

Abstract
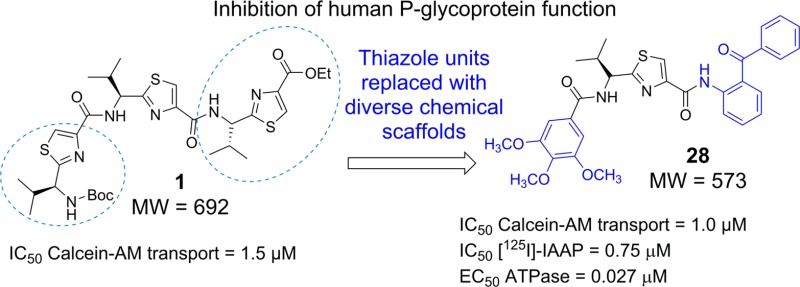
P-glycoprotein (P-gp) serves as a therapeutic target for the development of multidrug resistance reversal agents. In this study, we synthesized 21 novel compounds by peptide coupling at corresponding carboxyl and amino termini of (S)-valine-based bis-thiazole and monothiazole derivatives with diverse chemical scaffolds. Using calcein-AM efflux assay, we identified compound 28 (IC50 = 1.0 μM) carrying 3,4,5-trimethoxybenzoyl and 2-aminobenzophenone groups, respectively, at the amino and carboxyl termini of the monothiazole zwitter-ion. Compound 28 inhibited the photolabeling of P-gp with [125I]-iodoarylazidoprazosin with IC50 = 0.75 μM and stimulated the basal ATP hydrolysis of P-gp in a concentration-dependent manner (EC50 ATPase = 0.027 μM). Compound 28 at 3 μM reduced resistance in cytotoxicity assay to paclitaxel in P-gp-expressing SW620/Ad300 and HEK/ABCB1 cell lines. Biochemical and docking studies showed site-1 to be the preferable binding site for 28 within the drug-binding pocket of human P-gp.
Introduction
P-glycoprotein (P-gp, ABCB1 or MDR1) is a cell membrane ATP-binding cassette (ABC) transporter that is linked with the development of multidrug resistance (MDR) in cancer cells. Resistance to chemotherapy in cancer cells is a serious issue which can arise due to numerous mechanisms, including the overexpression of ABC drug transporters such as P-gp and breast cancer resistance protein (BCRP, ABCG2, or MXR). These transporters have been shown to contribute to decreased intracellular concentrations of chemotherapeutic drugs.1,2 P-gp utilizes energy derived from ATP hydrolysis to efflux an extremely diverse set of hydrophobic compounds typified by different chemical classes such as amphipathic, neutral, or weakly basic agents including a number of recently developed tyrosine kinase inhibitors.3 Human P-gp is composed of two transmembrane domains (TMDs), each containing six helices and two nucleotide-binding domains (NBDs). The TMDs house the drug/substrate-binding sites and a translocation conduit.4 Moreover, the TMD region contains a large hydrophobic drug-binding site that is capable of binding two to three molecules simultaneously.5,6 Drug transport process by P-gp is driven by ATP binding/hydrolysis to the drug-binding competent state which results in a rotation of the NBDs followed by adoption of a closed conformation and eventually NBD dimer returns to the open conformation after ATP hydrolysis.7 The closed conformation of NBD dimer is responsible for the substrate translocation in the TMDs’ drug-binding sites, thus initiating the release of the substrate to the extracellular side of the membrane.7
The ABC transporter-mediated MDR hinders the clinical cure of cancer by chemotherapy. Therefore, many research groups have been focusing on developing P-gp inhibitors to enhance the therapeutic concentration of chemotherapeutic drug inside cancer cells. Because of the unavailability of high-resolution 3D-crystal structure of human P-gp, we used the recently corrected crystal structure of mouse P-gp as a template to develop homology model of human P-gp,8 which was eventually used for understanding the binding mode of synthesized P-gp inhibitors. Numerous strategies such as random and focused screening, systematic chemical modifications, and combinatorial chemistry have been applied to develop the first three generations of P-gp inhibitors; however, they suffer from toxicity and drug–drug interactions. Although the first three generations of P-gp and/or ABCG2 modulators (valspodar, biricodar, laniquidar, zosuquidar, elacridar, and tariquidar) designed to reverse MDR failed in various stages of clinical trials, ABC transporters undoubtedly play a crucial role in the development of MDR, and researchers should not underestimate their importance. The clinical failures were only partly associated with ABC transporter modulation.9 They were also due to inadequate bioavailability at tumor sites,9 nonselective inhibition of P-gp across all tissues including the blood–brain barrier, simultaneous inhibition of the drug-metabolizing enzyme CYP3A4,10 and interference with the function of other ABC transporters that play a crucial role in antitumor immune response.11 Another problem was inappropriate selection of the patient population.12
With more carefully designed clinical trials, fourth generation P-gp modulators with low toxicity may be found to circumvent modulator-associated problems. New approaches leading to the development of fourth generation P-gp inhibitors exhibiting high P-gp selectivity and potency are highly desirable. One such approach is to append various chemical fragments that are frequently seen in P-gp inhibitors, to a novel chemotype such as thiazole amino acid. Using this approach, chemical fragments from reported potent P-gp inhibitors, as shown in Figure 1A, are inserted into chemically modified natural products.
Figure 1.

(A) Structures of the reported potent P-gp inhibitors containing chemical fragments such as methoxy substituted tetrahydroisoquinoline, various methoxy substituted aryl rings, biphenyl, and benzophenone. (B) Structures of compounds 1 and 1a–1d.
The first available cocrystal structures of murine P-gp bound to cyclic selenazole derivatives 1c (QZ59Se-SSS)13 and 1d (QZ59Se-RRR)13 provided significant insight into various drug-binding sites of P-gp transporter (Figure 1B). The cyclic trimer compound 1a (QZ59S-SSS), which is bioisosteric to compound 1c, exhibited an IC50 value of 2.7 μM, whereas 1b, an isostere of 1d, showed an IC50 of 8.4 μM against mouse P-gp efflux function (Figure 1B).14 Because of this distinguishable ability of P-gp for stereoisomers of cyclic peptides, we maintained (S) chirality for all the synthesized compounds. Moreover, several natural products have been shown to contain thiazole ring.15 Our recent study encompasses the investigation of the inhibitory effect of (S)-valine-based thiazole derivatives including compound 1a on human P-gp efflux activity.16 The results obtained from the exploration indicated the linear thiazole chain (linear trimer, 1; IC50 = 1.5 μM) to be equally effective as that of 1a (IC50 = 1.5 μM) in terms of inhibition of efflux activity of human P-gp. Also, we noticed that deviation from the trimer size (three consecutive thiazole units) of the thiazole chain had a detrimental effect on the inhibitory activity. Docking analysis of compound 1 within the drug-binding pocket of homology-modeled human P-gp showed interactions with certain key amino acid residues.16 The terminal thiazole fragments of compound 1 were found to capture hydrogen bonding and electrostatic interactions with the glutamine (Q990 and Q725) and tyrosine (Y307 and Y953) residues while being predominantly surrounded by hydrophobic amino acid side chains in the drug-binding pocket of P-gp. On the basis of these results, we decided to optimize compound 1 by replacing the terminal thiazole units with chemical scaffolds containing mono-, di-, and tri-methoxy aryl rings. For a few analogues, we also borrowed chemical fragments from the reported P-gp inhibitory structures that might increase the π–π interaction surface area within the drug-binding pocket of P-gp. Our choice of fragments was based on the chemical moieties which are frequently observed in various reported preclinical and clinical candidates such as tariquidar (XR9576),17 elacridar,18 LY402913,19 reserpine,20 XR9051,21 saracatinib,22 galloyl-based inhibitors,23 and benzophenone derivatives24 as illustrated in Figure 1A. Additionally, Didziapetris et al.25 estimated the “rule of fours” which states that the compounds with (N+O) ≥ 8, MW > 400, and acid pKa > 4 are likely to act as P-gp substrates. Therefore, we synthesized a series of (S)-valine derived thiazole analogues by extending the carboxyl and the amino termini of monomer (mono thiazole unit) as well as dimer (bis thiazole unit), taking into account the trimer size as well as the “rule of fours”. This optimization strategy was supported by the inhibitory activity of two representative compounds (2 and 3) mentioned in our recent report.16 Certain critical aspects were considered for the selection of fragments for extensions: (a) the coupled fragments must impart the desired hydrophobicity (ClogP = 3–6),26−28 (b) the selected fragments should present hydrogen bond acceptor atoms such as oxygen and nitrogen, and (c) both amino and carboxyl termini should be attached to various methoxy-substituted moieties and extended aromatic fragments to establish critical electrostatic interactions with hydrogen bond donor residues and the π-stacking interactions at the drug-binding pocket of the P-gp. Additionally in this regard, the presence of the methoxy groups has been shown previously to increase selective affinity toward P-gp.23 The synthesized derivatives were assessed for their inhibitory efficacies on the P-gp transport function by calcein-AM assay. Reversal of MDR due to ABCB1 inhibition in SW620/Ad300 and HEK/ABCB1 cell lines was also investigated using paclitaxel in the presence of compounds 28, 1a, and cyclosporine A. The effect of compound 28 was tested with biochemical studies including photolabeling with [125I]-iodoarylazidoprazosin (IAAP) and measurement of ATPase activity of P-gp using crude membranes of High Five insect cells expressing this transporter. Furthermore, to obtain insights into binding interactions of these analogues within the large drug-binding pocket, we performed docking of compounds on all the possible binding sites of the homology model of human P-gp.
