

Abstract
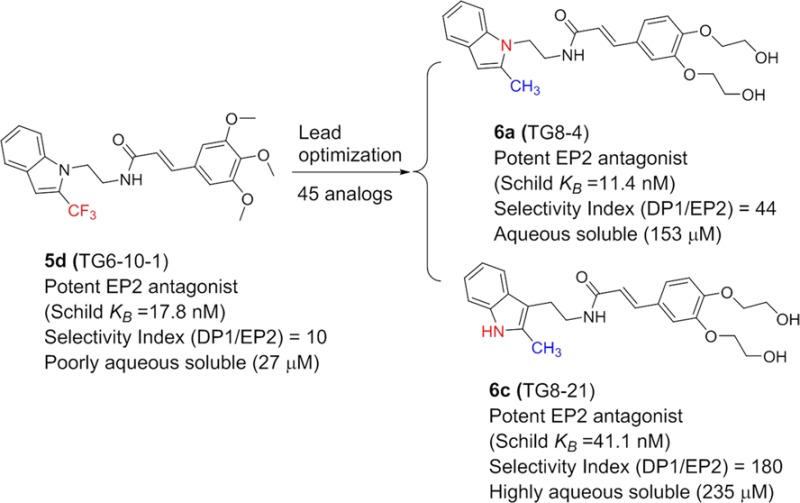
Prostanoid receptor EP2 can play a proinflammatory role, exacerbating disease pathology in a variety of central nervous system and peripheral diseases. A highly selective EP2 antagonist could be useful as a drug to mitigate the inflammatory consequences of EP2 activation. We recently identified a cinnamic amide class of EP2 antagonists. The lead compound in this class (5d) displays anti-inflammatory and neuroprotective actions. However, this compound exhibited moderate selectivity to EP2 over the DP1 prostanoid receptor (∼10-fold) and low aqueous solubility. We now report compounds that display up to 180-fold selectivity against DP1 and up to 9-fold higher aqueous solubility than our previous lead. The newly developed compounds also display higher selectivity against EP4 and IP receptors and a comparable plasma pharmacokinetics. Thus, these compounds are useful for proof of concept studies in a variety of models where EP2 activation is playing a deleterious role.
Introduction
Inflammation plays a pathogenic role in a variety of acute and chronic neurodegenerative diseases such as status epilepticus (SE), epilepsy, amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), Parkinson’s disease (PD), and traumatic brain injury (TBI).1−8 Cyclooxygenase 2 (COX-2) is induced during and after brain injury and is a major contributor to the inflammation and disease progression in a variety of central nervous system (CNS) diseases.9−12 COX-2 inhibitors have been widely explored for suppression of pain and inflammation in variety of peripheral diseases, for example, in patients with arthritis.13,14 However, COX-2 inhibitors cause adverse cardiovascular effects by reducing activation of a downstream prostanoid receptor subtype IP.15−18 As a result, two COX-2 inhibitors, rofecoxib (Vioxx) and valdecoxib (Bextra), were withdrawn from the U.S. market. Moreover, it is not yet clear that COX-2 inhibitors could provide a benefit to patients with chronic inflammatory neurodegenerative diseases such as epilepsy and AD.19−26 Thus, future anti-inflammatory therapy should be targeted through a specific proinflammatory prostanoid synthase or receptor to blunt the inflammation and neuropathology in CNS diseases rather than to block the entire COX-2 signaling.
COX-2 catalyzes the synthesis of prostaglandin-H2 (PGH2) from arachidonic acid, which is transformed into five prostanoids, PGD2, PGE2, PGF2, PGI2 and TXA2, by cell specific synthases. These prostanoids activate nine receptors, DP1, DP2, EP1, EP2, EP3, EP4, FP, IP, and TP. Each of these receptors can play protective as well as harmful roles in a variety of CNS and peripheral pathophysiologies.27−29 EP2 receptor has emerged as an important biological target for drug discovery to treat a variety of CNS and peripheral diseases.30,31 When activated by PGE2, EP2 stimulates adenylate cyclase resulting in elevation of cytoplasmic cAMP concentration, which initiates downstream events mediated by protein kinase A (PKA)32,33 or exchange protein activated by cAMP (Epac).34−36
The EP2 receptor is widely expressed in both neurons and glia in the brain and plays a “yin–yang” nature of protective as well as deleterious role.31 For example, in some chronic neurodegenerative disease models, EP2 activation appears to promote inflammation and neurotoxicity. Deletion of the EP2 receptor reduces oxidative damage and amyloid-β burden in a mouse model of AD.37 EP2 deletion also attenuates neurotoxicity by α-synuclein aggregation in mouse model of PD.38 Moreover, EP2 deletion improves motor strengths and the survival of the ALS mouse.39 Furthermore, mice lacking EP2 receptors have shown less cerebral oxidative damage produced by the activation of innate immunity.40 In vitro, microglia cultures from mice lacking EP2 have shown enhanced amyloid-β phagocytosis and are less sensitive to amyloid-β induced neurotoxicity.41 Despite a wealth of information available from EP2 gene knockout studies, results from pharmacological inhibition of EP2 are limited because the antagonists for EP2 receptors have only been created recently by Pfizer42 and us.43 Earlier, we reported identification of a cinnamic amide class of EP2 antagonists by using a high-throughput screening method.43 A limited structure–activity relationship study (SAR) concluded that this class of compounds displays high potency to EP2 receptor but moderate selectivity to EP2 over another prostanoid receptor, DP1. The lead compound in this class, 5d (aka TG6-10-1), displays about 10-fold selectivity to EP2 over DP1 and poor aqueous solubility (27 μM). However, 5d demonstrated robust neuroprotective and anti-inflammatory effects in a pilocarpine model of status epilepticus when administered in three doses beginning 4 h after mice entered into status epilepticus.44 A key to advance this class of compounds for preclinical studies in a variety of neurodegenerative disease models is to improve their EP2 selectivity, aqueous solubility, and in vivo pharmacokinetics. In the present study we report the synthesis of 45 new analogues and their structure–activity relationships and show that improvements are made in terms of selectivity, solubility, and metabolic stability in liver microsomes. Two compounds, 6a and 6c, display about 4- to 18-fold higher selectivity against DP1 receptor and 5- to 8-fold higher aqueous solubility than the previous best compound 5d.
Results and Discussion
First Generation Cinnamic Amide EP2 Antagonists Show Poor Aqueous Solubility, Poor in Vitro Liver Microsomal Stability, and Moderate Plasma Half-Life
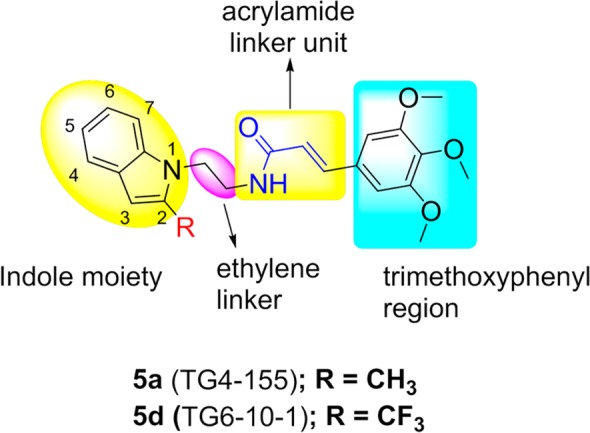
We previously synthesized 27 compounds around initial high-throughput screening hit 5a (aka TG4-155) (Figure 1) for structure–activity relationship study. Several derivatives from this set showed potent EP2 inhibition with Schild KB values at the low nanomolar level, and they also displayed excellent selectivity against EP4 and β-AR receptors.43 To examine the druglike properties within the class, 10 potent compounds (EP2 Schild KB < 20 nM) were selected and subjected to metabolic stability in microsomal fractions of mouse and human liver at two different concentrations (1 and 10 μM). A majority of these compounds were found to be labile in these liver fractions with <15 min half-life at 10 μM concentration43 except one compound 5d, which showed >15 min half-life in mouse and human liver microsomes at 10 μM (Table 3). Moreover, compound 5d showed improved brain-to-plasma ratio (1.7) and plasma half-life (1.7 h) in a pharmacokinetic (PK) study in C57BL/6 mice, in comparison to initial hit compound 5a.44 Although 5d has been used for initial proof of concept studies, the plasma half-life should be improved for testing in a wider variety of preclinical models.
Figure 1.
Optimization strategy. Structures of representative first generation EP2 antagonists. Regions marked are explored for SAR study.
