

Abstract
The peculiar biology of mitochondrial DNA (mtDNA) potentially has detrimental consequences for organismal health and lifespan. Typically, eukaryotic cells contain multiple mitochondria, each with multiple mtDNA genomes. The high copy number of mtDNA implies that selection on mtDNA functionality is relaxed. Furthermore, because mtDNA replication is not strictly regulated, within-cell selection may favour mtDNA variants with a replication advantage, but a deleterious effect on cell fitness. The opportunities for selfish mtDNA mutations to spread are restricted by various organism-level adaptations, such as uniparental transmission, germline mtDNA bottlenecks, germline selection and, during somatic growth, regular alternation between fusion and fission of mitochondria. These mechanisms are all hypothesized to maintain functional mtDNA. However, the strength of selection for maintenance of functional mtDNA progressively declines with age, resulting in age-related diseases. Furthermore, organismal adaptations that most probably evolved to restrict the opportunities for selfish mtDNA create secondary problems. Owing to predominantly maternal mtDNA transmission, recombination among mtDNA from different individuals is highly restricted or absent, reducing the scope for repair. Moreover, maternal inheritance precludes selection against mtDNA variants with male-specific effects. We finish by discussing the consequences of life-history differences among taxa with respect to mtDNA evolution and make a case for the use of microorganisms to experimentally manipulate levels of selection.
Keywords: mitochondria, mitochondrial DNA, evolution, genetic conflict, mitochondrial disease
1. Introduction
Mitochondria are membrane-enclosed organelles, responsible for bioenergy (adenosine triphosphate; ATP) production of eukaryotic cells. Because mitochondria cannot be synthesized de novo, they must originate from pre-existing ones, and they must be passed on from one generation to the next [1]. Ultimately, all extant mitochondrial lineages go back to an ancient endosymbiosis of an α-protobacterium into an archaebacterial host [2,3]. A small mitochondrial genome separate from the nuclear genome still testifies to the endosymbiotic origin of mitochondria. During the course of evolution, particularly in animals, most of the original α-protobacterial genes have either been lost, because their function was also coded for by nuclear genes, or transferred to the nucleus [4]. In animals, the only remaining subset of the genes on the circular mitochondrial genome (the mtDNA) encode core protein subunits of the oxidative phosphorylation system plus the ribosomal RNAs and tRNAs needed for the translation of the corresponding mRNAs [4]. The conventional view therefore is that the ancestor of the mitochondrion has been enslaved by its host cell and that the two genomes, that of the mitochondrion and that of the cell's nucleus, work in perfect harmony.
The reduction of the mitochondrial genome is consistent with the view that mitochondria are enslaved entities whose presence is only beneficial to the cell. But this argument can also be turned around. Given that each cell contains multiple copies of mitochondrial DNA (mtDNA) and mtDNA replicates independent of the cell cycle, it seems probably that shorter mtDNA molecules have a replication advantage. One could thus see the transmission of large parts of the mitochondrial genome into the nucleus as a strategy of selfish mtDNA to increase its ability to copy itself. Furthermore, under some circumstances, the multi-copy mtDNA molecules pursue their short-term selfish interests, at the cost of cell fitness. One of the best-characterized examples is the petite mutant found in yeast [5,6]. With a frequency of about 1%, yeast cells mutate to form mini colonies containing far fewer cells than the wild-type. The petite phenotype is caused by mtDNA mutations, giving a replication benefit to the mutated mtDNA, so that the mutated mtDNA molecules increase in frequency within the cell. Although yeast can meet their energetic needs by fermentation alone, respiration yields more energy. Therefore, mutated mtDNA molecules impose a cost to cell fitness and petite strains are selected against in competition with wild-type cells. Similarly, in animals in particular, including humans, selection at the level of mtDNA within cells can be detrimental to organismal fitness and contribute to ageing and several diseases [7]. Furthermore, mtDNA has a high mutation rate, possibly owing to its close proximity to the respiratory chains, which are the main cellular source of reactive oxygen species (ROS), which are highly mutagenic [8]. ROS production is particularly high during fatty acid metabolism, leading Speijer et al. [9] to hypothesize that protection against ROS-induced damage has been a major factor driving the evolution of peroxisomes. Lastly, because mtDNA is haploid and often non-recombining, the scope for repair is limited.