Results and Discussion
Chemistry
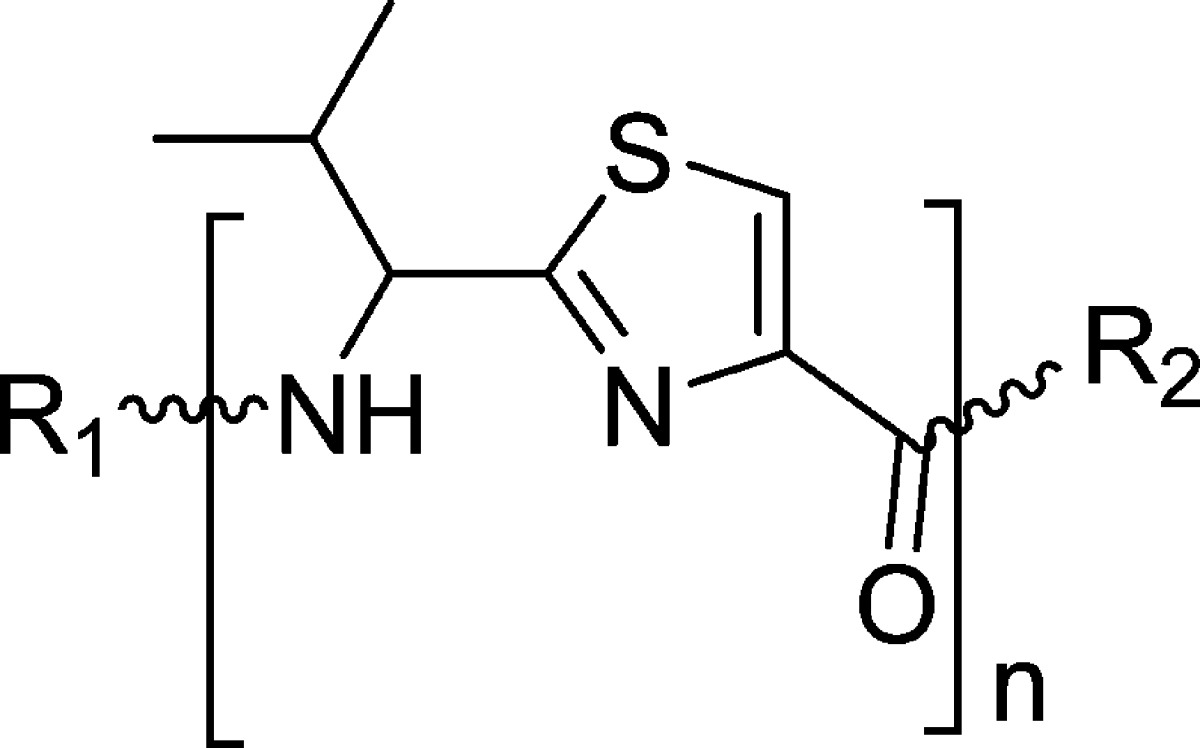
A library of 21 thiazole-based compounds targeted for P-gp efflux inhibition was synthesized as shown in Schemes 1–5. The synthesis of compounds 2 and 3 along with that of the required precursors (4, 6, 10, and 13) has been described in our recent report.16 Target compounds 5, 7–9, 11–12, and 14–15 were synthesized by peptide coupling reactions using thiazole acids (4 and 10) and thiazole amines (6 and 13) with various chemical scaffolds containing amine and acid derivatives, respectively. Schemes 1 and 2 shows the synthesis of monomer derivatives. Monomer acid 4 was reacted with a piperidine derivative (5a) to obtain compound 5 in the presence of the coupling agents HCTU, HOBt, and DIEA, as shown in Scheme 1. Monomer amine 6 was coupled with acid derivatives 7a and 8a to obtain compounds 7 and 8, respectively (Scheme 2). Alternatively, compound 9 was prepared by reacting trimethoxybenzoyl chloride (9a) with a monomer amine (6) using DIEA in THF. Our next objective was to synthesize dimer derivatives that could mimic the trimer structure using compounds 10 and 13. In Scheme 3, a dimer acid (10) was coupled with di- and tri-methoxy anilines (11a and 12a), respectively, to obtain compounds 11 and 12. Consequently, compounds 14 and 15 were synthesized by reaction of a dimer amine (13) with acid chlorides 14a and 15a using DIEA in THF (Scheme 4). Further, target compounds 17–29 were synthesized starting from compound 9 by use of the coupling reagents HCTU, HOBt, and DIEA in DMA (Scheme 5). These target compounds were the size of a trimer molecule, with both ends featuring chemical scaffolds borrowed from potent P-gp modulators. At first, the ethyl ester present in compound 9 was converted to acid 16 by aqueous basic hydrolysis. Compound 16 was then coupled with various aryl and arylalkyl amines to generate target compounds (17–29).
Scheme 1. Synthesis of Monomer Acid Derivative.

Reagents and conditions: (i) HCTU, HOBt, DIEA, DMA, rt, 18 h.
Scheme 5. Synthesis of Monomer Zwitter-ion Derivatives.

Reagents and conditions: (i) NaOH, THF/methanol/H2O (10:2:3), rt, 4 h; (ii) HCTU, HOBt, DIEA, DMA, rt, 18–24 h.
Scheme 2. Synthesis of Monomer Amine Derivatives.

Reagents and conditions: (i) HCTU, HOBt, DIEA, DMA, rt, 18 h; (ii) DIEA, THF, rt, 12 h.
Scheme 3. Synthesis of Dimer Acid Derivatives.

Reagents and conditions: (i) HCTU, HOBt, DIEA, DMA, rt, 18–24 h.
Scheme 4. Synthesis of Dimer Amine Derivatives.

Reagents and conditions: (i) DIEA, THF, rt, 12 h.
Structure–Activity Relationship
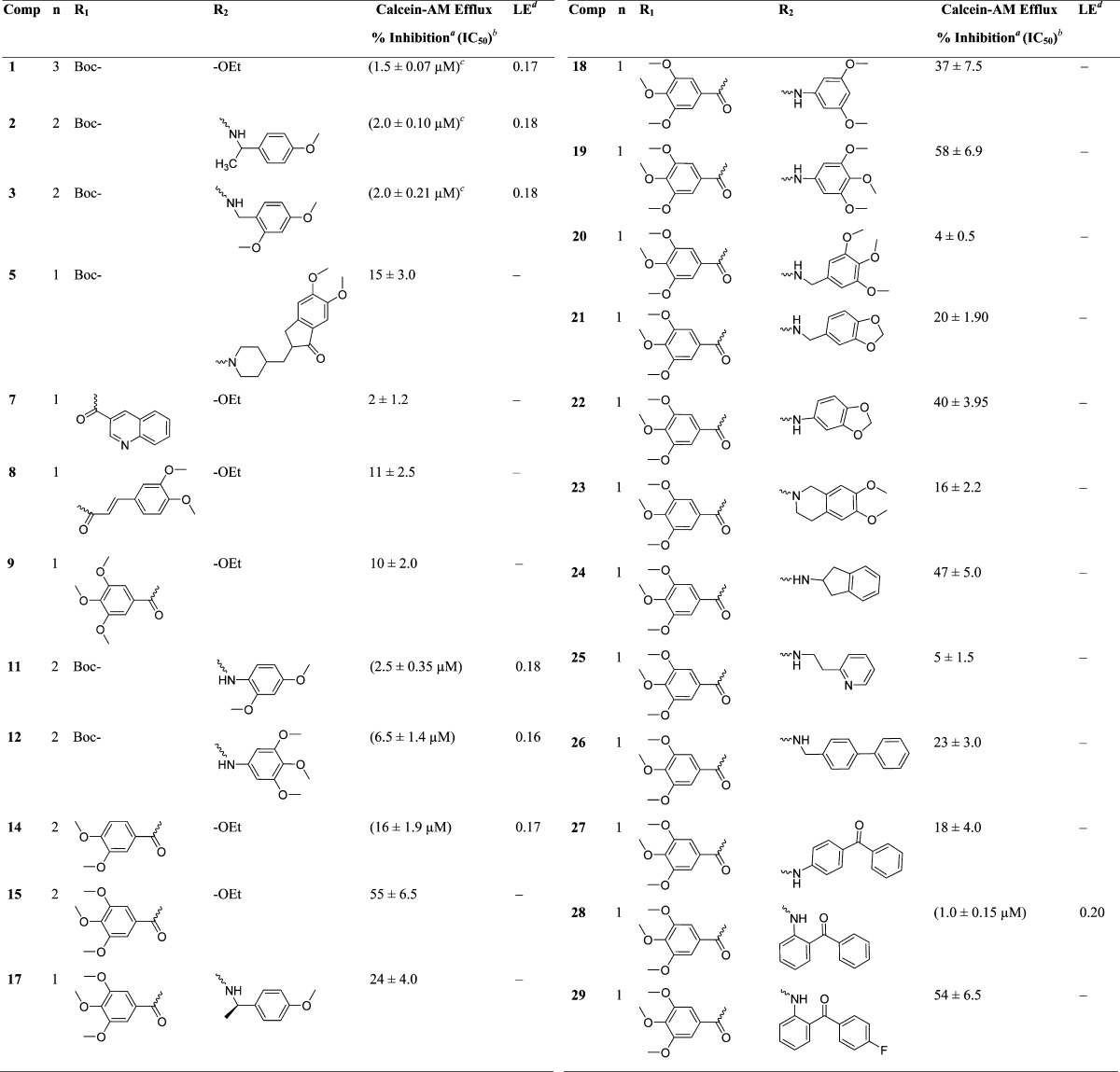
To optimize the efficacy of a linear trimer derivative (1, IC50 = 1.5 μM), 21 compounds (5, 7–9, 11–12, 14–15, and 17–29) were synthesized by appending the extremities of the monomer and dimer thiazole units with the selected fragments from the reported potent P-gp inhibitors. The target compounds were evaluated for their ability to inhibit transport function of human P-gp using BacMam-P-gp baculovirus-transduced HeLa cells overexpressing the multidrug transporter. The assay utilized for the evaluation involves measurement of fluorescence intensity imparted by calcein, which accumulates in the cells due to inhibition of P-gp efflux by the tested compounds (Table 1).
Table 1. (S)-Valine-Based Thiazole Derivatives As Inhibitors of P-gp Transport Function.
BacMam-P-gp baculovirus-transduced HeLa cells were incubated with 0.5 μM calcein-AM for 10 min at 37 °C under dark in the presence and absence of 10 μM (S)-valine-based thiazole derivatives. Percentage transport inhibition was derived by considering the level of inhibition obtained with the standard inhibitor, tariquidar at 1 μM, and the values ± SD shown are the average of two independent experiments done in triplicate at 10 μM concentrations of inhibitors.
IC50 values shown in brackets (±SD) were determined by at least two independent experiments done in triplicate.
Compounds reported in our recently published work.16
Ligand efficiency was calculated using formula, LE = (−RT ln IC50/number of non-hydrogen atoms); IC50 is in molar, R = 1.987 kcal K–1 mol–1, and T = 300 K.