Table 3. Liver Microsomal Stability and in Vivo Pharmacokinetic Properties of Selected Compoundsa.
| % of parent compound
remaining at 60 min vs T = 0 min |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| human liver microsomes |
mouse liver microsomes |
mouse in vivo pharmacokinetics properties |
|||||||
| compd | 1 μM | 10 μM | 1 μM | 10 μM | route of administration (dose, mg/kg) | Cmax (ng/mL) | AUClast (h·ng/mL) | T1/2 (plasma) (h) | B/P |
| 5a (TG4-155) | 0.1 | 10.6 | 0.2 | 4.8 | iv (3) | 2400 ± 350 | 749 ± 24 | 0.45 | 0.3 |
| ip (3) | 738 ± 207 | 457 ± 64 | 0.58 | ||||||
| 5c (TG7-98) | 20.9 | 24.0 | 18.3 | 13.0 | nd | ND | ND | ND | ND |
| 5d (TG6-10-1) | 2.3 | 39.9 | 2.0 | 23.1 | ip (5) | 115 ± 44 | 453 ± 49 | 1.6 | 1.8 |
| po (10) | 248 ± 61 | 475 ± 60 | 1.8 | 1.6 | |||||
| 5g (TG7-74) | 0.8 | 5.8 | 0 | 0.2 | nd | ND | ND | ND | ND |
| 5h (TG7-76) | 12.4 | 18.6 | 0.2 | 0.1 | nd | ND | ND | ND | ND |
| 5j (TG7-186) | 0.4 | 21.4 | 0.1 | 51.7 | nd | ND | ND | ND | ND |
| 6a (TG8-4) | 16.2 | 43.8 | 1.7 | 8.7 | ip (5) | 1510 ± 142 | 1050 ± 58 | 1.49 | <0.1 |
| po (10) | 128 ± 23 | 197 ± 26 | 1.44 | <0.1 | |||||
| 6c (TG8-21) | 58.5 | 70.4 | 7.1 | 19.3 | ND | ND | ND | ND | ND |
Male or female C57Bl/6 mice were used for in vivo pharmacokinetic study. Formulation used: 5% DMA, 50% PEG400, and 45% saline for compound 5a; 10% DMSO, 50% PEG400, 40% sterile water for 5d; and 2.5% DMA, 12.5% propylene glycol, and 85% phosphate-buffered saline (pH 7.4) for 6a. DMA = N,N-dimethylacetamide. Cmax is the maximum observed concentration that occurs at Tmax. T1/2 is the terminal half-life. AUC = area under the curve from time zero to the time of the last measurable observation (AUClast). B/P = brain to plasma ratio, calculated from drug concentrations in plasma and brain tissue at 1 and 2 h. ND = not determined.
The structural identity among the prostanoid receptor family is very limited. EP1, EP2, EP3, and EP4 share a common endogenous ligand PGE2 for their activation, but they only share 20–30% structural homology.45 In contrast, EP2 is more homologous to DP1 (44%) and IP receptors (40%).45 Earlier, compounds 5a and 5d were tested against other prostanoid receptors. Although they displayed high selectivity to EP2 over EP1, EP3, EP4, FP, IP, and TP receptors, they showed only moderate selectivity (∼10-fold) to DP1 receptor.44,46 None of the earlier set of 27 compounds were more selective over DP1 than 5a and 5d (not shown). Furthermore, compounds 5a and 5d displayed low aqueous solubility (45 and 27 μM, respectively) (Table 1). Thus, our initial goal was to identify compounds with enhanced selectivity and aqueous solubility.
Table 1. EP2 Bioactivity, DP1 Selectivity, and Aqueous Solubility of Cinnamic Amide Analoguesa.
|
KB (nM) |
|||||
|---|---|---|---|---|---|
| compd | EP2 | DP1 | SI (DP1/EP2) | solubility (μM) | |
| 5a | TG4-155 | 2.4 | 34.5 | 14.4 | 45 |
| 5b | TG7-23 | 3.4 | 83 | 24 | <25 |
| 5c | TG7-98 | 3.4 | 210 | 60 | <25 |
| 5d | TG6-10-1 | 17.8 | 166 | 9.3 | 27 |
| 5e | TG7-2 | 305 | ND | ND | ND |
| 5f | TG7-13 | 306 | ND | ND | ND |
| 5g | TG7-74 | 2.4 | 110 | 45 | 43 |
| 5h | TG7-76 | 4.9 | 255 | 52 | 41 |
| 5i | TG7-96 | 3.3 | 175 | 53 | <25 |
| 5j | TG7-186 | 11.3 | 900 | 80 | <25 |
| 5k | TG7-122 | 333 | ND | ND | ND |
| 5l | TG7-6 | >1000 | ND | ND | 91 |
| 5m | TG7-9 | >1000 | ND | ND | 180 |
| 5n | TG7-21 | >1000 | ND | ND | 75 |
| 5o | TG7-138 | >1000 | ND | ND | 110 |
| 5p | TG7-109 | >1000 | ND | ND | ND |
| 5q | TG7-91 | >1000 | ND | ND | ND |
| 5r | TG7-95 | 667 | ND | ND | ND |
| 5s | TG8-116 | >1000 | ND | ND | ND |
| 5t | TG7-133 | >1000 | ND | ND | ND |
| 5u | TG7-89 | >1000 | ND | ND | ND |
| 5v | TG4-156 | 214 | ND | ND | ND |
| 5w | TG7-149 | 410 | ND | ND | ND |
| 5x | TG7-128 | 680 | ND | ND | ND |
| 5y | TG7-97 | >1000 | ND | ND | ND |
| 5z | TG7-103 | >1000 | ND | ND | ND |
| 6a | TG8-4 | 11.4 | 505 | 44 | 153 |
| 6b | TG8-16 | 260 | 2820 | 10 | 67 |
| 6c | TG8-21 | 41.1 | 7450 | 181 | 235 |
| 6d | TG8-23 | 13.6 | 108 | 7.9 | 68 |
| 6e | TG8-32 | 11.8 | 67.1 | 5.6 | 66 |
| 6f | TG8-27 | 3.7 | 19.9 | 5.3 | 66 |
| 6g | TG8-30 | 58.3 | 198 | 3.4 | 35 |
| 6h | TG7-209 | 340 | ND | ND | ND |
| 6i | TG7-273 | 236 | ND | ND | ND |
| 6j | TG-109-1 | >1000 | ND | ND | ND |
| 6k | TG8-57 | 84.5 | 752 | 8.9 | 180 |
| 6l | TG8-53 | 74.6 | 283 | 3.8 | 306 |
| 6m | TG8-56 | 137 | 265 | 2 | 90 |
| 6n | TG7-291 | >1000 | ND | ND | ND |
| 6o | TG7-294 | >1000 | ND | ND | ND |
| 6p | TG8-17-1 | >1000 | ND | ND | ND |
| 6q | TG4-94-1 | 16.5 | 66 | 4 | ND |
| 6r | TG8-117 | 29.2 | ND | ND | ND |
| 6s | TG8-118 | 13.0 | ND | ND | ND |
| 6t | TG8-122 | >1000 | ND | ND | 152 |
Schild KB values are calculated using the formula log(dr – 1) = log XB – log KB, where dr (dose ratio) = fold shift in EC50 of PGE2 by the test compound, XB is antagonist concentration [1 μM]. KB value indicates a concentration required to produce a 2-fold rightward shift of PGE2 concentration–response curve. The values are the mean of two to four independent measurements run in duplicate. The solubility of the compounds is measured in PBS buffer (pH 7.4) with 1% DMSO by nephelometry.51 ND = not determined.
Synthesis and Further Structure–Activity Relationship Study on Cinnamic Amide Analogues
The scaffold 5a (Figure 1) possess four obvious sites for structural modification: (i) trimethoxyphenyl group, (ii) acrylamide moiety, (iii) ethylene linker, (iv) a methyl group on the indole ring. Earlier, we had designed a compound 5d with CF3 in place of CH3 on the indole ring, with a premise that the fluorine atom(s) often enhances ADME properties.47 Indeed, this transformation enhanced metabolic stability (Table 3) and brain and plasma PK properties.44 However, the CF3 analogue (5d) was about 7-fold less potent for EP2 in comparison to the CH3 analogue 5a (Table 2). In the present study, to examine whether three methoxyl groups on the phenyl ring are important for bioactivity, we have synthesized several derivatives that have reduced number of methoxyl groups or were completely substituted with other substituents as shown in Scheme 1. The synthesis is carried out starting from commercially available 2-methylindole or 2-trifluoromethylindoles (1a–c), which on treatment with bromoacetonitrile provided intermediates (2a–c), which then were subjected to lithium aluminum hydride to reduce cyanide to amine, providing advanced intermediates 3a–c in poor to moderate yields. In an effort to improve the yield of amines, we explored other methods of cyanide reduction using a variety of reducing agents (see Supporting Information Table S1). These methods provided limited success, and they often resulted in an unwanted indole-dimer product as a major constituent. The classical lithium aluminum hydride (LAH) reduction method provided only 32–57% yield of the required amine products (Supporting Information Table S1 and discussion in the Supporting Information text). These amines were coupled to 3,4,5-trimethoxycinnamic acid derivatives (4a–c) to provide final products (5a–f) (Scheme 1). As shown in Table 1, newly synthesized derivatives are tested by using a cAMP-derived TR-FRET assay48 at single concentration (1 μM) to observe a rightward shift of PGE2 (an EP2 agonist) concentration–response curve in a C6G cell line that overexpresses human EP2 receptors (see Experimental Methods for details). From this a Schild KB value (a concentration required to cause a 2-fold rightward shift of agonist EC50) is calculated assuming a Schild slope of 1.07, which is the mean slope determined from four concentration (0.1, 0.3, 1, and 3 μM) Schild plots carried out on a dozen compounds in this series. A similar procedure is carried out with human DP1 receptors at a single compound concentration of 10 μM and used to rank-order the analogues based on EP2 potency and selectivity against DP1.
