 and FLT357
| Kinase Inhibitor | FLT3a | MLK3a |
|---|---|---|
| BIBF-1120 | 3.8 | 700 |
| CEP-701 | 8.5 | 18 |
| EXEL-2880 | 0.9 | 340 |
| KW-2449 | 15 | 230 |
| Pazopanib | 1100 | 740 |
| PD-173955 | 150 | 550 |
| PKC-412 | 11 | 17 |
| R406 | 0.71 | 11 |
| Staurosporine | 2.9 | 20 |
| TAE-684 | 15 | 420 |
| VX-680 | 6.5 | 680 |
. ( ScanMax Binding Kd, nM)
FLT3 is a cytokine/growth factor receptor tyrosine kinase and is important for survival, proliferation, and differentiation of hematopoietic cells in bone marrow. Inhibitors of FLT3 are being developed for the treatment of acute myeloid leukemia (AML)55, and are also of interest for chronic neuroinflammatory and neurodegenerative conditions such as multiple sclerosis.56 It is therefore notable that all of the azaindole MLK3 inhibitors disclosed here exhibited significant FLT3 activity (Table 9).
Table 9.
IC50 (nanomolar) or percent inhibition at 1 μmolar, for FLT3 and LRRK2a
| Compound | FLT3 | LRRK2 |
|---|---|---|
| 7 | 88% | 2970 |
| 1 | 4.0 | 11 |
| 8 | 98% | 35 |
| 9 | 98% | 46 |
| 10 | 99% | 5 |
| 11 | 97% | ND |
| 12 | 98% | ND |
| 13 | 9.5 | ND |
. Average of two determinations
Global suppression of FLT3 is not expected to be benign, and potent FLT3 inhibitors such as compound 4 have caused suppression of platelet production.58,59 However, in practice it is difficult to achieve cellular and in vivo inhibition of FLT3. We explored the cellular activity of our inhibitors in a cell-based FLT3 phosphorylation assay, using a murine embryonal fibroblast (MEF) cell line that expresses a high level of exogenous full-length human FLT3-wt. Stimulation of these cells with human FLT3-Ligand results in FLT3 autophosphorylation. The IC50 for compound 1 against purified FLT3 was 4 nM, but in this cell-based assay, compound 1’s IC50 against FLT3 was 140-fold less (560 nM). The activity of compound 13 was similar (730 nM). These findings suggest that our azaindole MLK3 inhibitors exhibit only modest levels of activity against FLT3 in cells, and that FLT3 inhibition may not make a dominant contribution to the in vivo efficacy of compound 1.
LRRK2 Inhibition
Leucine-rich repeat kinase 2 (LRRK2) represents another interesting kinase implicated in neurodegenerative and inflammatory diseases, including familial Parkinson’s disease (which has been associated with the G2019S mutation in LRRK2) and Crohn’s disease.60 LRRK2 is potently inhibited by Compound 1 (IC50 = 11 nM), and 2 also showed 100% inhibition of LRRK2 binding and 98% inhibition of the LRRK2 (G2019S) mutant binding in the ScanMax assay at a concentration of 1 μM.
Interestingly, exposure of murine BV2 microglial cells to HIV-1 Tat results in phosphorylation of serine 935 on LRRK2, as well as release of pro-inflammatory cytokines and an increase in phagocytic activity. LRRK2 kinase inhibition attenuated Tat-induced cytokine release and phagocytosis, suggesting that LRRK2 may be a novel regulator of microglial inflammation in HAND.61
The role that kinases other than MLK3 play in the potent in vivo and in vitro activity of compound 1 in models of HAND is the subject of current investigation. Our hypothesis is that strong neuroprotective and anti-neuroinflammatory efficacy of compound 1 in our in vivo models for HAND are attributable to its ability to safely interfere with multiple kinase pathways that act cooperatively (or synergistically) to drive the pathogenesis of HAND. This has important implications for our understanding of HAND, and suggests that cooperative kinase-regulated gene networks may play a critical and previously under-appreciated role in this disease.
The knowledge we have gained from the structure activity relationships and requirements for metabolic stability and blood brain barrier penetration have allowed us to design second generation compounds not based on the 7-azaindole core, that are much more selective for MLK3 and are potently neuroprotective in rodent cells; but the details of biological activity deviate surprisingly from those exhibited by 2 and compound 1; the details of these studies will be published in due course.
Summary
Using potent and drug-like screening hits as a starting point, we employed a strategy to find CNS penetrant compounds based on low molecular weight, low polar surface area, limited number of hydrogen bond donors and well defined logD. By doing so, we identified a potent, orally bioavailable MLK3 inhibitor, with excellent pharmacokinetic properties and improved CNS penetration over previously developed MLK3 inhibitors. This compound has excellent activity in preclinical models for HIV associated neurocognitive disease1 and is undergoing safety testing for potential development. Compound 1 potently inhibits several key kinases involved in multiple inflammatory and neurodegenerative pathways - including MLK3 and LRRK2 - and is likely a ”selectively non-selective” kinase inhibitor which modulates cooperative kinase-regulated gene networks involved in the pathogenesis of HAND.
CHEMISTRY
Compounds were efficiently synthesized using sequential Suzuki couplings utilizing key intermediate 16. For routine synthesis of diverse analogs, best yields and purity were obtained by protecting the azaindole nitrogen as a tosyl group. The synthesis of compound 1 is illustrated in Scheme 1. 5-Bromo-1H-pyrrolo[2,3-b]pyridine 14 was iodinated with NIS to afford 15. The N-H was protected using TsCl and the resulting tosylate 16 was subjected to regioselective Suzuki coupling reaction using indole-5-boronic acid at room temperature to yield intermediate 17. The second Suzuki coupling with 4-formyl boronic acid with intermediate 17 gave the benzaldehyde 18. Reductive amination reaction with 1-methyl piperazine followed by hydrolysis of the tosyl group resulted in the desired product 1 in an overall efficient synthesis. Compounds 8, 9, 10, 11, and 12 were produced using the same synthetic approach.
Scheme 1.
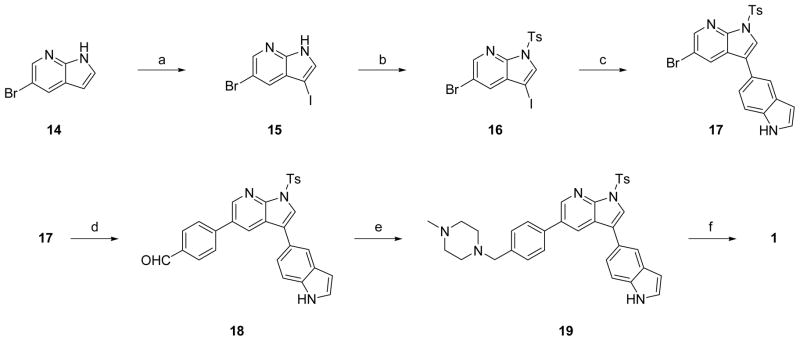
Synthesis of Compound 1a
aReagents and conditions: (a) NIS, acetone; (b) NaH, p-TSA, THF; (c) indole-5-boronic acid, PdCl2(PPh3)2, MeCN, aq. 2M Na2CO3; (d) 4-formylphenyl boronic acid, PdCl2(PPh3)2, MeCN, aq. 2M Na2CO3; (e) 1-methylpiperazine, Na(OAc)3BH, CH2Cl2; (f) aq. 5N NaOH, CH2Cl2, acetone.
Synthesis of compound 13 proceeded by similar sequential Suzuki couplings but required the protection of the side chain azaindole nitrogen to achieve best yields and purity. Suzuki coupling intermediate 16 with 7-aza-1H-indol-5-ylboronic acid provided 20, which was then tosylated to yield 21. The second Suzuki coupling produced intermediate aldehyde 22, which was reductively aminated and deprotected to yield compound 13.
Resynthesis of Screening Hit 7
Aminopyrazine 24 was brominated with NBS, and the resulting dibromo compound 25 underwent regioselective substitution with 5-aminoindole. Cyclization of 26 was accomplished by heating with carbonyl diimidazole. Suzuki coupling of 27, using standard conditions with the 3,4,5-trimethoxy substituted boronic acid provided compound 7.
