

Abstract

Inositol-requiring enzyme 1 (IRE-1) is a kinase/RNase ER stress sensor that is activated in response to excessive accumulation of unfolded proteins, hypoxic conditions, calcium imbalance, and other stress stimuli. Activation of IRE-1 RNase function exerts a cytoprotective effect and has been implicated in the progression of cancer via increased expression of the transcription factor XBP-1s. Here, we describe the synthesis and biological evaluation of novel chromenone-based covalent inhibitors of IRE-1. Preparation of a family of 8-formyltetrahydrochromeno[3,4-c]pyridines was achieved via a Duff formylation that is attended by an unusual cyclization reaction. Biological evaluation in vitro and in whole cells led to the identification of 30 as a potent inhibitor of IRE-1 RNase activity and XBP-1s expression in wild type B cells and human mantle cell lymphoma cell lines.
Introduction
The endoplasmic reticulum (ER) stress response is a cytoprotective mechanism activated in response to proteotoxic burden and is crucial for homeostatic regulation.1,2 Disruption in the stoichiometric balance, transport, or processing of intracellular proteins leads to the activation of three distinct pathways mediated by the ER stress sensor proteins IRE-1, ATF6, and PERK. IRE-1 is unique in that it contains a stress sensor domain in the lumen of the ER and a cytosolic serine/threonine kinase domain linked to an RNase domain. Multiple stress conditions can cause IRE-1 to oligomerize. Oligomerization brings the IRE-1 cytoplasmic kinase domains into proximity, allowing for autophosphorylation and activation IRE-1 RNase activity. The IRE-1 RNase domain is critical for the function of IRE-1 because it splices 26 nucleotides from the mRNA of X-box binding protein 1 (XBP-1), causing a frame shift in translation.3−5 The spliced XBP-1 mRNA encodes a functional 54 kDa XBP-1s protein in mammalian cells, which is a transcription factor that translocates into the nucleus and regulates ER stress response genes.
Since gene copy number amplifications and aberrant protein expression are hallmarks of cancer, many human tumors rely on a robust ER stress response for growth and survival.6,7 As a result, IRE-1 and related stress sensors have emerged as potential therapeutic targets for the treatment of cancer.8−10 IRE-1-mediated activation of XBP-1 has also been implicated in the evasion of virus-induced cytotoxicity11 as well as in the development of inflammatory arthritis.12,13 Small molecules capable of modulating IRE-1 RNase activity and XBP-1s transcription thus represent useful chemical tools and potential therapeutic agents.
Efforts to identify inhibitors of IRE-1 RNase function have relied primarily on high-throughput screening of large chemical libraries. This has led to the discovery of various salicylaldehydes with in vitro activity against IRE-1-mediated mRNA splicing.14−17 A limited number of nonelectrophilic inhibitors of IRE-1 RNase activity have also been reported (Figure 1).18,19 While aldehydes and related functional groups are generally considered undesirable with respect to chemical probe development, the recent FDA approval of various electrophilic drugs has renewed interest in covalent inhibitors.20 The importance of the aldehyde moiety for potent IRE-1 RNase inhibition by 5 (4μ8C)15 and related compounds has been rationalized by the formation of an unusually stable Schiff base with lysine 907 in the IRE-1 endonuclease domain.21 Although IRE-1 contains 25 lysine residues in its cytosolic domain, only covalent modification at K907 (and in some cases K599) is observed in vivo.15 This selectivity has been attributed to specific perturbation of the K907 ε-amino group pKa, resulting in enhanced nucleophilicity, increased rate of Schiff base formation with aldehyde inhibitors, and slow off-rate.
Figure 1.

Selected known inhibitors of IRE-1 RNase activity.
Here, we report the synthesis and biological evaluation of novel chromenone-based inhibitors of IRE-1 RNase activity. A tandem Duff formylation/annelation reaction en route to candidate inhibtors gave rise to fused tricyclic chromenopiperidine, chromenoazepane, and chromenoazecane scaffolds. Selected analogues based on a tetrahydrochromeno[3,4-c]pyridine core structure potently inhibit XBP-1 splicing in vitro and block the expression of XBP-1s in whole cells, making them useful compounds for interrogating IRE-1 RNase activity in biological systems.
Results and Discussion
FRET-Suppression Assay of Potential IRE-1 Inhibitors
To assess the in vitro activity of potential IRE-1 RNase inhibitors, we carried out the expression and purification of recombinant human IRE-1 for use in an in vitro FRET-suppression assay.17 The cytoplasmic kinase/RNase domain (aa 547–977) of human IRE-1 was expressed as a soluble puritin-His-tagged 59 kDa fusion protein in SF21 cells and purified by Ni-NTA affinity chromatography. To confirm that hIRE-1 exhibited a functional RNase domain, we evaluated its activity in vitro using a synthetic mRNA stem-loop corresponding to the XBP-1 substrate sequence. This stem-loop incorporates a Cy5 fluorophore on its 5′ end and the black hole quencher (BHQ) on its 3′ end, resulting in fluorescence only upon site-specific cleavage by the protein. Protein (5 nM) was incubated in a 96-well plate at room temperature with different concentrations of the XPB-1 stem loop for up to 2 h, and fluorescence was measured upon excitation and emission at 620 and 680 nm, respectively. Recombinant hIRE-1 exhibited functional RNase activity wth a Km value of 45 nM (see Supporting Information).
We first evaluated a small set of known IRE-1 inhibitors, synthetic analogues, and selected commercially available salicylaldehyde derivatives using the FRET-suppression assay (Figure 2). We recently reported the in vivo characterization of naphthaldehyde derivative 2 (A-I06), which was postulated to be the bioactive breakdown product of the known IRE-1 inhibitor 1 (STF-038010).22 When evaluated in our assay, 1 and 2 exhibit similar IC50 values (9.94 and 9.73 μM, respectively), while decomposition product 8 and reduced derivative 9 showed no appreciable inhibition at 20 μM. Interestingly, the salicylaldehyde moiety alone was not sufficient for IRE-1 RNase inhibition, as evidenced by the weak activity (>20 μM IC50) of compounds 10–13. Modification of the aldehyde or phenol functionalities also resulted in inactive compounds (14–16). Coumarin derivative 5, recently identified in a high-throughput screening effort,15 exhibited significantly enhanced potency against IRE-1 RNase function with an IC50 value of 206 nM in our FRET-suppression assay.
Figure 2.

Compounds evaluated for anti-IRE-1 RNase activity by FRET-suppression assay. IC50 and CI values are reported as the mean of four separate experiments.
Synthesis of Tricyclic Chromenones
In an effort toward functionalized derivatives of 5 for use in covalent tagging and pulldown experiments, we synthesized analogues 20a–d in four steps from the appropriate amino acids (Figure 3). Installation of the aldehyde moiety in each case relied on a Duff formylation carried out using hexamethylenetetramine (HMTA) in refluxing glacial acetic acid. Interestingly, when the reaction was carried out in refluxing TFA using intermediate 19b as a starting material, formylation was attended by an annelation involving the pendent carbamate nitrogen to give tetrahydrochromeno[3,4-c]pyridine 21b as the sole product. The structure and connectivity of this tricyclic scaffold were confirmed by HMBC NMR. As is typically the case for Duff formylations,23,24 complete consumption of 19 still resulted in low yields of 20 and 21 due to significant decomposition. However, the yield of 21b improved to 41% when the reaction was preceded by acetylation of the o-hydroxyl group.
Figure 3.

Synthesis of substituted bicyclic and tricyclic 8-formyl chromenones.
A plausible mechanism for the formation of 21b involves electrophilic aromatic substitution at position 3 of the chromenone core (Scheme 1). The reaction of electron rich aromatics with HMTA in organic acid occasionally results in aminomethylation in addition to formylation via decomposition of intermediates such as B.23,24 In the case of 21b, this decomposition is likely precluded by attack of the carbamate nitrogen onto the electrophilic methylene group in C. The interrupted Duff reaction at position 3 presumably occurs prior to formylation at position 8, as the use of only 1 equiv of HMTA in refluxing TFA afforded intermediate D as the major product from 19b. The concomitant annelation was not observed in the case of substrate 19a under any of the conditions listed in Figure 2. However, hexahydrochromeno[3,4-c]azepine 21c and hexahydrochromeno[3,4-c]azocine 21d were isolated as the sole products from 19c and 19d when TFA was used as the solvent.
Scheme 1. Proposed Mechanism of Cyclization during Duff Formylation.

Structure–Activity Relationships
When evaluated in the FRET-suppression assay, bicyclic derivatives 19a–d exhibited inhibitory activities in the 100–500 nM range (Figure 4). The constrained tricyclic derivative 21b consistently showed enhanced activity against IRE-1 RNase activity relative to the bicyclic compounds 20b and 5 in side-by-side experiments. Given the optimal in vitro potency and chemical yield of 21b, we carried out the synthesis of a family of analogues to assess the importance of the hydroxyl group and the distal N-substituent (Scheme 2). A potential covalent irreversible inhibitor 23 was obtained by chlorination of the reduced derivative 22. Compounds 24 and 25 were prepared by acid-catalyzed protection of the aldehyde in 21b as the 1,3-dioxane or dithiane derivative. Analogues 26 and 27 were prepared by O-alkylation of 24, followed by acidic hydrolysis of the dioxane. Compounds 29–34 were synthesized by reaction of intermediate 28 with various acylating or alkylating reagents, followed by acidolysis.
Figure 4.

In vitro inhibition of IRE-1 RNase activity by compounds 20 and 21. IC50 and CI values are reported as the mean of four separate experiments.
Scheme 2. Synthesis of O- and N-Substituted Analogues.

The presumed importance of the aldehyde functionality for IRE-1 RNase inhibition also prompted us to explore alternative electrophilic groups at the 8 position of the chromenone core. Scheme 3 depicts the synthesis of analogues 36–42 from compound 21b. Formation of the ketone in 36 via oxidation of the Grignard product required prior protection of the o-hydroxyl as methoxymethyl ether 35. Olefination of 35 and acetal hydrolysis afforded electrophilic analogues 37 and 38. Oxidized variants 40–42 were synthesized via Pinnick oxidation of 35.
Scheme 3. Synthesis of Analogues with Aldehyde Surrogates.

All compounds were evaluated by FRET-suppression assay in side-by-side experiments using 21b as a control inhibitor (Table 1). As anticipated, protection of the aldehyde group in 21b as the 1,3-dioxane or dithiane acetal (24 and 25) resulted in weaker IRE-1 inhibitory activity. Alkylation of the phenol oxygen (compounds 26, 27, and 35) resulted in a complete loss of potency below 20 μM. The N-acyl derivative 29 exhibited an IC50 value of 312 nM, while N-alkyl analogues 30–33 were found to be slightly more potent. Interestingly, N-benzyl analogue 31 was almost 3-fold more active than the corresponding fluorinated derivative 32. Guanidinylation to give 34 resulted in a notable increase in potency (IC50 = 47 nM) relative to the parent compound, though solubility significantly decreased. Ketone 36, vinyl sulfone 38, and Weinreb amide 42 showed no significant IRE-1 RNase inhibitory activity below 20 μM. However, electrophilic compounds 37, 40, and 41 displayed moderate potency (1–5 μM) in vitro. Also of note, 1,3-dioxane derivative 24 exhibited an in vitro IC50 of 3.1 μM, whereas the corresponding 1,3-dithiane analogue 25 displayed more than 5-fold weaker activity. To confirm that the enhanced inhibitory activity of 24 is not simply a function of a labile aldehyde masking group, we carried out stability studies in assay buffer and observed no significant decomposition of the 1,3-dioxane moiety over 12 h (see Supporting Information).