Table 2. EP2 Potency, Selectivity against EP4 and IP Receptors, and Cytotoxicity of Selected EP2 Antagonistsa.
| compd | KB(EP2), nM | KB(EP4), μM | selective index EP4/EP2 | KB(IP), μM | selective index IP/EP2 | cytotoxicity CC50, μM | therapeutic index CC50/KB(EP2) |
|---|---|---|---|---|---|---|---|
| 5a (TG4-155) | 2.4 | 11.4 | 4750 | 62 | 25800 | 172 | 71700 |
| 5c (TG7-98) | 3.4 | 41.0 | 12100 | 22.5 | 6630 | 368 | 108 000 |
| 5d (TG6-10-1) | 17.8 | 11.2 | 630 | 8.45 | 475 | 81 | 4550 |
| 5g (TG7-74) | 2.4 | 4.3 | 1790 | 12.7 | 5310 | 59.5 | 24800 |
| 5h (TG7-76) | 4.9 | 1.46 | 300 | 42.7 | 8720 | 246 | 50200 |
| 5j (TG7-186) | 11.3 | 3.5 | 310 | 21.7 | 1920 | 317 | 28000 |
| 6a (TG8-4) | 11.4 | 7.13 | 625 | 1.57 | 138 | 92.3 | 8100 |
| 6c (TG8-21) | 41.1 | 9.5 | 230 | 240 | 5840 | 126 | 3060 |
| 6d (TG8-23) | 13.6 | 7.58 | 560 | 30.0 | 2200 | 81.7 | 6000 |
| 6e (TG8-32) | 11.8 | 5.96 | 505 | 210 | 1780 | 36.6 | 3100 |
| 6f (TG8-27) | 3.7 | 7.49 | 2020 | 85.0 | 23000 | 43.3 | 11700 |
| 6g (TG8-30) | 58.3 | 7.93 | 136 | 95.9 | 164 | 31.2 | 535 |
EP2, EP4, and IP Schild KB values are average of two to three independent experiments run in duplicate. CC50 values are the average of two measurements run in triplicate. CC50 = critical concentration required to kill 50% cells.
Scheme 1. Synthesis of First Generation 1-Indole Cinnamic Amide EP2 Antagonists.

Reagents and conditions: (a) NaH, bromoacetonitrile, DMF, 75%; (b) lithium aluminum hydride (LAH), tetrahydrofuran (THF), 32–57%; (c) cinnamic acid drivative (4), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI), dimethylaminopyridine (DMAP), CH2Cl2, 75–80%.
The structure–activity relationship (SAR) study indicates a 3,5-dimethoxycinnamic amide derivative (5b) and a compound in which three methoxyls are substituted with two methyl groups and a fluorine (5c) display similar EP2 potency, in comparison to 5a. These derivatives show improved selectivity against DP1 (5b displays 24-fold, and 5c displays 60-fold) (Table 1). We have earlier shown that a single methoxycinnamic amide derivative (6q, Figure 2) exhibited 7-fold less potency (KB = 16.5 nM) on EP2 in comparison to parent 5a.43 We now synthesized compounds with single methoxyl group at ortho and meta positions. The m-methoxy derivative (6r, Figure 2) is about 2-fold less potent than p-methoxy derivative (6q), but the o-methoxy derivative (6s, Figure 2) displayed a similar potency to 6q (Table 1). All of these single methoxy derivatives are about 5- to 12-fold less potent than a trimethoxy derivative (5a) or a dimethoxy derivative (5b). Similar exercises on CF3 analogue 5d, for example, substitution of three methoxyls with a single methoxyl group (5e) or two methyl groups and a fluorine (5f), reduced the EP2 activity by 16-fold in comparison to 5d (Table 1). Taken together, these results indicate that three methoxyl groups on the phenyl ring are not absolutely essential for EP2 activity.
Figure 2.

List of additional active/inactive first generation cinnamic amide derivatives synthesized and used for SAR study.
In parallel to indole-1 derivatives (Scheme 1), we also explored synthesis of several indole-3 derivatives (Scheme 2) as positional isomers. As shown in Scheme 2, commercially available 2-(2-methyl-1H-indol-3-yl)ethanamines (3k, l) were coupled to cinnamic acid derivatives (4a,c,d) to synthesize compounds 5g–j. Isomeric 3-indole analogues displayed Schild potencies similar to those of the parent series shown in Scheme 1. In particular, compound 5g has equal potency to 5a against EP2, compound 5h with one less methoxyl group has equal potency to a previously described 1-indole isomer with two methoxyl derivative TG4-166,43 indicating that both indole positional isomers are equally active to pursue further. Compound 5j has about 3-fold less potency to equivalent 5c. Moreover, incorporation of two fluorine atoms on the indole phenyl ring to block the ortho and para (fifth, seventh) positions to the ring nitrogen (5i) maintained EP2 potency of the parent 5a. This result is consistent with our previous observation, where one fluorine atom at (fifth position) para to indole nitrogen on the phenyl ring yielded an equally potent compound to 5a.43 Nonetheless, all of these derivatives 5g,h showed EP2 Schild KB values less than that of the previous lead compound 5d. These 3-indole derivatives (5g–j) also displayed improved selectivity (45- to 80-fold) against DP1 (Table 1). To determine whether indole ring can be replaced with other structurally equivalent rings, a benzofuran derivative 5k was synthesized starting from 3-bromo-2-methylfuran as shown in Supporting Information Scheme S1. This analogue (5k) showed 138-fold reduced potency compared to its indole equivalents 5a and 5g. We then synthesized and tested an indazole derivative 6t (Figure 2), but this derivative completely lost EP2 potency. Moreover, we also examined other scaffolds such as indolin-2-one (5p) and phenyethyl and phenylpropyl groups (5q, 5r) (Figure 2). However, these derivatives showed very weak (300- to 1000-fold less) potencies (Table 1). In our earlier study we had shown that a 2-methylpiperidine ring (in place of indole ring) derivative 6j (Figure 2) displayed complete loss of activity.43 Taken together, these results suggest that the indole (1- or 3-positional isomers) ring is crucial for higher EP2 potency but substitution on the indole rings is allowable.
Scheme 2. Synthesis of Isomeric Indole-3 Cinnamic Amide Analogues for SAR Study.

Reagents and conditions: (a) cinnamic acid derivative (4a or 4c or 4d), EDCl, DMAP, CH2Cl2, 70–80%.
Although several compounds from Schemes 1 and 2 (5a–d, 5g–j) showed high EP2 potency and improved DP1 selectivity, they displayed poor aqueous solubility (Table 1). We explored two strategies to improve the aqueous solubility in this class of compounds. First, we functionalized the indole ring at second and third positions (see Figure 1 for number illustration) with more polar functional groups that should enhance the solubility of the scaffold. As shown in Scheme 3, the synthesis is initiated with 2-formylindole (1d), which on treatment with bromoacetonitrile provided 2d, which then on reductive amination49 with morpholine and 4-amino-1-methylpiperidine provided 2e and 2f. These compounds were reduced with lithium aluminum hydride to get 3e and 3f, which were then coupled to 3,4,5-trimethoxycinnamic acid (4a) to provide final products (5l, 5m). Similarly, 3-substituted indoles (5n–o) with more solubilizing functional groups were synthesized as shown in Supporting Information Scheme S2. We anticipated that these derivatives 5l–o could be transformed into their hydrochloride salt (HCl) forms to improve the solubility as needed. Indeed, these derivatives (5l–o) and their HCl salts forms (not shown) have improved solubility in the range of 75–180 μM (Table 1) compared to parents 5a and 5d, but they failed to show a strong EP2 antagonistic activity at 1 μM (Table 1), suggesting these regions on the indole ring are not flexible for structural modification.
Scheme 3. Synthesis of More Aqueous Soluble First Generation Cinnamic Amide EP2 Antagonists.