Negative effects caused by selection on mtDNA at the same time select for mechanisms at the level of the host cell to prevent or circumvent such effects. What are the mechanisms that maintain functional harmony between mitochondrial and nuclear genomes [10]? Here, we give a short overview of the levels of selection affecting mtDNA, identify potential conflicts between different levels of selection, and determine how conflicts are resolved or prevented. This article serves as the introduction to the special issue called ‘What cost mitochondria?’ in which these and other topics are discussed in greater detail. This issue broadly focuses on three aspects of mitochondrial biology: (i) mechanisms of mitochondrial functioning; (ii) theoretical and empirical studies on the evolution of mitochondria; and (iii) the application of evolutionary insights on the effect of mitochondria on health and disease. We hope that the combination of evolutionary biology and medical science will lead to better predictions regarding the role mitochondria play in certain diseases and ageing.
2. Mitochondrial DNA is exposed to natural selection at multiple hierarchical levels
Eukaryotic cells typically contain multiple mitochondria, and each mitochondrion contains multiple copies of mtDNA. For example, most human cell types contain 103–104 copies of mtDNA per cell, organized in a dynamic network of hundreds of fusing and dividing mitochondria [11,12]. The high copy number of mtDNA has two consequences. First, the efficiency of selection against mutations that negatively affect mitochondrial function is reduced [7]. As long as there are functional mtDNA copies, deleterious mtDNA mutations are sheltered from purifying selection. Typically, only when the copy number of mtDNA with deleterious mutations passes a critical threshold, and this threshold may differ between mutations and tissue type, will the cell suffer a fitness cost. Second, because mtDNA replication is not linked to cell division, competition between mtDNAs within a cell may favour those that have a within-cell replication or survival advantage, even if such variants reduce cell fitness [6,13,14].
The organization of mtDNA into dynamic networks within which they exchange proteins, lipids and mtDNA through regular fusion and fission, has profound effects on the way selection acts on mtDNA [15,16]. Fusion is thought to maintain homeostasis and to equilibrate nuclear- and mtDNA-encoded mitochondrial proteins [16,17]. An implication of frequent fusion is the possible loss of a direct link between mitochondrial genotype (mtDNA mutations) and phenotype (mitochondrion-specific energy and proton-gradient deficiencies) because each mitochondrion contains multiple mitochondrial genomes [18]. Thus, while there is competition among individual mtDNA to replicate faster, by reducing genome size, at a higher level the mitochondrion itself is the unit of selection with respect to functionality. However, to qualify as a unit of selection regular fission is required, followed by selective degradation or delayed fusion of non-functional mitochondria, to counteract the accumulation of fast-replicating mitochondrial genomes [16]. To establish a link between mitochondrial genotype and phenotype for selection to act on, Kowald & Kirkwood [16] suggested a physical attachment between mtDNA genomes and the oxidative phosphorylation complexes they encode [16,18].
3. Mitochondrial evolution: soma versus germline
In single-celled organisms, such as protists, yeast and some algae, the cell and the individual are the same, reducing selection on mtDNA to three levels: among individuals, among mitochondria within the cell and among mtDNA molecules within mitochondria (figure 1). In multicellular individuals, an extra level of selection is added: selection within a multicellular individual, i.e. among cells of the same individual. Importantly, only a small fraction of the cells, and the mtDNAs they contain, form the gametes, while the rest contributes only to somatic functions. This division of labour between the germline and soma creates the potential for reproductive conflict if cells are not genetically identical. A mutant mtDNA molecule that gains preferential access to the germline can increase in frequency even if this variant reduces the overall success of the individual [19]. One way to prevent such selfish lineages from arising is to enforce a germ-soma split early in development [20]. Now nuclear and mtDNA genomes of somatic cells can no longer be transmitted to the next generation so that these genomes can only increase their fitness indirectly, by helping copies of themselves present in germ cells [21] (figure 1).
Figure 1.