Table 1. In Vitro IRE-1 RNase Inhibition by Analogues of 21b.
| compd | IC50 (nM) | 95% CI (nM) |
|---|---|---|
| 21b | 111 | 76–162 |
| 22 | >20000 | |
| 23 | >20000 | |
| 24 | 3051 | 2031–4584 |
| 25 | 16210 | 12900–20360 |
| 26 | >20000 | |
| 27 | >20000 | |
| 28 | 1230 | 704–2148 |
| 29 | 312 | 222–439 |
| 30 | 200 | 149–268 |
| 31 | 113 | 62–207 |
| 32 | 303 | 181–500 |
| 33 | 255 | 183–354 |
| 34 | 47 | 35–64 |
| 35 | >20000 | |
| 36 | >20000 | |
| 37 | 1718 | 1289–2288 |
| 38 | >20000 | |
| 40 | 4109 | 3099–5448 |
| 41 | 5644 | 3902–8162 |
| 42 | >20000 |
Inhibition of XBP-1s Expression in Whole Cells
In order to determine whether our inhibitors could block the expression of XBP-1s in whole cells, we incubated LPS-stimulated B cells from the spleens of wild-type mice with 20 μM selected compounds for 24 h, lysed the cells, and analyzed the lysates for the expression of XBP-1s by immunoblots. Compounds 29 and 30 potently suppress the expression of XBP-1s at 20 μM in wild-type mouse B cells (Figure 5A). In addition, 5, 21b, and 24 exhibit strong inhibition of XBP-1s, as does treatment with 50 μM 2. Despite their activity in the FRET-suppression assay, compounds 31–34 did not effectively inhibit XBP-1s expression in whole cells, presumably because of poor cell permeability and solubility. Compounds 37, 40, and 41, which feature alternative electrophilic functional groups, similarly showed little to no inhibitory effect on XBP-1s expression in B cells at 20 μM. Consistent with previous results showing up-regulation of IRE-1 in response to XBP-1s deficiency25 and suppression,22 we observed an inverse correlation between pharmacological inhibition of XBP-1s and expression level of IRE-1 (Figure 5A).
Figure 5.

Inhibition of XBP-1s expression in whole cells. (A) B cells were purified from the spleens of wild-type mice, stimulated with LPS for 48 h, treated with the indicated inhibitors at 20 μM for 24 h, lysed, and analyzed for expression of the indicated proteins by immunoblots. (B) Mino and (C) Jeko cells were treated with the indicated inhibitors at 20 μM for 24 h, lysed, and analyzed for the expression of indicated proteins by immunoblots. (D) Mino and (E) Jeko cells were treated with the indicated inhibitors at various doses for 48 h, lysed, and analyzed for the expression of indicated proteins by immunoblots. (F) Mino and (G) Jeko dose–response curves and IC50 values for inhibition of XBP-1s expression by indicated inhibitors as determined by immunoblots and densitometry (N = 3).
The IRE-1/XBP-1 pathway is known to be critical for the survival multiple myeloma, malignancies derives from plasma cells.14,26 However, the functional role of the ER stress response in leukemia or lymphoma derived from mature B cells has been largely overlooked because leukemia and lymphoma cells do not expand their ER like that of multiple myeloma cells. We recently showed that chronic lymphocytic leukemia (CLL) growth and survival is highly dependent on the IRE-1/XBP-1 pathway and is inhibited by small molecules targeting IRE-1 RNase activity.22 Mantle cell lymphoma (MCL) is an incurable non-Hodgkin’s lymphoma developed from mantle zone-resident B cells. Since the role of the IRE-1/XBP-1 pathway in MCL is completely unknown, we examined the MCL cell lines Mino and Jeko for the expression of XBP-1s and discovered that XBP-1s is constitutively expressed by both. A subset of inhibitors was examined for inhibition of XBP-1s in these human MCL cell lines. As with wild-type mouse B cells, compounds 21b, 29, and 30 potently suppress the expression of XBP-1s and induce up-regulation of IRE-1 in Mino and Jeko cells. N-Isobutenyl derivative 33 also exhibits significant activity at 20 μM (Figure 5B and Figure 5C).
To establish the dependency of XBP-1s expression on inhibitor concentration, we used MCL cells to determine the whole cell IC50 values for 21b, 29, and 30, in comparison to 5, by immunoblots and densitometry (Figure 5D–G). Compound 30 proved to be the most potent inhibitor of XBP-1s expression in both Mino and Jeko cell lines (IC50 = 0.57 and 0.98 μM, respectively).
Lastly, we carried out XTT dose–response experiments to determine approximate GI50 concentrations for 30, our most potent inhibitor of XBP-1s expression. After a 48 h treatment, 30 exhibited GI50 values of 34 and 19 μM in Mino and Jeko cells, respectively (Figure 6A). Total growth inhibition by 30 was achieved between 55 and 66 μM for these cell lines. We confirmed that growth inhibition is the result of apoptosis by treating Mino and Jeko cells with 30 for 72 h and analyzing cell lysates for cleaved PARP. Consistent with its superior potency in the suppression of XBP-1s, compound 30 induced PARP cleavage more strongly than either 21b or 5 at 50 μM (Figure 6B). We also determined a GI50 value of ∼34 μM in LPS-stimulated wild-type mouse B cells after treatment with 30 for 72 h (see Supporting Information). As expected, this result suggests that the growth of antibody-secreting plasma cells is also sensitive to inhibition of IRE-1 RNase activity.
Figure 6.
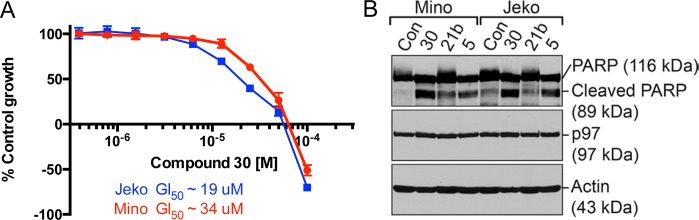
Growth inhibition and induction of apoptosis by 30. (A) Human Mino and Jeko cells were cultured in the presence of 30 at various concentrations for 48 h and subjected to XTT assay. Percentages of cell growth were calculated relative to DMSO-treated (control) groups. (B) Human Mino and Jeko cells were cultured for 72 h in the presence of DMSO (control), 30 (50 μM), 21b (50 μM), and 5 (50 μM). Cells were lysed for the analysis of the indicated proteins by immunoblot.
Conclusion
We have described the synthesis and biological characterization of novel inhibitors of IRE-1. Although various salicylaldehydes have been reported to inhibit IRE-1 RNase activity in vitro, our results confirm that the presence of an o-hydroxy aromatic aldehyde is not sufficient for biological activity. In an effort toward functionalized derivatives of potent chromenone-based inhibitors, we prepared a series of carbamate substituted 2H-chromene-2-ones for further derivatization. Duff formylation of these substrates resulted in a tandem annelation reaction, giving rise to novel fused tricyclic scaffolds. Tetrahydrochromeno[3,4-c]pyridine 21b served as a lead compound for the synthesis of a family of analogues.
Although replacement of the critical aldehyde group in 21b with electrophilic surrogates diminished potency, some compounds retained weak to moderate inhibitory activity in vitro. Modifications to the phenol group in 21b had a deleterious effect on potency in the FRET suppression assay, while changes at the distal N substituent were generally well tolerated. The ability of selected compounds to inhibit XBP-1s expression in wild-type B cells and human MCL cell lines highlights the importance of cell-based assays for this class of inhibitors, as a number of compounds with low- to mid-nanomolar activity in the FRET-suppression assay did not significantly reduce XBP-1s expression in whole cells. The N-methyl analogue 30 displayed an in vitro IRE-1 RNase IC50 value of 200 nM and potently inhibited the expression of XBP-1s in Mino and Jeko cells (IC50 = 0.57 and 0.98 μM, respectively). Compared to 21b, compound 30 is also more effective at inducing apoptosis in MCL cells. The described tricyclic chromenones thus represent useful tool compounds for suppressing IRE-1 RNase activity in whole cells and for probing the importance of the IRE-1/XBP-1 pathway of the ER stress response in biological systems.
Experimental Section
General Synthesis Notes
Unless stated otherwise, reactions were performed in flame-dried glassware under a positive pressure of argon or nitrogen gas using dry solvents. Commercial grade reagents and solvents were used without further purification except where noted. Diethyl ether, toluene, dimethylformamide dichloromethane, and tetrahydrofuran were purified by a Glass Contour column-based solvent purification system. Other anhydrous solvents were purchased directly from chemical suppliers. Thin-layer chromatography (TLC) was performed using silica gel 60 F254 precoated plates (0.25 mm). Flash chromatography was performed using silica gel (60 μm particle size). The purity of all compounds was judged by TLC analysis (single spot/two solvent systems) using a UV lamp, CAM (ceric ammonium molybdate), ninhydrin, or basic KMnO4 stain(s) for detection purposes. 1D and 2D NMR spectra were recorded on a Varian 400 MHz spectrometer. Proton chemical shifts are reported as δ values relative to residual signals from deuterated solvents (CDCl3, CD3OD, or DMSO-d6). The purity of all assayed compounds was determined by RP-HPLC using an analytical C18 column with MeCN/water (0.1% formic acid) as eluent (4 mm × 150 mm column, 1 mL/min flow rate). All final compounds were determined to be between 95% and 98% pure. Compounds 2, 5, 8, 10–12, and 14 were purchased from commercial sources. Compounds 1 and 9 were synthesized as described previously.22
Procedure for Synthesis of β-Ketoesters 18a–d
A solution of the appropriate (N-Alloc) amino acid 17 (23.9 mmol) in 100 mL of DCM at 0 °C was treated with 2,2-dimethyl-1,3-dioxane-4,6-dione (4.47 g, 31.0 mmol), 4-dimethylaminopyridine (2.92 g, 23.9 mmol), and diisopropylcarbodiimide (3.70 mL, 23.9 mmol). The mixture was stirred from 0 °C to room temperature over 4 h, then washed with 10% aqueous KHSO4 followed by brine. The organic layer was dried over Na2SO4 and concentrated. The resulting colorless liquid was dissolved in 50 mL of a 10:1 MeOH/toluene mixture and stirred at reflux for 15 h. After cooling, the mixture was concentrated under reduced pressure. Purification by flash column chromatography over silica gel (25%–60% EtOAc/hexanes) afforded 18a, 18b, and 18d as colorless oils. Alkylidene pyrrolidine 18c was obtained as a white solid.
