

Abstract
In the present study, we show that for one-electron oxidized A-T or G-C base pairs the singly occupied molecular orbital (SOMO) is located on A or G and is lower in energy than the doubly occupied highest-occupied molecular orbital (HOMO) localized to the pyrimidines, T or C. This directs second ionizations to the pyrimidine bases resulting in triplet state diradical dications, (A•+-T•+) and (G•+-C•+). On interbase proton transfer, the SOMO and HOMO levels switch and the second oxidation is redirected to G and A. For G-C, the doubly oxidized singlet G(-H)+-C(H+) is more stable than its triplet (G•+-C•+); however, for A-T, the triplet (A•+-T•+) lies lowest in energy. The study demonstrates that double ionization of the A-T base pair results in a triplet dication diradical, which is more stable than the proton-transferred triplet or singlet species; whereas, double ionization of the G-C base pair, the proton transferred doubly oxidized singlet, G(-H)+-C(H+), is more stable and has both oxidations on guanine. In DNA, with both A-T and G-C, multiple oxidations would transfer to the guanine base alone.
Introduction
For free radicals, the singly occupied molecular orbital (SOMO) is usually expected to be the highest occupied (half-filled) MO in accordance with the Aufbau principle.1,2 Violations of the Aufbau principle are rare but have been reported in a few studies in which the SOMO is found to be energetically lower in energy than the highest-occupied molecular orbital (HOMO).3−11 Using theory and experiment [photoelectron spectroscopy in the gas-phase and condensed-phase EPR (electron paramagnetic resonance)], Westcott et al.3 showed the non-Aufbau behavior in metalloporphyrins. SOMO–HOMO level inversion was also reported in 2,2,6,6-tetramethylpiperidine-l-oxy radical (TEMPO)-dithiolate complexed with Pt(II).7 Slipchenko et al.8 studied the triradical with an “open-shell” doublet ground state of 5-dehydro-m-xylylene. Sugawara et al.9 reported that the nitronyl nitroxide radical (NN•) bonded to TTF (tetrathiafulvalene) had the SOMO on the NN portion that lay lower in energy than the HOMO, which was on the TTF portion. Thus, one electron oxidation of this radical NN•-TTF removed an electron from the HOMO on the TTF portion and produced a triplet cation biradical, NN•-TTF•+.9,10 This was confirmed from the measured oxidation potential of NN•-TTF, which was similar to that of TTF and not of NN•. Using ab initio and DFT calculations and experiment, Coote and co-workers10,11 recently showed pH-induced SOMO–HOMO energy-level conversion in distonic radical anions. SOMO–HOMO energy level conversion in deprotonated DNA/RNA base radicals were also proposed by these authors.10
One-electron oxidized DNA bases [adenine (A), guanine (G), thymine (T), and cytosine (C)] and base pairs (A-T and G-C) produced by ionizing radiation have been extensively investigated by electron spin resonance (ESR) and pulse radiolysis experiments and theory.12−24 Both theory and experiment predicted that G and A have lower ionization potentials (IP) than thymine and cytosine and are therefore the most easily oxidized sites in DNA.12−21 On one-electron oxidation, all molecules, including DNA bases, become far more acidic in nature than their neutral state.14 The guanine radical cation (G•+) deprotonates to solvent from its N1–H site in nucleosides (pKa = 3.9)14 as well as in single-stranded DNA. But in the G•+-C base pair, G•+ partially transfers its N1–H proton to the N3 site of cytosine, forming proton transferred G(-H)•-C(H+) (see Scheme 1).12−14,18,20,21 Similarly, A•+ deprotonates from its N6–H site to solvent, and in an A•+-T base pair, it can transfer its N6 proton to O4 of thymine forming A(-H)•-T(H+) (see Scheme 1).12−14,22,23 These proton transfer (PT) reactions in one-electron oxidized DNA bases and base pairs control their electronic configuration and thus play an important role in determining their radical stability and reactivity. In this work, we provide the first report of the proton-induced switching of the SOMO and HOMO energy levels in one-electron oxidized A-T and G-C base pairs (A•+-T and G•+-C), using three different density functional methods.
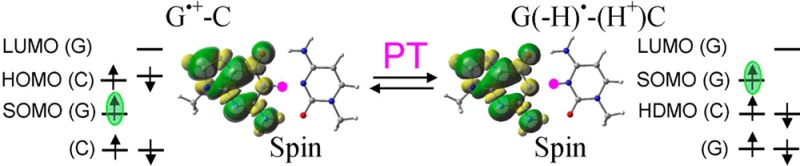
Scheme 1. DFT-Based Orbital Energy (Electronic Configuration) Diagram Showing the Proton Transfer Induced Switching of SOMO-to-HOMO in (a) A•+-T and (b) G•+-C base pairs.