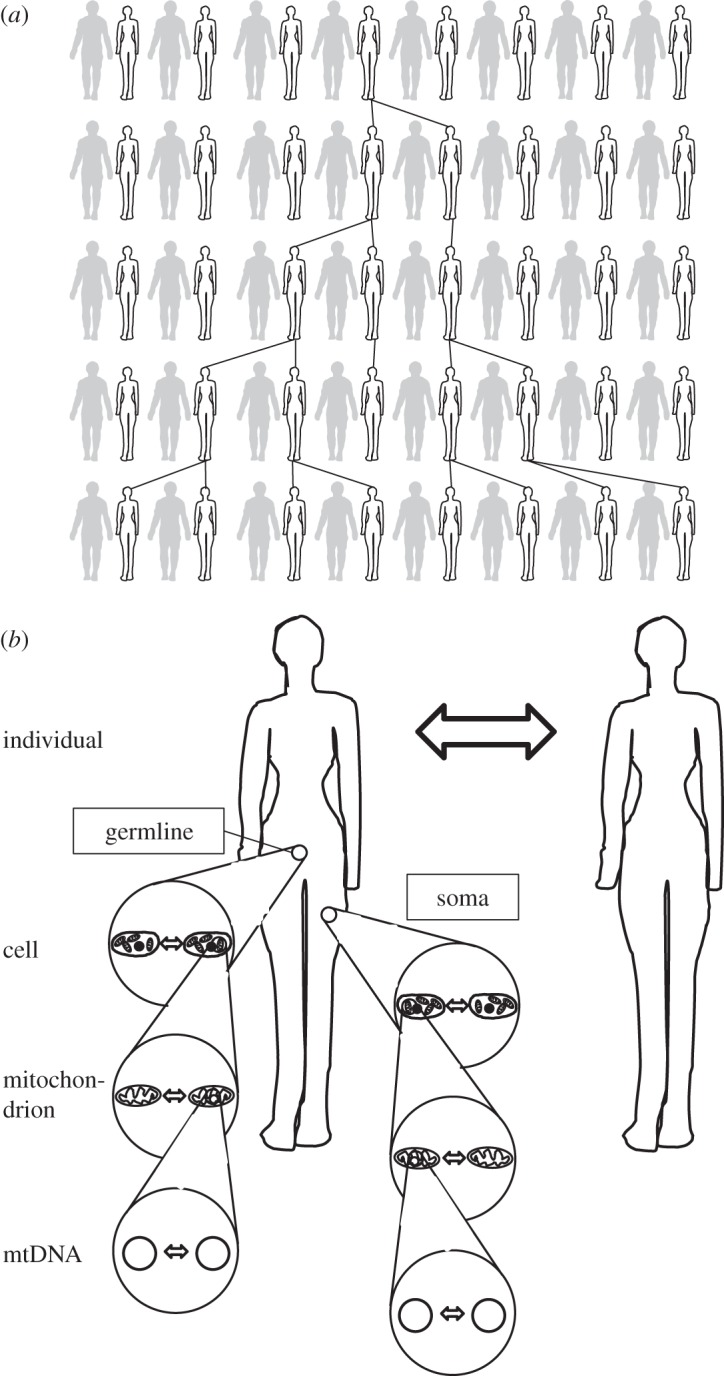
Levels of selection affecting mitochondrial evolution. (a) In the vast majority of sexually reproducing eukaryotes, mtDNA is inherited uniparentally, usually maternally. (b) Selection on mtDNA acts on two time scales: among generations, when mtDNA mutations in the germline are passed on to offspring, and within an individual, when mtDNA mutations accumulate during somatic growth. Thus, natural selection acts on mtDNA at four levels of selection: among individuals, among cells, among mitochondria within cells and among mtDNAs within mitochondria.
The lack of direct selection on somatic cells and the mitochondria they contain does not mean that the variation in mtDNA within and between somatic cells that arises through mutation, drift and among- and within-cell selection is unimportant. Selection still acts on mitochondria within the soma to ensure proper functioning of the organism, at least until reproductive age (‘disposable soma’ [22]). As the probability to reach high age is limited due to external mortality, the force of natural selection decreases with age [23]. Owing to the disposable soma, organismal adaptations to suppress the accumulation of deleterious mtDNA mutations during somatic growth may only be sufficient up to a certain age, depending on the life history of a species. As a result, the accumulation of mtDNA variants within the soma will increase with increasing age. Indeed, the accumulation of malfunctioning mitochondria contributes to diseases such as Alzheimer's and Parkinson's, the two most common age-related neurodegenerative diseases [24], and to diabetes and various cancers [25,26].