EXPERIMENTAL SECTION
The structures of compounds synthesized in the examples below were confirmed using the following procedures. LC-MS/UV/ELS analysis was performed on instrumentation consisting of Shimadzu LC-10AD vp series HPLC pumps and dual wavelength UV detector, a Gilson 215 autosampler, a Sedex 75c evaporative light scattering (ELS) detector, and a PE/Sciex API 150EX mass spectrometer 5.0 μL injections were performed for each sample, on a Phenomenex Gemini 5μm C18 column. Mobile phases consisted of 0.05% formic acid in both HPLC grade water (A) and HPLC grade acetonitrile (B). 5.0 μL injections were performed for each sample, using gradient elution from 5% B to 100% B in 4 min at a flow rate of 2.0 mL/min with a final hold at 100% B of 1.8 min. UV (220 nm and 254 nm) and ELS data is collected for 4.5 min. All final compounds exhibited > 95% purity. Routine one-dimensional NMR spectroscopy was performed on a 300 MHz Varian Mercury-Plus spectrometer. The samples were dissolved in deuterated solvents obtained from Cambridge Isotope Laboratories, Inc., and transferred to 5 mm ID NMR tubes. The spectra were acquired at 293 K. C, H, N, Pd combustion analysis provided by Robertson Mircrolit Laboratories, High Resolution Mass Spectra were obtained by the UCSD Department of Chemistry Agilent 6230 ESI-TOF mass spectrometer service.
3-(1H-Indol-5-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (1)
In a round bottom flask 19 (12.4 g, 21.5 mmol) was dissolved in CH2Cl2 (105 mL) and MeOH (105 mL), then added 5N NaOH (210 mL). The resulting mixture was heated at 50 °C for 60 min. After completion of the reaction, the product was extracted using EtOAc and purified on silica gel chromatography. After the purification all combined batches were again dissolved in DCM (250mL), MeOH (250 mL) and THF (250 mL) and added quadra pure TU thiourea resin (10 g) and stirred overnight on rotary evaporator.. After filtration, the solvents were removed and re-dissolved in EtOH (450 mL) then heated to 55 °C. After 2h, HPLC grade water was added and kept the resulting mixture in freezer to afford white solid was collected by filtration to afford 3-(1H-indol-5-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[ 2,3-b]pyridine 1 (6.4 g, 70%). 1H NMR (DMSO-d6, 300 MHz): δ 11.82 (bs, 1H), 11.07 (bs, 1H), 8.54 (d, J = 1.2 Hz, 1H), 8.41 (d, J = 1.2 Hz, 1H), 7.89 (s, 1H), 7.76 (d, J = 1.5 Hz, 1H), 7.69 (d, J = 5.1 Hz, 2H), 7.48 (m, 2H), 7.39 (d, J = 4.8 Hz, 2H), 7.35 (m, 1H), 6.49 (dd, J = 2.7, 1.2 Hz, 1H), 3.48 (s, 2H), 2.50 (m, 3H), 2.38 (m, 5H); ESI (m/z) 422 (M+H), calc. 421. HRMS (EI) m / z calcd for C27H28N5 (M+H)+ 422.2341, found 423.2329. CHN.
(9S,10R,12R)-9,12-Epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, 5,16-bis[(ethylthio)methyl]-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo methyl ester (2) was synthesized from K252a (LC Laboratories, Woburn, MA) according to the method of Kaneko.52 1H NMR (DMSO-d6, 300 MHz): δ 9.13 (s, 1H), 8.63 (s, 1H), 7.95 (s, 1H), 7.88 (d, J = 5.1 Hz, 1H), 7.84 (d, J = 5.1 Hz, 1H), 7.46 – 7.44 (m, 2H), 7.10 (dd, J = 3.0, 4.2 Hz, 1H), 6.33 (s, 1H), 5.02 (d, J = 10.2 Hz, 1H), 4.95 (d, J = 10.2 Hz, 1H), 3.98 (s, 2H), 3.94 (s, 2H), 3.92 (s, 3H), 3.38 – 3.35 (m, 1H), 2.50 – 2.46 (m, 4H), 2.13 (s, 3H), 1.99 (dd, J = 3.0, 8.4 Hz, 1H), 1.23 (t, J = 2.4 Hz, 6H); MS ESI (m/z): 616.3 (M+H)+, calc. 616.2. HRMS (EI) m / z calcd for C33H33N3O5Na (M+Na)+ 638.1754, found 638.1755.
1-(1H-Indol-5-yl)-6-(3,4,5-trimethoxyphenyl)-1H-imidazo[4,5-b]pyrazin-2(3H)-one (7)
To a solution of 24 (27 mg, 0.08 mmol) in CH3CN (1 mL) in a Personal Chemistry microwave reaction vial was added 3,4,5-trimethoxyphenylboronic acid (17 mg, 0.08 mmol), bis(triphenylphosphine)-palladium(II) dichloride (6.0 mg, 0.008 mmol), and 1 M Na2CO3 (1 mL). The resulting mixture was de-gassed with Ar for 10 min, after which it was heated at 150°C for 10 min in a Personal Chemistry Optimizer. The organic layer was separated, filtered, and concentrated in vacuo. The residue was purified by preparatory HPLC to yield 7 (6.5 mg, 19%). 1H NMR (DMSO-d6, 300 MHz): δ 12.18 (s, 1H), 11.28 (s, 1H), 8.57 (s, 1H), 7.83 (d, J = 1.8 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.42 (m, 1H), 7.37 (dd, J = 1.8, 8.4 Hz, 1H), 7.20 (s, 2H), 6.51 (m, 1H), 3.78 (s, 6H), 3.66 (s, 3H); HPLC retention time: 2.30 minutes; HR-MS (ESITOFMS) 418.1511 (M+H), calc 418.1511.
3-(1H-Indol-5-yl)-5-(3,4,5-trimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridine (8)
1H NMR (DMSO-d6, 300 MHz): δ 11.78 (s, 1H), 11.03 (s, 1 H), 8.51 (d, J = 2.1 Hz, 1H), 8.36 (d, J = 1.8 Hz, 1H), 7.86 (s, 1H), 7.72 (d, J = 2.4 Hz, 1H), 7.45 (s, 2H), 7.32 (m, 1H), 6.92 (s, 2H), 6.45 (m, 1 H), 3.85 (s, 6H), 3.70 (s, 3H); MS ESI (m/z): 400.4 (M+H) +, calc. 399. HRMS (EI) m / z calcd for C24H21N3O3Na (M+Na)+ 422.1475, found 422.1476.
5-(3-(1H-Indol-5-yl)-1H-pyrrolo[2,3-b]pyridin-5-yl)pyridin-2-amine (9)
1H NMR (DMSO-d6, 300 MHz): δ 11.73 (d, J = 1.8 Hz, 1H), 11.05 (s, 1 H), 8.43 (d, J = 2.4 Hz, 1H), 8.29 (d, J = 1.8 Hz, 1H), 8.27 (d, J = 2.1 Hz, 1H), 7.88 (s, 1H), 7.76 (dd, J = 2.4, 8.4 Hz, 1H), 7.46 (s, 2H), 7.33 (m, 1H), 6.55 (dd, J = 0.6, 8.7 Hz, 1H), 6.46 (m, 1 H), 5.99 (s, 2 H). HPLC retention time: 1.10 minutes. MS ESI (m/z): 326.2 (M+H)+, calc. 325. HRMS (EI) m / z calcd for C20H16N5 (M+H)+ 326.1400, found 326.1401.
5-(5-(3,4,5-Trimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)pyridin-2-amine (10)
1H NMR (DMSO-d6, 300 MHz): δ 11.82 (s, 1H), 8.53 (d, J = 1.8 Hz, 1H), 8.31 (d, J = 1.8, 1 H), 8.28 (d, J = 1.5 Hz), 7.76 (dd, J = 2.1, 8.4 Hz, 1 H), 7.70 (d, J = 2.4 Hz, 1H), 6.95 (s, 2H), 6.54 (d, J = 8.4 Hz, 1 H), 5.87 (s, 2H), 3.86 (s, 6H), 3.68 (s, 3H); MS ESI (m/z): 377.4 (M+H), calc. 376. HRMS (EI) m / z calcd for C21H21N4O3 (M+H)+ 377.1608, found 377.1607.
5-(3-(1H-Indol-5-yl)-1H-pyrrolo[2,3-b]pyridin-5-yl)pyrimidin-2-amine (11)
MS ESI (m/z): 327.2 (M+H), calc. 326; HRMS (EI) m / z calcd for C19H15N6 (M+H)+ 327.1353, found 327.1354.
5-(5-(3,4,5-Trimethoxyphenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-2-amine (12)
MS ESI (m/z): 378.4 (M+H), calc. 377; HRMS (EI) m / z calcd for C20H20N5O3 (M+H)+ 378.1561, found 378.1563.