Pink circle shows the location of the proton (H+) in the base pair. The SOMO is highlighted in green. For details of the electronic configuration, see the Supporting Information. PT = proton transfer.
Methods
The ground state structures of A-T and G-C base pairs in their neutral, one-electron oxidized, and proton-transferred state are fully optimized using the ωb97x,25 B3LYP,26 and M06-2x27 density functionals and the 6-31++G(D) basis set as implemented in Gaussian 09.28 To mimic the effect of deoxyribose (sugar), a methyl (CH3) group was attached at N9 of G and A and at N1 of C and T. In addition, a full sugar deoxyribose diphosphate was also considered. The present study clearly demonstrates that the electronic configuration of the one-electron oxidized A-T and G-C base pairs do not follow the Aufbau principle (see Figures 1–3 and Figures S1–S29 in the Supporting Information). The SOMO of an A•+-T base pair localized on A lies below the doubly occupied HOMO which localizes on T (see Scheme 1a). In the case of G•+-C, SOMO is localized on G and lies below the doubly occupied HOMO which localizes on C (see Scheme 1b). These DFT-based observations were further supported by the ab initio MP2/6-31++G(d) calculations of A•+-T and G•+-C base pairs and CASSCF(11,11) calculations for A•+-T (see the Supporting Information).
Figure 1.
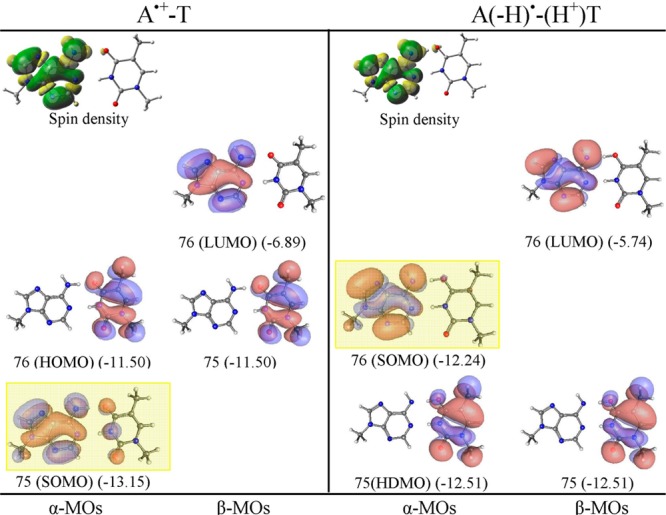
ωb97x/6-31++G(D) calculated spin density distribution and electronic configuration (α and β MOs distribution) of A•+-T and proton transferred A(-H)•-T(H+). Orbital energies are given in electronvolts in parentheses. SOMO is highlighted by yellow rectangle. See the Supporting Information for details and B3LYP, M06-2x, and MP2 results.
Figure 3.

ωb97x/6-31++G(D) based electronic configuration of one-electron oxidized A•+-T, A(-H)•-T(H+), G•+-C, and G(-H)•-C(H+). Spin-density distributions are shown for radicals, and MO plots are shown for the singlet state. Location of SOMO is green highlighted. ET = electron transfer.
Proton transfer from A•+ to T and G•+ to C restores the expected orbital ordering in which the SOMO lies highest in energy. This proton transfer induced reordering of the SOMO is important to redox processes in DNA as it directs the second electron loss from a base pair away from T and C and to the deprotonated G or A radicals. The effect of full solvation on the electronic configuration of A•+-T and G•+-C was also considered using PCM-ωb97x/6-31++G(D) method and show the same HOMO–SOMO switching as found in the gas phase results.
Results and Discussion
The theoretical IPs of bases and base pairs are well-studied and have been reviewed in several recent reviews.12,13,15,16 In this work, we assess the reliability of three density functionals used in the present study of base pairs by calculating the vertical and adiabatic ionization potentials (IPvert and IPadia) of A, T, G, and C for which the gas-phase experimental IPs are available in the literature.29,30 The calculated IPs along with the experimental values are presented in Table 1. From Table 1, it is evident that ωb97x and M06-2x calculated IP values of bases are very close to the experimental values having a maximum difference of ca. 0.1 eV, while B3LYP-calculated IP values have a maximum difference of ca. 0.27 eV. We note that CH3 substitution at N9 of A and G and at N1 of C and T lowers the IPs of the DNA bases (see Table 1). Also, in comparison to the B3LYP which severely suffers from self-interaction errors (SIE), ωb97x and M06-2x mitigate SIE and provide improved orbital descriptions of ionized states.31−34 We present ωb97x/6-31++G(D) results here and B3LYP and M06-2x results in the Supporting Information.