Methyl 5-(((Allyloxy)carbonyl)amino)-3-oxobutanoate (18a)
18a was obtained in 64% yield from 17a. 1H NMR (400 MHz, CDCl3) δ 5.88 (ddt, J = 16.2, 10.7, 5.6 Hz, 1H), 5.49 (s, 1H), 5.29 (d, J = 17.2 Hz, 1H), 5.20 (d, J = 10.5 Hz, 1H), 4.56 (d, J = 5.5 Hz, 2H), 4.18 (d, J = 5.1 Hz, 2H), 3.72 (s, 3H), 3.50 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 198.2. 167.0, 156.1, 132.5, 117.9, 66.0, 52.6, 50.8, 46.2; HRMS (ESI-TOF) m/z [M + H]+ calcd for C9H14NO5 216.0867, found 216.0862.
Methyl 5-(((Allyloxy)carbonyl)amino)-3-oxopentanoate (18b)
18b was obtained in 94% yield from 17b. 1H NMR (400 MHz, CDCl3) δ 5.97–5.82 (m, 1H), 5.37–5.12 (m, 3H), 4.53 (d, J = 5.6 Hz, 2H), 3.73 (s, 3H), 3.50–3.37 (m, 4H), 2.80 (t, J = 5.7 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 202.2, 167.3, 156.2, 132.8, 132.8, 117.6, 117.5, 65.4, 52.4, 52.4, 48.9, 42.8, 35.3; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C10H16NO5 230.10285, found 230.10297.
Allyl 2-(2-Methoxy-2-oxoethylidene)pyrrolidine-1-carboxylate (18c)
18c was obtained in 56% yield from 17c. 1H NMR (400 MHz, CDCl3) δ 6.52 (s, 1H), 5.94 (ddt, J = 17.2, 10.5, 5.7 Hz, 1H), 5.33 (d, J = 17.2 Hz, 1H), 5.25 (d, J = 10.4 Hz, 1H), 4.66 (d, J = 5.7 Hz, 2H), 3.73 (t, J = 7.2 Hz, 2H), 3.65 (s, 3H), 3.17 (t, J = 7.7 Hz, 2H), 1.91 (p, J = 7.5 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 169.2, 157.3, 152.6, 131.9, 118.5, 96.4, 66.6, 50.8, 49.5, 31.6, 21.1; HRMS (ESI-TOF) m/z [M + H]+ calcd for C11H16NO4 226.1074, found 226.1068.
Methyl 7-(((Allyloxy)carbonyl)amino)-3-oxoheptanoate (18d)
18d was obtained in 65% yield from 17d. 1H NMR (400 MHz, CDCl3) δ 5.89 (ddt, J = 16.2, 10.7, 5.4 Hz, 1H), 5.28 (dd, J = 17.2, 1.5 Hz, 1H), 5.19 (dd, J = 10.4, 1.1 Hz, 1H), 4.82 (s, 1H), 4.53 (d, J = 5.5 Hz, 2H), 3.72 (s, 3H), 3.43 (s, 2H), 3.16 (dd, J = 12.9, 6.5 Hz, 2H), 2.56 (t, J = 7.1 Hz, 2H), 1.68–1.57 (m, 2H), 1.56–1.43 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 202.4, 167.6, 156.3, 132.9, 117.6, 65.4, 52.4, 49.0, 42.4, 40.5, 29.1, 20.2; HRMS (ESI-TOF) m/z [M + H]+ calcd for C12H20NO5 258.1336, found 258.1326.
General Procedure for Synthesis of Coumarins 19a–d
A solution of the appropriate β-keto ester 18 (10.1 mmol) in 50 mL of methanesulfonic acid at 0 °C was treated with resorcinol (1.11 g, 10.1 mmol) and stirred for 3.5 h. The mixture was poured into ice cold water, and the resulting yellow mixture was filtered. The filtrate was extracted with EtOAc and combined with the solids. The combined organic layer was concentrated and purified by flash chromatography over silica gel (0–20% MeOH/CHCl3) to afford the pure coumarin derivatives 19a–d.
Allyl (2-(7-Hydroxy-2-oxo-2H-chromen-4-yl)methyl)carbamate (19a)
19a was obtained in 36% yield from 18a. 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 7.88 (t, J = 5.9 Hz, 1H), 7.64 (d, J = 8.7 Hz, 1H), 6.78 (d, J = 8.7 Hz, 1H), 6.73 (d, J = 2.3 Hz, 1H), 5.99 (s, 1H), 5.92 (ddt, J = 17.0, 10.6, 5.4 Hz, 1H), 5.29 (dd, J = 17.2, 1.6 Hz, 1H), 5.18 (d, J = 10.5 Hz, 1H), 4.52 (d, J = 5.3 Hz, 2H), 4.37 (d, J = 5.8 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 161.7, 160.8, 156.6, 155.4, 154.2, 134.0, 126.2, 117.6, 113.4, 110.3, 107.9, 102.8, 65.2, 41.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C13H14NO5 276.0867, found 276.0863.
Allyl (2-(7-Hydroxy-2-oxo-2H-chromen-4-yl)ethyl)carbamate (19b)
19b was obtained in 88% yield from 18b. 1H NMR (400 MHz, DMSO-d6) δ 10.55 (s, 1H), 7.68 (d, J = 8.8 Hz, 1H), 7.40 (m, 1H), 6.80 (dd, J = 8.7, 2.3 Hz, 1H), 6.71 (d, J = 2.3 Hz, 1H), 6.07 (s, 1H), 5.99–5.78 (m, 1H), 5.24 (m, 1H), 5.15 (m, 1H), 4.45 (m, 2H), 3.29 (m, 2H), 2.87 (t, J = 6.7 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 161.1, 160.3, 156.0, 155.2, 154.2, 133.8, 133.7, 126.3, 116.9, 113.0, 111.3, 110.5, 110.4, 102.5, 102.4, 64.3, 31.5, 23.4; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C16H16NO5 302.102 85, found 302.103 05.
Allyl (2-(7-Hydroxy-2-oxo-2H-chromen-4-yl)propyl)carbamate (19c)
19c was obtained in 88% yield yield from 18c. 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1H), 7.61 (d, J = 8.8 Hz, 1H), 7.33 (t, J = 5.5 Hz, 1H), 6.78 (d, J = 8.7, 1H), 6.69 (d, J = 2.4 Hz, 1H), 6.10 (s, 1H), 5.89 (ddt, J = 17.0, 10.6, 5.4 Hz, 1H), 5.25 (dd, J = 17.2, 1.6 Hz, 1H), 5.15 (d, J = 10.4 Hz, 1H), 4.45 (d, J = 5.3 Hz, 2H), 3.07 (q, J = 6.6 Hz, 2H), 2.72 (t, J = 7.6 Hz, 2H), 1.96–1.63 (m, 2H); 13C NMR (101 MHz, DMSO- d6) δ 161.5, 160.8, 157.0, 156.4, 155.6, 134.3, 126.7, 117.3, 113.3, 111.6, 109.9, 102.9, 64.6, 40.2, 28.7, 28.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C16H18NO5 304.1180, found 304.1172.
Allyl (2-(7-Hydroxy-2-oxo-2H-chromen-4-yl)butyl)carbamate (19d)
19d was obtained in 84% yield from 18d. 1H NMR (400 MHz, DMSO- d6) δ 10.50 (s, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.21 (t, J = 5.7 Hz, 1H), 6.76 (d, J = 8.7 Hz, 1H), 6.67 (d, J = 2.4 Hz, 1H), 6.05 (s, 1H), 5.86 (ddt, J = 17.2, 10.5, 5.3 Hz, 1H), 5.22 (dd, J = 17.2, 1.7 Hz, 1H), 5.11 (dd, J = 10.4, 1.6 Hz, 1H), 4.42 (d, J = 5.3 Hz, 2H), 3.00 (d, J = 6.1 Hz, 2H), 2.69 (t, J = 7.4 Hz, 2H), 1.62–1.51 (m, 2H), 1.51–1.42 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 161.5, 160.9, 157.5, 156.4, 155.5, 134.3, 126.8, 117.2, 113.3, 111.6, 109.7, 102.8, 64.5, 40.2, 31.0, 29.5, 25.8; HRMS (ESI-TOF) m/z [M + H]+ calcd for C17H20NO5 318.1336, found 318.1339.
Duff Reaction Condition A
The appropriate coumarin derivative 19 (0.73 mmol) in 9 mL of AcOH was treated with HMTA (255 mg, 1.82 mmol) and stirred for 18 h at 95 °C. The reaction mixture was concentrated, and the resulting slurry was dissolved in 12 mL of a 1:1 1 M aqueous HCl/EtOAc solution and stirred at 60 °C for 2 h. The organic layer was separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, dried with MgSO4, and concentrated. Purification by silica gel flash column chromatography (EtOAc/hexane) afforded the desired bicyclic formyl derivatives 20a–d.
Duff Reaction Condition B
The appropriate coumarin derivative 19 (0.73 mmol) in 3 mL of TFA was treated with HMTA (255 mg, 1.82 mmol) and stirred for 18 h at 75 °C. The reaction mixture was concentrated, and the resulting slurry was dissolved in 12 mL of a 1:1 1 M aqueous HCl/EtOAc solution and stirred at 60 °C for 2 h. The organic layer was separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, dried with MgSO4, and concentrated. Purification by silica gel flash column chromatography (EtOAc/hexane) afforded the desired bicyclic and tricyclic formyl derivatives.
Duff Reaction Condition C
The appropriate coumarin derivative 19 (0.47 mmol) in 15 mL of MeCN was treated with pyridine (18.5 mg, 0.23 mmol) and acetic anhydride (239 mg, 2.35 mmol). After being stirred for 6 h at room temperature, the mixture was diluted with brine and extracted with EtOAc. The organic layer was dried with MgSO4 and concentrated. The resulting crude product dissolved in 2 mL of TFA was treated with HMTA (164 mg, 1.17 mmol) and stirred for 18 h at 95 °C. The reaction mixture was concentrated, and the resulting slurry was dissolved in 12 mL of a 1:1 1 M aqueous HCl/EtOAc solution and stirred at 60 °C for 2 h. The organic layer was separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with water, dried with MgSO4, and concentrated. Purification by silica gel flash column chromatography (EtOAc/hexane) afforded the desired bicyclic and tricyclic formyl derivatives.
Allyl (2-(8-Formyl-7-hydroxy-2-oxo-2H-chromen-4-yl)methyl)carbamate (20a)
20a was obtained in 4% yield (methods A, B, and C) from 19a. 1H NMR (400 MHz, CDCl3) δ 12.24 (s, 1H), 10.60 (s, 1H), 7.73 (d, J = 9.0 Hz, 1H), 6.91 (d, J = 9.0 Hz, 1H), 6.32 (s, 1H), 5.94 (ddt, J = 16.5, 11.1, 5.8 Hz, 1H), 5.34 (d, J = 17.2 Hz, 1H), 5.27 (d, J = 10.3 Hz, 1H), 5.19 (t, J = 5.6 Hz, 1H), 4.64 (dt, J = 5.7, 1.4 Hz, 2H), 4.54 (d, J = 6.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 193.3, 165.4, 159.1, 156.3, 156.1, 152.1, 132.2, 131.8, 118.5, 114.7, 109.74, 109.71, 108.8, 66.4, 41.3; HRMS (ESI-TOF) m/z [M + H]+ calcd for C15H14NO6 304.0816, found 304.0820.