Reagents and conditions: (a) NaH, bromoacetonitrile, DMF, 75%; (b) morpholine or 4-amino-1-methylpiperidine, Na(OAc)3BH, AcOH, 60–75%; (c) LAH, THF, 3e, 55%, 3f, 20%; (d) cinnamic acid (4a), EDCl, DMAP, CH2Cl2, 75%.
The second strategy that we explored to improve the aqueous solubility of the scaffold 5a and 5d is a substitution of methoxyl groups with one or two hydroxyalkyl groups on the phenyl ring. The synthesis of these analogues is shown in Scheme 4. First commercially available 3,4-dihydroxycinnamic acid ethyl ester (2k) was subjected to Mitsunobu reaction50 with 2-tert-butyldimethylsilyloxyethanol to get bis-tert-butyldimethylsilyloxy ether (2l), which on treatment with 1 N NaOH in refluxing tetrahydrofuran and then quenching with 2 N HCl (in one pot) provided precursor acid (2m). This acid was coupled individually to indole amine 3a, 3b, or 3k to provide final products (6a–c) with two pendent hydroxyethyl ether moieties. As we predicted, compounds 6a and 6c have 3- to 5-fold higher aqueous solubility (Table 1) in comparison to parent compound 5a when measured by nephelometry in PBS buffer in 1% DMSO.51 Similarly, a trifluoromethylindole compound (6b) has shown 2.5-fold higher solubility (67 μM) in comparison to its parent compound 5d (Table 1). Moreover, we also synthesized several compounds with only one hydroxyethyl ether or hydroxypropropyl ether moieties (6d–g). These derivatives also showed about 1.2- to 1.4-fold higher solubility than the parents 5a and 5d (Table 1).
Scheme 4. Synthesis of Aqueous Soluble Hydroxyethyl Ether Cinnamic Amide Derivatives.

Reagents and conditions: (a) 2-tert-butyldimethylsilyloxyethanol, PPh3, DIAD,THF, reflux, 36 h, 70%; (b) 1 N NaOH, THF, 2 N HCl, 801%; (c) 3a, 3b, or 3k, EDCl, DMAP, DCM/DMF (5:1), 80%.
Some compounds with improved aqueous solubility have high EP2 potency. For example, compounds 6a, 6d, and 6e displayed similar EP2 potency (Schild KB of 11.4–13.6 nM). These derivatives are about 5-fold less potent than 5a but are slightly more potent than 5d. Among these three, 6a, a bis-hydroxyethyl ether compound, exhibited 44-fold selectivity to DP1, but monohydroxyethyl ether derivatives 6d, 6e, and 6f displayed <10-fold selectivity (Table 1). Likewise, compound 6c, which showed 17-fold less potency than 5a, 2.3-fold less than 5d, showed very high selectivity (180-fold) to DP1. This compound is the second most soluble in this whole class of compounds thus far. Moreover, compound 6f, which has two extra methoxyl groups in addition to a hydroxyethyl ether unit, has nearly equal EP2 potency to 5a, but a CF3 analogue 6g showed 3.2-fold less potency than its parent 5d (Table 1). In contrast to bis-hydroxyethyl ether derivatives 6a–c, the monohydroxyethyl ether derivatives 6d–g showed a modest selectivity (5- to 8-fold) over DP1 (Table 1).
We also synthesized compounds 6k–m containing a 2-dimethylaminoethoxy ether group on the phenyl ring as shown in Scheme 5. The qikprop (Schrodinger Inc.) predicted ADME properties (see Supporting Information Table S2) suggest that these derivatives may display similar solubilities (due to basic nitrogen) to hydroxyethyl ethers (6a–g) and may show improved metabolic stability and brain penetration properties because the pendent tertiary amine group is masked by hydrophobic methyl groups. Indeed, compounds 6k–m have enhanced solubility (180, 306, and 90 μM, respectively) (Table 1). However, these derivatives have reduced EP2 potencies (Table 1) in comparison to their hydroxyethyether equivalents 6d–f. Given their reduced potency and modest selectivity against DP1 (Table 1), they are not tested for liver metabolism and brain-permeation properties. A future study will address whether incorporation these basic amine functionality at meta or ortho positions improves EP2 potency and selectivity against DP1.
Scheme 5. Synthesis of 2-Dimethylaminoalkyl Ether Cinnamic Amide Derivatives.

Reagents and conditions: (a) MeOH, H2SO4 (drops), reflux, quantitative; (b) dimethylaminoethanol, PPh3, DIAD, THF, 70%; (c) 1 N NaOH, THF, reflux, quantitative (reagent grade salt); (d) 3a, EDCl, DMAP, DMF, 70%.
In our earlier study,43 we briefly explored the linker unit for structural modification and learned that extension of two-carbon ethylamino chain (see Figure 1) to three-carbon propylamino chain resulted in 775-fold less EP2 potency, and saturating the double bond of acrylamide as in 5v (Figure 2) reduced the potency by 90-fold compared with 5a (Table 1). In the present study, we synthesized compounds with one-carbon methylamino linker such as 5s,t (Figure 2), but these analogues showed complete loss of potency (Table 1). We also synthesized an analogue by reversing the amide (5u, Figure 2), which also killed EP2 potency. However, saturation of the double bond and then addition of an amino group (5w), or reducing the length of acrylamide to single methylphenyl (5x) (Figure 2), reduced the potency by 180- and 160-fold, respectively, in comparison to 5a. Given the limited availability of synthetic methods to modify the ethylene linker (Figure 1), only two derivatives with a carboxymethyl ester group on the ethylamine linker (5y,z) have been synthesized, but these compounds were inactive on EP2. Moreover, to determine whether the amide is absolutely essential for EP2 potency, we synthesized an ester analogue 6h (Figure 2). This analogue showed about 140-fold less potency than 5a, suggesting that the potency in the scaffold arises not just from the acrylamide in the linker. Furthermore, we synthesized a cyclopropylamide analogue 6i, which showed 100-fold less potency than 5a. Taken together, these results suggest an ethylamine linker, one side attached to the indole ring and other side attached to the acrylamide, is optimal for bioactivity, but acrylamide moiety is not solely responsible for the activity; thus, it may be expendable.
To minimize the conformational freedom arising from the ethylamine linker and to minimize the exposure of the linker unit to metabolizing enzymes, we synthesized derivatives with constrained and bulkier internal cyclic rings 6n–p as shown in Supporting Information Scheme S3. Compounds 6n−p were inactive suggesting that the acyclic ethylene amide is essential for high EP2 potency. It is worth mentioning that 6n–p are chiral compounds; we have synthesized only racemic forms, and we did not make any effort to make them in enantiomerically enriched form because of their weak or nil potency.
Overall, SAR indicates that in the 1- or 3-indole rings, a CH3 at second position is optimal for EP2 potency. A small structural change at the second position, for example, a CF3 group, reduces EP2 potency by 7–18 times (cf. 5a vs 5d; 5e vs 6q). An acrylamide group is optimal for high EP2 potency but may be removed. Modifications to the amide group and to the ethylamine linker reduce or eliminate EP2 potency. But three methoxyl groups on the phenyl ring could be substituted with a variety of other groups to maintain high EP2 potency.
Novel Analogues Show High EP2 Selectivity over Other Prostanoid Receptors
As indicated briefly in the previous section, there are nine prostanoid receptors in the family: DP1, DP2, EP1, EP2, EP3, EP4, FP, IP, and TP. These receptors are widely distributed in organs and cell types and are activated by endogenous prostanoids (PGD2, PGE2, PGF2, PGI2, and TXA2). Among these receptors, EP1, EP2, EP3, and EP4 share a common endogenous ligand PGE2 for their activation. EP2 and EP4 are positively coupled to cAMP signaling, whereas EP3 inhibits cAMP production and EP1 mediates cytosolic Ca2+ signaling, suggesting that these receptors could play different, often opposite, roles in pathophysiology.27−29 On the other hand, although DP1 receptor is not activated by PGE2, it has the highest structural homology to EP2 and is known to exert proinflammatory effects similar to those of EP2 in certain conditions.27−29 EP2 receptor also shares a 40% structural homology to the IP receptor. IP receptor activation is shown to play an important role in cardioprotection.15,17 Thus, it is crucial to establish selectivity for the novel antagonists to EP2 over DP1, EP4, and IP, for preclinical and clinical studies. Previously synthesized first generation analogues43,44and several other newly synthesized derivatives showed modest selectivity to DP1 (Table 1). But derivatives 5c, 5g–j, 6a, 6c showed >44-fold selectivity to EP2 over DP1. So we selected these derivatives for selectivity testing against EP4 and IP receptors. We created cell lines that overexpress EP4 receptors, or IP receptors on C6-glioma cells, and developed a cAMP-derived TR-FRET assay using agonists PGE2 (for EP4) and iloprost (for IP), similar to EP2 assay (see Experimental Methods for details). The results show that the new analogues display micromolar Schild KB values for EP4 and IP receptors (Table 2), with high selectivity indexes. For example, 5c displayed 12100-fold selectivity against EP4 and over 6000-fold selectivity against IP receptor. Compound 5g also displayed high selectivity to EP2 over EP4 (1790-fold) and IP (5310-fold). Likewise compounds 5h and 5j showed 300- and 310-fold selectivity to EP4 and 8720- and 1920-fold selectivity to IP receptor (Table 2). However, these derivatives showed weak aqueous solubility (<25 μM). Compounds with improved aqueous solubility, for example, 6a, displayed 625-fold selectivity against EP4 and 138-fold selectivity against IP; 6c displayed 230-fold selectivity to EP4 and greater than 5000-fold selectivity against IP. Likewise, compounds 6d–f also displayed good selectivity against EP4 and IP receptor (Table 2), but these latter derivatives showed poor selectivity against DP1 (Table 1). Compound 6g has slightly less selectivity to EP4 (136-fold) and IP (164-fold) (Table 2). We also tested these selective antagonists in a cell viability assay against C6 glioma cells, and these derivatives have insignificant toxicity with in vitro therapeutic indexes over several orders of magnitude (Table 2).