Table 1. Calculated IPvert and IPadia of DNA Bases, Base Pairs and DNA Model Systems with phosphates: (5′-G-3′)·(3′-C-5′) and (5′-A-3′)·(3′-T-5′) in electronvolts.
| bases
and base pairs |
DNA models |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ωb97x/6-31++G(D) |
experimentb |
B3LYP/6-31++G(D) |
M06-2x/6-31++G(D) |
ωb97x/6-31G(D)g |
||||||
| molecule | IPvert | IPadia | IPvert | IPadia | IPvert | IPadia | IPvert | IPadia | IPvert | IPadia |
| guanine (G) | 8.14a (7.86) | 7.71a (7.53) | 8.24 | 7.77 | 7.99a (7.70) | 7.64a (7.46) | 8.10 (7.95) | 7.79 (7.64) | ||
| adenine (A)f | 8.42a (8.27) | 8.16a (7.99) | 8.44 | 8.26 | 8.24a (8.08) | 8.05a (7.87) | 8.50 (8.34) | 8.25 (8.07) | ||
| thymine (T) | 9.13a (8.81) | 8.81a (8.52) | 9.14 | 8.87 | 8.98a (8.65) | 8.74a (8.44) | 9.23 (8.88) | 8.94 (8.61) | ||
| cytosine (C) | 8.86a (8.59) | 8.74a (8.44) | 8.94 | 8.68 | 8.67a (8.40) | 8.56a (8.30) | 8.95 (8.67) | 8.82 (8.53) | ||
| G-C | 7.24 | 6.81 | – | – | 7.06 | 6.72 | 7.33 | 6.91 | 6.90 | 6.30 |
| G-C (PT) | – | 6.86c | – | – | – | 6.76c | – | 6.97c | – | 6.35 |
| A-T | 7.99 (8.01)f | 7.62 | – | – | 7.58 | 7.43 | 8.08 | 7.72 | 7.64 | 7.12 |
| A-T (PT) | – | 7.80c | – | – | – | 7.63c | – | 7.80c | – | 7.26 |
| G•+-C (PT)d | 11.26 | 10.76 | 10.54 | 9.71 | ||||||
| G•+-Ce | 11.67 | 11.26 | 11.48 | 11.13 | 11.77 | 11.34 | 10.97 | 9.92 | ||
| A•+-T (PT)d | 12.67 | 11.50 | – | – | – | – | 12.23 | 11.41 | 11.22 | 10.46 |
| A•+-Te | 11.12 | 10.72 | – | – | 10.91 | 10.69 | 11.23 | 10.81 | 10.41 | 9.34 |
Ref (32) and methylated base IPs in parentheses.
IPadia calculated with respect to neutral G-C and A-T base pairs.
Singlet state.
Triplet state.
Ref (34). EOM-IP-CCSD/6-311+G(d,p) calculation. IPvert (A) = 8.35 eV.
(5′-G-3′)·(3′-C-5′) and (5′-A-3′)·(3′-T-5′). See Figures S23–S28 in the Supporting Information.
The electronic configuration (orbital distribution) of neutral A-T base pair calculated by the ωb97x, B3LYP, and M06-2x methods are shown in Figures S1–S3, and a PCM (polarized continuum model) using ωb97x is shown in Figure S22 in the Supporting Information. As expected, all the methods predict HOMO as π in nature and localizing on A in the A-T base pair. This preliminary inspection of the electronic configuration of the A-T base pair clearly shows that the initial oxidation will take place from the HOMO localizing on A. This is also supported by the experiment as A has a lower reduction potential than T.14 The calculated IPvert of the A-T base pair by ωb97x, B3LYP, and M06-2x methods are 7.99, 7.58, and 8.08 eV, while the corresponding IPadia are 7.62, 7.43, and 7.72 eV, respectively (see Table 1). The ωb97x/6-31++G(D) calculated IPvert (7.99 eV) is in excellent agreement (8.01 eV) with those calculated by Krylov and co-workers34 using the EOM-IP-CCSD/6-311+G(d,p) method (see Table 1). The spin density distribution and electronic configuration of A•+-T and proton transferred A(-H•)-T(H+) is shown in Figure 1 and in Figures S1–S3 in the Supporting Information. The methods ωb97x, B3LYP, and M06-2x predict that A•+-T is more stable than A(-H•)-T(H+) by 4.15, 4.44, and 1.98 kcal/mol, respectively (see Figure S7 in the Supporting Information). From the electronic configuration, we see that the SOMO which is localized on A in A•+-T lies below the HOMO localizing on T, in violation of the Aufbau principle, see Figure 1 and Figures S1–S3 and MP2 calculated electronic configuration in Figure S29 in the Supporting Information.