5-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl)-1Hpyrrolo[2,3-b]pyridine (13)
To a solution of 23 (4.00 g, 5.46 mmol) in MeOH (20 mL) was added NaOH (723 mg, 16.4 mmol). The resulting mixture was heated at 50 °C for 60 min. After completion of the reaction, the product was extracted using 250 mL IPA:CHCl3 (1:3) and water. The organic portions were dried and concentrated in vacuo. The crude product was purified on a silica gel column to afford 13 (1.57 g, 68%). 1H NMR (DMSO-d6, 300 MHz): δ 11.96 (s, 1H), 11.63 (s, 1H), 8.61 (d, J = 1.5 Hz, 1H), 8.56 (d, J = 1.2 Hz, 1H), 8.41 (d, J = 0.9 Hz, 1H), 8.31 (d, J = 1.2 Hz, 1H), 7.89 (d, J = 1.5 Hz, 1H), 7.73 (d, J = 4.8 Hz, 2H), 7.49 – 7.48 (m, 1H), 7.40 (d, J = 4.8 Hz, 2H), 6.51 – 6.49 (m, 1H), 3.49 (s, 2H), 2.38 – 2.36 (m, 4H), 2.21 (s, 3H); ESI (m/z) 423.4 (M+H)+, calc. 422; HRMS (EI) m / z calcd for C26H27N6 (M+H)+ 423.2292, found 423.2295.
5-Bromo-3-iodo-1H-pyrrolo[2,3-b]pyridine (15)
In a 3 liter round bottom flask 5-bromo-1H-pyrrolo[2,3-b]pyridine (63g, 319 mmol) was dissolved in acetone 1500 mL. To the stirred mixture was added NIS (79.1g, 351 mmol) and the resulting mixture was stirred for 1.5 hour; the precipitated solid was collected by filtration and washed with cold acetone (400mL) to afford 15 (89.8 g, 88% yield) as white solid. 1H NMR (DMSO-d6, 300MHz) δ 12.35 (bs, 1H), 8.31 (d, J = 1.5 Hz, 1H), 7.86 (dd, J = 1.2 Hz, 0.3 Hz, 1 H), 7.80 (d, J = 1.5 Hz, 1H); MS ESI (m/z): 322/324 (M+H)+, calc. 323.
5-Bromo-3-iodo-1-tosyl-1H-pyrrolo[2,3-b]pyridine (16)
To a stirred solution of 15 (45.0g, 139 mmol) in 700 mL of anhydrous THF cooled to 0°C with an ice bath was added NaH [60% dispersion in mineral oil] (8.3 g, 208 mmol). The reaction mixture was stirred for 20 min at 0°C, after which p-toluenesulfonyl chloride (29.1 g, 153 mmol) was added. The resulting mixture was stirred at 0 °C for 1.5 hr, after confirmation of completion by LCMS and TLC, the reaction mixture was warmed to room temperature, evaporated to dryness and quenched with water. The crude product was mixed with EtOAc (1000 mL) refluxed for 1h and then hexane (500mL) was added to precipitate the product 16 (60 g, 90% yield) as a light yellow powder. 1H NMR (DMSO-d6, 300MHz) δ 8.50 (d, J = 1.5 Hz, 1H), 8.21 (s, 1H), 8.00 (d, J = 4.8 Hz, 0.3 Hz, 2H), 7.98 (d, J = 1.5 Hz, 1H), 7.44 (dd, J = 4.8 Hz, 0.3 Hz, 2H), 2.34 (s, 3H); MS ESI (m/z): 477.0/479.0 (M+1)+, calc. 477.
5-Bromo-3-(1H-indol-5-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (17)
To a stirred suspension of 16 (60 g, 125 mmol) and 1H-indol-5-ylboronic acid (22.2 g, 138 mmol) in CH3CN (625 mL) was added 1 M Na2CO3 (312 mL) followed by bis(triphenylphosphine)palladium(II) dichloride (4.4 g, 6.2 mmol). The resulting mixture was stirred at room temperature for 1 hour. After complete consumption of starting materials the mixture was extracted with EtOAc and evaporated to dryness in vacuo, it was dissolved in CH2Cl2 (50 mL), absorbed onto Celite, and dried. The residue was purified via silica gel chromatography using CH2Cl2 as the eluent to obtain 17 (38.6 g, 65% yield). 1H NMR (DMSO-d6, 300 MHz): δ 11.21 (bs, 1H), 8.52 (d, J = 1.2 Hz, 1H), 8.47 (d, J = 1.5 Hz, 1H), 8.13 (s, 1H), 8.05 (d, J = 5.1 Hz, 2H), 7.92 (s, 1H), 7.51 (d, J = 4.8 Hz, 1H), 7.46 (dd, J = 5.1, 1.2 Hz, 1H), 7.43 (d, J = 5.1 Hz, 1H), 7.40 (dd, J = 3.9, 1.8 Hz, 2H), 6.52 (dd, J = 2.7, 1.2 Hz,1 H), 2.33 (s, 3H); MS ESI (m/z): 466.2/468.2 (M+H)+, calc. 466.
4-(3-(1H-Indol-5-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (18)
To a solution of 17 (29.5 g, 63.3 mmol) in CH3CN (315 mL) in a round bottom flask was added 4-formylphenylboronic acid (11.4 g, 76 mmol), bis(triphenylphosphine)-palladium(II) dichloride (4.4 g, 6.3 mmol), and 1 M Na2CO3 (160 mL). The resulting mixture was heated to reflux for 2.5 hours. The reaction was cooled to room temperature; the precipitated product was filtered and dried. The organic layer was extracted with EtOAc and washed with brine and evaporated to dryness to afford more crude material. The filtered solid and crude and material from evaporation were re-dissolved in CH2Cl2, absorbed on Celite and purified via silica gel chromatography using CH2Cl2 as the eluent to afford 18 (38.6 g, 65% yield). 1H NMR (DMSO-d6, 300 MHz): δ 11.21 (bs, 1H), 10.07 (s, 1H), 8.81 (d, J = 1.2 Hz, 1H), 8.53 (d, J = 1.2 Hz, 1H), 8.13 (s, 1H), 8.08 (d, J = 5.1 Hz, 2H), 8.02 (m, 5H), 7.53 (dd, J = 5.1 Hz, 2H), 7.45 (d, J = 5.1 Hz, 2H), 7.46 (dd, J = 3.0, 1.5 Hz, 1H), 6.52 (dd, J = 2.7, 1.2 Hz,1 H), 2.34 (s, 3H); MS ESI (m/z): 492 (M+H)+, calc. 491.
3-(1-Tosyl-1H-indol-5-yl)-5-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-pyrrolo[2,3-b]pyridine (19)
To a solution of 18 (22 g, 44.8 mmol) in CH2Cl2 (448 mL) was added 1-methylpiperazine (8.9 g, 89 mmol) and sodium triacetoxyborohydride (14.2 g, 67.2 mmol). The reaction mixture was stirred for 1 hr at room temperature, after which it was partitioned between CH2Cl2 and brine. The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The crude product was purified on silica gel column to give 15 (17 g, 68% yield). 1H NMR (DMSO-d6, 300 MHz): δ 11.23 (bs, 1H), 8.70 (d, J = 1.2 Hz, 1H), 8.40 (d, J = 1.5 Hz, 1H), 8.09 (d, J = 0.9 Hz, 2H), 8.07 (s, 1H), 7. 98 (s, 1H), 7.71 (d, J = 5.1 Hz, 2H), 7.52 (s, 2H), 7.41 (m, 5H), 7.24 (d, J = 4.5 Hz, 1H), 7.16 (dd, J = 10.8, 4.5 Hz, 2H), 6.52 (dd, J = 2.7, 1.2 Hz, 1H), 3.53 (s, 2H), 2.57 (m, 3H), 2.34 (m, 5H) 2.29 (s, 3H); MS ESI (m/z): 576 (M+H)+, calc. 575.