4. Transmission of mitochondrial DNA across generations
The distribution of allelic variants of nuclear genes among gametes is strictly regulated during gamete formation: a ‘fair’ meiosis guarantees that each allele has a 50% chance to enter a gamete [27]. Not so for mtDNA. During gamete formation, some variants may have an increased probability to be transmitted to the gametes, for example because they replicate faster. This is where genome size becomes important because mtDNA variants with reduced genome size can have a replication advantage [28]. However, genome size is not the only way by which mtDNA can gain a replication advantage; in yeast, petite mutants are not caused by small genome size but by tandem repeats of a fragment of the mitochondrial genome [29].
Within-cell competition will be more severe when the competing mtDNA molecules are non-clonal, simply because there will then be more variation for selection to act on. Thus, the mixing of two genetically different cytoplasms during sexual reproduction opens the door to the spread of selfish mtDNA variants [30–33]. More generally, the mixing of cytoplasm and the organelles it contains may incur a cost to the cell and thus to the organism. Such costs may be due to interactions between organelle lineages [31]. For example, the fitness of the cell may be reduced due to the destruction of genetically different cytoplasmic organelles, or because the presence of multiple organelle lineages triggers diversion of resources towards competition between them.
(a). Uniparental transmission
In the vast majority of sexually reproducing organisms, mtDNA is inherited almost exclusively via one parent, usually the mother, although some level of paternal leakage has been demonstrated in many organisms [34]. Uniparental transmission not only reduces the scope for selection for selfish mtDNA variants, but it also prevents their spread if they do arise because the fate of the mtDNA is now coupled with that of the transmitting sex. Selfish mtDNA variants can then be selected against via among-host selection if their presence negatively affects the host lineage they are in. Even if mutations are not deleterious, uniparental inheritance will prevent their spread, thus maintaining cells that contain only one mtDNA lineage [35]. Several theoretical studies have mathematically analysed the exact conditions under which uniparental transmission is expected to evolve [36–40]. An important question is if uniparental transmission provides a selected (long-term) benefit, or an immediate benefit by avoiding cytoplasmic mixing [31,32]. Most models explaining the evolution of uniparental inheritance assume that it increases the variance in mtDNA among individuals, while it decreases variation within individuals. Thus, uniparental inheritance provides the among-individual variation natural selection can act on, but it seems likely that many generations are necessary before this transmission mode goes to fixation. If, on the other hand, there are immediate benefits to restricting cytoplasmic mixing, the evolution of uniparental transmission becomes much easier to understand [31,32,36]. A recent study by Sharpley et al. [41] on mice, which contained either one or two types of mtDNA (originating from two different individuals), showed that those carrying mtDNA from two individuals suffered a severe reduction in fitness, thus providing experimental support for the hypothesis that carrying different types of mtDNA is costly.
(b). Mitochondrial DNA bottlenecks
Uniparental transmission alone is insufficient to completely remove the scope for selection within cells. The high copy number of mtDNA per cell and the high mutation rate of mtDNA leave ample opportunity for selection within cells [42]. Thus, additional mechanisms are necessary to reduce conflict among mitochondrial genomes. Probably the most important mechanism is the genetic bottleneck during egg-cell formation when only a subset of mtDNA genomes is transmitted to the oocyte [43–45]. Because of this bottleneck genetic variation among mtDNAs within oocytes is reduced, but not necessarily among oocytes. Owing to uniparental inheritance, competition on mtDNA will therefore take place at the level of the individual and not so much within the individual, facilitating the removal of deleterious mtDNA variants. Theoretical models support the idea that a germline bottleneck in mtDNA is important for the long-term maintenance of functional mtDNA [35].
(c). Germline selection of mitochondrial DNA
Both maternal transmission and a strong bottleneck during egg-cell formation facilitate selection at the level of the individual. However, uniparental clonal transmission creates a secondary problem. Owing to uniparental transmission and haploidy, mtDNA has a much smaller effective population size than nuclear DNA (nDNA; a quarter of that of nDNA; [46]), reducing the efficiency of natural selection at the level of the individual. Perhaps a counter mechanism is found in strong purifying selection that has recently been shown to occur within the female germline [4]. The exact timing of selection is unknown; selection could occur at the level of zygotes, primordial germ cells, or earlier at the level of mitochondria or mtDNA [47]. Germline selection of egg cells or zygotes with functional mtDNA could be an example of a ‘selection arena’: an individual produces an excess number of offspring and invests only in the most promising ones [48].