The electronic configuration of A•+-T clearly predicts that further ionization will take place from T not from A, and this is confirmed from the calculated IP of A•+ (Figure S11 in the Supporting Information). The calculated IPvert of A•+ by ωb97x, B3LYP, and M06-2x are 13.51, 13.39, and 13.50 eV, respectively, and the corresponding IPadia of A•+ are 13.08, 13.06, and 13.10 eV, respectively, which are much larger than the IP of thymine, ca. 9 eV (see Table 1 and Figure S11 in the Supporting Information). As mentioned above, the ionized A-T (A•+-T) becomes far more acidic in nature and the pKa of A•+ was predicted to be ca. <1–4 by experiment and theory.12−15,35 Thus, A•+ in the base pair can transfer its N6 proton to O4 of thymine. Although it is energetically uphill in our work, a recent QM/MM simulation by Conwell and co-workers23 suggests proton transfer is likely. The electronic configuration of A(-H•)-T(H+) (Figure 1) shows that upon proton transfer the SOMO which is localized on A(-H)• is highest in energy and lying above the highest doubly occupied molecular orbital (HDMO) localized on O4-protonated T [T(H+)]. The electronic configuration of A(-H•)-T(H+) thus shows that further ionization will occur from A(-H)•. The calculated IPvert(IPadia) of A(-H)• by ωb97x, B3LYP, and M06-2x are 8.81(8.39), 8.54(8.27), and 8.73(8.32) eV, respectively, and the calculated IPvert and IPadia of T(H+) by ωb97x, B3LYP, and M06-2x lie in the range of 13.28–13.71 eV, respectively (see Figures S10 and S13 in the Supporting Information).
The electronic configuration of the neutral G-C base pair is shown in the Supporting Information in Figures S4–S6 and in Figure S21 using PCM (ωb97x). In the G-C base pair, the HOMO is localized on guanine, while the lowest unoccupied molecular orbital (LUMO) localizes on cytosine; these are π in nature. Thus, the electronic configuration of the G-C base pair depicts that ionization will take place on guanine. The ωb97x, B3LYP, and M06-2x calculated IPvert of the G-C base pair are 7.24, 7.06, and 7.33 eV, respectively, and the corresponding IPadia are 6.81, 6.72, and 6.91 eV, respectively. The IP of the G-C base pair is lower than the A-T base pair, and it is well-known that guanine is the prime site for hole localization in DNA.12−23 From the electronic configuration of G•+-C, shown in Figure 2, we see that SOMO localized on guanine is no longer the highest energy orbital and lies below the doubly occupied HOMO localized on cytosine. This is also supported by MP2 calculations presented in Figure S29 in the Supporting Information. Thus, in G•+-C, cytosine is available for the oxidation. The calculated IPvert and IPadia of G•+ (Figure S18 in the Supporting Information) lie in the range of 12.62–13.11 eV, which is larger than the IP (ca. 9 eV) of cytosine (see Table 1). Steenken14 estimated the pKa of one-electron oxidized deoxyguanosine (dG•+) as 3.9 and proposed that a facile PT can take place from N1 of G to N3 of C in a G•+-C base pair, which has been confirmed by ESR experiments and DFT calculations.18,20,21 From the present calculation, G•+-C was found to be more stable than the proton transferred G(-H)•-C(H+) by 1.06, 0.91, and 1.23 kcal/mol using the ωb97x, B3LYP, and M06-2x calculations (see Figure S14 in the Supporting Information). We note that surrounding solvation plays an important role in the stability of G•+-C and G(-H)•-C(H+), and other work shows that the inclusion of the first hydration layer around these structures make G(-H)•-C(H+) more stable (ca. 1 kcal/mol) than G•+-C.21
Figure 2.