New Selective EP2 Antagonists Show Improved Microsomal Stability
Having several potent and selective EP2 antagonists in hand, we asked whether any of these compounds show improved metabolic stability in human and mouse liver microsomes in comparison to previous lead compound 5d. Compound 5a, which showed 0.2% remaining at 60 min (at 1 μM concentration) in mouse liver microsomes, exhibited an in vivo plasma half-life of ∼30 min. Compound 5d with 2% remaining at 60 min had 1.6 h in vivo plasma half-life in mouse (Table 3), suggesting that in vitro liver metabolism may be correlated to in vivo plasma half-life in this class. Thus, we examined novel compounds that showed enhanced selectivity in comparison to previous lead 5d for liver microsomal stability. Compound 5c showed high stability in both liver fractions, but this compound exhibited poor aqueous solubility; thus, it was not selected for further exploration. Likewise, 3-indole isomeric derivatives 5g–j also showed poor stability in liver fractions (Table 3). Interestingly, a 5-fold more aqueous soluble compound 6a showed nearly similar stability in both liver fractions in comparison to 5d. Furthermore, compound 6c which is about 8-fold more soluble than 5d displayed 3-fold improved stability in mouse liver fractions. It is also more stable in human liver microsomal fractions (Table 3), suggesting that these two compounds are suitable for in vivo pharmacokinetic study.
ADME Characterization and Pharmacokinetic Studies of Selected EP2 Antagonists
We examined a number of these derivatives to estimate their ADME properties by qikprop software (Schrodinger Inc.). As shown in Supporting Information Table S2, compounds 6a, 6c, and 6f possess solubility and permeability properties in the suggested range for 95% of the known drugs. However, because of the free hydroxyl group, they may encounter some resistance in crossing the blood–brain barrier (BBB) because the predicted values for 6a and 6c are lower in comparison to 5d, a compound experimentally determined to be highly brain permeable. Nonetheless, we have selected compound 6a and subjected it to in vivo pharmacokinetics study in C57Bl6 mice. As shown in Table 3, this compound displayed more than an hour plasma half-life. However, its brain penetration property is poor compared to previous lead 5d (Table 3), consistent with qikprop predictions (Supporting Information Table S2).
Blood–brain barrier (BBB) is composed of a network of endothelial cells, astroglia, pericytes, and a basal lamina. The capillary of endothelium of the brain is sealed by tight junctions, produced by the interaction of several transmembrane proteins.52,53 Interaction of these junctional proteins blocks the entry of polar solutes from blood along the paracellular pathways and so denies access to brain interstitial fluid. However, small molecules with less than 500 molecular weight and high lipophilicity can pass through this barrier by passive transport mechanism. Small molecules could also enter into brain by other mechanisms (e.g., active transport).54,55 Endothelial cells also express a variety of efflux pumps on their surface, which play a role in export of small molecules into brain. Compound 5d (our earlier lead) and 6a display <500 molecular weight, but 5d is more lipophilic than 6a based on its poor aqueous solubility (Table 1) and predicted log P (Table 2). On the other hand, compound 6a is more polar (5-fold more aqueous soluble) with two free hydroxyl groups readily available to form hydrogen bonds. However, we do not yet know whether low levels of 6a in brain are due to poor passive diffusion or extrusion by efflux pumps. A future study will address this question by synthesis and testing of additional hydroxyl group masked derivatives (e.g., methoxy ethers). Nevertheless, 6a displayed a 2-fold higher potency and 4-fold higher selectivity against DP1 and 4-fold higher aqueous solubility than 5d; thus, this compound should be useful for exploring in in vitro and in vivo proof of concept studies in a variety of peripheral disease models where EP2 plays a deleterious role.56−60
Novel Analogues Show Competitive Mechanism of Inhibition
We previously demonstrated that compounds 5a and 5d and other analogues in this class exhibit a competitive antagonism of EP2.43,44 All of these derivatives had only methoxyl groups on the phenyl ring. In this study, to determine whether the compounds containing hydroxyethyl ether moieties (6a–g) also exhibit a similar mechanism of action, we selected three derivatives 6a, 6c, and 6f and tested them in concentration response against PGE2 EC50 on EP2 receptors. As illustrated in Figure 3D, a linear regression of log(dr – 1) on log XB with slope of unity characterizes a competitive antagonism. Schild KB values are derived by the equation log(dr −1) = log XB – log KB, where dr = dose ratio (i.e., the fold shift in EC50), XB is [antagonist], and KB indicates the antagonist concentration required for a 2-fold rightward shift in the PGE2 concentration–response curve. A lower KB value indicates a higher inhibitory potency. The selected three compounds induced a concentration-dependent, parallel rightward shift in the PGE2 concentration–response curve (Figure 3A–C). Schild regression analyses demonstrated that these compounds have a competitive mechanism of antagonism on EP2 with Schild KB 14.8 nM for 6a, 47.1 nM for 6c, and 6.7 nM for 6f. Thus, the mechanism is competitive in general for this class of EP2 antagonists presented in this study.
Figure 3.

Competitive antagonism of EP2 receptor by novel acrylamide analogues. (A–C) Compounds 6a (TG8-4), 6c (TG8-21), and 6f (TG8-27) inhibited PGE2-induced human EP2 receptor activation in a concentration dependent manner. (D) Schild regression analysis is performed to determine the modality of antagonism by these compounds. Schild KB values for each compound are shown in inset of part D. Data were normalized as percentage of maximum response; points represent the mean ± SEM (n = 4). We observed about 1.1- to 1.8-fold higher KB values from dose–response test in comparison to KB values derived from single concentration (1 μM) tests (presented in Table 2). These changes are within the limits of assay variability.
In conclusion, we have synthesized 45 new cinnamic amide EP2 antagonists to optimize the selectivity against DP1 and IP receptors and to improve aqueous solubility and pharmacokinetics properties. Two compounds, namely, 6a (TG8-4) and 6c (TG8-21), emerged as selective EP2 antagonists (with 44- to 180-fold selectivity against DP1), with more aqueous solubility (153 and 235 μM) and more stability in vitro in pooled human and mouse liver microsomes in comparison to previous lead compound 5d (TG6-10-1). But in vivo pharmacokinetics properties still need to be optimized within the class to be useful for in vivo preclinical studies. However, the new analogues 6a and 6c could serve as tools for in vitro proof of concept studies.
Experimental Methods
Chemistry General
Proton NMR spectra were recorded in solvent in CDCl3 on a Varian Inova 400 (400 MHz) instrument. Thin layer chromatography was performed on precoated, aluminum-backed plates (silica gel 60 F254, 0.25 mm thickness) from EM Science and was visualized by UV lamp. Column chromatography was performed with silica gel cartridges on Teledyne-ISCO machine. An Agilent LCMS instrument was used to measure purity of the products. Elemental analyses were performed by Atlantic Microlab Inc. (Norcross, GA). Chemicals and drugs PGE2, BW245C, iloprost, and rolipram were purchased from Cayman Chemical.
General Procedure for Synthesis of 2-(2-Substituted-1H-indol-1-yl)acetonitriles (2) from Indoles (1)61
A solution of 2-(trifluoromethyl)-1H-indole (1b) (0.5 g, 2.7 mmol) in DMF (2.5 mL) was added to a suspension of NaH (160 mg, 1.5 equiv) in DMF (3 mL) at 0 °C, and the resulting reaction mixture was stirred for 30 min. Then bromoacetonitrile (0.27 mL, 1.5 equiv) in DMF (2.5 mL) was introduced into the above mixture at 0 °C, and then the mixture was brought to room temperature overnight. Water (20 mL) was added to quench the reaction. Then the product was extracted with ethyl ether (30 mL × 3). Organics were washed with water, brine, dried over Na2SO4, and concentrated. The crude mass on silica gel chromatography, eluting with 0–10% ethyl acetate, furnished 2b (865 mg, 71% yield; 85% based on recovered starting material).