5. Uniparental transmission sets the stage for a new set of problems
Uniparental transmission, in combination with an mtDNA copy number bottleneck and germline selection, allows the selection of functional mtDNA molecules. But biology would not be so interesting if some of these organism-level adaptations did not set the stage for new problems. First, uniparental transmission implies the absence of recombination between genetically different mtDNA variants. The absence of recombination, in combination with the high mutation rate of mtDNA, leads to the accumulation of deleterious mutations in a ‘Muller's ratchet’ [49]. Germline selection against deleterious mtDNA mutations will counteract or at least slow down this process. Second, if cytoplasmic elements are transmitted by only one sex, all that matters for their evolution is their effect on the fitness of that sex. Thus, if transmission occurs only via females, mutations harmful to males will not be selected against, if they are neutral to females, or even be selected for if they are beneficial to females (‘Mother's Curse’; [50,51]). Empirical evidence for ‘Mother's Curse’ comes from fruit flies in which mtDNA mutations have a disproportional effect on gene expression of males compared with females [52] leading to faster ageing of male flies [53]. In humans, a relatively common mitochondrial haplotype is associated with reduced sperm motility (asthenozoospermia) [54]. As females do not produce sperm, there is no equivalent trait in females for selection to act on. Similar detrimental consequences of uniparental inheritance are found in flowering plants, including many agricultural crops (reviewed in [55]) and fungi [56]. Often, mutations that are only detrimental to one sex are hidden, as natural selection will favour compensatory mutations in the nuclear genome [51,57–59].
6. Coevolution between nuclear and mitochondrial DNA
So far, we have treated mtDNA and nDNA as two genomes engaged in a tug of war. Whereas natural selection is predicted to favour mtDNA variants with increased transmission rates even if they impose a cost to the cell and organism, nuclear genes will be selected to counteract the negative consequences of selfish mtDNA. It is therefore unlikely that nDNA is a passive bystander, allowing mtDNA to pursue its selfish interests. In addition, cell function requires a high level of cooperation between the two genomes. The transfer of most of the mitochondrial genes to the nuclear genome means that mitochondrial function (ATP production via oxidative phosphorylation) depends on the concerted interaction between these two genomes. Thus, even though mtDNA mutations with deleterious effects on the organism can rise to high frequencies, these deleterious mtDNA mutations will exert strong selection for compensatory mutations at interacting nuclear loci. Nuclear genes most probably contribute to adaptive compensatory mutations that maintain co-adapted states between mtDNA and nDNA [57–59]. In addition to avoiding selection for selfish mtDNA, uniparental inheritance of mitochondria facilitates coevolution between mtDNA and nDNA [60]. A myriad of studies have shown adverse effects of the break-up of co-evolved mtDNA and nDNA lineages (reviewed in [61]), indicating that the two genomes indeed co-evolve despite experiencing divergent selection pressures.
7. Mitochondrial DNA in health and disease
The year 1988 saw the first discovery of disease-associated mtDNA mutations [62–64]. This discovery not only paved the way for the identification of numerous additional mtDNA disease mutations, and more widely to diseases associated with oxidative phosphorylation defects, it also spearheaded the study of mtDNA genetics. These studies were, and still are, driven by two main aims: the desire to understand mtDNA inheritance, somatic segregation and tissue specificity of the effects of mtDNA mutations, and the prospect of ultimately being able to predict levels of mutant mtDNA and the prevention of disease.
Levels of selection are essential in understanding mtDNA disease genetics. For heritable mitochondrial diseases, we need to understand how selection within the germline affects the transmission of mtDNA mutations from one generation to the next. For functional decline linked to the accumulation of faulty mitochondria in somatic cells (e.g. in ageing), we need to understand how selection within mitochondrial fission–fusion networks and among somatic cells can act against mtDNA mutations. Here, we will highlight a few recent advances in our understanding of mtDNA genetics in relation to human disease and ageing, linking where possible to some of the papers in this special issue.