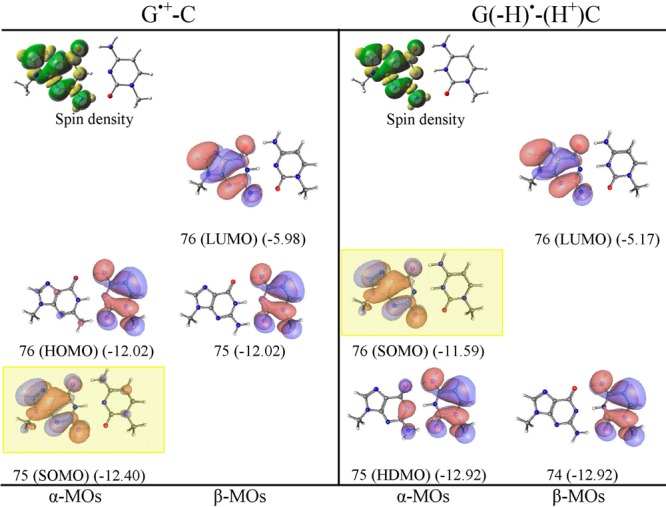
ωb97x/6-31++G(D) calculated spin density distribution and electronic configuration (α and β MOs distribution) of G•+-C and proton transferred G(-H)•-C(H+). MO energies are given in eV in parentheses. SOMO is highlighted by a yellow rectangle. See the Supporting Information for details and B3LYP, M06-2x, and MP2 results.
From the electronic configuration of proton transferred G(-H)•-C(H+) (shown in Figure 2), it is evident that the SOMO localizing on G(-H)• is highest in energy and the HDMO localized on cytosine lies below the SOMO. Thus, in G(-H)•-C(H+), the oxidation takes place from G(-H)•, which is confirmed from the calculated IP of G(-H)• (Figure S17 in the Supporting Information). The calculated IPs of G(-H)• are in the range of 8.11–8.56 eV, while the IPs of C(H+) lie in the range of 13.36–13.74 eV (see Figures S17 and S20 in the Supporting Information).
The electronic configuration of species formed by one-electron oxidation of A•+-T, A(-H)•-T(H+), G•+-C, and G(-H)•-C(H+), based on ωb97x/6-31++G(D) calculations, are shown in Figure 3. One-electron oxidation of A•+-T yields A•+-T•+, which is found to be the most stable in the triplet state.36 Thus, in the triplet state, both A and T are oxidized to the diradical dication form (A•+-T•+) with one electron spin localized on each base (Figure 3). The calculated IPvert of A•+-T by ωb97x, B3LYP, and M06-2x methods are 11.12, 10.91, and 11.23 eV, respectively, and the corresponding IPadia are 10.72, 10.69, and 10.81 eV, respectively (see Table 1).
Our calculation predicts that one-electron oxidation of proton-transferred A(-H)•-T(H+) removes an electron from A and yields, A(-H)+-T(H+). The singlet state of this species [A(-H)+-T(H+)] is found to be more stable than its triplet form. The HOMO in the singlet state lies on T(H+) (see Figure 3). The calculated IPadia of A(-H)•-T(H+) by ωb97x and M06-2x are 11.50 and 11.41 eV, respectively. Most importantly, we note that the A•+-T•+ (triplet state) is more stable than the A(-H)+-T(H+) (singlet state) by ca. 18 kcal/mol (see Figure S8 in the Supporting Information). Also, the IP of A•+-T•+ in the triplet state is lower than A(-H)+-T(H+) in the singlet state, thus, the present calculations show that on double oxidation both A and T will be oxidized in the A-T base pair. Recently, Peluso and co-workers37 measured the oxidation potential of a single strand 5′-TTAATT-3′ sequence and measured two peaks centered at 0.97 and 1.35 V. The first anodic peak (0.97 V) was assigned to the oxidation of two stacked adenines, and the second peak (1.35 V) was assigned to the two stacked thymines.
One-electron oxidation of G•+-C produces the triplet diradical (G•+-C•+), and one-electron oxidation of proton transferred G(-H)•-C(H+) produces singlet G(-H)+-C(H+), the latter species is the most stable structure overall (see Figure 3 and Figure S15 in the Supporting Information). The SOMO in triplet G•+-C•+ places an unpaired electron on both G and C (Figure 3). Calculations using ωb97x/6-31++G(D) predict singlet G(-H)+-C(H+) to be more stable than the triplet diradical G•+-C•+ by ca. 11 kcal/mol (Figure S15 in the Supporting Information). The ωb97x/6-31++G(D) calculated IP of G•+-C in the triplet state is 11.67 eV (IPvert) and 11.26 eV (IPadia), and the IP of G(-H)•-C(H+) in the singlet state is 11.26 eV (IPvert) and 10.76 eV (IPadia). Thus, in a G-C base pair, the sequential two-electron oxidation will occur only on guanine.