Allyl (2-(8-Formyl-7-hydroxy-2-oxo-2H-chromen-4-yl)ethyl)carbamate (20b)
20b was obtained in 10% yield (method A) from 19b. 1H NMR (400 MHz, CDCl3) δ 12.24 (s, 1H), 10.60 (s, 1H), 7.92 (d, J = 9.1 Hz, 1H), 6.93 (d, J = 9.0 Hz, 1H), 6.19 (s, 1H), 5.90 (m, 1H), 5.39–5.15 (m, 2H), 5.03 (bs, 1H), 4.58 (m, 2H), 3.49 (m, 2H), 2.99 (t, J = 7.2 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 193.5, 193.4, 165.5, 159.2, 156.6, 156.5, 153.4, 133.1, 132.6, 118.2, 114.8, 112.2, 112.1, 111.1, 109.0, 66.0, 40.1, 32.8; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C16H16NO6 318.097 77, found 318.097 46.
Allyl (2-(8-Formyl-7-hydroxy-2-oxo-2H-chromen-4-yl)propyl)carbamate (20c)
20c was obtained in 13% yield (method A) from 19c. 1H NMR (400 MHz, CDCl3) δ 12.20 (s, 1H), 10.58 (s, 1H), 7.72 (d, J = 9.0 Hz, 1H), 6.88 (d, J = 9.0 Hz, 1H), 6.19 (s, 1H), 5.90 (ddt, J = 16.8, 11.1, 5.6 Hz, 1H), 5.29 (dd, J = 17.2, 1.5 Hz, 1H), 5.20 (dd, J = 10.4, 1.2 Hz, 1H), 4.98 (t, J = 5.2 Hz, 1H), 4.56 (d, J = 5.4 Hz, 2H), 3.33 (q, J = 6.5 Hz, 2H), 2.98–2.59 (m, 2H), 1.90 (tt, J = 13.7, 6.9 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 193.4, 165.2, 159.3, 156.4, 156.3, 155.6, 132.7, 132.5, 117.9, 114.4, 111.0, 110.9, 108.8, 65.7, 40.4, 29.1, 28.5; HRMS (ESI-TOF) m/z [M + H]+ calcd for C17H18NO6 332.1129, found 332.1128.
Allyl (2-(8-Formyl-7-hydroxy-2-oxo-2H-chromen-4-yl)butyl)carbamate (20d)
20d was obtained in 15% yield (method A) from 19d. 1H NMR (400 MHz, CDCl3) δ 12.22 (s, 1H), 10.60 (s, 1H), 7.74 (d, J = 9.0 Hz, 1H), 6.89 (d, J = 9.0 Hz, 1H), 6.17 (s, 1H), 5.90 (ddt, J = 16.1, 10.8, 5.7 Hz, 1H), 5.29 (dd, J = 17.2, 1.6 Hz, 1H), 5.20 (dd, J = 10.4, 1.3 Hz, 1H), 4.80 (s, 1H), 4.55 (d, J = 5.6 Hz, 2H), 3.26 (q, J = 6.4 Hz, 2H), 2.93–2.56 (m, 2H), 1.78–1.56 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 139.4, 165.2, 159.4, 156.38, 156.37, 156.1, 132.8, 132.7, 117.8, 114.4, 111.1, 111.0, 109.8, 65.6, 40.3, 31.5, 29.9, 25.2; HRMS (ESI-TOF) m/z [M + H]+ calcd for C18H20NO6 346.1285, found 346.1288.
Allyl 7-Formyl-8-hydroxy-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (21b)
21b was obtained in 22% (method B) and 41% (method C) yield from 19b. 1H NMR (400 MHz, CDCl3) δ 12.15 (s, 1H), 10.61 (s, 1H), 7.68 (d, J = 8.4 Hz, 1H), 6.92 (d, J = 9.0 Hz, 1H), 5.94 (m, 1H), 5.33 (m, 1H), 5.24 (m, 1H), 4.64 (d, J = 5.7 Hz, 2H), 4.47 (m, 2H), 3.81 (t, J = 5.8 Hz, 2H), 2.86 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 193.3, 164.9, 158.4, 155.2, 154.7, 146.4, 132.7, 131.8, 118.3, 117.2, 114.8, 111.2, 108.7, 66.7, 41.9, 39.2, 24.9; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C17H16NO6 330.097 21, found 330.096 24.
Allyl 8-Formyl-9-hydroxy-6-oxo-2,3,5,6-tetrahydrochromeno[3,4-c]azepine-4(1H)-carboxylate (21c)
21c was obtained in 18% (method B) and 17% (method C) yield from 19c. 1H NMR (400 MHz, CDCl3) δ 12.17 (s, 1H), 10.61 (s, 1H), 7.79 (d, J = 9.2 Hz, 1H), 6.90 (d, J = 8.9 Hz, 1H), 5.87 (ddt, J = 16.3, 10.8, 5.3 Hz, 1H), 5.28 (dd, J = 17.2, 1.5 Hz, 1H), 5.16 (d, J = 10.8 Hz, 1H), 4.65 (s, 2H), 4.55 (d, J = 5.1 Hz, 2H), 3.98–3.57 (m, 2H), 3.08–2.96 (m, 2H), 2.13–2.00 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 193.4, 164.8, 159.3, 155.7, 155.0, 152.2, 132.54, 132.50, 122.0, 117.3, 114.4, 111.9, 108.6, 66.3, 47.8, 42.9, 27.6, 24.6; HRMS (ESI-TOF) m/z [M + H]+ calcd for C18H18NO6 344.1129, found 344.1137.
Allyl 9-Formyl-10-hydroxy-7-oxo-2,3,5,6-tetrahydro-1H-chromeno[3,4-d]azocine-4(7H)-carboxylate (21d)
21d was obtained in 3% (method B) and 9% (method C) yield from 19d. 1H NMR (400 MHz, CDCl3) δ 12.18 (s, 1H), 10.61 (s, 1H), 7.78 (d, J = 8.6 Hz, 1H), 6.91 (d, J = 8.9 Hz, 1H), 5.95 (ddt, J = 16.9, 10.8, 5.6 Hz, 1H), 5.32 (d, J = 17.1 Hz, 1H), 5.21 (d, J = 10.3 Hz, 1H), 4.66 (s, 2H), 4.64 (d, J = 4.7 Hz, 2H), 3.62–3.49 (m, 2H), 3.07–2.96 (m, 2H), 1.92–1.80 (m, 2H), 1.80–1.69 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 139.3, 164.8, 159.1, 155.8, 155.5, 152.0, 132.9, 132.6, 119.5, 117.6, 114.5, 111.3, 108.7, 66.5, 46.2, 44.4, 26.1, 25.7, 25.1; HRMS (ESI-TOF) m/z [M + H]+ calcd for C19H20NO6 358.1285, found 358.1290.
Allyl 8-Hydroxy-7-(hydroxymethyl)-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (22)
Compound 21b (24 mg, 73 μmol) in 2 mL of MeOH at 0 °C was treated with sodium borohydride (3.0 mg, 73 μmol) and stirred for 40 min. The reaction was quenched with 1 M aqueous HCl and extracted with EtOAc. The organic layers were dried over Na2SO4 and concentrated. Purification by flash column chromatography over silica gel (40% −50% EtOAc/hexane) afforded 22 as a white foam (16 mg, 66%). 1H NMR (400 MHz, CDCl3) δ 9.65 (bs, 1H), 7.35 (d, J = 8.4 Hz, 1H), 6.85 (d, J = 8.7 Hz, 1H), 5.94 (m, J = 11.1, 5.6 Hz, 1H), 5.33 (m, 3H), 5.24 (m, 1H), 4.64 (m, 2H), 4.40 (s, 2H), 3.77 (t, J = 5.7 Hz, 2H), 2.84 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 180.6, 160.4, 149.8, 147.8, 132.7, 123.5, 118.2, 114.7, 111.8, 111.1, 66.7, 59.0, 41.8, 39.3, 24.9; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C17H18NO6 332.113 42, found 332.114 73.
Allyl 8-Hydroxy-7-chloro-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (23)
Compound 22 (17 mg, 51 μmol) in 2 mL of DCM at room temperature was treated with thionyl chloride (19 μL, 257 μmol) and stirred for 5.5 h. The reaction was diluted with DCM and washed with saturated aqueous NH4Cl, dried over Na2SO4, and concentrated under reduced pressure. The resulting white solid 23 was suffiently pure by NMR and HPLC analysis for further use (12 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 9.42 (m, 0.5H), 7.69 (m, 0.5H), 7.38 (m, 1H), 6.89 (m, 1H), 5.95 (m, 1H), 5.30 (m, 3H), 4.91 (s, 1H), 4.65 (m, 2H), 4.44 (d, J = 17.7 Hz, 2H), 3.79 (m, 2H), 2.85 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 160.3, 157.9, 151.5, 149.9, 132.7, 124.7, 123.6, 118.3, 114.5, 113.1, 112.7, 112.3, 111.9, 111.2, 66.8, 58.9, 42.0, 39.4, 34.3, 29.8, 24.8; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C17H16ClNO5 350.078 98, found 346.128 50 (observed mass corresponds to the 7-methoxymethyl derivative, resulting from displacement of the chloride with methanol during LCMS).
Allyl 7-(1,3-Dioxan-2-yl)-8-hydroxy-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (24)
A solution of 21b in (150 mg, 455 μmol) in 4 mL of benzene was treated with 1,3-propanediol (99.0 μL, 1.40 mmol) and p-toluenesulfonic acid monohydrate (4.3 mg, 23 μmol) and stirred for 2 h. The reaction was quenched with 2 drops of NEt3, diluted with EtOAc, and washed with brine. The organic layer was dried over Na2SO4 and concentrated. Purification by flash column chromatography over silica gel (30%–50% EtOAc/hexanes eluent) afforded 24 as a yellow solid (157 mg, 89%). 1H NMR (400 MHz, CDCl3) δ 8.82 (s, 1H), 7.36 (d, J = 8.2 Hz, 1H), 6.79 (d, J = 8.8 Hz, 1H), 6.28 (s, 1H), 5.91 (m, 1H), 5.30 (m, 1H), 5.20 (m, 1H), 4.61 (d, J = 5.6 Hz, 2H), 4.39 (s, 2H), 4.28 (dd, J = 11.6, J = 4.6 Hz, 2H), 4.09 (m, 2H), 3.74 (t, J = 5.8 Hz, 2H), 2.79 (m, 2H), 2.26 (m, 1H), 1.53 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 159.5, 159.3, 155.2, 150.5, 146.6, 132.8, 125.3, 118.0, 116.3, 114.5, 111.8, 109.9, 98.1, 67.9, 66.5, 41.8, 39.3, 25.8, 24.7; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C20H22NO7 388.139 08, found 388.138 10.