In many instances, deleterious mtDNA mutations are selected against during germline development and, as a result, are poorly transmitted to the next generation. Point mutations, such as the MELAS 3243 tRNALeu(UUR) A–G transition [65–67] and the MERFF 8344 tRNALys A–G [68,69] are transmitted maternally, as predicted. This is not necessarily the case for single large-scale deletions [67,70,71]. In addition, deleterious mtDNA variants can be selected against during oocyte development, at least in mice [4,72]. Interestingly both in mice and humans, disease-causing tRNA mutations are frequently transmitted from mother to offspring indicating that they often escape a putative selective sieve (as discussed in [4]). By contrast, missense mutations (non-synonymous point mutations) in the 13 protein-coding mitochondrial genes are less frequently transmitted to offspring, suggesting the selective sieve is effective against certain mutations but not others. The nature and mechanism of the selective sieve is currently unknown. It could be that selection acts on properties of mtDNA molecules that carry the mutation, for example, via selective replication or segregation. However, it is more probably that selection acts on the phenotypic expression of the mutants during germline development.
In this issue, Bush et al. [18] suggest a possible explanation for the difference in transmission between mutations in tRNA and missense mutations in protein coding genes. They suggest, in the context of somatic mtDNA mutations, that tRNA molecules are more mobile thus allowing wild-type and mutant tRNAs to mix resulting in wild-type tRNAs functionally complementing mutant tRNAs. By contrast, missense mtDNA mutations might result in local respiratory defects within the mitochondrial network and elicit a degradation response that in the process also destroys the mutant mtDNA [18].
In general, both mitochondrial disease and ageing are linked to the accumulation of mutations in mtDNA. To complicate matters, the accumulation of mutations is not uniform across tissues. Different tissues within an organism can contain multiple mtDNA variants (heteroplasmy), and different variants can be transmitted from one generation to the next [73–76]. In order to understand the causes of mtDNA-related diseases and ageing, we thus need to know what determines the accumulation and segregation of mutations in the soma. Why do some mutations accumulate with age, often in a mosaic- and tissue-specific pattern, while others do not? Long-lived (e.g. postmitotic) cells that undergo few divisions and/or cells with high energetic demands that require large numbers of mitochondria are predicted to be most sensitive to accumulating deleterious mitochondrial mutations [77]. In line with this, mtDNA abnormalities are most frequently found in tissues, such as brain, heart and muscle [78,79].
8. Life-history, organism-level adaptations and the scope for selfish mitochondrial DNA
What causes the dramatic variation among organisms, tissues and cell types, in their propensity to accumulate deleterious mtDNA mutations during somatic growth? Organisms with indeterminate growth, such as single-celled organisms, or filamentous fungi, appear to be highly resilient to the accumulation of deleterious mtDNA mutations. By contrast, such accumulation is common in organisms with determinate growth, such as animals. This difference most probably can be attributed to differences in the degree of competition between cells: in organisms with indeterminate growth, there is continuous selection at the level of the cell, and any mtDNA variant that reduces cell performance will be selected against [6,14]. Such cell-level competition usually does not occur in organisms with determinate growth, where cell competition is disfavoured in order to restrict opportunities for the emergence of cancer [80].
There is a striking difference in genome architecture of mtDNA among plants, animals and fungi [81,82]. Animals typically have highly streamlined genomes, i.e. small genomes encoding few proteins and rDNA and with few intergenic sequences. By contrast, plant genomes are large and encode a higher number of proteins, and have introns and larger intergenic sequences. Fungi are intermediate between these extremes [81]. Lynch [82] attributed these differences to differences in the mutation rate between these groups, with the mutation rate of animal mtDNA being two orders of magnitude higher than that of plants [82]. Introns increase the mutational target of their host genomes, which must maintain specific nucleotide sequences for splice-site recognition during mRNA processing. A high mutation rate in mtDNA thus would favour intron-free genes, and this is indeed observed in animal mtDNAs. Kowald & Kirkwood [16] proposed a different explanation for the difference in mtDNA genome size. They argue that mtDNA genome architecture is a consequence of the presence of mtDNA recombination in plants and fungi, and its absence in animals. In the presence of mtDNA recombination, deletion mutants can be repaired via recombination, making deletion mutants potentially short lived. Thus, animal mtDNA genomes have been reduced due to the accumulation of deletion mutations in the absence of repair mechanisms.