Initially, four compounds (5, 7, 8, and 9) comprising substitutions on either side of the monomer unit were synthesized and evaluated for P-gp transport inhibition. The calcein-AM inhibition assay data (≤15% inhibition at 10 μM) suggests that straight monomer module substitutions were insufficient for any perceptible activity even with extended appendages such as 5,6-dimethoxyindanonemethylpiperidine, quinoline, and 3,4-dimethoxycinnamoyl, as seen in compounds 5, 7, and 8, respectively. This might be due to the lack of appropriate orientation of these derivatives within the P-gp drug-binding site, resulting in ineffective interactions. These results imply that a compound similar to a trimer in terms of length as well as shape is critical for effective P-gp efflux inhibitory activity for this class of compounds. In view of this substantiation, dimer derivatives 11–12 and 14–15 containing methoxy-substituted aryl moieties were prepared and tested for inhibitory potencies against P-gp transport function. Compounds 11 (IC50 = 2.5 μM) and 12 (IC50 = 6.5 μM), both dimer acid derivatives, were found to possess appreciable inhibition, comparable to that of compounds 2 and 3. Likewise, the dimer amine derivatives 14 (IC50 = 16 μM) and 15 (maximum 55% inhibition at 10 μM) were moderate inhibitors of the P-gp mediated efflux process. These outcomes show a significant improvement in P-gp efflux inhibition efficiency of the compounds on advancing from dimer to trimer structural size. Further, according to our strategy, we required concomitant incorporation of chemical scaffolds on either end of the mono-thiazole (monomer) unit. To achieve this, we decided to maintain the trimethoxybenzoyl fragment at the amino terminus because the presence of a trimethoxybenzoyl group has been shown to increase the potency as well as selectivity toward P-gp inhibition.23 To this end, 13 compounds (17–29) were synthesized and examined in the calcein-AM assay. Compounds 17 and 18 containing 4-methoxyphenylethyl amine and 3,5-dimethoxyaniline fragments, respectively, were poor to moderately active (24% and 37% inhibition at 10 μM, respectively), whereas compound 19 comprising a 3,4,5-trimethoxyaniline fragment showed improvement with 58% inhibition at 10 μM. It appears that an increase in the number of methoxy groups on the phenyl ring of the compounds enhances the binding affinity for P-gp. However, compound 20, with a 3,4,5-trimethoxybenzyl amine fragment, lost the P-gp inhibitory activity (4% inhibition at 10 μM). Compounds 21 and 22 with methylenedioxybenzyl amine and methylenedioxy aniline showed 20% and 40% inhibition of P-gp, respectively. Comparing compounds 19 with 20 and 21 with 22, the insertion of a methylene spacer between the aryl and the amine group proved detrimental for the P-gp inhibitory activity. This finding suggests potential steric clashes within the drug-binding pocket of P-gp for compounds 20 and 21 resulting from the introduction of the methylene spacer group. The 6,7-dimethoxytetrahydroisoquinoline group containing compound 23 was found to be devoid of P-gp inhibitory activity (16% at 10 μM). Furthermore, incorporation of a 2-aminoindane substitution resulted in moderate activity of compound 24 (47% inhibition at 10 μM); however, incorporation of 2-aminoethylpyridine (25) and 4-phenylbenzyl amine (26) were found to have a detrimental effect on P-gp inhibitory activity (5% and 23% inhibition at 10 μM, respectively), supporting our previous observation of the unfavorable effect of an alkyl spacer group. Weak inhibition of calcein-AM transport by compounds 22, 23, and 24 indicates a potential steric hindrance by the bicyclic ring structure at the drug-binding pocket of P-gp. Compound 27, containing a 4-aminobenzophenone substitution, lacks any significant inhibitory activity (18% at 10 μM), while compound 28 with a 2-aminobenzophenone substitution was found to have efficient P-gp inhibitory activity with IC50 value of 1 μM. Also, compound 29 showed appreciable inhibition (54% inhibition at 10 μM) of P-gp transport activity. Compound 27, with a benzoyl group at the para-position, might be similar to compounds 22, 23, and 24 with respect to steric hindrance. Moreover, effective inhibitory data for compounds 28 and 29 indicates that it is essential for inhibitors to have an angular shape to increase the contact surface with P-gp. Compound 28, bearing the trimethoxybenzoyl group at the amino terminus and 2-aminobenzophenone at the carboxyl terminus, proved to be the most effective P-gp inhibitor (IC50 = 1.0 μM) among the synthesized compounds, as evident from the dose–response curve shown in Figure 2B. Panel A of Figure 2 shows a representative histogram with low, moderate, and high inhibitory activity of selected thiazole derivatives, whereas panel B shows the concentration-dependent inhibition of calcein-AM efflux by compounds 11, 12, 14, and 28. We attempted to correlate the pIC50 values of compounds 1, 2, 3, 11, 12, 14, and 28 with their respective ClogP values 5.81, 5.22, 4.93, 5.47, 5.87, 5.03, and 5.49 in order to appraise the function of lipophilicity for P-gp inhibition. However, we were unable to find any significant correlation within the narrow range of ClogP values, which could be a result of a small sample size. Among various parameters that govern P-gp inhibition, one such parameter is ClogP, which should be in the range of 3–6 as mentioned earlier. Therefore, apart from ClogP other parameters, such as a molecular framework including hydrogen bonding ability and interaction surface area, also play critical role in inhibition of P-gp transport function. Ligand efficiency calculations showed a marginal improvement for compound 28 (0.20) as compared to compound 1 (0.17) (Table 1). In addition, the molecular weight decreased by 119 Da from compound 1 to benzophenone analogue 28.
Figure 2.

Inhibition of calcein-AM transport. BacMam P-gp baculovirus-transduced HeLa cells were assayed for calcein-AM transport (A) in the presence of selected derivatives at 10 μM and (B) in the presence of increasing concentrations of 11, 12, 14, and 28. (A) Representative histograms show low, moderate, and high P-gp inhibitory activity of selected thiazole derivatives. Tariquidar (1 μM), a known inhibitor of P-gp, was used for comparison. The values shown are the average of two independent experiments each done in triplicate. (B) Concentration-dependent inhibition of calcein-AM efflux by selected derivatives was studied. The average values from two independent experiments each done in triplicate were plotted and the IC50 values for compounds 11 (filled squares), 12 (filled diamonds), 14 (filled triangles), and 28 (filled circles) are 2.5, 6.5, 16, and 1 μM, respectively.
Effect of Compounds 28 and 1a on ABCB1-Mediated Resistance to Paclitaxel in ABCB1 Overexpressing Drug-Selected Cell Lines
Does compound 28 reverse resistance to anticancer drugs in P-gp overexpressing cell lines? To address this question, we performed MDR reversal experiments. To select a nontoxic or relatively low drug concentration for compounds 28 and 1a, cytotoxicity assays were performed on parental and P-gp-overexpressing cell lines (data not shown). On the basis of these results, concentrations of 10 μM for 28 and 3 μM for 1a demonstrated >85% cell survival.
To determine whether 28 and 1a could reverse P-gp-mediated MDR, cell survival assays were performed in the presence or absence of 28 and 1a, using the parental SW620 and drug-selected SW620/Ad300 cell lines. The drug-selected SW620/Ad300 cell line showed 688.2-fold resistance to paclitaxel, as compared to the parental SW620 cell line (Table 2). Compound 1a, at 0.3, 1, and 3 μM, significantly decreased the resistance of the SW620/Ad300 cell line to paclitaxel from 669.8-fold to 117.7-, 56.7-, and 20.8-fold, respectively, compared to SW620 cell line (Table 2). Compound 28, at 1, 3, and 10 μM, further reduced the resistance of SW620/Ad300 cell line to paclitaxel from 669.8-fold to 31.0-, 8.5-, and 1.8-fold, respectively, compared to SW620 cell line (Table 2). Moreover, at 10 μM, compound 28 can almost completely reverse the ABCB1-mediated drug resistance in SW620/Ad300 cells. As expected, a known modulator of P-gp, cyclosporine A (3 μM) significantly decreased the resistance of SW620/Ad300 to 13.2-fold for paclitaxel as compared to the control SW620 cell line (Table 2). Interestingly, at 3 μM, compound 28 showed more potent reversal activity than cyclosporine A and 1a (Figure 3A). These results suggest that compound 28 and 1a have the potential to enhance the sensitivity of P-gp-overexpressing drug-selected cell lines to anticancer drug substrates.
Table 2. Reversal Effect of Compounds 28, 1a, and Cyclosporine A on the Cytotoxicity of Paclitaxel to SW620, SW620/Ad300, HEK293, and HEK/ABCB1 Cell Lines.
| SW620 |
SW620/Ad300 |
HEK293 |
HEK/ABCB1 |
|||||
|---|---|---|---|---|---|---|---|---|
| compd | IC50 ± SEMa (μM) | FRb | IC50 ± SEMa (μM) | FRb | IC50 ± SEMa (μM) | FRb | IC50 ± SEMa (μM) | FRb |
| paclitaxel | 0.006 ± 0.001 | [1.0] | 4.019 ± 0.215 | [669.8] | 0.049 ± 0.004 | [1.0] | 4.138 ± 0.132 | [84.4] |
| + 28(1 μM) | 0.007 ± 0.001 | [1.2] | 0.186 ± 0.016 | [31.0] | 0.048 ± 0.003 | [1.0] | 0.633 ± 0.042 | [12.9] |
| + 28 (3 μM) | 0.006 ± 0.001 | [1.0] | 0.051 ± 0.005 | [8.5] | 0.047 ± 0.003 | [1.0] | 0.154 ± 0.015 | [3.1] |
| + 28 (10 μM) | 0.006 ± 0.001 | [1.0] | 0.011 ± 0.001 | [1.8] | 0.048 ± 0.004 | [1.0] | 0.098 ± 0.004 | [2.0] |
| + 1a (0.3 μM) | 0.006 ± 0.001 | [1.0] | 0.706 ± 0.068 | [117.7] | 0.049 ± 0.003 | [1.0] | 1.230 ± 0.063 | [25.1] |
| + 1a (1 μM) | 0.006 ± 0.001 | [1.0] | 0.340 ± 0.035 | [56.7] | 0.048 ± 0.005 | [1.0] | 0.780 ± 0.047 | [15.9] |
| + 1a (3 μM) | 0.007 ± 0.001 | [1.2] | 0.125 ± 0.011 | [20.8] | 0.048 ± 0.004 | [1.0] | 0.256 ± 0.024 | [5.2] |
| + CsA (3 μM) | 0.006 ± 0.001 | [1.0] | 0.079 ± 0.007 | [13.2] | 0.050 ± 0.003 | [1.0] | 0.205 ± 0.017 | [4.2] |
IC50, concentration of indicated compound required for 50% inhibition of cell survival were calculated from the killing curves shown in Figure 3. Mean values (±SEM) are from four independent experiments, each performed in triplicate.
FR: fold-resistance was calculated by dividing the IC50 value for paclitaxel of SW620 (or HEK293) and SW620/Ad300 (or HEK/ABCB1) cells in the absence or presence of compounds 28, 1a, and cyclosporine A by IC50 value for paclitaxel of SW620 (or HEK293) cells. CsA, cyclosporine A.
Figure 3.

Effect of compounds 28, 1a, and cyclosporine A (CsA) on ABCB1-mediated resistance to paclitaxel in ABCB1 overexpressing drug selected (A) and transfected (B) cell lines. (A) Concentration-dependent curves of paclitaxel with or without compounds 28, 1a, and CsA at 3 μM in parental SW620 and ABCB1 overexpressing SW620/Ad300 cells. The IC50 values of SW620/Ad300 cell line were compared with those of parental SW620 cells (see Table 2). (B) Concentration-dependent curves of paclitaxel with or without compounds 28, 1a, and CsA at 3 μM in parental HEK293/pcDNA3.1 and ABCB1 overexpressing HEK/ABCB1 cells. The IC50 values of HEK/ABCB1 cell line were compared with those of parental HEK293 cells (see Table 2). Points with error bars represent the mean ± SEM. The figure is a representative of four independent experiments, each done in triplicate.
Effect of Compounds 28 and 1a on P-gp-Mediated Resistance to Paclitaxel in P-gp-Overexpressing Transfected Cell Lines
There are multiple factors contributing to the drug resistance mechanisms in drug-selected cell lines. Therefore, we directly determined whether compounds 28 and 1a reverse P-gp-mediated MDR by performing cell survival assays in the presence or absence of compounds 28 and 1a, using the parental HEK293 cell line and ABCB1-transfected HEK/ABCB1 cell line. The transfected HEK/ABCB1 cell line showed 84.5-fold resistance to paclitaxel, as compared to the parental HEK293 cell line (Table 2). Compound 1a, at 0.3, 1, and 3 μM, significantly decreased the resistance of HEK/ABCB1 cell line to paclitaxel from 84.4-fold to 25.1-, 15.9-, and 5.2-fold, respectively, compared to HEK293 cell line (Table 2). Compound 28, at 1, 3, and 10 μM, further reduced the resistance of the HEK/ABCB1 cell line to paclitaxel to 12.9-, 3.1-, and 2.0-fold, respectively, compared to HEK293, for which resistance was not significantly altered by compound 28 (Table 2). At 10 μM concentration, compound 28 can almost completely reverse ABCB1-mediated drug resistance in HEK/ABCB1 cells. Being a positive control, cyclosporine A (3 μM) significantly decreased the resistance of HEK/ABCB1 to 4.2-fold for paclitaxel as compared to HEK293, for which drug sensitivity was not significantly altered (Table 2). Interestingly, at 3 μM, compound 28 demonstrated more potent reversal activity than cyclosporine A and 1a (Figure 3B). These results suggest that compounds 28 and 1a enhance the sensitivity of P-gp-overexpressing transfected cell lines to paclitaxel.