Allyl 7-(1,3-Dithian-2-yl)-8-hydroxy-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (25)
Compound 21b (39.0 mg, 118 μmol) and 1,3-propanedithiol (13.0 μL, 130 μmol) in 2.5 mL of DCM at room temperature were treated with BF3·OEt2 (6.0 μL, 47 μmol) and stirred for 17 h. The reaction was quenched with saturated aqueous NaHCO3, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification by flash column chromatography over silica gel (25–50% EtOAc/hexane) afforded 25 as a white foam (39 mg, 79%). 1H NMR (400 MHz, CDCl3) δ 7.55 (s, 1H), 7.43 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 8.9 Hz, 1H), 6.26 (s, 1H), 5.95 (m, 1H), 5.33 (m, 1H), 5.24 (m, 1H), 4.64 (m, 2H), 4.47 (m, 2H), 3.78 (t, J = 5.8 Hz, 2H), 3.17 (m, 2H), 2.91 (m, 4H), 2.24 (m, 1H), 1.94 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 159.5, 155.3, 149.8, 146.9, 132.8, 124.8, 118.1, 116.7, 114.9, 112.5, 111.1, 110.6, 77.5, 77.2, 76.8, 66.6, 42.0, 39.2, 37.4, 31.3, 24.9, 24.7, 23.0, 14.3, 14.3. HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C20H22NO5S2 420.093 99, found 420.092 48.
Allyl 7-Formyl-8-methoxy-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (26)
A solution of 24 (20 mg, 52 μmol) in 1 mL of DMF was treated with K2CO3 (36 mg, 258 μmol) followed by iodomethane (10 μL, 155 μmol). After being stirred at room temperature for 18 h, the mixture was diluted with saturated aqueous NH4Cl, extracted with DCM, and concentrated to dryness. The residue was taken up in 500 μL of dioxane, treated with 2 mL of 4 M aqueous HCl, and stirred at room temperature for 30 min. The mixture was diluted with water and extracted with DCM. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification by flash column chromatography over silica gel (0–10% MeOH/CHCl3) afforded 26 as a white powder (12 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 10.68 (s, 1H), 7.71 (d, J = 8.7 Hz, 1H), 6.98 (d, J = 9.0 Hz, 1H), 5.95 (m, 1H), 5.33 (m, 1H), 5.24 (m, 1H), 4.65 (m, 2H), 4.48 (s, 2H), 4.01 (s, 3H), 3.82 (t, J = 5.8 Hz, 2H), 2.87 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 187.2, 162.6, 158.6, 157.2, 155.3, 145.7, 132.8, 132.7, 132.7, 129.7, 118.3, 118.2, 112.9, 112.7, 108.2, 66.7, 56.8, 42.0, 39.3, 29.9, 24.9; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C18H18NO6 344.113 41, found 344.114 32.
Allyl 7-Formyl-8-benzyloxy-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (27)
A solution of 24 (20 mg, 52 μmol) in 1 mL of DMF was treated with K2CO3 (36 mg, 260 μmol) followed by benzyl bromide (9.0 μL, 78 μmol). After being stirred at room temperature for 18 h, the mixture was diluted with saturated aqueous NH4Cl, extracted with DCM, and concentrated to dryness. The residue was taken up in 500 μL of dioxane, treated with 2 mL of 4 N aqueous HCl, and stirred at room temperature 30 min. The mixture was diluted with water and extracted with DCM. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification by flash column chromatography over silica gel (0–10% MeOH/CHCl3) afforded 27 as a white powder (18 mg, 72%). 1H NMR (400 MHz, CDCl3) δ 10.72 (d, J = 5.4 Hz, 1H), 7.67 (d, J = 8.6 Hz, 1H), 7.58–7.31 (m, 5H), 7.01 (d, J = 9.0 Hz, 1H), 5.95 (m, 1H), 5.35 (m, 0.5H), 5.30 (m, 2.5H), 5.24 (m, 1H), 4.65 (m, 2H), 4.47 (s, 2H), 3.81 (t, J = 5.8 Hz, 2H), 2.87 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 187.1, 161.7, 158.7, 154.0, 145.6, 135.4, 132.7, 129.5, 128.9, 128.5, 127.0, 118.3, 113.2, 113.1, 109.6, 71.2, 66.7, 51.3, 42.0, 39.2, 29.8, 24.8; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C24H22NO6 420.144 71, found 420.145 29.
7-(1,3-Dioxan-2-yl)-8-hydroxy-3,4-dihydro-1H-chromeno[3,4-c]pyridin-5(2H)-one (28)
A solution of 24 (70 mg, 180 μmol) in 4 mL of DCM at room temperature was treated with phenylsilane (67 mg, 540 μmol) and tetrakis(triphenylphosphine)palladium(0) (10 mg, 9.0 μmol) and stirred at room temperature for 25 min. The mixture was concentrated and the residue purified by flash chromatography over silica gel (0%–10% MeOH/CHCl3) to afford 28 as a yellow solid (54 mg, 98%). 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 8.8 Hz, 1H), 6.78 (d, J = 8.8 Hz, 1H), 6.28 (s, 1H), 4.24 (m, 2H), 4.06 (m, 2H), 3.75 (m, 2H), 3.11 (t, J = 5.8 Hz, 2H), 2.70 (m, 2H), 2.36–2.11 (m, 1H), 1.92 (bs, 1H), 1.50 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 160.2, 159.0, 150.6, 146.8, 135.0, 125.1, 119.0, 114.3, 112.5, 109.9, 98.3, 68.0, 43.4, 42.0, 25.9, 25.3; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C16H18NO5 304.117 95, found 304.117 82.
3-Acetyl-8-hydroxy-5-oxo-2,3,4,5-tetrahydro-1H-chromeno[3,4-c]pyridine-7-carbaldehyde (29)
A solution of 28 (20 mg, 66 μmol) in 1 mL of DCM was treated with pyridine (11 μL, 130 μmol) and acetyl chloride (7.0 μL, 99 μmol), then stirred at room temperature for 20 min. After concentration under reduced pressure, the residue was taken up in 500 μL of dioxane, treated with 2 mL of 4 M aqueous HCl, and stirred at room temperature for 30 min. The mixture was diluted with water and extracted with DCM. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification by flash column chromatography over silica gel (0–10% MeOH/CHCl3) afforded 29 as a white powder (17 mg, 90%). 1H NMR (400 MHz, DMSO-d6) δ 11.84 (s, 1H), 10.46 (s, 1H), 7.90 (m (rotomer), 1H), 7.00 (d, J = 8.9 Hz, 1H), 4.32 (m, 2H), 3.73 (t, J = 5.7 Hz, 2H), 2.96 (m, 2H), 2.83 (m, 1H), 2.10 (m (rotomer), 3H); 13C NMR (101 MHz, DMSO-d6) δ 191.1, 191.0, 168.9, 163.2, 163.1, 158.2, 153.5, 147.0, 146.8, 132.2, 116.7, 116.5, 113.9, 111.1, 109.1, 104.6, 43.2, 41.4, 36.3, 25.0, 24.3, 21.8, 21.3; HRMS (ESI-TOF) (m/z) [M + H]+ calcd C15H14NO5 288.087 20, found 288.086 54.
8-Hydroxy-3-methyl-5-oxo-2,3,4,5-tetrahydro-1H-chromeno[3,4-c]pyridine-7-carbaldehyde (30)
A solution of 28 (50.0 mg, 165 μM) in 2 mL of 1:1 dioxane/THF was treated with 37% aqueous formaldehyde (27.0 μL, 330 μM), 10% Pd/C (40 mg), placed under H2 atmosphere, and stirred at room temperature for 3 h. The mixture was filtered through Celite with MeOH rinsing and concentrated to afford the crude methylamine. The residue was taken up in 500 μL of dioxane, treated with 2 mL of 4 M aqueous HCl, and stirred at room temperature 30 min. The mixture was diluted with water and extracted with DCM. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification by flash column chromatography over silica gel (0–10% MeOH/CHCl3) afforded 30 as a white powder (35 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 12.15 (s, 1H), 10.61 (s, 1H), 7.66 (d, J = 9.0 Hz, 1H), 6.92 (d, J = 9.0 Hz, 1H), 3.59 (s, 2H), 3.03–2.97 (m, 2H), 2.97–2.90 (m, 2H), 2.64 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.2, 164.8, 158.4, 154.6, 145.5, 131.7, 117.3, 114.6, 111.0, 108.6, 51.6, 50.2, 45.0, 25.3; HRMS (ESI-TOF) m/z [M + H]+ calcd for C14H14NO4 260.0917, found 260.0915.
3-Benzyl-8-hydroxy-5-oxo-2,3,4,5-tetrahydro-1H-chromeno[3,4-c]pyridine-7-carbaldehyde (31)
A solution of 28 (20 mg, 66 μmol) in 1.5 mL of DMF at room temperature was treated with NEt3 (10 mg, 99 μmol) and benzyl bromide (12 mg, 73 μmol). After being stirred for 5 h, the mixture was concentrated and treated with 4 mL of 4 M aqueous HCl and stirred for 1 h. The mixture was adjusted to pH 7 with 10% aqueous Na2CO3, extracted with DCM, dried over MgSO4, and concentrated. Purification by silica gel flash column chromatography (MeOH/CHCl3) gave 31 as a white solid (15.4 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 12.15 (s, 1H), 10.61 (s, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.42–7.30 (m, 5H), 6.90 (d, J = 9.0 Hz, 1H), 3.87 (s, 2H), 3.59 (s, 2H), 2.94 (s, 4H); 13C NMR (101 MHz, CDCl3) δ 193.3, 164.5, 158.8, 154.5, 146.2, 137.1, 131.7, 129.2, 128.5, 127.6, 119.0, 114.3, 111.5, 108.5, 62.3, 50.1, 48.0, 26.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C20H18NO4 336.1230, found 336.1224.
3-(4-Fluorobenzyl)-8-hydroxy-5-oxo-2,3,4,5-tetrahydro-1H-chromeno[3,4-c]pyridine-7-carbaldehyde (32)
A solution of 28 (20 mg, 66 μmol) in 1.5 mL of DMF at room temperature was treated with NEt3 (10 mg, 99 μmol) and 4-fluorobenzyl bromide (12 mg, 73 μmol). After being stirred for 5 h, the mixture was concentrated and treated with 4 mL of 4 M aqueous HCl and stirred for 1 h. The mixture was adjusted to pH 7 with 10% aqueous Na2CO3, extracted with DCM, dried over MgSO4, and concentrated. Purification by silica gel flash column chromatography (MeOH/CHCl3) gave 32 as a pale yellow solid (12 mg, 49%). 1H NMR (400 MHz, CDCl3) δ 12.15 (s, 1H), 10.62 (s, 1H), 7.66 (d, J = 9.0 Hz, 1H), 7.36 (m, 2H), 7.06 (m, 2H), 6.86 (m, 1H), 3.70 (bs, 2H), 3.51 (m, 2H), 2.86 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 193.4, 164.7, 158.8, 156.3, 154.7, 146.2, 143.8, 131.9, 131.0, 127.9, 125.4, 115.7, 115.5, 114.6, 114.0, 111.5, 108.7, 68.7, 68.0, 61.5, 50.4, 48.1, 31.2, 29.9, 26.0; HRMS (ESI-TOF) m/z [M + H]+ calcd for C20H17FNO4 354.114 16, found 354.114 38.