2-(2-Trifluoromethyl-1H-indol-1-yl)acetonitrile (2b)
1H NMR (CDCl3): δ 7.70 (d, J = 8 Hz, 1H), 7.44 (m, 2H), 7.28 (t × d, J = 7.2, 1.6 Hz, 1H), 7.04 (s, 1H), 5.1 (s, 2H). LCMS (ESI): >95% purity at λ = 254 nm. MS m/z, 225 [M + H]+. See Supporting Information for synthesis and characterization data for compounds 2a and 2c–h.
General Procedure for Synthesis 2-(2-Substituted-1H-indol-1-yl)ethanamines (3) from Acetonitriles (2)
To a solution of 2b (855 mg, 3.81 mmol) in THF (30 mL) was added LAH (1 M, 9.54 mmol, 2.5 equiv), dropwise at 0 °C, and the resulting reaction mixture was brought to room temperature overnight. Methanol (2 mL) was slowly added to quench the reaction at −78 °C, followed by 1 N NaOH (3 mL) at room temperature. The product was extracted with ethyl ether (30 mL × 3). Organics were washed with water, brine and dried over Na2SO4 and concentrated. The crude mass was subjected to silica gel chromatography, eluting with 0–5% methanol in dichloromethane to provide 3b (490 mg, 56% yield).
2-(2-(Trifluoromethyl)-1H-indol-1-yl)ethanamine (3b)
1H NMR (CDCl3): δ 7.66 (d, J = 8 Hz, 1H), 7.44 (dd, J = 8.4, 0.8 Hz, 1H), 7.34 (t × d, J = 7.6, 0.8 Hz, 1H), 7.17 (t × d, J = 7.4, 1.2 Hz, 1H) 6.94 (s, 1H), 4.28 (t, J = 6.8 Hz, 2H) 3.12 (t, J = 6.8 Hz, 2H) 2.45 (s, 3H). LCMS (ESI): >97% purity at λ = 254 nm. MS m/z, 229 [M + H]+. See Supporting Information for synthesis and characterization data for 3a and 3c–h.
General Procedures for Synthesis of Cinnamic Amide Final Products
To a solution of 3b (480 mg, 2.1 mmol) in dichloromethane (10 mL) were added (E)-3-(3,4,5-trimethoxyphenyl)acrylic acid (4a) (504 mg, 1 equiv), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDCI) (523 mg, 1.3 equiv), and N,N-dimethylaminopyridine (10 mg), and resulting reaction mixture was stirred at room temperature for 8 h. The reaction was quenched with water (10 mL), and the product was extracted with ethyl acetate (20 mL × 3). Organics were washed with 1% HCl (10 mL), saturated NaHCO3 (10 mL), water (20 mL), brine solution (20 mL) and dried over Na2SO4. The crude product was purified by silica gel chromatography, eluting with 0–35% ethyl acetate in hexane to provide 5d (700 mg, 74% yield).
(E)-N-(2-(2-(Trifluoromethyl)-1H-indol-1-yl)ethyl)-3-(3,4,5-trimethoxyphenyl)acrylamide (5d)
1H NMR (CDCl3): δ 7.59 (d, J = 8 Hz, 1H), 7.54 (d, J = 8.4, Hz, 1H), 7.50 (d, J = 15.2 Hz, 1H), 7.27 (q, J = 7.2 Hz, 1H), 7.1 (t, J = 7. 2 Hz, 1H), 6.89 (s, 1H), 6. 63 (s, 2H), 6.4 (t, J = 6 Hz, 1H), 6. 25 (d, J = 15.2 Hz, 1H), 4.4 (t, J = 6.4 Hz, 1H), 3.8 (s, 3H), 3.76 (s, 6H), 3.69 (q, J = 6.4 Hz, 2H). LCMS (ESI): >95% purity at λ = 254 nm. MS m/z, 449 [M + H]+. Anal. Calcd for C23H23F3N2O4: C, 61.60; H, 5.17; N, 6.25. Found: C, 61.34; H, 5.10; N, 6.16. See Supporting Information for characterization of 5a–c,e–z.
General Synthesis for 2-Hydroxyethyl- Or 2-Dimethylaminoethylcinnamic Acids
Step 1. To a solution of ethyl-3,4-dihydroxycinnamate (2k) (460 mg, 2.21 mmol), 2-tert-butyldimethylsilyloxyethanol (2 mL, 9.52 mmol 4.3 equiv), and triphenylphosphine (3.43 g, 13 mmol, 5.8 mmol) in THF (40 mL) was added diisopropyl azodicarboxylate (2.4 mL, 12 mmol, 5.3 equiv) dropwise at 0 °C. Then the resulting solution was refluxed for 36 h. The volatiles were removed under vacuum and the crude product was subjected to silica gel chromatography, eluting with 0–20% ethyl acetate in hexane to furnish 2l (775 mg, 67%).
Ethyl (E)-3-(3,4-Bis(2-((tert-butyldimethylsilyl)oxy)ethoxy)phenyl)acrylate (2l)
1H NMR (CDCl3): δ 7.60 (d, J = 15.6 Hz, 1H), 7.07 (dd, J = 6.8, 1.6 Hz, 1H), 7.04 (d, J = 2 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 16 Hz, 1H) 4.24 (q, J = 6.8 Hz, 2H), 4.07 (q, J = 5.6 Hz, 4H), 3.97 (t, J = 5.6 Hz, 4H), 1.30 (t, J = 7.2 Hz, 3H), 0.88 (two singlets, 18H), 0.08 (two singlets, 12H). LCMS (ESI): >95% purity at λ = 254 nm. MS m/z, 525 [M + H]+.
Step 2.
To a solution of 2l (375 mg, 0.71 mmol) in THF (10 mL) was added 1 N NaOH (2.13 mL, 2.13 mmol, 3 equiv), and the resulting reaction was refluxed for 48 h. The reaction mixture was cooled and neutralized with 1 N HCl (10 mL) to pH 4. Then the product was extracted with ethyl acetate (25 mL × 3). Organics were dried over Na2SO4 and concentrated to dryness under vacuum to furnish 2m (190 mg, quantitative yield), which was used for next step without further purification.
(E)-3-(3,4-Bis(2-hydroxyethoxy)phenyl)acrylic Acid (2m)
1H NMR (DMSO-d6): δ 12.0 (bs, 1H), 7.46 (d, J = 15.6 Hz, 1H), 7.29 (d, J = 2 Hz, 1H), 7.14 (m, 1H), 6.96 (d, J = 8 Hz, 1H), 6.38 (d, J = 16 Hz, 1H) 4.0 (m, 4H), 3.68 (bs, 4H). LCMS (ESI): >95% purity at λ = 254 nm. MS m/z, 267 [M – H]. See Supporting Information for synthesis and characterization for other carboxylic acid derivatives 2o–q.
(E)-3-(3,4-Bis(2-hydroxyethoxy)phenyl)-N-(2-(2-methyl-1H-indol-1-yl)ethyl)acrylamide (6a, TG8-4)
This compound was prepared from 2m and 3a in 80% yield by the method described for 5d. 1H NMR (CDCl3): δ 7.46 (d, J = 8.8 Hz, 1H), 7.43 (d, J = 16 Hz, 1H), 7.26 (d, J = 9.6 Hz, 1H), 7.07 (t, J = 6.8 Hz, 1H), 7.04 (t × d, J = 8.4, 2 Hz, 2H), 6.81 (d, J = 8 Hz, 1H), 6.12 (d, J = 15.6 Hz, 1H), 4.25 (t, J = 6 Hz, 2H), 4.04 (q, J = 4 Hz, 4H), 3.87 (q, J = 4.4 Hz, 4H), 3.62 (t, J = 5.6 Hz, 2H), 2.34 (s, 3H). LCMS (ESI): >97% purity at λ = 254 nm. MS m/z, 425 [M + H]+. HRFABMS: calcd for C24H28N2O5Na, 447.189 04; found 447.189 76.
(E)-3-(3,4-Bis(2-hydroxyethoxy)phenyl)-N-(2-(2-methyl-1H-indol-3-yl)ethyl)acrylamide (6c, TG8-21)
This compound was prepared from 2m and 3k in 80% yield by the method described for 5d. 1H NMR (CDCl3 + MeOH-d4): δ 7.41 (d, J = 7.2 Hz, 1H), 7.34 (d, J = 15.6 Hz, 1H), 7.19 (d × t, J = 8.4, 0.8 Hz, 1H), 6.92 (m, 4H), 6.77 (d, J = 8 Hz, 1H), 6.10 (d, J = 16 Hz, 1H), 3. 99 (t, J = 4 Hz, 4H), 3.81 (q, J = 3.6 Hz, 4H), 3.48 (t, J = 6.8 Hz, 2H), 2.87 (t, J = 7.2 Hz, 2H), 2.27 (s, 3H). LCMS (ESI): >97% purity at λ = 254 nm. MS m/z, 425 [M + H]+. HRFABMS calcd for C24H28N2O5Na, 447.189 04; found 447.188 89. See Supporting Information for synthesis and characterization data for remaining compounds 6b,d–p.