The second ionization of A-T and G-C base pairs lies in the range of 10.72–12.67 eV (Table 1), which is close to the ionization potential of phosphate (ca., 11–12 eV),38 thus in DNA, phosphate may be the prime site for a second oxidation. To test this possibility, we calculated the first and second ionization potentials (Table 1 last column) and electronic configurations of double stranded (5′-G-3′)·(3′-C-5′) and (5′-A-3′)·(3′-T-5′) using the ωb97x/6-31G(D) method. The calculations clearly show that the second oxidation occurs only on the bases and not on the phosphate in DNA (see MOs in Figures S23–S28 in the Supporting Information). Also, from Table 1, it is evident that the IPs of bases in the DNA decrease substantially in comparison to the IPs of isolated bases and base pairs. We note that in the present calculations, the phosphates groups are protonated. In aqueous solution, phosphate groups are neutralized by solvated proximate cations. The energy differences between the protonated and anionic system with counterions in an aqueous media is relatively small.38 Thus, only a modest change in base ionization potential is expected on phosphate protonation, but protonation will have a greater effect on the phosphate IPs.
Conclusions
In conclusion, we find that PT in one-electron oxidized DNA base pairs plays an important role in altering the IPs of individual bases in the base pair, induces HOMO–SOMO level switching, and thus directs multiple ionizations to a single base G or A. For the A–T base pair, we find PT is not favored within A•+-T and the HOMO lies on T so that the second ionization oxidizes both A and T forming triplet A•+-T•+, which is more stable than the singlet A(-H)+-T(H+). For the G-C base pair, PT is favored within G•+-C forming G(-H)•-(H+)C, thus G(-H)• becomes available for further oxidation resulting in doubly oxidized G. PT reactions in one-electron oxidized DNA base pairs are an expected phenomenon. Since one electron-oxidation of a base makes it more acidic, oxidation induces a shift in the prototropic equilibrium between the two bases as proposed by Steenken.14 This interbase PT is able to quench the SOMO–HOMO level inversion in the one-electron oxidized A-T and G-C base pair as observed for distonic anion radicals after protonation from the solvent.10,11 High-energy radiation often produces two one electron ionizations within a short-range.39,40 Two-electron oxidation at sites within DNA were proposed earlier by Bernhard to account for products formed in nonradical processes.41,42 The present work shows that the A-T base pair and G-C base pair would respond differently to double oxidation. In a doubly oxidized G-C, the double oxidation would reside only on G (in the singlet state), whereas in a doubly oxidized A-T, the oxidations would be shared between A and T in the triplet state. Subsequent rapid hole transfers and deprotonation processes in DNA then lead to the most stable double-oxidized G. Finally, we note that since spectroscopic studies of gas phase base pairs are now well-established,43 tests of predictions made in this work are quite feasible.
Acknowledgments
The authors thank the NCI of the NIH for support under Grant R01CA045424. We thank Prof. A. W. Bull for valuable editorial suggestions.
Supporting Information Available
Complete ref (26), optimized geometries, electronic configuration, and ionization potentials of A-T and G-C base pairs calculated using DFT methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- IUPAC. Compendium of Chemical Terminology (the “Gold Book”), 2nd ed.; McNaught A. D., Wilkinson A., Eds.; Blackwell Scientific Publications: Oxford, 1997. (http://goldbook.iupac.org/AT06996.html). [Google Scholar]
- Scerri E. R.The Trouble with the Aufbau Principle. Educ. Chem. 2013. (November), 24–26. [Google Scholar]
- Westcott B. L.; Gruhn N. E.; Michelson L. J.; Lichtenberger D. L. Experimental Observation of Non-Aufbau Behavior: Photoelectron Spectra of Vanadyloctaethylporphyrinate and Vanadylphthalocyanine. J. Am. Chem. Soc. 2000, 122, 8083–8084. [Google Scholar]