Effect of Compound 28 on IAAP Photoaffinity Labeling of P-gp
To assess the interaction of compound 28 at the drug-binding pocket of P-gp, its effect on biochemical assays including photolabeling of P-gp with [125I]-IAAP and ATP hydrolysis by P-gp was determined. Both compounds 28 and 1a inhibited the photoaffinity labeling of P-gp with [125I]-IAAP in a concentration-dependent manner, reaching a maximum of 90% inhibition at concentrations higher than 5 μM (see Figure 4A, data shown only for compound 28).16 The concentration required for 50% inhibition of photolabeling by compound 28 is 0.72 μM (Figure 4A). These results clearly show that compound 28, similar to the other modulators, binds at the drug-binding pocket located in the transmembrane domains of human P-gp.
Figure 4.

Effect of compound 28 on the photoaffinity labeling and ATPase activity of P-gp. (A) Compound 28 inhibits the photoaffinity labeling of P-gp with [125I]-IAAP. Crude membranes of High Five insect cells expressing P-gp (65 μg protein/100 μL) in 50 mM MES-Tris pH 6.8 were incubated with increasing concentrations of compound 28 (0–20 μM), at 37 °C for 10 min. Samples were then transferred to 4 °C bath, and 4–5 nM [125I]-IAAP was added under subdued light. The samples were photo-cross-linked with [125I]-IAAP as described previously.36 A representative autoradiogram from one of two independent experiments is shown. In the graph, points represent the average of two independent experiments. The data were fitted (R2 = 0.94) with a one-phase decay equation using GraphPad Prism 6.01. (B) Compound 28 stimulates the basal ATPase activity of ABCB1. Crude membranes of High Five insect cells expressing P-gp (10 μg protein/100 μL) were incubated with increasing concentrations of compound 28 (0–10 μM), in the presence and absence of sodium orthovanadate (0.3 mM), in ATPase assay buffer as described previously.37 The data obtained with compound 28 up to 2.5 μM concentration are shown in (B). Points with error bars represent the mean and SEM of three independent experiments. The data were fitted (R2 = 0.92) with the Michaelis–Menten equation using GraphPad Prism 6.01.
Effect of Compound 28 on the Basal ATPase Activity of P-gp
The effect of compound 28 on the basal ATPase activity of P-gp was investigated in crude membranes of High Five cells expressing this transporter. Compound 28 stimulated the ATP hydrolysis by P-gp up to 2-fold (see Figure 4B), at concentrations ranging from 0.5 to 2.5 μM. At higher concentrations, although basal ATPase activity was still stimulated, the extent of the stimulation was slightly lower, approximately 1.8-fold. The apparent affinity or concentration required for 50% stimulation of ATP hydrolysis was calculated with a concentration up to 2.5 μM, and the EC50 value was 0.027 μM. These data demonstrate that compound 28 is a potent stimulator of the ATPase activity of P-gp.
Glide-XP (Schrödinger, LLC., New York, NY, 2013) docking experiments were performed to understand the molecular interactions of these compounds within the drug-binding sites of P-gp. Docking experiments were targeted to all possible binding sites of P-gp, as proposed by Shi et al. (site-1, site-2, site-3, and site-4) using “Extra Precision” (XP) glide mode.29 Analysis of the binding energy data indicated site-1 as the preferred site of binding. For closer inspection of the possible binding conformation of compound 28 within site-1, induced-fit docking was performed to emulate the flexible receptor binding model, as suggested by Loo et al.30 It may be noted that the actual binding mode can be discerned through site-directed mutagenesis and/or cocrystal structural studies of human P-gp in the presence of compound 28.
Docking Interaction of Compound 28 with Homology-Modeled Human P-gp
The binding interactions of compound 28 were analyzed within site-1 of homology-modeled human P-gp (Figure 5A,B). Compound 28 is stabilized through specific interactions such as hydrogen bonding and nonspecific interactions such as hydrophobic interactions with residues in the drug-binding pocket of P-gp. The hydrogen bond acceptor oxygen atom at the para-position of the trimethoxyphenyl moiety showed hydrogen bonding interaction with the side chain of Q990 (H3CO---HN-Q990). The amide bond between the trimethoxyphenyl ring and the thiazole ring forms a hydrogen bond with Q725 (NH---O=CNH2-Q725). The two phenyl rings of the benzophenone group interacts with F336 through π–π stacking in which the phenyl group attached to the thiazole ring via amide linkage does so in a parallel displaced (offset) setting while the distal benzoyl group forms an edge-to-face contact. Additionally, the benzoyl group forms similar π–π stacking interactions with the side chain phenyl ring of Y310 and F335. The amide bond connecting the benzophenone and the thiazole group is in close proximity to S979. Compound 28 was able to establish some nonspecific interactions with the surrounding hydrophobic residues. The trimethoxyphenyl, benzophenone, isopropyl, and thiazole groups are mainly stabilized through hydrophobic contacts within the large hydrophobic pocket formed by the side chains of M69, F72, F303, I306, Y307, F314, L332, L339, I340, F343, A729, F732, F759, Y953, F957, F978, F983, M986, and A987.
Figure 5.
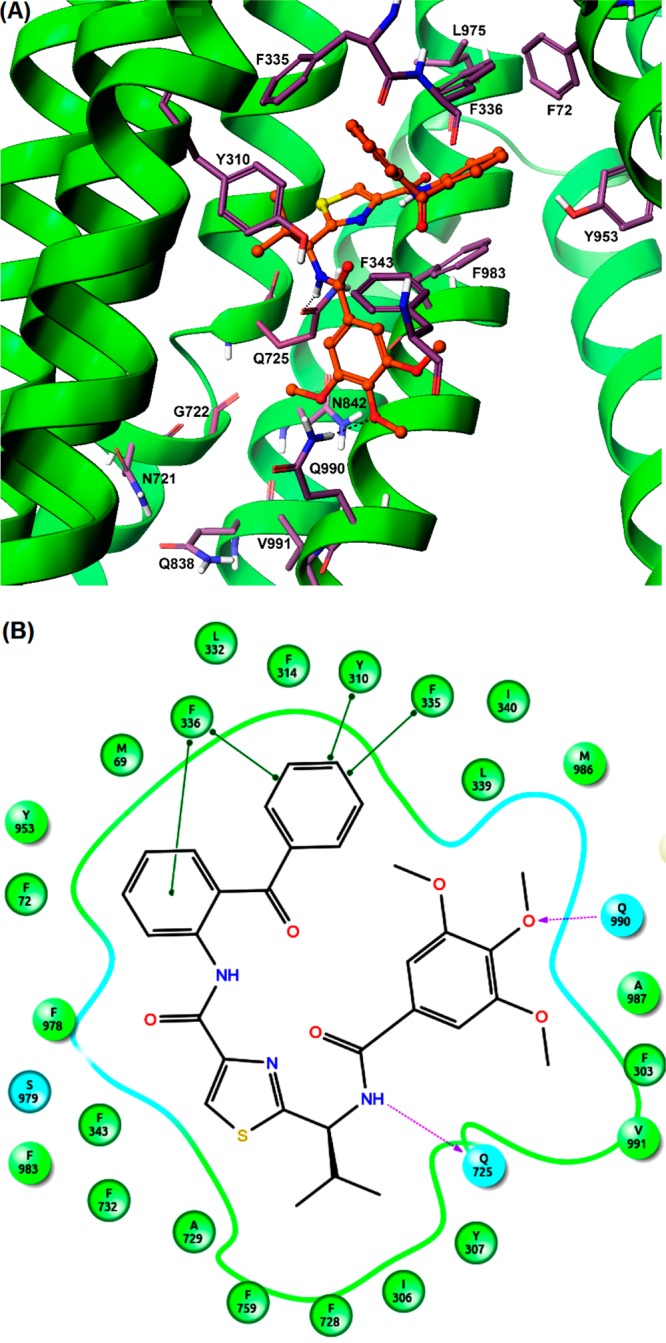
Induced-fit docking model of compound 28 at site-1 of human P-gp homology model. (A) A portion of the transmembrane region of homology modeled human P-gp is shown in ribbon presentation. Selected amino acids are depicted as sticks with the atoms colored as carbon, purple–blue; hydrogen, white; nitrogen, blue; oxygen, red; sulfur, yellow), whereas the inhibitor is shown as a ball and stick model with the same color scheme as above except carbon atoms are represented in orange. Hydrogen bonds are shown as black dashes. The ribbon representation for portions of TM3 and TM6 was undisplayed for better view. (B) A two-dimensional ligand–receptor interaction diagram with important interactions observed in the docked complex of compound 28 with the drug-binding site residues of human P-gp is shown. The amino acids within 5 Å are shown as colored bubbles, cyan indicates polar, and green indicates hydrophobic residues. Hydrogen bonds are shown by purple dotted arrows, while π-stacking aromatic interactions are shown by green lines.
Analysis of the binding model of 28 at site-1 of P-gp attempts to rationalize its effective P-gp inhibitory activity as well as provides a potential possibility for further optimization with respect to the residues around it and substitution pattern on N-terminal benzoyl and C-terminal benzophenone moieties. For example, the N-benzoyl group may be scanned for various combinations of hydroxyl and methoxy groups to capture electrostatic interactions with proximal polar residues such as N721, Q838, N839, N842, and Q990. Because the C-terminal benzophenone group occupied a subsite formed by side chains of aromatic residues, the benzoyl portion of the benzophenone moiety may be similarly scanned for substitutions with small electron donating/withdrawing groups to capture aromatic interactions through inductive increase in π–π stacking.
Conclusions
We synthesized a series of (S)-valine derived thiazole analogues that are comparable in size to a trimer length, composed of various chemical scaffolds to inhibit human P-gp efflux activity. This investigation resulted in identification of compound 28 bearing a trimethoxyphenyl ring and a benzophenone moiety at the amino and carboxyl termini of the monothiazole zwitter-ion, respectively. Intracellular accumulation of calcein (from calcein-AM) in P-gp-transduced HeLa cells clearly demonstrated that compound 28 inhibits the transport activity of P-gp. Compound 28 was found to be superior to 1a and cyclosporine A for reversal of resistance to paclitaxel in both SW620/Ad300 and HEK/ABCB1 cell lines. Moreover, compound 28 did not exhibit any toxicity up to 10 μM concentration in the cell lines studied in this report. Future in vivo reversal effects of 28 will prove its worth as an effective MDR reversal agent for combination with conventional chemotherapy. The inhibition of IAAP-labeling and stimulation of basal ATP hydrolysis of P-gp provide further evidence that the effect observed in intact cells is mediated by the specific interaction of compound 28 at the drug-binding pocket of P-gp. Consistent with cell- and membrane-based assays, docking analysis revealed interactions of compound 28 within site-1 in the drug-binding pocket of homology-modeled human P-gp. Further efforts will be focused on optimization of the substitution pattern on the terminal phenyl ring of the benzophenone moiety to obtain not only highly potent P-gp inhibitors but also to develop photoactivable derivatives for identification of residues in site-1 that interact with the inhibitor.