8-Hydroxy-3-(2-methylallyl)-5-oxo-2,3,4,5-tetrahydro-1H-chromeno[3,4-c]pyridine-7-carbaldehyde (33)
A solution of 28 (20 mg, 67 μmol) in 1.5 mL of DMF at room temperature was treated with NEt3 (10 mg, 99 μmol) and 3-bromo-2-methylpropene (9.9 mg, 74 μmol). After being stirred for 5 h, the mixture was concentrated and treated with 4 mL of 4 M aqueous HCl and stirred for 1 h. The mixture was adjusted to pH 7 with 10% aqueous Na2CO3, extracted with DCM, dried over MgSO4, and concentrated. Purification by silica gel flash column chromatography (MeOH/CHCl3) gave 33 as a yellow solid (15 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 12.14 (s, 1H), 10.61 (s, 1H), 7.66 (d, J = 9.0 Hz, 1H), 6.90 (d, J = 9.0 Hz, 1H), 5.01 (s, 2H), 3.52 (s, 2H), 3.22 (s, 2H), 2.94 (s, 2H), 2.87 (s, 2H), 1.81 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.2, 164.6, 158.6, 154.6, 146.0, 140.5, 131.7, 115.3, 114.5, 111.3, 108.5, 105.0, 64.4, 50.2, 48.0, 25.6, 20.8; HRMS (ESI-TOF) m/z [M + H]+ calcd for C17H18NO4 300.1230, found 300.1223.
7-Formyl-8-hydroxy-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboximidamide (34)
A solution of 28 (20 mg, 66 μmol) in 1 mL of DCM was treated with NEt3 (28 μL, 198 μmol) followed by 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine (58 mg, 146 μmol) and stirred at room temperature for 18 h. The mixture was diluted with saturated aqueous NH4Cl and extracted with DCM. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Purification by flash column chromatography over silica gel (40% EtOAc/hexanes) gave the guanidinylated intermediate as a glassy solid. The material was then treated with 2 mL of a 1:1 TFA/DCM solution and stirred at room temperature for 4 h. The mixture was concentrated to remove TFA, and the resulting solid was washed with three portions of DCM. Drying of the solid under vacuum afforded 34 (12 mg, 63%), which was pure by NMR. 1H NMR (400 MHz, DMSO-d6) δ 11.93 (s, 1H), 10.46 (s, 1H), 7.94 (d, J = 9.0 Hz, 1H), 7.64 (m, 3H), 7.02 (d, J = 9.0 Hz, 1H), 4.31 (s, 2H), 3.71 (t, J = 5.7 Hz, 2H), 2.99 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 190.8, 163.5, 158.1, 156.3, 153.5, 146.7, 132.3, 115.3, 114.1, 110.9, 109.3, 104.7, 43.2, 41.0, 24.2; HRMS (ESI-TOF) m/z [M + H]+ calcd for C14H14N3O4 288.098 43, found 288.098 81.
Allyl 7-Formyl-8-(methoxymethoxy)-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (35)
Compound 21b (301 mg, 914 μmol) in 5 mL of DCM at 0 °C was treated with DIEA (790 μL, 4.57 mmol) and chloromethyl methyl ether (347 μL, 4.57 mmol). The mixture was stirred for 30 h, quenched with saturated aqueous NH4Cl, and the organic layer was washed with saturated aqueous NH4Cl. The organic layer was dried over Na2SO4 and concentrated. Purification by flash column chromatography over silica gel (35%–70% EtOAc/hexanes) afforded 35 as a white solid (226 mg, 67%). 1H NMR (400 MHz, CDCl3) δ 10.68 (s, 1H), 7.67 (d, J = 8.9 Hz, 1H), 7.20 (d, J = 9.0 Hz, 1H), 5.95 (m, 1H), 5.35 (m, 2.5H), 5.30 (m, 0.5H), 5.24 (m, 1H), 4.64 (d, J = 5.7 Hz, 2H), 4.48 (s, 2H), 3.81 (t, J = 5.8 Hz, 2H), 3.53 (s, 3H), 2.87 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 186.8, 160.2, 158.4, 155.0, 153.4, 145.7, 132.6, 129.4, 118.2, 117.9, 113.5, 113.3, 111.4, 94.9, 66.4, 56.8, 41.7, 39.1, 24.7; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C19H20NO7 374.123 43, found 374.123 10.
Allyl 7-Acetyl-8-hydroxy-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (36)
Compound 35 (50.0 mg, 134 μmol) in 2 mL of THF at −78 °C under Ar was treated with 3 M MeMgBr in Et2O (134 μL, 402 μmol). After 3 h at −78 °C, the reaction was carefully quenched. Then the mixture was diluted with saturated aqueous NH4Cl, warmed to room temperature, and partitioned with EtOAc. The organic layer was dried over Na2SO4 and concentrated under reduced pressure to give the crude alcohol as an oil.
The above alcohol was dissolved in 3 mL of DCM and treated with Dess–Martin periodinane (123 mg, 291 μmol) and stirred at room temperature for 3 h. The reaction was quenched with 10% aqueous Na2S2O3 and washed with brine. The organic layer was dried over Na2SO4, concentrated, and the residue was purified by flash column chromatography over silica gel (35–70% EtOAc/hexane) to give the intermediate ketone as a gum (34 mg, 66%, two steps). 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.8 Hz, 1H), 7.13 (d, J = 8.9 Hz, 1H), 5.95 (m, 1H), 5.34 (m, 1H), 5.30 (m, 1H), 5.25 (m, 2.5H), 5.21 (m, 0.5H), 4.64 (d, J = 5.7 Hz, 2H), 4.45 (m, 2H), 3.80 (t, J = 5.8 Hz, 2H), 3.48 (s, 3H), 2.86 (m, 2H), 2.61 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 199.1, 158.9, 155.6, 149.3, 145.8, 133.5, 132.7, 125.0, 120.6, 118.2, 114.0, 111.2, 108.6, 94.8, 66.7, 56.7, 42.0, 39.3, 32.7, 29.8, 24.8; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C20H22NO7 388.139 08, found 388.139 45.
The ketone above (9.0 mg, 23 μmol) in 1.5 mL of 33% TFA/DCM solution was stirred for 1.5 h at room temperature. The mixture was concentrated under reduced pressure and the resulting residue was purified by flash column chromatography over silica gel (30% EtOAc/hexane) to afford 36 as a white foam (6.0 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 13.54 (s, 1H), 7.63 (d, J = 8.8 Hz, 1H), 6.95 (d, J = 9.0 Hz, 1H), 5.96 (m, 1H), 5.33 (m, 2H), 5.24 (ddd, J = 10.4, 2.5, 1.2 Hz, 1H), 4.65 (dt, J = 5.7, 1.3 Hz, 2H), 4.47 (m, 2H), 3.81 (t, J = 5.8 Hz, 2H), 2.98 (s, 3H), 2.87 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 204.4, 166.3, 158.6, 155.3, 153.8, 132.8, 130.2, 118.2, 116.6, 115.7, 111.3, 109.5, 66.7, 41.8, 39.3, 34.2, 25.1; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C18H18NO6 344.112 86, found 344.111 16.
Allyl 7-(3-Ethoxy-3-oxoprop-1-en-1-yl)-8-hydroxy-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (37)
Compound 35 (40.0 mg, 107 μmol) in 2 mL of DCM at room temperature was treated with triethylphosphonoacetate (48.0 mg, 139 μmol) and stirred for 20 h. The mixture was concentrated under reduced pressure. Purification by flash column chromatography over silica gel (20%–40% EtOAc/hexanes) afforded the intermediate ethyl enoate as a white solid (46 mg, 96%). 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 16.4 Hz, 1H), 7.46 (m, 1H), 7.16 (d, J = 9.0 Hz, 1H), 7.05 (d, J = 16.4 Hz, 1H), 5.96 (m, 1H), 5.34 (m, 2.5H), 5.30 (m, 0.5H), 5.23 (m, 1H), 4.64 (dt, J = 5.6, 1.2 Hz, 2H), 4.46 (s, 2H), 4.28 (q, J = 7.1 Hz, 2H), 3.79 (t, J = 5.8 Hz, 2H), 3.50 (s, 3H), 2.85 (m, 2H), 1.35 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 167.8, 158.5, 151.8, 132.8, 132.7, 132.2, 132.1, 132.1, 132.0, 128.7, 128.5, 125.2, 124.4, 118.1, 113.8, 112.4, 110.9, 94.8, 66.6, 60.7, 56.8, 56.8, 42.0, 39.3, 24.9, 14.5; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C23H26NO8 444.165 29, found 444.165 76.
The above ethyl enoate (20 mg, 45 μmol) in 2 mL of MeOH/CHCl3 (3:1) at room temperature was treated with 2 mL of 4 N aqueous HCl and stirred for 18 h. The reaction was extracted with EtOAc, dried over Na2SO4, and concentrated under reduced pressure. Purification by flash column chromatography over silica gel (40%–70% EtOAc/hexanes) afforded 37 as a white solid (15 mg, 83%). 1H NMR (400 MHz, DMSO-d6) δ 11.57 (m, 1H), 8.01 (d, J = 16.3 Hz, 1H), 7.62 (d, J = 8.8 Hz, 1H), 6.99 (m, 2H), 5.96 (ddt, J = 17.2, 10.6, 5.3 Hz, 1H), 5.31 (m, 1H), 5.22 (m, 1H), 4.59 (dt, J = 5.3, 1.4 Hz, 2H), 4.34–4.12 (m, 4H), 3.68 (m, 2H), 2.87 (m, 2H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 167.0, 160.3, 158.7, 154.3, 151.7, 146.9, 133.3, 133.2, 126.7, 121.5, 117.4, 112.9, 111.3, 108.2, 65.6, 60.1, 41.3, 24.4, 14.3; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C21H22NO7 400.139 08, found 400.139 92.