- Gary J. B.; Buda C.; Johnson M. J. A.; Dunietz B. D. Accessing Metal-Carbide Chemistry. A Computational Analysis of Thermodynamic Considerations. Organometallics 2008, 27, 814–826. [Google Scholar]
- Zuo T.; Xu L.; Beavers C. M.; Olmstead M. M.; Fu W.; Crawford T. D.; Balch A. L.; Dorn H. C. M2@C79N (M = Y, Tb): Isolation and Characterization of Stable Endohedral Metallofullerenes Exhibiting M-M Bonding Interactions inside Aza[80]fullerene Cages. J. Am. Chem. Soc. 2008, 130, 12992–12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bester G.; Reuter D.; He L.; Zunger A.; Kailuweit P.; Wieck A. D.; Zeitler U.; Maan J. C.; Wibbelhoff O.; Lorke A. Experimental Imaging and Atomistic Modeling of Electron and Hole Quasiparticle Wave Functions in InAs/GaAs Quantum Dots. Phys. Rev. B 2007, 76, 075338. [Google Scholar]
- Kusamoto T.; Kume S.; Nishihara H. Realization of SOMO–HOMO Level Conversion for a TEMPO-Dithiolate Ligand by Coordination to Platinum(II). J. Am. Chem. Soc. 2008, 130, 13844–13845. [DOI] [PubMed] [Google Scholar]
- Slipchenko L. V.; Munsch T. E.; Wenthold P. G.; Krylov A. I. 5-Dehydro-1,3-quinodimethane: A Hydrocarbon with an Open-shell Doublet Ground State. Angew. Chem., Int. Ed. 2004, 43, 742–745. [DOI] [PubMed] [Google Scholar]
- Sugawara T.; Komatsu H.; Suzuki K. Interplay Between Magnetism and Conductivity Derived from Spin-polarized Donor Radicals. Chem. Soc. Rev. 2011, 40, 3105–3118. [DOI] [PubMed] [Google Scholar]
- Gryn’ova G.; Marshall D. L.; Blanksby S. J.; Coote M. L. Switching Radical Stability by pH-induced Orbital Conversion. Nat. Chem. 2013, 5, 474–481. [DOI] [PubMed] [Google Scholar]
- Gryn’ova G.; Coote M. L. Origin and Scope of Long-Range Stabilizing Interactions and Associated SOMO–HOMO Conversion in Distonic Radical Anions. J. Am. Chem. Soc. 2013, 135, 15392–15403. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Sevilla M. D. Proton-Coupled Electron Transfer in DNA on Formation of Radiation-Produced Ion Radicals. Chem. Rev. 2010, 110, 7002–7023and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Sevilla M. D. DFT Treatment of Radiation Produced Radicals in DNA Model Systems. Adv. Quantum Chem. 2007, 52, 59–87. [Google Scholar]
- Steenken S. Purine Bases, Nucleosides, and Nucleotides: Aqueous Solution Redox Chemistry and Transformation Reactions of Their Radical Cations and e- and OH Adducts. Chem. Rev. 1989, 89, 503–520. [Google Scholar]
- Kumar A.; Sevilla M. D. In Radical and Radical Ion Reactivity in Nucleic Acid Chemistry; Greenberg M., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2009; pp 1–40. [Google Scholar]
- Close D. M. In Radiation Induced Molecular Phenomena in Nucleic Acids; Shukla M. K., Leszczynski J., Eds.; Challenges and Advances in Computational Chemistry and Physics; Springer Science: Dordrecht, The Netherlands, 2008; Vol. 5, pp 493–529. [Google Scholar]
- Sevilla M. D.; Becker D.; Yan M.; Summerfield S. R. Relative Abundances of Primary Ion Radicals in γ-Irradiated DNA: Cytosine vs Thymine Anions and Guanine vs Adenine Cations. J. Phys. Chem. 1991, 95, 3409–3415. [Google Scholar]
- Adhikary A.; Khanduri D.; Sevilla M. D. Direct Observation of the Hole Protonation State and Hole Localization Site in DNA Oligomers. J. Am. Chem. Soc. 2009, 131, 8614–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hole E. O.; Nelson W. H.; Close D. M.; Sagstuen E. ESR and ENDOR Study of the Guanine Cation: Secondary Product in 5-dGMP. J. Chem. Phys. 1987, 86, 5218–5219. [Google Scholar]
- Adhikary A.; Kumar A.; Becker D.; Sevilla M. D. The Guanine Cation Radical: Investigation of Deprotonation States by ESR and DFT. J. Phys. Chem. B 2006, 110, 24171–24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Sevilla M. D. Influence of Hydration on Proton Transfer in the Guanine–Cytosine Radical Cation (G•+–C) Base Pair: A Density Functional Theory Study. J. Phys. Chem. B 2009, 113, 11359–11361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran J.; Oliva A.; Rodríguez-Santiago L.; Sodupe M. Single versus Double Proton-Transfer Reactions in Watson-Crick Base Pair Radical Cations. A Theoretical Study. J. Am. Chem. Soc. 1998, 120, 8159–8167. [Google Scholar]