Experimental Section
General Synthesis
Chemicals were purchased from Aldrich Chemical Co. (Milwaukee, WI), AK scientific (Union City, CA), Oakwood Products (West Columbia, SC), Alfa Aesar (Ward Hill, MA), and TCI America (Portland, OR) and were used as received. All compounds were checked for homogeneity by TLC using silica gel as a stationary phase. Melting points were determined on a Thomas–Hoover capillary melting point apparatus and were uncorrected. NMR spectra were recorded on a Bruker 400 Avance DPX spectrometer (1H at 400 MHz) outfitted with a z-axis gradient probe. The chemical shifts for 1H NMR were reported in parts per million (δ ppm) downfield from tetramethylsilane (TMS) as an internal standard. The 1H NMR data are reported as follows: chemical shift, multiplicity s (singlet), d (doublet), t (triplet), dd (doublet of doublets), m (multiplet), and bs (broad singlet). Flash chromatography was performed using silica gel (0.060–0.200 mm) obtained from Dynamic adsorbents. The purity of all target compounds was assessed using an Agilent 1260 Infinity HPLC system. The column used was a C18 reverse phase column (Phenomenex-Kinetex, 150 mm × 4.6 mm, 5 μ, 100 Å, serial no. 660057-3) eluting with an isocratic mobile phase (acetonitrile/water 60:40) at a flow rate of 1.0 mL/min, and samples were monitored at UV = 254 nm. All tested compounds were confirmed to be ≥95% pure based on the area of the major peak when compared to the total combined area.
Synthesis
The synthesis and characterization data for compounds 2, 3, 4, 6, 10, and 13 was described in our recent report.16
Method A. General Procedures for Peptide Coupling of Thiazole Amino Acid Units to Linear Oligomers.16
Di-isopropylethylamine (DIEA) (1.5 equiv) was added to a well-stirred suspension of carboxylic acid derivatives in anhydrous N,N-dimethylacetamide (DMA). Upon cooling the reaction mixture to 0 °C, HCTU (1.5 equiv) and HOBt (1.5 equiv) were sequentially added. The reaction mixture was allowed to stir at 0 °C for 10 min and then treated with a precooled solution of the corresponding amines (1.2 equiv) in DMA. The reaction mixture was stirred at rt, and after completion as monitored by TLC, the reaction mass was concentrated in vacuo. The solution was partitioned between ethyl acetate and aqueous citric acid (10% w/v), and the separated aqueous phase was extracted again with ethyl acetate. The organic extracts were combined and washed sequentially with saturated aqueous sodium bicarbonate, water, and brine, then dried over sodium sulfate, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica gel using n-hexane–ethyl acetate (1:1) as eluent to provide desired peptide products.
Method B. General Procedures for Peptide Coupling of Thiazole Amines with Acyl Chlorides
To the cooled suspension of thiazole amine derivatives (0 °C) in anhydrous THF were added DIEA (1.5 equiv) and commercially available acyl chlorides (1.2–1.5 equiv). The reaction mixture was then allowed to warm to rt and stirred for 12 h. The solvent was then removed by evaporation, and the resulting reaction mass was diluted with ethyl acetate. The ethyl acetate layer was then washed with aqueous citric acid (10% w/v), saturated sodium bicarbonate, water, and brine. The organic fractions were dried over anhydrous sodium sulfate and evaporated under vacuum. The crude product was purified by flash chromatography on silica gel using n-hexane–ethyl acetate (1:1) as eluent to obtain the required peptides.
(1-(4-(4-(5,6-Dimethoxy-1-oxo-2,3-dihydro-1H-inden-2-yl)methyl)piperidine-1-carbonyl)thiazol-2-yl)-(S)-2-methylpropylcarbamic Acid tert-Butyl Ester (5)
Compounds 4 (0.30 g, 0.99 mmol) and 5a (0.65 g, 1.99 mmol) were reacted together by following method A to obtain compound 5 as a white solid (0.638 g, 85%); mp 80–84 °C; Rf = 0.25 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 7.91 (s, 1H), 7.71 (d, 1H, J = 6.6 Hz), 7.09 (s, 1H), 7.06 (s, 1H), 4.61 (t, 1H, J = 7.1 Hz), 3.86 (s, 3H), 3.79 (s, 3H), 3.21–3.28 (m, 3H), 2.66–2.76 (m, 4H), 2.45–2.47 (m, 1H), 1.71–1.78 (m, 4H), 1.40 (s, 9H), 1.24–1.31 (m, 3H), 0.88 (dd, 6H, J = 17.7 Hz, J = 5.5 Hz). m/z (ESI-MS) 594.33 (C30H41N3O6SNa requires 594.27, [M + Na]+). HPLC tR = 4.7 min, purity 100%.
(S)-2-{2-Methyl-1-[(quinoline-3-carbonyl)amino]propyl}thiazole-4-carboxylic Acid Ethyl Ester (7)
Compounds 6 (0.30 g, 1.31 mmol) and 7a (0.453 g, 2.62 mmol) were reacted together according to method A to obtain compound 7 as yellow foam (0.443 g, 88%); Rf = 0.40 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 9.38 (s, 1H), 8.68 (s, 1H), 8.16 (d, 1H, J = 2.8 Hz), 8.13 (s, 1H), 7.96 (d, 1H, J = 8.2 Hz), 7.83 (t, 1H, J = 7.5 Hz), 7.64 (t, 1H, J = 7.5 Hz), 7.55 (d, 1H, J = 8.48 Hz), 5.50 (t, 1H, J = 8.68 Hz) 4.44 (q, 2H, J = 5.8 Hz), 2.53–2.59 (m, 1H), 1.44 (t, 3H, J = 5.8 Hz), 1.13 (d, 6H, J = 6.5 Hz). m/z (ESI-MS) 384.33 (C20H22N3O3S requires 384.13, [M + H]+). HPLC tR = 3.3 min, purity 99%.
(E)-2-{1-[3-(3,4-Dimethoxyphenyl)acryloylamino]-(S)-2-methylpropyl}thiazole-4-carboxylic Acid Ethyl Ester (8)
Compounds 6 (0.30 g, 1.31 mmol) and 8a (0.328 g, 1.57 mmol) were reacted together as per method A to obtain compound 8 as a light-yellow foam (0.462 g, 84%); Rf = 0.50 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 8.30 (s, 1H), 7.90 (1H, s), 7.53 (d, 1H, J = 15.68 Hz), 7.15–7.16 (m, 1H), 7.13 (d, 1H, J = 8.3 Hz), 6.95 (d, 1H, J = 8.2 Hz), 6.68 (d, 1H, J = 15.64 Hz), 5.23 (d, 1H, J = 6.7 Hz), 4.37 (q, 2H, J = 7.12 Hz), 3.85 (s, 3H), 3.84 (s, 3H), 2.44–2.42 (m, 1H), 1.38 (t, 3H, J = 7.0 Hz), 1.03 (d, 3H, J = 6.7 Hz), 0.98 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 441.25 (C21H26N2O5SNa requires 441.16, [M + Na]+). HPLC tR = 3.4 min, purity 99%.
(S)-2-[2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid Ethyl Ester (9)
Compounds 6 (2 g, 8.77 mmol) and 9a (2.42 g, 10.52 mmol) were reacted together by following method B to obtain compound 9 as a white solid (3.03 g, 82%); mp 144–150 °C; Rf = 0.60 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 9.03 (d, 1H, J = 8.12 Hz), 8.45 (s, 1H), 7.23 (s, 2H), 5.07 (t, 1H, J = 8.16 Hz), 4.30 (q, 2H, J = 6.4 Hz), 3.83 (s, 6H), 3.70 (s, 3H), 2.47–2.55 (m, 1H), 1.31 (t, 3H, J = 5.8 Hz), 1.05 (d, 3H, J = 6.1 Hz), 0.90 (d, 3H, J = 6.24 Hz). m/z (ESI-MS) 445.33 (C20H26N2O6SNa requires 445.15, [M + Na]+). HPLC tR = 3.7 min, purity 99%.
[1-(4-{1-[4-(2,4-Dimethoxyphenylcarbamoyl)thiazol-2-yl]-(S)-2-methylpropylcarbamoyl}thiazol-2-yl)-(S)-2-methylpropyl]carbamic Acid tert-Butyl Ester (11)
Compounds 10 (0.10 g, 0.21 mmol) and 11a (0.065 g, 0.42 mmol) were reacted together as per method A to obtain compound 11 as a white solid (0.104 g, 81%); mp 70–72 °C; Rf = 0.55 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 9.10 (s, 1H), 8.13 (s, 1H), 8.08 (s, 1H), 7.90 (d, 1H, J = 9.12 Hz), 7.61 (s, 1H), 7.27 (s, 1H), 7.09 (d, 1H, J = 8.76 Hz), 6.89 (d, 1H, J = 8.6 Hz), 5.41–5.36 (m, 1H), 5.13 (bs, 1H), 3.96 (s, 3H), 3.91 (s, 3H), 2.60–2.55 (m, 1H), 2.39 (bs, 1H), 1.48 (s, 9H), 1.10 (d, 6H, J = 6.2 Hz), 1.04 (d, 3H, J = 6.68 Hz), 0.98 (t, 3H, J = 7.12 Hz). m/z (HRMS) 640.2236 (C29H39N5O6S2Na requires: 640.2239, [M + Na]+). HPLC tR = 7.6 min, purity 95%.
[2-Methyl-1-(4-{2-methyl-1-[4-(3,4,5-trimethoxyphenylcarbamoyl)thiazol-2-yl]-(S)-propylcarbamoyl}thiazol-2-yl)-(S)-propyl]carbamic Acid tert-Butyl Ester (12)
Compounds 10 (0.10 g, 0.21 mmol) and 12a (0.077 g, 0.42 mmol) were reacted together following method A to obtain compound 12 as oil (0.11 g, 71%); Rf = 0.45 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 8.09 (s, 1H), 8.06 (s, 1H), 7.84 (d, 1H, J = 9.4 Hz), 7.65 (s, 1H), 7.29 (s, 1H), 6.62 (s, 1H), 6.61(s, 1H), 5.36 (m, 1H), 4.66 (m, 1H), 3.88 (s, 6H), 3.78 (s, 3H), 2.54–2.49 (m, 1H), 2.30 (bs, 1H), 1.48 (s, 9H), 1.05–0.90 (m, 12H). m/z (HRMS) 670.2348 (C30H41N5O7S2Na requires: 670.2345, [M + Na]+). HPLC tR = 8.0 min, purity 95%.
2-[1-({2-[1-(3,4-Dimethoxybenzoylamino)-(S)-2-methylpropyl]thiazole-4-carbonyl}amino)-(S)-2-methylpropyl]thiazole-4-carboxylic Acid Ethyl Ester (14)
Compounds 13 (0.1 g, 0.24 mmol) and 14a (0.058 g, 0.29 mmol) were reacted together according to method B to obtain compound 14 as a white foam (0.096 g, 69%); Rf = 0.40 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 8.12 (d, 1H, J = 8.5 Hz), 8.05 (d, 1H, J = 3.2 Hz), 7.96 (t, 1H, J = 6.2 Hz), 7.49 (d, 1H, J = 5.8 Hz), 7.40 (dd, 1H, J = 8.3 Hz, 2.0 Hz), 6.92 (dd, 1H, J = 8.3 Hz, 4.4 Hz), 6.80 (m, 1H), 5.40 (m, 1H), 5.31 (m, 1H), 3.95–3.93 (m, 8H), 2.64–2.58 (m, 1H), 2.54–2.47 (m, 1H), 1.25 (bs, 3H), 1.98–0.97 (m, 12H). m/z (HRMS) 597.1826 (C27H34N4O6S2Na requires: 597.1817, [M + Na]+). HPLC tR = 3.5 min, purity 95%.