5-(5-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-1H-pyrrolo[2,3-b]pyridine (20)
To a stirred suspension of 16 (60 g, 125 mmol) and 7-aza-1H-indol-5-ylboronic acid (22.2 g, 138 mmol) in CH3CN (625 mL) was added 1 M Na2CO3 (312 mL) followed by bis(triphenylphosphine)palladium(II) dichloride (4.4 g, 6.2 mmol). The resulting mixture was stirred at room temperature for 1 hour. After complete consumption of starting materials the mixture was extracted with EtOAc and evaporated to dryness in vacuo, it was dissolved in CH2Cl2 (50 mL), absorbed onto Celite, and dried. The residue was purified via silica gel chromatography using CH2Cl2 as the eluent to obtain 20 (38.6 g, 65% yield). 1H NMR (DMSO-d6, 300 MHz): δ 11.21 (bs, 1H), 8.52 (d, J = 1.2 Hz, 1H), 8.47 (d, J = 1.5 Hz, 1H), 8.13 (s, 1H), 8.05 (d, J = 5.1 Hz, 2H), 7.92 (s, 1H), 7.51 (d, J = 4.8 Hz, 1H), 7.46 (dd, J = 5.1, 1.2 Hz, 1H), 7.43 (d, J = 5.1 Hz, 1H), 7.40 (dd, J = 3.9, 1.8 Hz, 2H), 6.52 (dd, J = 2.7, 1.2 Hz,1 H), 2.33 (s, 3H); MS ESI (m/z): 466.2/468.2 (M+H)+, calc. 466.
5-(5-Bromo-1-tosyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (21)
To a stirred solution of 20 (1.01 g, 2.17 mmol) in 20 mL of anhydrous DMF was added NaH [60% dispersion in mineral oil] (130 mg, 3.25 mmol). The reaction mixture was stirred for 20 min at room temperature, after which p-toluene sulfonyl chloride (538 mg, 2.82 mmol) was added. The resulting mixture was stirred at room temperature for 2 hr. The mixture was evaporated to dryness, quenched with water. The crude product was mixed with EtOAc (100 mL) and extracted. The organic solution was dried and evaporated in vacuo. The residue was purified via silica gel chromatography eluting with 20% EtOAc in hexanes to obtain 21 as a white solid (1.21 g, 90%). 1H NMR (DMSO-d6, 300MHz) δ 8.76 (d, J = 1.2 Hz, 1H), 8.56 (d, J = 1.2 Hz, 1H), 8.54 (d, J = 1.2 Hz, 1H), 8.45 (d, J = 1.5 Hz, 1H), 8.39 (s, 1H), 8.04 – 8.01 (m, 4H), 7.96 (d, J = 2.4 Hz, 1H), 7.43 (d, J = 4.5 Hz, 4H), 6.87 (d, J = 2.4 Hz, 1H), 2.34 (s, 6H); MS ESI (m/z): 622.4/624.1 (M+1)+, calc. 621.
4-(1-Tosyl-3-(1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (22)
To a solution of 21 (1.32 g, 2.12 mmol) in CH3CN (20 mL) in a round bottom flask was added 4-formylphenylboronic acid (349 mg, 2.33 mmol), bis(triphenylphosphine)-palladium(II) dichloride (149 mg, 0.212 mmol), and 1 M Na2CO3 (20 mL). The resulting mixture was heated to reflux for 3 hours and then cooled to room temperature. The precipitated product was filtered and dried. The organic layer was extracted with EtOAc and washed with brine and evaporated to dryness to afford more crude material. The filtered solid and crude material from evaporation were re-dissolved in CH2Cl2, absorbed on Celite and purified on silica gel column chromatography to yield 22 as a white solid (822 mg, 60% yield). MS ESI (m/z): 647.2 (M+H)+, calc. 646.
5-(5-(4-((4-Methylpiperazin-1-yl)methyl)phenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-1-tosyl-1H-pyrrolo[2,3-b]pyridine (23)
To a solution of 22 (3.35 g, 5.18 mmol) in CH2Cl2 (50 mL) was added 1-methylpiperazine (1.15 mL, 10.4 mmol) and sodium triacetoxyborohydride (2.19 g, 10.4 mmol). The reaction mixture was stirred for 1 hr at room temperature, after which it was partitioned between CH2Cl2 and brine. The organic layer was separated, dried over MgSO4, and concentrated in vacuo. The crude product was purified on silica gel column to afford 23 (2.65 g, 70% yield). 1H NMR (DMSO-d6, 300 MHz): δ 8.83 (d, J = 1.2 Hz, 1H), 8.71 (d, J = 1.2 Hz, 1H), 8.52 (d, J = 1.5 Hz, 1H), 8.44 (d, J = 1.2 Hz, 1H), 8.36 (s, 1H), 8.07 (d, J = 5.1 Hz, 2H), 8.02 (d, J = 5.1 Hz, 2H), 7.96 (d, J = 2.4 Hz, 1H), 7.71 (d, J = 4.8 Hz, 2H), 7.43 (d, J = 5.1 Hz, 4H), 7.38 (d, J = 4.8 Hz, 2H), 6.87 (d, J = 2.4 Hz, 1H), 3.48 (s, 2H), 2.40 – 2.25 (m, 8H), 2.14 (s, 3H); MS ESI (m/z): 731.4 (M+H)+, calc. 730.
3,5-Dibromopyrazin-2-amine (25)
To a stirred solution of aminopyrazine 24 (8.21 g, 86.4 mmol) in anhydrous methylene chloride (215 mL) cooled to 0°C was added N-bromosuccinimide (32.3 g, 181 mmol) in portions over a six hour period, during which time the temperature of the reaction was kept below 0°C. The resulting mixture was stored at 4°C overnight, after which it was stirred vigorously and quenched with H2O (100 mL). The organic layer was separated, after which it was washed with saturated aqueous NaHCO3, washed with brine, dried over MgSO4, filtered, and evaporated in vacuo to yield a residue that was triturated with 20% EtOAc in hexanes to yield 25 (10.3 g, 47%) as a yellow/brown powder. 1H NMR (CDCl3, 300MHz) δ 8.02 (s, 1H), 5.05 (bs, 2H); HPLC retention time: 1.99 minutes; MS ESI (m/z): 252.0/254.0/256.2 (M+1)+, calc. 251.
6-Bromo-N2-(1H-indol-5-yl)pyrazine-2,3-diamine (26)
To a stirred suspension of 25 (3.48 g, 13.7 mmol) and 1H-indol-5-amine (2.00 g, 15.0 mmol) in EtOH (3.5 mL) was added diisopropylethylamine (2.60 mL, 15.0 mmol). The resulting mixture was stirred for 48 hr at 80°C, after which it was partitioned between EtOAc and H2O. The organic layer was separated, after which it was washed with brine, dried over Na2SO4, filtered, and evaporated in vacuo to yield a residue that was purified via silica gel chromatography eluting with 1:1 EtOAc:hexanes to yield 26 (1.75 g, 42%) as a red/brown solid. 1H NMR (DMSO-d6, 300 MHz): δ 10.98 (s, 1H), 8.22 (s, 1H), 7.83 (s, 1H), 7.31–7.28 (m, 3H), 7.19 (d, J = 8.7 Hz, 1H), 6.43 (s, 2H), 6.36 (s, 1H); HPLC retention time: 2.07 minutes; MS ESI (m/z): 304.2/306.2 (M+H)+, calc. 303.
6-Bromo-1-(1H-indol-5-yl)-1H-imidazo[4,5-b]pyrazin-2(3H)-one (27)
To a solution of 26 (0.450 g, 1.48 mmol) in THF (5 mL) was added carbonyldiimidazole (1.20 g, 7.40 mmol). The resulting mixture was heated at 65°C for 48 hr, after which it was concentrated in vacuo and partitioned between EtOAc and H2O. The organic layer was separated, dried over MgSO4, filtered, and concentrated in vacuo to yield a residue that was purified via silica gel chromatography eluting with EtOAc to yield 27 (0.20 g, 41%). HPLC retention time: 2.07 minutes; MS ESI (m/z): 330.2/332.2 (M+1) +, calc. 329.
LPS-induced TNFα release from microglial BV2 Cells
A TNFα release assay in LPS-stimulated BV-2 cells was performed essentially as described.62 Briefly, mouse microglial BV-2 cells were treated with test compounds followed by LPS (100ng/ml final concentration), and culture supernatants were harvested 8 hours thereafter for TNFα ELISA.
Screening PK (iv dosing) in Mice
Three mice were used for each time point. Male C57/BL/6 mice were dosed iv (10 mg/mL) by tail vein injection of a solution of 2 mg/mL in solutions containing the indicated compound and vehicles. (Compounds 1, 9, 10, 11 and 13: 5% DMSO, 40% PEG-400, 55% saline; Compounds 8 and 12: 5% DMSO, 40% PEG-400, 55% H2O containing 20% HP-βCD). Blood samples of approximately 0.30 μL were collected from each mouse (n = 3 mice per time point) by retro-orbital bleed while the animals were anesthetized with isoflurane. Blood samples were collected in tubes containing sodium heparin as the anticoagulant, predose and at 0.083, 0.25, 0.5, 1, 2, 4, 6, 8 and 24 h postdose. Samples were centrifuged within 1 h of collection and plasma was collected and stored at −20 °C until analysis. Total concentrations of the compound were determined by liquid chromatography–tandem mass spectrometry (LC-MS/MS), following plasma protein precipitation with acetonitrile and injection of the supernatant onto the column (XTerra®MS C18, 5 μm, 4.6 × 50 mm). The LC system comprised an Agilent (Agilent Technologies Inc., USA) 1100 series liquid chromatography equipped with G1379A degasser, G1311A Quantpump, G1313A autosampler and G1316A Column Oven. Mass spectrometric analysis was performed using an API4000 (triple-quadrupole) instrument from AB Inc. (Canada) with an ESI interface.