Bioactivity Testing
Cell Culture
The rat C6 glioma (C6G) cells stably expressing human DP1, EP2, EP4, or IP receptors were created in the laboratory43,44,48 and grown in Dulbecco’s modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS) (Invitrogen), 100 U/mL penicillin and 100 μg/mL streptomycin (Invitrogen), and 0.5 mg/mL G418 (Invitrogen).
Cell-Based cAMP Assay
Intracellular cAMP was measured with a cell-based homogeneous time-resolved fluorescence resonance energy transfer (TR-FRET) method (Cisbio Bioassays), as previously described.43,44 The assay is based on generation of a strong FRET signal upon the interaction of two molecules, an anti-cAMP antibody coupled to a FRET donor (cryptate), and cAMP coupled to a FRET acceptor (d2). Endogenous cAMP produced by cells competes with labeled cAMP for binding to the cAMP antibody and thus reduces the FRET signal. Cells stably expressing human DP1, EP2, EP4, or IP receptors were seeded into 384-well plates in 30 μL of complete medium (4000 cells/well) and grown overnight. The medium was carefully withdrawn, and 10 μL of Hanks’ buffered salt solution (HBSS) (Hyclone) containing 20 μM rolipram was added into the wells to block phosphodiesterases. The cells were incubated at room temperature for 0.5–1 h and then treated with vehicle or test compound for 10 min before addition of increasing concentrations of appropriate agonist: BW245C for DP1, PGE2 for EP2 and EP4, or iloprost for IP. The cells were incubated at room temperature for 40 min and then lysed in 10 μL of lysis buffer containing the FRET acceptor cAMP-d2, and 1 min later another 10 μL of lysis buffer with anti-cAMP-cryptate was added. After a 60–90 min incubation at room temperature, the FRET signal was measured by an Envision 2103 multilabel plate reader (PerkinElmer Life Sciences) with a laser excitation at 337 nm and dual emissions at 665 and 590 nm for d2 and cryptate (50 μs delay), respectively. The FRET signal was expressed as (F665/F590) × 104.
Acknowledgments
This work was supported by Alzheimer’s Drug Discovery Foundation (T.G.), NIH/NINDS Grants K99/R00NS082379 (to J.J.) and U01NS058158 (to R.D.), NARSAD Young Investigator Grant (to J.J.), and the Epilepsy Foundation (to J.J.).
Supporting Information Available
Synthesis schemes for compounds 5k,o,p, and 6n–p; a table of the reagents and reaction conditions tested to improve yield of starting materials 3 from 2; a table of qikprop calculations on selected compounds; NMR and MS characterization data for remaining compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
T.G. and R.D. designed the research. T.G., J.J., and M.-S.Y. performed the research. T.G. wrote the manuscript. R.D. helped with the writing.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Akiyama H.; Barger S.; Barnum S.; Bradt B.; Bauer J.; Cole G. M.; Cooper N. R.; Eikelenboom P.; Emmerling M.; Fiebich B. L.; Finch C. E.; Frautschy S.; Griffin W. S.; Hampel H.; Hull M.; Landreth G.; Lue L.; Mrak R.; Mackenzie I. R.; McGeer P. L.; O’Banion M. K.; Pachter J.; Pasinetti G.; Plata-Salaman C.; Rogers J.; Rydel R.; Shen Y.; Streit W.; Strohmeyer R.; Tooyoma I.; Van Muiswinkel F. L.; Veerhuis R.; Walker D.; Webster S.; Wegrzyniak B.; Wenk G.; Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M. T.; O’Banion M. K. Inflammatory processes in Alzheimer’s disease. J. Neuroimmunol. 2007, 184, 69–91. [DOI] [PubMed] [Google Scholar]
- McGeer P. L.; McGeer E. G. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 459–470. [DOI] [PubMed] [Google Scholar]
- Nagamoto-Combs K.; McNeal D. W.; Morecraft R. J.; Combs C. K. Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury. J. Neurotrauma 2007, 24, 1719–1742. [DOI] [PubMed] [Google Scholar]
- Przedborski S. Inflammation and Parkinson’s disease pathogenesis. Mov. Disord. 2010, 25Suppl. 1S55–S57. [DOI] [PubMed] [Google Scholar]
- Ramlackhansingh A. F.; Brooks D. J.; Greenwood R. J.; Bose S. K.; Turkheimer F. E.; Kinnunen K. M.; Gentleman S.; Heckemann R. A.; Gunanayagam K.; Gelosa G.; Sharp D. J. Inflammation after trauma: microglial activation and traumatic brain injury. Ann. Neurol. 2011, 70, 374–383. [DOI] [PubMed] [Google Scholar]
- Vezzani A.; Aronica E.; Mazarati A.; Pittman Q. J. Epilepsy and brain inflammation. Exp. Neurol. 2013, 244, 11–21. [DOI] [PubMed] [Google Scholar]
- Vezzani A.; French J.; Bartfai T.; Baram T. Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L.; Pieroni C.; Winger D.; Purohit D. P.; Aisen P. S.; Pasinetti G. M. Regional distribution of cyclooxygenase-2 in the hippocampal formation in Alzheimer’s disease. J. Neurosci. Res. 1999, 57, 295–303. [DOI] [PubMed] [Google Scholar]
- Lucas S. M.; Rothwell N. J.; Gibson R. M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147Suppl. 1S232–S240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minghetti L. Role of inflammation in neurodegenerative diseases. Curr. Opin. Neurol. 2005, 18, 315–321. [DOI] [PubMed] [Google Scholar]
- Pasinetti G. M.; Aisen P. S. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer’s disease brain. Neuroscience 1998, 87, 319–324. [DOI] [PubMed] [Google Scholar]
- Cannon G. W.; Caldwell J. R.; Holt P.; McLean B.; Seidenberg B.; Bolognese J.; Ehrich E.; Mukhopadhyay S.; Daniels B. Rofecoxib, a specific inhibitor of cyclooxygenase 2, with clinical efficacy comparable with that of diclofenac sodium: results of a one-year, randomized, clinical trial in patients with osteoarthritis of the knee and hip. Rofecoxib Phase III Protocol 035 Study Group. Arthritis Rheum. 2000, 43, 978–987. [DOI] [PubMed] [Google Scholar]
- Clemett D.; Goa K. L. Celecoxib: a review of its use in osteoarthritis, rheumatoid arthritis and acute pain. Drugs 2000, 59, 957–980. [DOI] [PubMed] [Google Scholar]
- Abraham N. S.; El-Serag H. B.; Hartman C.; Richardson P.; Deswal A. Cyclooxygenase-2 selectivity of non-steroidal anti-inflammatory drugs and the risk of myocardial infarction and cerebrovascular accident. Aliment. Pharmacol. Ther. 2007, 25, 913–924. [DOI] [PubMed] [Google Scholar]
- Egan K. M.; Lawson J. A.; Fries S.; Koller B.; Rader D. J.; Smyth E. M.; FitzGerald G. A. COX-2-derived prostacyclin confers atheroprotection on female mice. Science 2004, 306, 1954–1957. [DOI] [PubMed] [Google Scholar]
- Grosser T.; Fries S.; FitzGerald G. A. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest. 2006, 116, 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosser T.; Yu Y.; Fitzgerald G. A. Emotion recollected in tranquility: lessons learned from the COX-2 saga. Annu. Rev. Med. 2010, 61, 17–33. [DOI] [PubMed] [Google Scholar]
- Aisen P. S.; Schafer K. A.; Grundman M.; Pfeiffer E.; Sano M.; Davis K. L.; Farlow M. R.; Jin S.; Thomas R. G.; Thal L. J. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA, J. Am. Med. Assoc. 2003, 289, 2819–2826. [DOI] [PubMed] [Google Scholar]
- Akula K. K.; Dhir A.; Kulkarni S. K. Rofecoxib, a selective cyclooxygenase-2 (COX-2) inhibitor increases pentylenetetrazol seizure threshold in mice: possible involvement of adenosinergic mechanism. Epilepsy Res. 2008, 78, 60–70. [DOI] [PubMed] [Google Scholar]
- Breitner J. C.; Baker L. D.; Montine T. J.; Meinert C. L.; Lyketsos C. G.; Ashe K. H.; Brandt J.; Craft S.; Evans D. E.; Green R. C.; Ismail M. S.; Martin B. K.; Mullan M. J.; Sabbagh M.; Tariot P. N. Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimer’s Dementia 2011, 7, 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir A.; Naidu P. S.; Kulkarni S. K. Effect of naproxen, a non-selective cyclo-oxygenase inhibitor, on pentylenetetrazol-induced kindling in mice. Clin. Exp. Pharmacol. Physiol. 2005, 32, 574–584. [DOI] [PubMed] [Google Scholar]
- Holtman L.; van Vliet E. A.; Edelbroek P. M.; Aronica E.; Gorter J. A. Cox-2 inhibition can lead to adverse effects in a rat model for temporal lobe epilepsy. Epilepsy Res. 2010, 91, 49–56. [DOI] [PubMed] [Google Scholar]