- Kinz-Thompson C.; Conwell E. Proton Transfer in Adenine–Thymine Radical Cation Embedded in B-Form DNA. J. Phys. Chem. Lett. 2010, 1, 1403–1407. [Google Scholar]
- Wetmore S. D.; Boyd R. J.; Eriksson L. A. Comparison of Experimental and Calculated Hyperfine Coupling Constants. Which Radicals Are Formed in Irradiated Guanine?. J. Phys. Chem. B 1998, 102, 9332–9343. [Google Scholar]
- Chai J.-D.; Head-Gordon M. Systematic Optimization of Long-range Corrected Hybrid Density Functionals. J. Chem. Phys. 2008, 128, 084106. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Zhao Y.; Truhlar D. G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- Hush N. S.; Cheung A. S. Ionization Potentials and Donor Properties of Nucleic Acid Bases and Related Compounds. Chem. Phys. Lett. 1975, 34, 11–13. [Google Scholar]
- Orlov V. M.; Smirnov A. N.; Varshavsky Y. M. Ionization Potentials and Electron Donor Ability of Nucleic Acid Bases and Their Analogues. Tetrahedron Lett. 1976, 48, 4377–4378. [Google Scholar]
- Mantz Y. A.; Gervasio F. L.; Laino T.; Parrinello M. Charge Localization in Stacked Radical Cation DNA Base Pairs and the Benzene Dimer Studied by Self-Interaction Corrected Density-Functional Theory. J. Phys. Chem. A 2007, 111, 105–112. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Pottiboyina V.; Sevilla M. D. One-Electron Oxidation of Neutral Sugar Radicals of 2′-Deoxyguanosine and 2′-Deoxythymidine: A Density Functional Theory (DFT) Study. J. Phys. Chem. B 2012, 116, 9409–9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Sevilla M. D. Density Functional Theory Studies of the Extent of Hole Delocalization in One-Electron Oxidized Adenine and Guanine Base Stacks. J. Phys. Chem. B 2011, 115, 4990–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravaya K. B.; Epifanovsky E.; Krylov A. I. Four Bases Score a Run: Ab Initio Calculations Quantify a Cooperative Effect of H-Bonding and π-Stacking on the Ionization Energy of Adenine in the AATT Tetramer. J. Phys. Chem. Lett. 2012, 3, 2726–2732. [DOI] [PubMed] [Google Scholar]
- Close D. M. Calculated pKa’s of the DNA Base Radical Ions. J. Phys. Chem. A 2013, 117, 473–480. [DOI] [PubMed] [Google Scholar]
- ωb97x and M06-2x predict proton transfer from N6 of A to O4 of T during optimization in the singlet state while B3LYP shows no proton transfer in the singlet state. However, A•+-T•+ (triplet state) was found to be the most stable than the proton transferred structure A(-H)+-T(H+) in the singlet state (see Figure S8 in the Supporting Information).
- Capobianco A.; Caruso T.; Celentano M.; D’Ursi A. M.; Scrima M.; Peluso A. Stacking Interactions Between Adenines in Oxidized Oligonucleotides. J. Phys. Chem. B 2013, 117, 8947–8953. [DOI] [PubMed] [Google Scholar]
- Colson A.-O.; Besler B.; Sevilla M. D. Ab Initio Molecular Orbital Calculations on DNA Radical Ions. 3. Ionization Potentials and Ionization Sites in Components of the DNA Sugar Phosphate Backbone. J. Phys. Chem. 1993, 97, 8092–8097. [Google Scholar]
- Pimblott S. M.; Mozumder A. Structure of Electron Tracks in Water. 2. Distribution of Primary Ionizations and Excitations in Water Radiolysis. J. Phys. Chem. 1991, 95, 7291–7300. [Google Scholar]
- LaVerne J. A.; Pimblott S. M. Electron Energy-Loss Distributions in Solid, Dry DNA. Radiat. Res. 1995, 141, 208–215. [PubMed] [Google Scholar]
- Close D. M.; Nelson W. H.; Bernhard W. A. DNA Damage by the Direct Effect of Ionizing Radiation: Products Produced by Two Sequential One-Electron Oxidations. J. Phys. Chem. A 2013, 117, 12608–12615. [DOI] [PubMed] [Google Scholar]
- Bernhard W. A. In Radical and Radical Reactivity in Nucleic Acid Chemistry; Greenberg M., Ed.; John Wiley and Sons: Hoboken, NJ, 2010; pp 41–68. [Google Scholar]
- Nir E.; Kleinermanns K.; de Vries M. S. Pairing of Isolated Nucleic-acid Bases in the Absence of the DNA Backbone. Nature 2000, 408, 949–951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.