The aqueous mobile phase was water with 0.1% formic acid, and the organic mobile phase was methanol with 0.1% formic acid. The lower and upper limits of quantitation of the assay were 2.5 ng/mL and 5000 ng/mL based on known standards, respectively. Brains were collected from three different animals at each time point, rinsed with ice-cold saline, weighed, and stored at −80 °C until analysis. For compound quantitation, mouse brains were homogenized in 5 volumes of water. The homogenates were extracted by protein precipitation with acetonitrile. LC-MS/MS analysis was conducted as described for the plasma. Brain homogenate concentrations were converted to brain concentrations for the calculations of brain to plasma ratios.
Brain Penetration Comparison for Compounds 1, 2, and 13
Compounds were dissolved in 5% DMSO, 40% PEG-400 and 55% saline to yield a nominal concentration of 2 mg/mL (pH = 8) and were dosed at 10 mg/kg in C57BL/6 mice by tail vein injection. Samples were collected as described above at 30, 60 and 180 minutes post-dose. Mice were anesthetized, sacrificed and blood collected by retro-orbital bleeds, and brains collected. (3 time points over 24 hours, 3 mice per time point). Concentration of compounds was determined in plasma and brain samples as described above.
Detailed Oral PK Study for Compound 1 in C57/Bl/6 mice
I.V. Dosing for %F determination: Male C57/BL/6 mice were dosed iv (2.5 mg/Kg ) by tail vein injection of a solution of 2 mg/mL of compound 1 dissolved in 5% DMSO, 40% PEG-400, 55% saline. Blood samples of approximately 0.30 μL were collected from each mouse (n = 3 mice per time point) by retro-orbital bleed while the animals were anesthetized with isoflurane. Blood samples were collected in tubes containing sodium heparin as the anticoagulant, pre-dose and at 0.083, 0.25, 0.5, 1, 2, 4, 7and 24h post-dose. Samples were centrifuged within 1 h of collection and plasma was collected and stored at −80 °C until analysis.
Oral Dosing for %F determination: Compound was dosed by oral gavage (10 mg/ kg) as a suspension of 1 mg/mL of 1 in a solution of 0.5% hydroxypropyl methylcellulose, 0.4 % Tween-80 in pH 7.4 PBS buffer. Blood samples were collected in tubes containing sodium heparin as the anticoagulant, pre-dose and at 0.25, 0.5, 1, 2, 4 6, 8 and 24 hrs post dose. Total concentrations of the compound were determined by liquid chromatography–tandem mass spectrometry (LC-MS/MS) as described above.
Stability in Human Liver Microsomes
Stability of the test compound was determined in human liver microsomes by Cerep Inc., using standard assay conditions of 1 micromolar concentration of the test compound. Compound concentration was determined by HPLC, and reported values are the average of duplicate values.
Stability in Hepatocytes
Stability of the test compound was determined in cryo-preserved human, cynomolgus monkey, and CD-1 mice hepatocytes, by Cerep Inc., using standard assay conditions of 1 micromolar concentration of the test compound. Compound concentration was determined by HPLC, and reported values are the average of tripicate values.
CYP2C9, 2D6 and 3A4 Inhibition
Test compound was incubated with pooled human liver microsomes at 37 °C in 0.1 M Tris buffer, pH 7.4, and its effect on the metabolism of probe substrates for CYP enzymes determined (2D6: Diclofenac, 3A4:Dextromethorphan, 2C9: Midazolam). The compound was tested at 6 concentrations ranging from 0.12 μM to 30 μM. Conditions of incubation in this assay have been optimized to maintain first order reaction conditions and to minimize the potential for non-specific binding of probe or study compound. Reactions were terminated with acetonitrile containing analytical internal standard (carbamazepine), samples centrifuged to remove microsomal protein and analysed using optimized HPLC and MS conditions. The MS responses for the solvent control samples were taken as the 100% reference values against which the inhibition of metabolism was measured. IC50 values were calculated using a sigmoidal dose-response equation within GraphPad Prism.
Inhibition of HIV Tat-induced cytokine release in primary human monocytes
This assay was performed essentially as described.14,63 Briefly, human monocytes were isolated from freshly collected whole blood using CD14 immunomagnetic beads (Miltenyi-Biotec), plated in 24-well plates and incubated with the specified compounds at the indicated concentrations (100, 3000, 1000 nM); in control wells, no compound was added. 30 min later, HIV-1 Tat was added to a final concentration of 50 nM; the cells were incubated for 8 hours (in control wells, nothing was added [NT]). Cell supernatants were then collected, centrifuged to remove debris, transferred to new microcentrifuge tubes and frozen at −20°C. A Luminex bead array assay was then performed, to quantitate the indicated chemokines and cytokines. Results were measured in triplicate or quadruplicate, and data are presented as mean values; error bars denote the standard deviation. Note that similar results were obtained with monocytes derived from multiple (n>5) different donors, as well as in terminally differentiated monocyte-derived macrophages (data not shown). * = p < 0.05; one-way ANOVA with Bonferroni’s correction (when compared to Tat only control).
Inhibition of JNK phosphorylation in HIV-Tat treated mice
Wild-type C57BL/6 mice were either pretreated with 3 doses of 10 mg/kg Compound 1 spaced 12 hours apart (n=6) or were left untreated (n=6). Treatment with Compound 1 continued every 12 hours throughout the duration of the experiment. Half of the mice (3 pretreated with Compound 1, 3 untreated) received a stereotactic injection of 3μl of 3μg/μl HIV-1 Tat1–72 in PBS into the somatosensory cortex at the coordinates 1.0mm posterior to Bregma, 1.0mm lateral left of Bregma, and 0.7mm ventral to the pial surface. The remaining mice received an injection of 3μl sterile PBS at the same coordinates. A 35-gauge needle with a 10μl Hamilton syringe controlled by a micro-syringe pump (Micro4 World Precision Instruments) was used to perform the injections, which were delivered at a flow rate of 80 nl/minute to minimize brain injury occurring as a result of injection pressure. The needle and syringes were coated with Sigmacote (Sigma SL-2) to prevent the Tat from sticking to the inside of the syringe. 24 hours after injection, the mice were sacrificed by pentobarbital overdose; animals were then briefly transcardially perfused with icecold saline, and the brains were removed, sectioned, and flash frozen on dry ice.
The brain tissue of interest was homogenized in a glass homogenizer in a solution of 1X Tris-buffered saline pH 7.4 with 0.05% Tween and protease and phosphatase inhibitors (#161280; Thermo Fisher Scientific). The homogenate was spun down at 13,000 rcf for 15 minutes to remove insoluble debris and the lysis supernatant was collected. The protein concentrations were measured and normalized using a Bradford assay. 12 μg of protein sample was then mixed with loading dye, boiled for 5 minutes, and run on a 4% to 15% SDS-PAGE gel (BioRad 456–1086) at 100 V. The gel was transferred onto a nitrocellulose membrane at 100 V for 66 minutes on ice. The membranes were blocked in 5% milk in 1X Tris-buffered saline with 0.05% Tween (TBS-T) for 1 hour at room temperature with shaking. The membranes were washed 3 times in TBS-T for 10 minutes a wash. The primary antibody against phosphorylated JNK (Cell Signaling 4668P) was applied overnight at 4°C at a concentration of 1:2000 in TBS-T with 5% milk. The next day, the membranes were washed 3 times in TBS-T for 10 minutes a wash. The horseradish peroxidase (HRP)-conjugated secondary antibody (BioRad 170-6515) was applied at a concentration of 1:11,000 in 5% milk TBS-T for 45 minutes at room temperature with shaking. The membranes were washed 3 times in TBS-T and enhanced chemiluminescence (ECL) substrate (Thermo 34076, Thermo 34080) was applied for 3 minutes. The membranes were developed on film (Thermo 34091). In order to control for variations in protein loading, the membranes were stripped (Millipore 2504) and then blocked in 5% milk TBS-T for 30 minutes. The mouse anti-α-tubulin (Sigma T5168) was applied in 5% milk TBS-T overnight with shaking. The process of washing, secondary application, and developing was repeated in order to obtain the α-tubulin loading control blot. The optical density measurements used for quantification were obtained using ImageJ. * = p < 0.05; one-way ANOVA with Bonferroni’s correction.