2-[(S)-2-Methyl-1-({2-[-(S)-2-methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carbonyl}amino)propyl]thiazole-4-carboxylic Acid Ethyl Ester (15)
Compounds 13 (0.1 g, 0.24 mmol) and 15a (0.063 g, 0.29 mmol) were reacted together following method B to obtain compound 15 as a yellow solid (0.106 g, 72%); mp 58–62 °C; Rf = 0.40 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 8.12 (d, 1H, J = 7.3 Hz), 8.07 (d, 1H, J = 2.16 Hz), 8.02–7.94 (m, 1H,), 7.11 (s, 1H), 7.08 (s, 1H), 6.89 (dd, 1H, J = 12.7 Hz, 9.1 Hz), 5.42–5.31 (m, 2H), 3.96–3.89 (m, 11H), 2.61–2.47 (m, 2H), 1.27 (t, 3H, J = 5.8 Hz), 1.12–0.97 (m, 12H). m/z (ESI-MS) 627.28 (C28H36N4O7S2Na requires 627.19, [M + Na]+). HPLC tR = 3.6 min, purity 99%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid (16)
Compound 9 (2.2 g, 5.20 mmol) was added to the solvent mixture [THF:methanol:water (10:2:3)], and cooled to 0 °C. Sodium hydroxide (10 equiv) was added, and the mixture was stirred at rt for 12 h. The reaction mixture was then concentrated in vacuo and partitioned between ethyl acetate (30 mL) and water (20 mL). The aqueous phase containing compound was collected and acidified to pH 4 with 10% potassium hydrogen sulfate and then extracted with ethyl acetate (3 × 20 mL). Organic fractions were dried over sodium sulfate and concentrated under reduced pressure to yield the intermediate compound 16 as a white solid (1.88 g, 92%); Rf = 0.20 (MeOH/CH2Cl2 5:95). 1H NMR (400 MHz; DMSO-d6) δ 12.34 (s, 1H), 8.10 (s, 1H), 7.27 (s, 1H), 7.08 (s, 2H), 5.36 (t, 1H, J = 7.1 Hz), 3.93 (s, 6H), 3.88 (s, 3H), 2.47–2.55 (m, 1H), 1.08 (d, 3H, J = 8.0 Hz), 0.99 (d, 3H, J = 6.5 Hz).
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid [1-(4-Methoxyphenyl)ethyl]amide (17)
Compounds 16 (0.1 g, 0.25 mmol) and 17a (0.046 g, 0.30 mmol) were reacted together following method A to obtain compound 17 as a light-yellow solid (0.098 g, 74%); mp 50–54 °C; Rf = 0.45 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 8.01 (s, 1H), 7.41 (d, 1H, J = 7.7 Hz), 7.32 (d, 1H, J = 8.3 Hz), 7.01–7.07 (m, 2H), 6.86–6.89 (m, 2H), 6.67–6.79 (m, 2H), 5.38–5.26 (m, 2H), 3.90 (s, 6H), 3.87 (s, 3H), 3.80 (s, 3H), 2.47–2.45 (m, 1H), 1.59 (d, 3H, J = 6.4 Hz), 1.05 (d, 6H, J = 7.2 Hz). m/z (ESI-MS) 550.17 (C27H33N3O6SNa requires 550.21, [M + Na]+). HPLC tR = 3.9 min, purity 97%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid (3,5-Dimethoxyphenyl)amide (18)
Compounds 16 (0.1 g, 0.25 mmol) and 18a (0.046 g, 0.30 mmol) were reacted together using method A to obtain compound 18 as a light-yellow solid (0.099 g, 74%); mp 124–130 °C; Rf = 0.55 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 9.05 (s, 1H), 8.12 (s, 1H), 7.06 (s, 2H), 6.93 (s, 2H), 6.66 (t, 1H, J = 8.7 Hz), 6.28 (s, 1H), 5.41 (dd, 1H, J = 9.33 Hz, 6.6 Hz), 3.89–3.92 (m, 9H), 3.81 (s, 6H), 2.49–2.58 (m, 1H), 1.06–1.10 (m, 6H). m/z (HRMS) 552.1770 (C26H31N3O7SNa requires: 552.1780, [M + Na]+). HPLC tR = 4.3 min, purity 100%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid (3,4,5-Trimethoxyphenyl)amide (19)
Compounds 16 (0.1 g, 0.25 mmol) and 19a (0.055 g, 0.30 mmol) were reacted together following method A to obtain compound 19 as off-white solid (0.106 g, 75%); mp 58–62 °C; Rf = 0.40 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 9.03 (s, 1H), 8.15 (s, 1H), 7.06 (s, 2H), 7.02 (s, 2H), 6.61 (d, 1H, J = 8.68 Hz), 5.45 (dd, 1H, J = 9.3 Hz, 6.6 Hz), 3.94 (s, 6H), 3.91 (s, 9H), 3.86 (s, 3H), 2.52–2.60 (m, 1H), 1.09–1.12 (m, 6H). m/z (HRMS) 582.1900 (C27H33N3O8SNa requires: 582.1886, [M + Na]+). HPLC tR = 3.3 min, purity 99%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid 3,4,5-Trimethoxybenzylamide (20)
Compounds 16 (0.1 g, 0.25 mmol) and 20a (0.059 g, 0.30 mmol) were reacted together by following method A to obtain compound 20 as a light-yellow solid (0.104 g, 72%); mp 76–80 °C; Rf = 0.30 (EtOAc/n-hexane 1:1). 1H NMR (400 MHz; CDCl3; TMS) δ 8.07 (s, 1H), 7.57 (t, 1H, J = 5.8 Hz), 7.01 (s, 2H), 6.68 (d, 1H, J = 8.9 Hz), 6.58 (s, 2H), 5.37 (dd, 1H, J = 8.9 Hz, 6.5 Hz), 4.56 (t, 2H, J = 5.0 Hz), 3.88 (s, 9H), 3.84 (s, 6H), 3.83 (s, 3H), 2.48–2.43 (m, 1H), 1.05 (d, 3H, J = 6.8 Hz), 1.01 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 596.25 (C28H35N3O8SNa requires 596.21, [M + Na]+). HPLC tR = 2.7 min, purity 100%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid (Benzo[1,3]dioxol-5-ylmethyl)amide (21)
Compounds 16 (0.1 g, 0.25 mmol) and 21a (0.045 g, 0.30 mmol) were reacted together following method A to obtain compound 21 as a dark-brown solid (0.105 g, 79%); mp 54–58 °C; Rf = 0.35 (EtOAc/n-hexane 3:2). 1H NMR (400 MHz; CDCl3; TMS) δ 8.07 (s, 1H), 7.49 (t, 1H, J = 5.8 Hz), 7.00 (s, 2H), 6.85 (s, 1H), 6.81 (s, 1H), 6.76 (d, 1H, J = 8.1 Hz), 6.62 (d, 1H, J = 8.8 Hz), 5.95 (s, 2H), 5.35 (dd, 1H, J = 9.1 Hz, 6.4 Hz), 4.55 (t, 2H, J = 8.1 Hz), 3.88 (s, 9H), 2.41–2.49 (m, 1H), 1.04 (d, 3H, J = 6.8 Hz), 1.00 (d, 3H, J = 6.8 Hz). m/z (ESI-MS) 550.25 (C26H29N3O7SNa requires 550.17, [M + Na]+). HPLC tR = 3.1 min, purity 100%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid Benzo[1,3]dioxol-5-ylamide (22)
Compounds 16 (0.1 g, 0.25 mmol) and 22a (0.041 g, 0.30 mmol) were reacted together following method A to obtain compound 22 as a light-yellow solid (0.087 g, 67%); mp 60–64 °C; Rf = 0.50 (EtOAc/n-hexane 3:2). 1H NMR (400 MHz; CDCl3; TMS) δ 9.00 (s, 1H), 8.12 (s, 1H), 7.42 (s, 2H), 7.02 (s, 1H), 6.97 (d, 1H, J = 6.72 Hz), 6.80 (d, 1H, J = 8.3 Hz), 6.62 (d, 1H, J = 8.7 Hz), 5.98 (s, 2H), 5.40 (dd, 1H, J = 8.8 Hz, 6.4 Hz), 3.92 (s, 6H), 3.90 (s, 3H), 2.48–2..57 (m, 1H), 1.08 (t, 6H, J = 6.9 Hz). m/z (HRMS) 536.1478 (C25H27N3O7SNa requires: 536.1467, [M + Na]+). HPLC tR = 3.5 min, purity 99%.
N-{1-[4-(6,7-Dimethoxy-3,4-dihydro-1H-isoquinoline-2-carbonyl)thiazol-2-yl]-(S)-2-methylpropyl}-3,4,5-trimethoxybenzamide (23)
Compounds 16 (0.1 g, 0.25 mmol) and 23a (0.058 g, 0.30 mmol) were reacted together as per method A to obtain compound 23 as a light-yellow solid (0.116 g, 81%); mp 62–64 °C; Rf = 0.30 (EtOAc/n-hexane 3:2). 1H NMR (400 MHz; CDCl3; TMS) δ 7.83 (s, 1H), 7.06 (s, 2H), 6.90 (d, 1H, J = 8.64), 6.65 (s, 1H), 6.59 (s, 1H), 5.44–5.48 (m, 1H), 4.80–4.88 (m, 2H), 3.89 (s, 9H), 3.86 (s, 6H), 2.83–2.88 (m, 2H), 2.46–2.52 (m, 1H), 1.8 (s, 2H), 1.03–1.07 (m, 6H). m/z (ESI-MS) 592.17 (C29H35N3O7SNa requires 592.22, [M + Na]+). HPLC tR = 2.8 min, purity 100%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid Indan-2-ylamide (24)
Compounds 16 (0.1 g, 0.25 mmol) and 24a (0.040 g, 0.30 mmol) were reacted together following method A to obtain compound 24 as a light-yellow solid (0.117 g, 91%); mp 74–78 °C; Rf = 0.50 (EtOAc/n-hexane 3:2). 1H NMR (400 MHz; CDCl3; TMS) δ 8.04 (s, 1H), 7.40 (d, 1H, J = 7.84), 7.17–7.24 (m, 4H), 6.99 (s, 2H), 6.55 (d, 1H, J = 8.64 Hz), 5.32 (dd, 1H, J = 8.9 Hz, 6.6 Hz), 4.89–4.95 (m, 1H), 3.89 (s, 6H), 3.88 (s, 3H), 3.46 (m, 2H), 2.98 (m, 2H), 2.49 (m, 1H), 1.04 (d, 3H, J = 6.76 Hz), 1.04 (d, 3H, J = 6.68 Hz). m/z (HRMS) 532.1893 (C27H31N3O5SNa requires: 532.1882, [M + Na]+). HPLC tR = 4.1 min, purity 99%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid (2-Pyridin-2-yl-ethyl)amide (25)
Compounds 16 (0.1 g, 0.25 mmol) and 25a (0.037 g, 0.30 mmol) were reacted together by using method A to obtain compound 25 as a light-yellow oil (0.119 g, 94%); Rf = 0.20 (EtOAc/n-hexane 3:2). 1H NMR (400 MHz; CDCl3; TMS) δ 8.40 (s, 1H), 8.14 (s, 1H), 7.92 (d, 1H, 10.3 Hz), 7.59 (td, 1H, J = 7.7 Hz, 6.4 Hz), 7.38 (d, 1H, J = 8.1 Hz), 7.14–7.18 (m, 3H), 7.08–7.11 (m, 1H), 5.35–5.39 (m, 1H) 3.87–3.89 (m, 9H), 3.77–3.82 (m, 2H), 3.06 (t, 2H, J = 6.4 Hz), 2.43–2.51 (m, 1H), 1.07 (d, 3H, J = 6.8 Hz), 1.02 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 499.42 (C25H31N4O5S requires 499.19, [M + H]+). HPLC tR = 2.2 min, purity 99%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid (Biphenyl-4-ylmethyl)amide (26)
Compounds 16 (0.1 g, 0.25 mmol) and 26a (0.055 g, 0.30 mmol) were reacted together following method A to obtain compound 26 as a white solid (0.120 g, 85%); mp 70–74 °C; Rf = 0.50 (EtOAc/n-hexane 3:2). 1H NMR (400 MHz; CDCl3; TMS) δ 8.11 (s, 1H), 7.57–7.60 (m, 5H), 7.42–7.47 (m, 4H), 7.38 (d, 1H, J = 7.4 Hz), 7.02 (s, 2H), 6.66 (d, 1H, J = 7.6 Hz), 5.34–5.38 (m, 1H), 4.67–4.69 (m, 2H), 3.88 (s, 9H), 2.41–2.49 (m, 1H), 1.01–1.05 (m, 6H). m/z (ESI-MS) 560.17 (C31H34N3O5S requires 560.21, [M + H]+). HPLC tR = 6.3 min, purity 100%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid (4-Benzoylphenyl)amide (27)
Compounds 16 (0.1 g, 0.25 mmol) and 27a (0.060 g, 0.30 mmol) were reacted together following method A to obtain compound 27 as a white solid (0.118 g, 81%); mp 72–76 °C; Rf = 0.50 (EtOAc/n-hexane 3:2). 1H NMR (400 MHz; DMSO-d6) δ 10.49 (s, 1H), 9.00 (d, 1H, J = 8.4 Hz), 8.46 (s, 1H), 8.06 (d, 2H, J = 8.7 Hz), 7.81 (d, 2H, J = 8.6 Hz), 7.75 (d, 2H, J = 7.1 Hz), 7.65–7.70 (m, 1H), 7.57 (t, 2H, J = 7.6 Hz), 7.24 (s, 2H), 5.24 (t, 1H, J = 8.2 Hz), 3.85 (s, 6H), 3.71 (s, 3H), 2.38–2.46 (m, 1H), 1.08 (d, 3H, J = 6.6 Hz), 0.98 (d, 3H, J = 6.7 Hz). m/z (ESI-MS) 574.17 (C31H32N3O6S requires 574.19, [M + H]+). HPLC tR = 6.8 min, purity 96%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid (2-Benzoylphenyl)amide (28)
Compounds 16 (0.1 g, 0.25 mmol) and 28a (0.060 g, 0.30 mmol) were reacted together using method A to obtain compound 28 as a light-yellow solid (0.097 g, 67%); mp 78–82 °C; Rf = 0.45 (EtOAc/n-hexane 3:2). 1H NMR (400 MHz; DMSO-d6) δ 11.73 (s, 1H), 9.02 (d, 1H, J = 7.9 Hz), 8.52 (d, 1H, J = 8.5 Hz), 8.39 (s, 1H), 7.65–7.73 (m, 4H), 7.55 (t, 3H, J = 7.7 Hz), 7.24–7.29 (m, 3H), 5.17 (t, 1H, J = 8.3 Hz), 3.83 (s, 6H), 3.71 (s, 3H), 2.38–2.46 (m, 1H), 1.09 (d, 3H, J = 6.7 Hz), 1.02 (d, 3H, J = 6.7 Hz). m/z (HRMS) 596.1851 (C31H31N3O6SNa requires: 596.1831, [M + Na]+). HPLC tR = 9.4 min, purity 96%.