Allyl 8-Hydroxy-7-(2-(methylsulfonyl)vinyl)-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (38)
LiCl (8.0 mg, 20 μmol) and diethyl(methylsulfonylmethyl) phosphonate (54.0 g, 236 μmol) in 2.5 mL of acetonitrile at room temperature was treated with DBU (24.0 μL, 157 μmol) and stirred for 10 min. Compound 35 (43.0 mg, 131 μmol) in 2 mL of acetonitrile was cannulated into the mixture and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl and extracted with EtOAc. Purification by flash column chromatography over silica gel (0–5% MeOH/CHCl3) afforded the intermediate vinyl sulfone as a white solid (41 mg, 79%). 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 15.8 Hz, 1H), 7.68 (d, J = 15.8 Hz, 1H), 7.56 (d, J = 8.9 Hz, 1H), 7.20 (d, J = 9.0 Hz, 1H), 5.95 (m, 1H), 5.34 (m, 2.5H), 5.30 (m, 0.5H), 5.23 (m, 1H), 4.64 (m, 2H), 4.46 (s, 2H), 3.80 (t, J = 5.8 Hz, 2H), 3.51 (s, 3H), 3.06 (s, 3H), 2.86 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 158.8, 158.6, 155.3, 152.2, 146.1, 132.7, 131.7, 131.5, 126.8, 118.2, 113.8, 110.9, 109.9, 95.1, 66.6, 57.0, 43.3, 41.9, 39.3, 24.9; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C21H24NO8S 450.121 72, found 450.123 90.
The above vinyl sulfone (38 mg, 84 μmol) in 2.5 mL of acetonitrile/CHCl3 (2:1) was treated with 2.5 mL of 4 N aqueous HCl and stirred for 20 h at room temperature. The mixture was concentrated under reduced pressure. The resulting white solid was washed with DCM/Et2O and the resulting solid dried to afford pure 38 (32 mg, 92%). 1H NMR (400 MHz, DMSO-d6) δ 11.78 (s, 1H), 7.87 (d, J = 15.7 Hz, 1H), 7.68 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 15.7 Hz, 2H), 7.01 (d, J = 8.9 Hz, 1H), 5.97 (m, 1H), 5.31 (m, 1H), 5.21 (m, 1H), 4.59 (dt, J = 5.3, 1.5 Hz, 2H), 4.29 (s, 2H), 3.84 (s, 3H), 3.15 (s, 3H), 2.88 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 160.5, 158.6, 154.3, 151.8, 146.9, 133.3, 130.8, 130.1, 127.6, 117.4, 112.9, 111.3, 106.5, 65.6, 42.6, 41.3, 24.2; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C19H20NO7S 406.095 50, found 406.095 00.
3-((Allyloxy)carbonyl)-8-(methoxymethoxy)-5-oxo-2,3,4,5-tetrahydro-1H-chromeno[3,4-c]pyridine-7-carboxylic Acid (39)
Compound 35 (80.0 mg, 214 μmol) and 2-methyl-2-butene (272 μL, 2.57 mmol) in 3.5 mL of t-BuOH/H2O/CH3CN (3:3:1) at 0 °C were treated with a solution of sodium chlorite (145 mg, 1.29 mmol) and sodium monophosphate (265 mg, 1.93 mmol) in water, dropwise. After 30 min the reaction was quenched with 5% aqueous Na2S2O3. The pH of the solution was adjusted to 6, and the aqueous portion was extracted with EtOAc. The organic layer was dried over Na2SO4 and concentrated. Purification by flash column chromatography over silica gel (70–100% EtOAc/hexanes) afforded 39 as a thick oil (64 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 8.2 Hz, 1H), 7.19 (d, J = 9.0 Hz, 1H), 6.07 (bs,12H), 5.96 (m, 1H), 5.33 (m, 2.5H), 5.24 (m, 1H), 4.65 (m, 2H), 4.48 (s, 2H), 3.82 (t, J = 5.7 Hz, 2H), 3.52 (s, 3H), 2.90 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 166.5, 163.5, 162.5, 159.9, 156.9, 156.4, 155.4, 150.3, 149.7, 146.7, 132.7, 125.5, 118.3, 113.7, 113.5, 111.5, 111.1, 99.9, 95.0, 94.7, 91.8, 66.8, 56.9, 56.7, 41.9, 41.7, 39.3, 24.8; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C19H20NO8 390.118 35, found 390.117 50.
3-((Allyloxy)carbonyl)-8-hydroxy-5-oxo-2,3,4,5-tetrahydro-1H-chromeno[3,4-c]pyridine-7-carboxylic Acid (40)
Compound 39 (32 mg, 82 μmol) in 1 mL of MeOH was treated with 2 mL of 4 N aqueous HCl and stirred for 20 h at room temperature. The mixture was concentrated under reduced pressure. Purification by semipreparative RP-HPLC (C18 column, 0–70% MeCN/H2O gradient over 20 min) and subsequent lyophilization afforded compound 40 as a white solid (12 mg, 56%). 1H NMR (400 MHz, CD3CN) δ 12.39–11.57 (m, 1H), 7.75 (d, J = 9.0 Hz, 0.7H), 7.69 (rotamer: d, J = 8.9 Hz, 0.3H), 6.95 (dd, J = 9.0, 1.0 Hz, 0.7H), 6.85 (rotamer: d, J = 8.9 Hz, 0.3H), 5.99 (m, 1H), 5.32 (m, 1H), 5.21 (m, 1H), 4.61 (m, 2H), 4.31 (s, 2H), 3.73 (m, 2H), 2.86 (m, 2H); 13C NMR (101 MHz, CD3CN) δ 171.4, 165.9, 159.7, 153.7, 148.9, 147.7, 134.3, 131.4, 130.4, 117.6, 116.1, 115.0, 112.9, 102.5, 66.8, 42.4, 42.3, 25.6; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C17H16NO7 346.092 13, found 346.091 98.
3-Allyl 7-Methyl-8-hydroxy-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3,7(2H)-dicarboxylate (41)
Compound 39 (20 mg, 51 μmol) in 2 mL of acetone at room temperature was treated with potassium carbonate (10 mg, 77 μmol) and methyl iodide (5.0 μL, 77 μmol) and stirred for 24 h. The mixture was diluted with EtOAc, washed with brine, and dried over Na2SO4. Purification by flash column chromatography over silica gel (50–70% EtOAc/hexanes) afforded the intermediate methyl ester as a thick oil (10 mg, 48%). 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 8.2 Hz, 1H), 7.13 (d, J = 9.0 Hz, 1H), 5.94 (m, 1H), 5.37–5.18 (m, 4H), 4.63 (m, 2H), 4.44 (s, 2H), 3.98 (s, 3H), 3.78 (t, J = 5.8 Hz, 2H), 3.48 (m, 3H), 2.85 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 164.4, 163.5, 158.8, 156.3, 150.1, 145.6, 132.8, 125.6, 125.4, 118.2, 113.9, 113.4, 111.2, 94.8, 91.9, 66.7, 56.7, 53.2, 42.2, 39.3, 24.8; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C20H22NO8 404.133 99, found 404.134 65.
The above ester (10 mg, 25 μmol) was treated with 1.5 mL of 33% TFA/DCM at room temperature and stirred for 1 h. The excess TFA was removed under reduced pressure to afford 41 as a semisolid (8.0 mg, 90%). 1H NMR (400 MHz, CDCl3) δ 11.96 (bs, 1H), 7.63 (d, J = 8.5 Hz, 1H), 7.01 (d, 8.9 Hz, 1H), 5.95 (m, 1H), 5.33 (m, 1H), 5.25 (m, 1H), 4.66 (m, 2H), 4.49 (s, 2H), 4.08 (s, 3H), 3.80 (m, 2H), 2.88 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 170.4, 165.3, 152.8, 152.6, 146.8, 132.5, 129.5, 118.5, 115.1, 111.9, 102.2, 101.0, 67.0, 53.5, 41.8, 39.5, 25.0; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C18H18NO7 360.107 78, found 360.107 59.
Allyl 8-Hydroxy-7-(methoxy(methyl)carbamoyl)-5-oxo-4,5-dihydro-1H-chromeno[3,4-c]pyridine-3(2H)-carboxylate (42)
Compound 39 (91.0 mg, 276 μmol) and 2-methyl-2-butene (350 μL, 3.31 mmol) in 3.5 mL of CH3CN/H2O (1:1) at 0 °C was treated with a solution of sodium chlorite (187 mg, 1.66 mmol) and sodium monophosphate (343 mg, 2.48 mmol) in water, dropwise. After the mixture was stirred for 1 h, the reaction was quenched with 5% aqueous Na2S2O3 solution in water. The pH of the solution was adjusted to 6 and the aqueous portion extracted with EtOAc. The organic layer was dried over Na2SO4 and concentrated under reduced pressure.
The resulting thick oil was dissolved in 4 mL of DCM and treated with 4-N-methylmorpholine (60 μL, 540 μmol), N,O-dimethylhydroxylamine hydrochloride (27 mg, 280 μmol), and EDC (53 mg, 280 μmol). The mixture was stirred for 20 h at room temperature, diluted with DCM, and washed with 1 M aqueous HCl. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. Purification by flash column chromatography over silica gel (3–6% MeOH/CHCl3) gave the intermediate Weinreb amide as a gum (61 mg, 51%, two steps). 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.6 Hz, 1H), 7.14 (t, J = 7.8 Hz, 1H), 5.94 (m, 1H), 5.28 (m, 4H), 4.63 (d, J = 5.6 Hz, 2H), 4.42 (m, 2H), 3.96 (s, 0.5H), 3.73 (m, 2H), 3.48 (m, 5.5H), 3.43 (m, 2.5H), 3.14 (s, 0.5H), 2.87 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 164.3, 159.1, 155.8, 155.3, 149.5, 145.8, 132.8, 125.4, 124.7, 118.2, 115.0, 113.8, 111.1, 94.7, 66.6, 61.8, 61.2, 56.7, 42.1, 39.3, 35.8, 32.4, 24.8; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C21H25N2O8 433.160 54, found 433.158 86.
The above amide (15 mg, 35 μmol) was treated with 1.5 mL of 33% TFA/DCM at room temperature and stirred for 2 h. The excess TFA was removed under reduced pressure to afford pure 42 as a semisolid (13 mg, 96%). 1H NMR (400 MHz, CDCl3) δ 7.49 (bs, 1H), 6.96 (d, J = 7.2 Hz, 1H), 6.88–6.35 (bs 1H), 5.94 (m, 1H), 5.33 (m, 1H), 5.24 (m, 1H), 4.65 (m, 2H), 4.45 (m, 2H), 3.80 (m, 2H), 3.75–3.50 (bs, 3H), 3.39 (s, 3H), 2.87 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 159.2, 158.9, 155.4, 150.0, 146.6, 132.6, 126.4, 118.3, 116.9, 114.2, 112.0, 108.8, 76.6, 76.5, 66.8, 61.8, 41.9, 39.4, 24.9; HRMS (ESI-TOF) (m/z) [M + H]+ calcd for C19H21N2O7 389.134 33, found 389.133 65.