Discover RX ScanMax Kinome Binding Scan
Compounds that bind the kinase active site directly (sterically) or indirectly (allosterically) prevent kinase binding to the immobilized ligand, and will reduce the amount of kinase captured on a solid support. Conversely, test molecules that do not bind the kinase have no effect on the amount of kinase captured on the solid support. Screening “hits” are identified by measuring the amount of kinase captured in test versus control samples by using a quantitative qPCR method that detects the associated DNA label to determine a Kd for ligand binding. Compounds 1 and 2 were tested at 1 μM concentration against a panel of 442 known protein kinases. Data are presented as percent of control activity remaining. (0% indicates very tight binders, 100% indicates no binding). Complete data are presented in the supporting information section.
Reaction Biology Wild Type Human Kinome Inhibition Scan
Compound 1 was tested against 342 wild type human kinases for inhibition of protein phosphorylation using the Reaction Biology Inc. Hot Spot™ P33 radio binding assay.51 The compound was tested in single dose duplicate mode at a concentration of 1 μM. Control Compound was tested in 10-dose IC50 mode with 3-fold serial dilution starting at 20 μM. Reactions were carried out at 10 μM ATP. Complete data are available in the supporting information section.
Kinase Specificity Inhibition Data
Kinase Inhibition IC50 values were determined from 10 point curves. IC50s for various kinases in specificity screens were conducted by Reaction Biology, Inc. (Malvern, Pennsylvania) as described.51
Radiometric filter plate MLK3 assay (Method A)
200ng (130nM) MLK3 (Dundee, DU8313) was incubated with 1μM unactive MKK7b (Dundee, DU703) in the presence of 2μM cold ATP (Km) and 0.5μCi/assay 33P ATP and appropriate concentrations of compounds. After a twenty-minute incubation, the reactions were washed through filter plates and read on a scintillation counter.
Biochemical assay for the inhibition of kinase activity for MLK3 (Method B)
Myelin basic protein (20 μM final concentration) was dissolved in 20 mM Hepes (pH 7.5) containing 10 μM MgCl2, 1 μM EGTA, 0.02% Brij35, 0.02 mg/ml BSA, 0.1 μM Na3VO4, 2 mM DTT, and 1% DMSO. Activated MLK3 was added and mixed (20 nM final concentration), and inhibitors were added in DMSO. 33P-ATP (specific activity 500 μCi/μL) was delivered into the reaction mixture to initiate the reaction (ATP concentration: 10 μM) and the mixture was incubated at room temperature for 20 minutes. % Activity was determined using a proprietary HOTSPOT™ microfluidic filter binding technology.51
FLT3 cellular assay
This was performed by Proqinase GMBH, Freiburg, Germany. Briefly, this assay uses a murine embryonal fibroblast (MEF) cell line, which expresses a high level of exogenously introduced full-length human, wild-type FLT3. Stimulation of these cells with human FLT3-Ligand results in receptor tyrosine autophosphorylation. MEF-FLT3-wt cells were plated in DMEM supplemented with 10% FCS in multiwell cell culture plates. After serum-starvation overnight, cells were incubated with compounds in serum-free medium. After 90 min incubation at 37°C, cells were stimulated with FLT3-L at 250 ng/mL for 5 minutes. Quantification of substrate phosphorylation was assessed in 96 well plates via sandwich ELISA using a substrate specific capture antibody and an anti-phosphotyrosine detection antibody. Raw data were converted into percent substrate phosphorylation relative to controls (incubated with FLT3L alone), which were set to 100%. IC50 values were determined using GraphPad Prism 5.01 software using a nonlinear regression curve fit with variable hill slope. The equation is a four-parameter logistic equation.
Plasma Protein Binding
Protein binding in human plasma was assessed by performing equilibrium dialysis with plasma containing test compounds (10 μM) against 0.1 M PBS (pH 7.4). Following incubation (6 hours at 37 °C), the parent compound was measured in both plasma and buffer compartments by LC-MS and the percentage of compound bound to plasma proteins determined. Test compounds (10 μM) were added to plasma (n=2) and dialysed against 0.1 M phosphate buffered saline (pH 7.4) for 6 hours at 37 °C. After incubation the contents of each plasma and buffer compartment were removed and mixed with equal volumes of control buffer or plasma as appropriate to maintain matrix equivalence for analysis. Plasma proteins were then precipitated by the addition of acetonitrile containing carbamazepine as analytical internal standard, centrifuged and the supernatant removed for analysis by mass spectrometry (LC-MS/MS).
Molecular Modeling and Ligand Docking
The 3DTC.PDB structure of MLK1 was used as a surrogate for MLK3 to evaluate the binding mode for the series. The compounds contained a conserved scaffold that was hand docked into MLK1 and protein and ligand were energy minimized using the molecular modeling suite MOE. It was hypothesized that the compounds acted as type 1 kinase inhibitor so the scaffold was positioned to ensure that the hydrogen bond donor and acceptor of the 7-azaindole core interacted with the corresponding contacts on the hinge. This provided two binding mode hypotheses for evaluation. The binding mode that fit the SAR data allowed the scaffold to donate and accept a hydrogen bond to and from Glu221 and Ala223, respectively. This placed the side-chain indole group to the rear of the ATP site with the N-methyl-piperazine group positioned towards the solubilizing pocket.
Supplementary Material
Fig 4.

Inhibition of HIV Tat Stimulated Cytokine Release in Human Monocytes with Compound 1 at 100, 300 and 1000 nM concentrations, (NT = no treatment).
Scheme 2.

Synthesis of compound 13a
aReagents and conditions: (a) 7-azaindole-5-boronic acid pinacol ester, PdCl2(PPh3)2, MeCN, aq. 1M Na2CO3; (b) NaH, p-toluene sulfonyl chloride, DMF; (c) 4-formylphenyl boronic acid, PdCl2(PPh3)2, MeCN, aq. 1 M Na2CO3; (d) 1-methyl piperazine, Na(OAc)3BH, CH2Cl2; (e) NaOH, MeOH.
Scheme 3.

Synthesis of Screening Hit 7.
aReagents and Conditions: (a) NBS, CH2Cl2, 0 °C; (b) EtOH, DIEA, 80 °C; (c) CDI, THF, 65 °C; (d) PdCl2(PPh3)2, CH3CN, H2O, 150 °C.
Acknowledgments
Funding Sources
This work was funded in part by grants from the National Institutes of Health, Division of Mental Health, NIH Grant P01MH064570 (Gelbard PI) and a Qualifying Therapeutic Discovery Project Grant to Califia Bio, Inc.
We would like to thank John Harris, John McCall, Angus MacLeod, and Sanjay B. Maggirwar for helpful guidance for this project and the National Institutes of Health for continuing funding of these efforts.