- Holtman L.; van Vliet E. A.; van Schaik R.; Queiroz C. M.; Aronica E.; Gorter J. A. Effects of SC58236, a selective COX-2 inhibitor, on epileptogenesis and spontaneous seizures in a rat model for temporal lobe epilepsy. Epilepsy Res. 2009, 84, 56–66. [DOI] [PubMed] [Google Scholar]
- Jung K. H.; Chu K.; Lee S. T.; Kim J.; Sinn D. I.; Kim J. M.; Park D. K.; Lee J. J.; Kim S. U.; Kim M.; Lee S. K.; Roh J. K. Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neurobiol. Dis. 2006, 23, 237–246. [DOI] [PubMed] [Google Scholar]
- Polascheck N.; Bankstahl M.; Loscher W. The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. Exp. Neurol. 2010, 224, 219–233. [DOI] [PubMed] [Google Scholar]
- Breyer R. M.; Bagdassarian C. K.; Myers S. A.; Breyer M. D. Prostanoid receptors: subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 661–690. [DOI] [PubMed] [Google Scholar]
- Hirata T.; Narumiya S. Prostanoid receptors. Chem. Rev. 2011, 111, 6209–6230. [DOI] [PubMed] [Google Scholar]
- Narumiya S.; Sugimoto Y.; Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol. Rev. 1999, 79, 1193–1226. [DOI] [PubMed] [Google Scholar]
- Ganesh T.Prostanoid receptor EP2 as a therapeutic target. J. Med. Chem. [Online early access]. DOI: 10.1021/jm401431x. Published Online: Nov 26, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Dingledine R. Prostaglandin receptor EP2 in the crosshairs of anti-inflammation, anti-cancer, and neuroprotection. Trends. Pharmacol. Sci. 2013, 34, 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino H.; Salvi S.; Regan J. W. Differential regulation of phosphorylation of the cAMP response element-binding protein after activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. Mol. Pharmacol. 2005, 68, 251–259. [DOI] [PubMed] [Google Scholar]
- Regan J. W. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003, 74, 143–153. [DOI] [PubMed] [Google Scholar]
- Bos J. L. Epac: a new cAMP target and new avenues in cAMP research. Nat. Rev. Mol. Cell Biol. 2003, 4, 733–738. [DOI] [PubMed] [Google Scholar]
- Quan Y.; Jiang J.; Dingledine R. EP2 receptor signaling pathways regulate classical activation of microglia. J. Biol. Chem. 2013, 288, 9293–9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J.; Zwartkruis F. J.; Verheijen M. H.; Cool R. H.; Nijman S. M.; Wittinghofer A.; Bos J. L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [DOI] [PubMed] [Google Scholar]
- Liang X.; Wang Q.; Hand T.; Wu L.; Breyer R. M.; Montine T. J.; Andreasson K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J. Neurosci. 2005, 25, 10180–10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J.; Shie F. S.; Liu J.; Wang Y.; Davis J.; Schantz A. M.; Montine K. S.; Montine T. J.; Zhang J. Prostaglandin E2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated alpha-synuclein. J. Neuroinflammation 2007, 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.; Wang Q.; Shi J.; Lokteva L.; Breyer R. M.; Montine T. J.; Andreasson K. The prostaglandin E2 EP2 receptor accelerates disease progression and inflammation in a model of amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine T. J.; Milatovic D.; Gupta R. C.; Valyi-Nagy T.; Morrow J. D.; Breyer R. M. Neuronal oxidative damage from activated innate immunity is EP2 receptor-dependent. J. Neurochem. 2002, 83, 463–470. [DOI] [PubMed] [Google Scholar]
- Shie F. S.; Breyer R. M.; Montine T. J. Microglia lacking E prostanoid receptor subtype 2 have enhanced Abeta phagocytosis yet lack Abeta-activated neurotoxicity. Am. J. Pathol. 2005, 166, 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- af Forselles K. J.; Root J.; Clarke T.; Davey D.; Aughton K.; Dack K.; Pullen N. In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP(2) receptor antagonist. Br. J. Pharmacol. 2011, 164, 1847–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Ganesh T.; Du Y.; Quan Y.; Serrano G.; Qui M.; Speigel I.; Rojas A.; Lelutiu N.; Dingledine R. Small molecule antagonist reveals seizure-induced mediation of neuronal injury by prostaglandin E2 receptor subtype EP2. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 3149–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Quan Y.; Ganesh T.; Pouliot W. A.; Dudek F. E.; Dingledine R. Inhibition of the prostaglandin receptor EP2 following status epilepticus reduces delayed mortality and brain inflammation. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 3591–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y.; Narumiya S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Dingledine R. Role of prostaglandin receptor EP2 in the regulations of cancer cell proliferation, invasion, and inflammation. J. Pharmacol. Exp. Ther. 2013, 344, 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagmann W. K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Ganesh T.; Du Y.; Thepchatri P.; Rojas A.; Lewis I.; Kurtkaya S.; Li L.; Qui M.; Serrano G.; Shaw R.; Sun A.; Dingledine R. Neuroprotection by selective allosteric potentiators of the EP2 prostaglandin receptor. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 2307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter E. W.; Reitz A. B. Reductive aminations of carbonyl compounds with borohydride and borane reducing agents. Org. React. 2002, 59, 1–70. [Google Scholar]
- Mitsunobu O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural-products. Synthesis 1981, 1–28. [Google Scholar]
- Bevan C. D.; Lloyd R. S. A high-throughput screening method for the determination of aqueous drug solubility using laser nephelometry in microtiter plates. Anal. Chem. 2000, 72, 1781–1787. [DOI] [PubMed] [Google Scholar]
- Alavijeh M. S.; Chishty M.; Qaiser M. Z.; Palmer A. M. Drug metabolism and pharmacokinetics, the blood–brain barrier, and central nervous system drug discovery. NeuroRx 2005, 2, 554–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge W. M. The blood–brain barrier: bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabathuler R. Approaches to transport therapeutic drugs across the blood–brain barrier to treat brain diseases. Neurobiol. Dis. 2010, 37, 48–57. [DOI] [PubMed] [Google Scholar]
- Loscher W.; Potschka H. Blood–brain barrier active efflux transporters: ATP-binding cassette gene family. NeuroRx 2005, 2, 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu S. K.; Lee J.; Speights V. O. Jr.; Starzinski-Powitz A.; Arosh J. A. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 induces apoptosis of human endometriotic cells through suppression of ERK1/2, AKT, NFkappaB, and beta-catenin pathways and activation of intrinsic apoptotic mechanisms. Mol. Endocrinol. 2009, 23, 1291–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnini S.; Finetti F.; Solito R.; Terzuoli E.; Sacchetti A.; Morbidelli L.; Patrignani P.; Ziche M. EP2 prostanoid receptor promotes squamous cell carcinoma growth through epidermal growth factor receptor transactivation and iNOS and ERK1/2 pathways. FASEB J. 2007, 21, 2418–2430. [DOI] [PubMed] [Google Scholar]
- Honda T.; Segi-Nishida E.; Miyachi Y.; Narumiya S. Prostacyclin-IP signaling and prostaglandin E2-EP2/EP4 signaling both mediate joint inflammation in mouse collagen-induced arthritis. J. Exp. Med. 2006, 203, 325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheibanie A. F.; Khayrullina T.; Safadi F. F.; Ganea D. Prostaglandin E2 exacerbates collagen-induced arthritis in mice through the inflammatory interleukin-23/interleukin-17 axis. Arthritis Rheum. 2007, 56, 2608–2619. [DOI] [PubMed] [Google Scholar]
- Sung Y. M.; He G.; Hwang D. H.; Fischer S. M. Overexpression of the prostaglandin E2 receptor EP2 results in enhanced skin tumor development. Oncogene 2006, 25, 5507–5516. [DOI] [PubMed] [Google Scholar]
- Roy S.; Eastman A.; Gribble G. W. Synthesis of N-alkyl substituted bioactive indolocarbazoles related to Go6976. Tetrahedron 2006, 62, 7838–7845. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.