Recombinant Human IRE-1 Expression and Purification
Expression of 59.2 kDa polyhistidine-tagged puritin-hIRE-1 fusion protein was carried out in SF21 cells using the Bac to Bac expression system (Invitrogen) according to manufacturer’s specifications. An 8×-His-puritin sequence was fused to the N-terminal end of the cytoplasmic kinase/RNase domain of human IRE-1 (aa 547–977) in the pFastbacDual-PBL expression vector and included a PreScission protease cleavage site in the linker. Frozen insect cell paste (1 g) was suspended in 8 mL of lysis buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 5 mM βME, 10 mM imidazole) containing one protease inhibitor tablet and lysed using sonication. After removal of the cell debris via centrifugation, the supernatant was applied to a Ni(NTA) column (5 mL). After the untagged protein was washed by flushing with 10 column volumes of lysis buffer, the target protein was eluted using a linear imidazole gradient (15 column volumes, 10–300 mM). Fractions were analyzed via SDS–PAGE. Pooled protein-containing fractions were concentrated and rebuffered into 50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM DTT via ultrafiltration. Typically, 1 L of insect cell culture yielded 3 mg of recombinant 8×-His-puritin-hIRE-1 following Ni(NTA) column purification.
In Vitro IRE-1 RNase FRET-Suppression Assay
The endoribonuclease activity of recombinant hIRE-1 was assayed by incubation of 50 μL of 10 nM hIRE-1 and 50 μL of various concentrations (0.01–1 μM) of fluorescently tagged XBP-1 RNA stem loop (5′-Cy5-CAGUCCGCAGCACUG-BHQ-3′, obtained from Sigma-Aldrich Co.) in assay buffer (20 mM HEPES, pH 7.5, 50 mM KOAc, 0.5 mM MgCl2, 3 mM DTT, 0.4% PEG, and 5% DMSO) for up to 2 h at room temperature in a black 96-well plate. Fluorescence was read at various time points using a Biotek Synergy H1 plate reader with excitation and emission at 620 and 680 nm, respectively. The Km of purified recombinant hIRE-1 was determined to be 45 nM using the Michaelis–Menten kinetic model. Inhibition of RNA cleavage by small molecules was determined by preincubation of 40 μL of 15 nM hIRE-1 with various concentrations of compounds (40 μL) in assay buffer for 30 min at room temperature. A 150 nM solution of fluorescent XBP-1 RNA (40 μL) was then added to each well and the reaction allowed to proceed for 2 h before reading fluorescence as described above. Final concentrations of hIRE-1 and XBP-1 RNA were 5 and 50 nM, respectively. All fluorescence readings were corrected using background values from wells containing only 120 μL of 50 nM XBP-1 RNA. Dose–response experiments were carried out a minimum of 4 times on different days and IC50 values calculated from the mean inhibition value at each concentration.
Antibodies and Reagents
Antibodies against IRE-1 (Cell Signaling), PARP (Cell Signaling), XBP-1s (Santa Cruz), p97 (Fitzgerald), and actin (Sigma), were obtained commercially.
Cell Culture
Primary B cells were purified from wild-type mouse spleens by negative selection using anti-CD43 magnetic beads (Miltenyi Biotech). These cells as well as the human mantle cell lymphoma (MCL) cell lines Mino and Jeko were cultured in RPMI 1640 medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/mL penicillin G sodium, 100 μg/mL streptomycin sulfate, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, and 0.1 mM β-mercaptoethanol (β-ME).
Protein Isolation and Immunoblotting
Cells were lysed using RIPA buffer (10 mM Tris-HCl, pH 7.4; 150 mM NaCl; 1% NP-40; 0.5% sodium deoxycholate; 0.1% SDS; 1 mM EDTA) supplemented with protease inhibitors (Roche). Protein concentrations were determined by BCA assays (Pierce). Samples were boiled in SDS–PAGE sample buffer (62.5 mM Tris-HCl, pH 6.8; 2% SDS; 10% glycerol; 0.1% bromophenol blue) with β-ME and analyzed by SDS–PAGE. Proteins were transferred to nitrocellulose membranes, blocked in 5% nonfat milk (wt/vol in PBS), and immunoblotted with indicated primary antibodies and appropriate horseradish peroxidase-conjugated secondary antibodies. Immunoblots were developed using Western Lighting chemiluminescence reagent (PerkinElmer).
Cell Proliferation XTT Assays
Appropriate numbers of cells were suspended in phenol red-free culture medium, seeded in 96-well cell culture plates, and treated with indicated IRE-1 inhibitors. After 48 h, cells were spun down and proliferation was assessed by XTT assays (Roche) according to the manufacturer’s instructions. Briefly, 50 μL of XTT labeling reagent, 1 μL of electron-coupling reagent, and 100 μL of phenol red-free culture medium were combined and applied to each well of the 96-well plates. Cells were then incubated for 4 h in a CO2 incubator to allow for the yellow tetrazolium salt XTT to be cleaved by mitochondrial dehydrogenases of metabolic active cells to form the orange formazan dye, which can be quantified at 492 nm using a BioTek Synergy H1 microplate reader.
Acknowledgments
This work was supported by the National Institutes of Health (Grant R01CA163910), the American Cancer Society (Grants IRG9309214 and IRG9303216), a Moffitt Team Science award, and the Miles for Moffitt Foundation.
Glossary
Abbreviations Used
- XBP-1s
X-box binding protein 1 spliced form
- FRET
fluorescence resonance energy transfer
- HMBC
heteronuclear multiple bond correlation
- CI
confidence interval
Supporting Information Available
1H NMR spectra for assayed inhibitors, kinetic RNase activity data for recombinant IRE-1, FRET-suppression assay dose–response curves for all active inhibitors, stability data for compound 24, and XTT assay with 30 in the presence of mouse B cells. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Due to a production error, the IC50 curve and Western Blot gel in the abstract graphic were omitted from the version published on May 2, 2014. The revised version was reposted on May 2, 2014.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Rutkowski D. T.; Hegde R. S. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J. Cell Biol. 2010, 189, 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P.; Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Shen X.; Ellis R. E.; Lee K.; Liu C. Y.; Yang K.; Solomon A.; Yoshida H.; Morimoto R.; Kurnit D. M.; Mori K.; Kaufman R. J. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 2001, 107, 893–903. [DOI] [PubMed] [Google Scholar]
- Yoshida H.; Matsui T.; Yamamoto A.; Okada T.; Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [DOI] [PubMed] [Google Scholar]
- Calfon M.; Zeng H.; Urano F.; Till J. H.; Hubbard S. R.; Harding H. P.; Clark S. G.; Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [DOI] [PubMed] [Google Scholar]
- Wang S.; Kaufman R. J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.; Solimini N. L.; Elledge S. J. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman D. E.; Chauhan V.; Koong A. C. The unfolded protein response: a novel component of the hypoxic stress response in tumors. Mol. Cancer Res. 2005, 3, 597–605. [DOI] [PubMed] [Google Scholar]
- Romero-Ramirez L.; Cao H.; Nelson D.; Hammond E.; Lee A. H.; Yoshida H.; Mori K.; Glimcher L. H.; Denko N. C.; Giaccia A. J.; Le Q. T.; Koong A. C. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [DOI] [PubMed] [Google Scholar]
- Hetz C.; Chevet E.; Harding H. P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discovery 2013, 12, 703–719. [DOI] [PubMed] [Google Scholar]
- Yu C. Y.; Hsu Y. W.; Liao C. L.; Lin Y. L. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J. Virol. 2006, 80, 11868–11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F.; Chen X.; Lee A. H.; Glimcher L. H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd D. J.; McHeyzer-Williams L. J.; Kowal C.; Lee A. H.; Volpe B. T.; Diamond B.; McHeyzer-Williams M. G.; Glimcher L. H. XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J. Exp. Med. 2009, 206, 2151–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I.; Denko N. C.; Olson M.; Van Melckebeke H.; Lust S.; Tam A.; Solow-Cordero D. E.; Bouley D. M.; Offner F.; Niwa M.; Koong A. C. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 2011, 117, 1311–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross B. C.; Bond P. J.; Sadowski P. G.; Jha B. K.; Zak J.; Goodman J. M.; Silverman R. H.; Neubert T. A.; Baxendale I. R.; Ron D.; Harding H. P. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Nat. Acad. Sci. U.S.A 2012, 109, E869–E878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura N.; Fulciniti M.; Gorgun G.; Tai Y. T.; Cirstea D.; Santo L.; Hu Y.; Fabre C.; Minami J.; Ohguchi H.; Kiziltepe T.; Ikeda H.; Kawano Y.; French M.; Blumenthal M.; Tam V.; Kertesz N. L.; Malyankar U. M.; Hokenson M.; Pham T.; Zeng Q.; Patterson J. B.; Richardson P. G.; Munshi N. C.; Anderson K. C. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann K.; Lucas J. L.; Vuga D.; Wang X.; Brumm D.; Stiles C.; Kriebel D.; Der-Sarkissian A.; Krishnan K.; Schweitzer C.; Liu Z.; Malyankar U. M.; Chiovitti D.; Canny M.; Durocher D.; Sicheri F.; Patterson J. B. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ri M.; Tashiro E.; Oikawa D.; Shinjo S.; Tokuda M.; Yokouchi Y.; Narita T.; Masaki A.; Ito A.; Ding J.; Kusumoto S.; Ishida T.; Komatsu H.; Shiotsu Y.; Ueda R.; Iwawaki T.; Imoto M.; Iida S. Identification of toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J. 2012, 2, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Perera B. G.; Hari S. B.; Bhhatarai B.; Backes B. J.; Seeliger M. A.; Schurer S. C.; Oakes S. A.; Papa F. R.; Maly D. J. Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012, 8, 982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- Tomasio S. M.; Harding H. P.; Ron D.; Cross B. C.; Bond P. J. Selective inhibition of the unfolded protein response: targeting catalytic sites for Schiff base modification. Mol. BioSyst. 2013, 9, 2408–2416. [DOI] [PubMed] [Google Scholar]
- Kriss C. L.; Pinilla-Ibarz J. A.; Mailloux A. W.; Powers J. J.; Tang C. H.; Kang C. W.; Zanesi N.; Epling-Burnette P. K.; Sotomayor E. M.; Croce C. M.; Del Valle J. R.; Hu C. C. Overexpression of TCL1 activates the endoplasmic reticulum stress response: a novel mechanism of leukemic progression in mice. Blood 2012, 120, 1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata Y.; Kawasaki A.; Sugiura F. Kinetics and mechanism of the Duff reaction. Tetrahedron 1968, 24, 5001–5010. [Google Scholar]
- Blazzevic N.; Kolbah D.; Belin B.; Sunjic V.; Kajfez F. Hexamethylenetetramine, a versatile reagent in organic synthesis. Synthesis 1979, 161–176. [Google Scholar]
- Hu C. C.; Dougan S. K.; McGehee A. M.; Love J. C.; Ploegh H. L. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. 2009, 28, 1624–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura N.; Fulciniti M.; Gorgun G.; Tai Y.-T.; Cirstea D.; Santo L.; Hu Y.; Fabre C.; Minami J.; Ohguchi H.; Kiziltepe T.; Ikeda H.; Kawano Y.; French M.; Blumenthal M.; Tam V.; Kertesz N. L.; Malyankar U. M.; Hokenson M.; Pham T.; Zeng Q.; Patterson J. B.; Richardson P. G.; Munshi N. C.; Anderson K. C. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.