ABBREVIATIONS
- HAND
HIV-1-associated neurocognitive disorders
- HIV-1
human immunodeficiency virus type 1
- LPS
lipopolysaccharide
- TNF-α
tumor necrosis factor alpha
- MLK
mixed lineage kinase
- CNS
central nervous system
- BBB
blood brain barrier
- CSF
cerebrospinal fluid
- hERG
human Ether-à-go-go-Related Gene
- JNK
c-Jun N-terminal kinase
- FLT3
fms-related tyrosine kinase 3
- LRRK2
leucine-rich repeat kinase 2
- MOE
Molecular Operating Environment
- NIS
N-iodosuccinimide
- MeCN, CH3CN
acetonitrile
- DIEA
diisopropylethylamine
- THF
tetrahydrofuran
- CDI
1,1′-Carbonyldiimidazole
- EtOAC
ethyl acetate
- DMSO
dimethylsulfoxide
- CH2CL2, DCM
dichloromethane
- PK
pharmacokinetic
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript
Supporting Information. Kinase inhibition profiles for 442 kinases for compound 1 and 2 performed by DiscoverRx; inhibition profile using radio ligand binding assay for 342 wild type human kinases performed by Reaction Biology Corp for compound 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Marker DF, Tremblay ME, Puccini JM, Barbieri J, Gantz Marker MA, Loweth CJ, Muly EC, Lu SM, Goodfellow VS, Dewhurst S, Gelbard HA. The new small-molecule mixed-lineage kinase 3 inhibitor URMC-099 is neuroprotective and anti-inflammatory in models of human immunodeficiency virus-associated neurocognitive disorders. J Neurosci. 2013;33:9998–10010. doi: 10.1523/JNEUROSCI.0598-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I CHARTER Group; HNRC Group. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates nature and predictors. J Neurovirol. 2011;17:3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gelbard HA, Dewhurst S, Maggirwar SB, Kiebala M, Polesskaya O, Gendelman HE. Rebuilding synaptic architecture in HIV-1 associated neurocognitive disease: a therapeutic strategy based on modulation of mixed lineage kinase. Neurotherapeutics. 2010;7:392–398. doi: 10.1016/j.nurt.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 pathways. Nat Rev Mol Cell Biol. 2002;9:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 5.Silva RM, Kuan CY, Rakic P, Burke RE. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: a new therapeutic target in Parkinson’s disease. Mov Disord. 2005;20:653–664. doi: 10.1002/mds.20390. [DOI] [PubMed] [Google Scholar]
- 6.Wang LH, Besirli CG, Johnson EM., Jr Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu Rev Pharmacol Toxicol. 2004;44:451–474. doi: 10.1146/annurev.pharmtox.44.101802.121840. [DOI] [PubMed] [Google Scholar]
- 7.Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, Xu Z, Meyer SL, Savage MJ, Greene LA, Scott RW, Vaught JL. Cep-1347 (KT7515), A semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem. 2001;276:25302–25308. doi: 10.1074/jbc.M011601200. [DOI] [PubMed] [Google Scholar]
- 8.Handley ME, Rasaiyaah J, Barnett J, Thakker M, Pollara G, Katz DR, Chain BM. Expression and function of mixed lineage kinases in dendritic cells. Int Immunol. 2007;19:923–933. doi: 10.1093/intimm/dxm050. [DOI] [PubMed] [Google Scholar]
- 9.Handley ME, Rasaiyaah J, Chain BM, Katz DR. Mixed lineage kinases (MLKs): a role in dendritic cells, inflammation and immunity? Int J Exp Pathol. 2007;88:111–126. doi: 10.1111/j.1365-2613.2007.00531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaeschke A, Davis RJ. Metabolic stress signaling mediated by mixed-lineage kinases. Mol Cell. 2007;27:498–508. doi: 10.1016/j.molcel.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sathyanarayana P, Barthwal MK, Kundu CN, Lane ME, Bergmann A, Tzivion G, Rana A. Activation of the drosophila MLK by ceramide reveals TNF-alpha and ceramide as agonists of mammalian MLK3. Mol Cell. 2002;10:1527–1533. doi: 10.1016/s1097-2765(02)00734-7. [DOI] [PubMed] [Google Scholar]
- 12.Leung IW, Lassam N. Dimerization via tandem leucine zippers is essential for the activation of the mitogen-activated protein kinase kinase kinase, MLK-3. J Biol Chem. 1998;273:32408–32415. doi: 10.1074/jbc.273.49.32408. [DOI] [PubMed] [Google Scholar]
- 13.Leung IW, Lassam N. The kinase activation loop is the key to mixed lineage kinase-3 activation via both autophosphorylation and hematopoietic progenitor kinase 1 phosphorylation. J Biol Chem. 2001;276:1961–1967. doi: 10.1074/jbc.M004092200. [DOI] [PubMed] [Google Scholar]
- 14.Sui Z, Fan S, Sniderhan L, Reisinger E, Litzburg A, Schifitto G, Gelbard HA, Dewhurst S, Maggirwar SB. Inhibition of mixed lineage kinase 3 prevents HIV-1 Tat-mediated neurotoxicity and monocyte activation. J Immunol. 2006;177:702–711. doi: 10.4049/jimmunol.177.1.702. [DOI] [PubMed] [Google Scholar]
- 15.New DR, Maggirwar SB, Epstein LG, Dewhurst S, Gelbard HA. HIV-1 Tat induces neuronal death via tumor necrosis factor-alpha and activation of non-N-methyl-D-aspartate receptors by a NFkappaB-independent mechanism. J Biol Chem. 1998;273:17852–17858. doi: 10.1074/jbc.273.28.17852. [DOI] [PubMed] [Google Scholar]
- 16.Dewhurst S, Maggirwar SB, Schifitto G, Gendelman HE, Gelbard HA. Glycogen synthase kinase 3 beta (GSK-3 beta) as a therapeutic target in neuroAIDS. J Neuroimmune Pharmacol. 2007;2:93–96. doi: 10.1007/s11481-006-9051-1. [DOI] [PubMed] [Google Scholar]
- 17.Mishra R, Barthwal MK, Sondarva G, Rana B, Wong L, Chatterjee M, Woodgett JR, Rana A. Glycogen synthase kinase-3beta induces neuronal cell death via direct phosphorylation of mixed lineage kinase 3. J Biol Chem. 2007;282:30393–30405. doi: 10.1074/jbc.M705895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sweeney ZK, Lewcock JW. ACS Chemical Neuroscience Spotlight on CEP-1347. ACS Chem Neurosci. 2011;2:3–4. doi: 10.1021/cn1000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 20.Falsig J, Pörzgen P, Lotharius J, Leist M. Specific modulation of astrocyte inflammation by inhibition of mixed lineage kinases with CEP-1347. J Immunol. 2004;173:2762–2770. doi: 10.4049/jimmunol.173.4.2762. [DOI] [PubMed] [Google Scholar]
- 21.Hudkins RL, Diebold JL, Tao M, Josef KA, Park CH, Angeles TS, Aimone LD, Husten J, Ator MA, Meyer SL, Holskin BP, Durkin JT, Fedorov AA, Fedorov EV, Almo SC, Mathiasen JR, Bozyczko-Coyne D, Saporito MS, Scott RW, Mallamo JP. Mixed-lineage kinase 1 and mixed-lineage kinase 3 subtype-selective dihydronaphthyl [3,4-a]pyrrolo[3,4-c]carbazole-5-ones: optimization, mixed-lineage kinase 1 crystallography, and oral in vivo activity in 1-methyl-4-phenyltetraphydropyridine models. J Med Chem. 2008;51:5680–5689. doi: 10.1021/jm8005838. [DOI] [PubMed] [Google Scholar]
- 22.Lotharius J, Falsig J, van Beek J, Payne S, Dringen R, Brundin P, Leist M. Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J Neurosci. 2005;25:6329–6342. doi: 10.1523/JNEUROSCI.1746-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saporito MS, Brown EM, Miller MS, Carswell S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons in vivo. J Pharmacol Exp Ther. 1999;288:421–427. [PubMed] [Google Scholar]
- 24.Mathiasen JR, McKenna BA, Saporito MS, Ghadge GD, Roos RP, Holskin BP, Wu ZL, Trusko SP, Connors TC, Maroney AC, Thomas BA, Thomas JC, Bozyczko-Coyne D. Inhibition of mixed lineage kinase 3 attenuates MPP+-induced neurotoxicity in SH-SY5Y cells. Brain Res. 2004;1003:86–97. doi: 10.1016/j.brainres.2003.11.073. [DOI] [PubMed] [Google Scholar]
- 25.Bodner A, Toth PT, Miller RJ. Activation of c-Jun N-terminal kinase mediates gp120IIIB- and nucleoside analogue-induced sensory neuron toxicity. Exp Neurol. 2004;188:246–253. doi: 10.1016/j.expneurol.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 26.Bodner A, Maroney AC, Finn JP, Ghadge G, Roos R, Miller RJ. Mixed lineage kinase 3 mediates gp120IIIB-induced neurotoxicity. J Neurochem. 2002;82:1424–1434. doi: 10.1046/j.1471-4159.2002.01088.x. [DOI] [PubMed] [Google Scholar]
- 27.Eggert D, Dash PK, Gorantla S, Dou H, Schifitto G, Maggirwar SB, Dewhurst S, Poluektova L, Gelbard HA, Gendelman HE. Neuroprotective activities of CEP-1347 in models of neuroAIDS. J Immunol. 2010;184:746–756. doi: 10.4049/jimmunol.0902962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parkinson Study Group PRECEPT Investigators. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2007;69:1480–1490. doi: 10.1212/01.wnl.0000277648.63931.c0. [DOI] [PubMed] [Google Scholar]