2-[(S)-2-Methyl-1-(3,4,5-trimethoxybenzoylamino)propyl]thiazole-4-carboxylic Acid [2-(4-Fluorobenzoyl)phenyl]amide (29)
Compounds 16 (0.1 g, 0.25 mmol) and 29a (0.065 g, 0.30 mmol) were reacted together following method A to obtain compound 29 as a light-yellow solid (0.095 g, 64%); mp 90–94 °C; Rf = 0.40 (EtOAc/n-hexane 3:2). 1H NMR (400 MHz; CDCl3; TMS) δ 12.29 (s, 1H), 8.81 (d, 1H, J = 8.2 Hz), 8.20 (s, 1H), 7.63–7.68 (m, 3H), 7.57 (d, 1H, J = 7.1 Hz), 7.50 (d, 1H, J = 8.8 Hz), 7.29 (s, 1H), 7.20 (s, 2H), 7.15 (t, 2H, J = 8.2 Hz), 5.51 (dt, 1H, J = 6.2 Hz), 3.91 (s, 3H), 3.85 (s, 6H), 2.55–2.47 (m, 1H), 1.11 (d, 3H, J = 6.8 Hz), 1.06 (d, 3H, J = 6.7 Hz). m/z (HRMS) 614.1736 (C31H30FN3O6SNa requires: 614.1737, [M + Na]+). HPLC tR = 9.5 min, purity 100%.
Biological Procedures
Chemicals
Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, and trypsin 0.25% were products of Hyclone, Thermo Scientific (Logan, UT). Phosphate buffered saline (PBS) 20× concentrate (PH 7.5) was purchased from AMRESCO (Solon, OH). Paclitaxel, cyclosporine A, paraformaldehyde, 3-(4,5-dimethylthiazol-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), and other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO). [125I]-Iodoarylazidoprazosin (2200 Ci/mmol) was obtained from PerkinElmer Life Sciences (Wellesley, MA). OPSYS microplate reader was purchased from Dynex Technologies (Chantilly, VA).
Cell Lines and Cell Culture
HeLa cells were cultured in DMEM media supplemented with 10% FBS, 1% glutamine, and 1% penicillin. The human colon cancer cell line SW620 and its doxorubicin-selected P-gp-overexpressing subline SW620/Ad30031 were cultured at 37 °C, 5% CO2, with DMEM containing 10% FBS and 1% penicillin/streptomycin. The human embryo kidney parental cell line HEK293 and HEK/ABCB1 transfected with human ABCB1 cDNA were maintained in the DMEM with G418 (2 mg/mL) and cultured in a manner similar to that of the above cell lines. All cells were grown as adherent monolayers in drug-free culture media for more than 2 weeks before the assay.
Transduction of HeLa Cells with BacMam-P-gp Baculovirus
HeLa cells were transduced with BacMam WT-P-gp virus, which was added at a titer of 10–15 viral particles per cell.32 After an hour, DMEM medium was added and the cells were incubated further. Then 10 mM butyric acid was added after 3–4 h, and the cells were grown overnight at 37 °C. The cells were trypsinized, washed, counted, and analyzed by flow cytometry for inhibition of transport function of WT P-gp by the derivatives.
Calcein-AM Efflux Assay
The ability of the synthesized derivatives to inhibit the transport function of P-gp was checked using flow cytometry as described previously.33,34 Briefly, the transfected cells were trypsinized and incubated with various concentrations of the derivatives followed by calcein-AM (0.5 μM).32 The cells were analyzed after washing with cold PBS. Fluorescence produced by calcein was measured on a FACSort flow cytometer equipped with a 488 nm argon laser and 530 nm bandpass filter. The results are reported as an average of two independent experiments, each done in triplicates. The IC50 of these derivatives was calculated using GraphPad Prism 5.0.
Cytotoxicity Determination by MTT Assay
We used a modified MTT colorimetric assay to detect the sensitivity of cells to anticancer drugs in vitro.35 Briefly, cells were seeded in 180 μL of medium in 96-well plates in triplicate at 5000–6000 cells/well and incubated at 37 °C, 5% CO2, for 24 h to allow the cells to attach to the wells. Cells in 96-well plates were preincubated with or without the reversal agents (20 μL/well) for 2 h, and then different concentrations of chemotherapeutic drugs (20 μL/well) were added into designated wells. After 72 h of incubation at 37 °C, 20 μL of MTT solution (4 mg/mL) was added to each well. The plates were further incubated at 37 °C for 4 h, allowing viable cells to change the yellow-colored MTT into dark-blue formazan crystals. Subsequently, the MTT/medium was removed from each well without disturbing the cells, and 100 μL of DMSO was added into each well. Plates were placed on a shaking table to thoroughly mix the formazan into the solvent. Finally, the absorbance was determined at 570 nm by Opsys microplate reader (Dynex Technologies, Chantilly, VA). The MTT assays were performed four times independently, and each independent experiment was done in triplicate.
Photoaffinity Labeling of ABCB1 with [125I]-Iodoarylazidoprazosin
ABCB1-transfected High Five insect cell membranes expressing ABCB1 (P-gp) (65 μg protein/100 μL) were incubated with varying concentrations of compound 28 (0–20 μM) for 10 min at 37 °C in 50 mM MES-Tris pH 6.8. [125I]-IAAP (2200 Ci/mmol; 4–6 nM) was added and membranes incubated for an additional 5 min with minimal exposure to light. The samples were then illuminated with a 365 nm UV lamp for 10 min and photoaffinity labeling of P-gp with [125I]-IAAP was determined as previously described.36 Results from two independent experiments are reported (Figure 4A).
ATP Hydrolysis by P-gp
The vanadate-sensitive ATPase activity of P-gp in crude membranes of High Five insect cells, in the presence of concentrations of compound 28 ranging from 0 to 10 μM, was measured as previously described.37 Three independent experiments in duplicates were carried out and results are reported as mean ± SEM (Figure 4B).
Molecular Modeling
Ligand Preparation
The structures of the linear thiazole derivatives were built using the fragment dictionary of Maestro v9.5 and energy minimized by Macromodel program v10.1 (Schrödinger, LLC, New York, NY, 2013). LigPrep v2.7 tool was used to generate low energy 3D conformers of the minimized structures. Default settings were used, but the “Generate Tautomers” option was not selected. The resultant ligand structures were eventually docked at all four grids (site-1 to site-4) generated on homology-modeled human P-gp.
Homology Modeling
The refined crystal structure of mouse P-gp in complex with compounds 1c (PDB ID: 4M2T)8 and 1d (PDB ID: 4M2S)8 served as templates to generate ligand-bound homology models of human P-gp. The alignment of human P-gp and mouse P-gp sequences resulted in 87% sequence identity and 93% similarity. The protocol for homology modeling using the default parameters of Prime v3.3 implemented in Maestro v9.5 (Schrödinger, LLC, New York, NY, 2013) is essentially the same as reported earlier.29 Validation of the generated homology models was performed using Ramachandran plot analysis, which suggested more than 91% residues in the core allowed region, 5–8% residues in the allowed regions, and <0.7% residues in the sterically disallowed regions. The backbone root-mean-square deviation (RMSD) was calculated for the homology models from the corresponding experimental structures. The RMSD was found to be less than 0.33 Å for all of the generated models, which is not surprising considering that mouse P-gp and human P-gp share high (93%) sequence similarity. The homology modeled human P-gp generated from the mouse model in complex with 1c and 1d were used as templates for generating grids for site-2 and site-1, respectively. The human P-gp homology model based on mouse P-gp in apoprotein state was generously provided by Dr. S. Aller and was used to generate grids for site-3 and site-4.
Protein Preparation and Grid Generation
All homology models were refined by default parameters in Protein Preparation Wizard implemented in Maestro v9.5 and Impact program v6.0 (Schrödinger, LLC, New York, NY, 2013), in which the protonation states of the ionizable residues were adjusted to the dominant ionic forms at physiological pH. On the basis of refined human P-gp homology model, different receptor grids were generated by selecting 1c (site-2) and 1d (site-1) bound ligands, all amino acid residues that are known to contribute to verapamil binding (site-3), and two residues (Phe728 and Val982) common in the previous three sites (site-4).
Docking Protocol
To determine the probable binding site for compound 28, the LigPrep derived ligand structure was docked at each of the generated grids (site-1 to site-4) of P-gp using the “Extra Precision” (XP) mode of Glide program v6.0 (Schrödinger, LLC, New York, NY, 2013) with the default parameters. The analysis based on the glide scores revealed site-1 as the preferred site of binding for these ligands. For appropriate prediction of the binding conformation of compound 28 at site-1, induced-fit docking was carried out using the default parameters in the protocol “IFD” implemented in Schrödinger Suite 2013-2. The refined human P-gp homology model generated from 1d bound mouse P-gp and the LigPrep-derived ligand structure for compound 28 was used as input for grid generation and ligands to be docked, respectively. In this, the centroid of the ligand in the refined protein structure was selected to generate grid. The top scoring conformation of 28 at site-1 of human P-gp was used for graphical analysis. Computational work was carried out on a Dell Precision 490n dual processor with the Linux OS (Ubuntu 12.04 LTS).
Acknowledgments
This research was supported by the Department of Pharmaceutical Sciences of St. John’s University and St. John’s University Seed Grant no. 579-1110 to T.T.T. Drs. N.R.P., E.E.C., and S.V.A. were supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. Financial support from Indian Council of Medical Research, New Delhi, in the form of International Fellowship for Young Indian Biomedical Scientists to Dr. N.R.P. is gratefully acknowledged (Indo/FRC/452(Y-19)/2012-13IHD). We acknowledge Drs. Susan E. Bates and Robert W. Robey (NCI, NIH, Bethesda, MD) for providing SW620 and SW620/Ad300 cell lines. We thank Dr. Stephen Aller (The University of Alabama at Birmingham, Birmingham, AL) for generously providing human apo-state P-gp homology model. We thank George Leiman for editorial assistance. S.S. and T.T.T. are thankful to Dr. Sanjai Kumar and Dibyendu Dana (Queens College—CUNY, Queens, NY) for assisting in ESI-MS and HRMS analyses.
Glossary
Abbreviations Used
- ABC
ATP-binding cassette
- calcein-AM
calcein acetoxymethylester
- HCTU
O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HOBt
1-hydroxybenzotriazole
- IAAP
iodoarylazidoprazosin
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- MDR
multidrug resistance
- MES
2-(N-morpholino)ethanesulfonic acid
- P-gp
P-glycoprotein
- TM
transmembrane
Author Present Address
§ For N.R.P.: Department of Biochemistry and Biotechnology, Annamalai University, Annamalainagar 608 002, Tamilnadu, India.
Author Contributions
The manuscript was written through contributions of all the authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Gottesman M. M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [DOI] [PubMed] [Google Scholar]
- Gottesman M. M.; Fojo T.; Bates S. E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nature Rev. Cancer 2002, 2, 48–58. [DOI] [PubMed] [Google Scholar]
- Ambudkar S. V.; Dey S.; Hrycyna C. A.; Ramachandra M.; Pastan I.; Gottesman M. M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [DOI] [PubMed] [Google Scholar]
- Ernst R.; Kueppers P.; Stindt J.; Kuchler K.; Schmitt L. Multidrug efflux pumps: substrate selection in ATP-binding cassette multidrug efflux pumps—first come, first served?. FEBS J. 2010, 277, 540–549. [DOI] [PubMed] [Google Scholar]
- Dey S.; Ramachandra M.; Pastan I.; Gottesman M. M.; Ambudkar S. V. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 10594–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski S.; Mir L. M.; Belehradek J. Jr; Garrigos M. Effects of steroids and verapamil on P-glycoprotein ATPase activity: progesterone, desoxycorticosterone, corticosterone and verapamil are mutually non-exclusive modulators. Biochem. J. 1996, 317Pt 2515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins C. F. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007, 446, 749–757. [DOI] [PubMed] [Google Scholar]
- Li J.; Jaimes K. F.; Aller S. G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert J.; Jarry C. Multidrug resistance reversal agents. J. Med. Chem. 2003, 46, 4805–4817. [DOI] [PubMed] [Google Scholar]
- Wandel C.; Kim R. B.; Kajiji S.; Guengerich P.; Wilkinson G. R.; Wood A. J. P-glycoprotein and cytochrome P-450 3A inhibition: dissociation of inhibitory potencies. Cancer Res. 1999, 59, 3944–3948. [PubMed] [Google Scholar]
- Bohme M.; Buchler M.; Muller M.; Keppler D. Differential inhibition by cyclosporins of primary-active ATP-dependent transporters in the hepatocyte canalicular membrane. FEBS Lett. 1993, 333, 193–196. [DOI] [PubMed] [Google Scholar]
- Shukla S.; Ohnuma S.; Ambudkar S. V. Improving cancer chemotherapy with modulators of ABC drug transporters. Curr. Drug Targets 2011, 12, 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; Zhuo R.; Harrell P. M.; Trinh Y. T.; Zhang Q.; Urbatsch I. L.; Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H.; Weng Y.; Zhuo R.; Chang G.; Urbatsch I. L.; Zhang Q. Design and synthesis of Selenazole-containing peptides for cocrystallization with P-glycoprotein. ChemBioChem 2011, 12, 868–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley M. C.; Dale J. W.; Merritt E. A.; Xiong X. Thiopeptide antibiotics. Chem. Rev. 2005, 105, 685–714. [DOI] [PubMed] [Google Scholar]
- Singh S.; Prasad N. R.; Kapoor K.; Chufan E. E.; Patel B. A.; Ambudkar S. V.; Talele T. T. Design, Synthesis, and Biological Evaluation of (S)-Valine Thiazole-Derived Cyclic and Noncyclic Peptidomimetic Oligomers as Modulators of Human P-Glycoprotein (ABCB1). ChemBioChem 2014, 15, 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly R. J.; Draper D.; Chen C. C.; Robey R. W.; Figg W. D.; Piekarz R. L.; Chen X.; Gardner E. R.; Balis F. M.; Venkatesan A. M.; Steinberg S. M.; Fojo T.; Bates S. E. A Pharmacodynamic Study of Docetaxel in Combination with the P-glycoprotein Antagonist Tariquidar (XR9576) in Patients with Lung, Ovarian, and Cervical Cancer. Clin. Cancer Res. 2011, 17, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardelmeijer H. A.; Beijnen J. H.; Brouwer K. R.; Rosing H.; Nooijen W. J.; Schellens J. H.; van Tellingen O. Increased oral bioavailability of paclitaxel by GF120918 in mice through selective modulation of P-glycoprotein. Clin. Cancer Res. 2000, 6, 4416–4421. [PubMed] [Google Scholar]
- Norman B. H.; Gruber J. M.; Hollinshead S. P.; Wilson J. W.; Starling J. J.; Law K. L.; Self T. D.; Tabas L. B.; Williams D. C.; Paul D. C.; Wagner M. M.; Dantzig A. H. Tricyclic isoxazoles are novel inhibitors of the multidrug resistance protein (MRP1). Bioorg. Med. Chem. Lett. 2002, 12, 883–886. [DOI] [PubMed] [Google Scholar]
- Pearce H. L.; Safa A. R.; Bach N. J.; Winter M. A.; Cirtain M. C.; Beck W. T. Essential features of the P-glycoprotein pharmacophore as defined by a series of reserpine analogs that modulate multidrug resistance. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 5128–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry P.; Plumb J.; Eccles S.; Watson S.; Dale I.; Ryder H.; Box G.; Charlton P.; Templeton D.; Bevan P. B. In vivo efficacy of XR9051, a potent modulator of P-glycoprotein mediated multidrug resistance. Br. J. Cancer 1999, 79, 1672–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K. J.; He J. H.; Su X. D.; Sim H. M.; Xie J. D.; Chen X. G.; Wang F.; Liang Y. J.; Singh S.; Sodani K.; Talele T. T.; Ambudkar S. V.; Chen Z. S.; Wu H. Y.; Fu L. W. Saracatinib (AZD0530) is a potent modulator of ABCB1-mediated multidrug resistance in vitro and in vivo. Int. J. Cancer 2013, 132, 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicani R. Z.; Stefanachi A.; Niso M.; Carotti A.; Leonetti F.; Nicolotti O.; Perrone R.; Berardi F.; Cellamare S.; Colabufo N. A. Potent galloyl-based selective modulators targeting multidrug resistance associated protein 1 and P-glycoprotein. J. Med. Chem. 2012, 55, 424–436. [DOI] [PubMed] [Google Scholar]
- Jabeen I.; Pleban K.; Rinner U.; Chiba P.; Ecker G. F. Structure–activity relationships, ligand efficiency, and lipophilic efficiency profiles of benzophenone-type inhibitors of the multidrug transporter P-glycoprotein. J. Med. Chem. 2012, 55, 3261–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didziapetris R.; Japertas P.; Avdeef A.; Petrauskas A. Classification analysis of P-glycoprotein substrate specificity. J. Drug Target 2003, 11, 391–406. [DOI] [PubMed] [Google Scholar]
- Klopman G.; Shi L. M.; Ramu A. Quantitative structure–activity relationship of multidrug resistance reversal agents. Mol. Pharmacol. 1997, 52, 323–334. [DOI] [PubMed] [Google Scholar]
- Crivori P.; Reinach B.; Pezzetta D.; Poggesi I. Computational models for identifying potential P-glycoprotein substrates and inhibitors. Mol. Pharmaceutics 2006, 3, 33–44. [DOI] [PubMed] [Google Scholar]
- Pajeva I. K.; Globisch C.; Wiese M. Combined pharmacophore modeling, docking, and 3D QSAR studies of ABCB1 and ABCC1 transporter inhibitors. ChemMedChem 2009, 4, 1883–1896. [DOI] [PubMed] [Google Scholar]
- Shi Z.; Tiwari A. K.; Shukla S.; Robey R. W.; Singh S.; Kim I. W.; Bates S. E.; Peng X.; Abraham I.; Ambudkar S. V.; Talele T. T.; Fu L. W.; Chen Z. S. Sildenafil reverses ABCB1- and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. 2011, 71, 3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T. W.; Bartlett M. C.; Clarke D. M. Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2003, 278, 39706–39710. [DOI] [PubMed] [Google Scholar]
- Bates S. E.; Lee J. S.; Dickstein B.; Spolyar M.; Fojo A. T. Differential modulation of P-glycoprotein transport by protein kinase inhibition. Biochemistry 1993, 32, 9156–9164. [DOI] [PubMed] [Google Scholar]
- Shukla S.; Schwartz C.; Kapoor K.; Kouanda A.; Ambudkar S. V. Use of baculovirus BacMam vectors for expression of ABC drug transporters in mammalian cells. Drug Metab. Dispos. 2012, 40, 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiberghien F.; Loor F. Ranking of P-glycoprotein substrates and inhibitors by a calcein-AM fluorometry screening assay. Anticancer Drugs 1996, 7, 568–578. [DOI] [PubMed] [Google Scholar]
- Robey R. W.; Steadman K.; Polgar O.; Morisaki K.; Blayney M.; Mistry P.; Bates S. E. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004, 64, 1242–1246. [DOI] [PubMed] [Google Scholar]
- Shi Z.; Peng X. X.; Kim I. W.; Shukla S.; Si Q. S.; Robey R. W.; Bates S. E.; Shen T.; Ashby C. R. Jr.; Fu L. W.; Ambudkar S. V.; Chen Z. S. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007, 67, 11012–11020. [DOI] [PubMed] [Google Scholar]
- Sauna Z. E.; Ambudkar S. V. Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar S. V. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 1998, 292, 504–514. [DOI] [PubMed] [Google Scholar]