- 29.Ma Q, Gelbard HA, Maggirwar SB, Dewhurst S, Gendelman HE, Peterson DR, Difrancesco R, Hochreiter JS, Morse GD, Schifitto G. Pharmacokinetic interaction of CEP-1347 and atazanavir in HIV-infected patients. J Neurovirol. 2013;3:254–60. doi: 10.1007/s13365-013-0172-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.http://www.optibrium.com/stardrop/
- 31.Dash PK, Gorantla S, Gendelman HE, Knibbe J, Casale GP, Makarov E, Epstein AA, Gelbard HA, Boska MD, Poluektova LY. Loss of neuronal integrity during progressive HIV-1 infection of humanized mice. J Neurosci. 2011;31:3148–3157. doi: 10.1523/JNEUROSCI.5473-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, Keane RW, Resnick L, Mizrachi Y, Volsky DJ, Epstein LG, Gendelman HE. Cytokins and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–1718. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wesselingh SL, Power C, Glass JD, Tyor WR, McArthur JC, Farber JM, Griffin JW, Griffin DE. Intracerebral cytokine messenger RNA expression in acquired immunodeficiency syndrome dementia. Ann Neurol. 1993;33:576–582. doi: 10.1002/ana.410330604. [DOI] [PubMed] [Google Scholar]
- 34.Tyor WR, Glass JD, Griffin JW, Becker PS, McArthur JC, Bezman L, Griffin DE. Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Nuerol. 1992;31:349–360. doi: 10.1002/ana.410310402. [DOI] [PubMed] [Google Scholar]
- 35.Wesselingh SL, Glass J, McArthur JC, Griffin JW, Griffin DE. Cytokine dysregulation in HIV-associated neurological disease. Adv Neuroimmunol. 1994;4:199–206. doi: 10.1016/s0960-5428(06)80258-5. [DOI] [PubMed] [Google Scholar]
- 36.Chico LK, Van Eldik LJ, Watterson DM. Targeting protein kinases in central nervous system disorders. Nat Rev Drug Discov. 2009;8:892–909. doi: 10.1038/nrd2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.http://www.cerep.fr/cerep/users/pages/downloads/Documents/Marketing/Pharmacology%20&%20ADME/Application%20notes/Cardiac%20toxicity%20hERG-2012.pdf
- 38.Weiss JM, Nath A, Major EO, Berman JW. HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood-brain barrier and up-regulates CCR5 expression on human monocytes. J Immunol. 1999;163:2953–2959. [PubMed] [Google Scholar]
- 39.Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, Kulkarni H, Bamshad MJ, Telles V, Anderson SA, Walter EA, Stephan KT, Deucher M, Mangano A, Bologna R, Ahuja SS, Dolan MJ, Ahuja SK. HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci US A. 2002;99:13795–13800. doi: 10.1073/pnas.202357499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mankowski JL, Queen SE, Clements JE, Zink MC. Cerebrospinal fluid markers that predict SIV CNS disease. J Neuroimmunol. 2004;157:66–70. doi: 10.1016/j.jneuroim.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 41.Li CJ, Ueda Y, Shi B, Borodyansky L, Huang L, Li YZ, Pardee AB. Tat protein induces self-perpetuating permissivity for productive HIV-1 infection. Proc Natl Acad Sci US A. 1997;94:8116–8120. doi: 10.1073/pnas.94.15.8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ganju RK, Munshi N, Nair BC, Liu ZY, Gill P, Groopman JE. Human immunodeficiency virus tat modulates the Flk-1/KDR receptor, mitogen-activated protein kinases, and components of focal adhesion in Kapsi’s sarcoma cells. J Virol. 1998;72:6131–6137. doi: 10.1128/jvi.72.7.6131-6137.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar A, Manna SK, Khawan S, Aggarwal BB. HIV-Tat protein activates c-Jun N-terminal kinase and activator protein-1. J Immunol. 1998;161:776–781. [PubMed] [Google Scholar]
- 44.Singh IN, El-Hage N, Campbell ME, Lutz SE, Knapp PE, Nath A, Hauser KF. Differential involvement of p38 and JNK MAP kinases in HIV-1 Tat and gp120-induced apoptosis and neurite degeneration in striatal neurons. Neuroscience. 2005;135:781–790. doi: 10.1016/j.neuroscience.2005.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davies C, Tournier C. Exploring the function of the JNK (c-Jun N-terminal kinase) signaling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans. 2012;40:85–89. doi: 10.1042/BST20110641. [DOI] [PubMed] [Google Scholar]
- 46.Santiago V, Giorgio C, Stefano M. Medicinal Chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem. 2008;8:1555–1572. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- 47.Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, Qu K, Herlaar E, Lau A, Young C, Wong BR, Lovel S, Sun T, Park G, Argade A, Jurcevic S, Pine P, Singh R, Grossbard EB, Payan DG, Masuda ES. R406 an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther. 2006;319:998–1008. doi: 10.1124/jpet.106.109058. [DOI] [PubMed] [Google Scholar]
- 48.Bamborough P, Brown MJ, Christopher JA, Chung C, Mellow GW. Selectivity of kinase inhibitor fragments. J Med Chem. 2011;54:5131–5143. doi: 10.1021/jm200349b. [DOI] [PubMed] [Google Scholar]
- 49.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhard DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. NatBiotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 50.Fabian MA, Biggs WH, 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 51.Ma H, Deacon S, Horiuchi K. The challenge of selecting protein kinase assays for lead discovery optimization. Expert Opin Drug Discov. 2008;3:607–621. doi: 10.1517/17460441.3.6.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaneko M, Saito Y, Saito H, Matsumoto T, Matsuda Y, Vaught JL, Dionne CA, Angeles TS, Glicksman MA, Neff NT, Rotella DP, Kauer JC, Mallamo JP, Hudkins RL, Murakata C. Neurotrophic 3, 9-bis[(alkylthio)methyl]-and-bis(alkoxymethyl)-K-252a derivatives. J Med Chem. 1997;40:1863–1869. doi: 10.1021/jm970031d. [DOI] [PubMed] [Google Scholar]
- 53.Olaharski AJ, Gonzaludo N, Bitter H, Goldstein D, Kirchner S, Uppal H, Kolaja K. Identification of a kinase profile that predicts chromosome damage induced by small molecule kinase inhibitors. PLoS Comput Biol. 2009;5:e1000446. doi: 10.1371/journal.pcbi.1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morphy R. Selectively nonselective kinase inhibition: striking the right balance. J Med Chem. 2010;53:1413–1437. doi: 10.1021/jm901132v. [DOI] [PubMed] [Google Scholar]
- 55.Grunwald MR, Levis MJ. FLT3 inhibitors for acute myeloid leukemia: a review of their efficacy and mechanisms of resistance. Int J Hematol. 2013;97:683–694. doi: 10.1007/s12185-013-1334-8. [DOI] [PubMed] [Google Scholar]
- 56.DeBoy CA, Rus H, Tegla C, Cudrici C, Jones MV, Pardo CA, Small D, Whartenby KA, Calabresi PA. FLT-3 expression and function on microglia in multiple sclerosis. Exp Mol Pathol. 2010;89:109–116. doi: 10.1016/j.yexmp.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 58.Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G, Kennedy D, Extrov Z, Cortes J, Verstovsek S. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood. 2010;115:1131–1136. doi: 10.1182/blood-2009-10-246363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olaharski AJ, Bitter H, Gonzaludo N, Kondru R, Goldstein DM, Zabka TS, Lin H, Singer T, Kolaja K. Modeling bone marrow toxicity using kinase structural motifs and the inhibition profiles of small molecular kinase inhibitors. Toxicol Sci. 2010;118:226–275. doi: 10.1093/toxsci/kfq258. [DOI] [PubMed] [Google Scholar]
- 60.Tsika E, Moore DJ. Mechanisms of LRRK2-mediated neurodegeneration. Curr Neurol Neurosci Rep. 2012;12:251–260. doi: 10.1007/s11910-012-0265-8. [DOI] [PubMed] [Google Scholar]
- 61.Marker DF, Puccini JM, Mockus TE, Barbieri J, Lu SM, Gelbard HA. LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J Neuroinflammation. 2012;9:261. doi: 10.1186/1742-2094-9-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kiebala M, Maggirwar SB. Ibudilast a pharmacologic phosphodiesterase inhibitor prevents human immunodeficiency virus-1 Tat-mediated activation of microglial cells. PLoS One. 2011;6:e18633. doi: 10.1371/journal.pone.0018633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sui Z, Sniderhan LF, Schifitto G, Phipps RP, Gelbard HA, Dewhurst S, Maggirwar SB. Functional synergy between CD40 ligand and HIV-1 Tat contributes to inflammation: implication in HIV type 1 dementia. J Immunol. 2007;178:3226–3236. doi: 10.4049/jimmunol.178.5.3226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.