We propose yet another alternative hypothesis for the differences in genome architecture between plants, animals and fungi. In contrast to plants and fungi, most animals have an early sequestration of the germline, thus removing direct selection on somatic mtDNA mutations. In the absence of early sequestration of the germline, mtDNA mutations are continuously exposed to selection at the level of the organism. This would thus generate stronger selection in modular organisms against selfish genetic elements arising in the soma. It might follow then that strategies to restrict such elements, such as mtDNA genome streamlining by transferring mitochondrial genes to the nuclear genome, is relaxed. However, this novel hypothesis is not mutually exclusive with the two other hypotheses and it remains to be established how other differences among plant, animal and fungal mtDNA, such as a different mutation rate and the presence of mtDNA recombination and fusion–fission cycles, are related to mtDNA genome architecture.
9. Experimental studies with microorganisms: playing with the levels of selection
In this introduction, we have argued that mtDNA is exposed to different levels of selection. We used a levels-of-selection approach to outline under what conditions selection on mtDNA can cause detrimental effects on host fitness, health and ageing. We also discussed mechanisms that allow the host to counterbalance selection on mtDNA when such selection would negatively affect the nuclear genome and, by extension, the organism. In many instances, the predicted outcomes of such conflicts are hypothetical. Has uniparental inheritance evolved to minimize conflict among mtDNA lineages? If so, as theoretical studies have made clear, we would expect immediate costs of the mixing of mtDNA from different individuals [31,32,36]. However, an alternative hypothesis that uniparental inheritance selects for cytonuclear compatibility also predicts a direct cost of the mixing of genomic lineages. Both hypotheses are supported by theoretical models (conflict hypothesis: [36–40]; ‘cytonuclear cooperation’ hypothesis: [60]) but they are difficult to tease apart experimentally simply because organisms in which the basic assumptions can be tested are hard to find.
So far the only experimental support for the notion that within-cell selection selects for variants that have a replication advantage comes from baker's yeast [6,33] and from filamentous fungi [14]. By experimentally relaxing among-cell selection, Taylor et al. [6] showed that the change from among-cell selection to within-cell selection results in the accumulation of mtDNA variants that are detrimental to the host. Yeast is an important model organism not only because one can manipulate the levels of selection, but also because, contrary to the general rule, mitochondria are inherited biparentally [34]. A recent study empirically demonstrated that the mixing of mtDNA lineages in yeast indeed selects for fast-replicating variants [33]. Under natural conditions, such ‘cheating’ mtDNA variants are not likely to occur because yeast is highly inbred, resulting in mtDNA lineages that are closely related. Furthermore, following sex, yeast mtDNAs quickly segregate during vegetative growth, resulting in largely homoplasmic individual cells and high variation between cells, facilitating efficient selection against deleterious mtDNA. Thus, despite biparental inheritance, the scope for selection on cheater genotypes is reduced. When forced to outbreed, however, thereby increasing the variation among mtDNA lineages, cheating mtDNA variants rapidly emerged.
As with yeast, Bastiaans et al. [14] showed experimentally that in species of the ascomycete fungus Neurospora within-cell relatedness among mtDNA is essential to prevent selection for dysfunctional mtDNA variants. They varied mtDNA relatedness by manipulating the size of the genetic somatic bottleneck and the opportunities for somatic fusion between individuals. A different study, also in this issue, using the fungus Podospora anserina, a multicellular model organism for ageing studies and mitochondrial deterioration, describes a link between dietary restriction and autophagy [83]. Both carbon and nitrogen restriction extended lifespan in P. anserina, and the size of this effect varied with the amount and type of restricted nutrient. Those two studies nicely illustrate the usefulness of filamentous fungi as model organisms to study mtDNA evolution.
10. This issue
‘What cost mitochondria?’ may seem an unusual title. Our aim for this special issue was to bring together researchers from diverse fields who are united in their interest in the evolution and function of mitochondria and their effects on ageing and diseases. While an understanding of the clinical aspects of ageing and diseases linked to mitochondria requires an intimate knowledge of the molecular processes that occur within cells, the appreciation of the causes of such processes should be found in evolutionary biology. In this issue, we focus on both aspects, allowing us to answer not only how mitochondria are costly but also why we expect them to be.
Acknowledgements
We thank the Lorentz Centre in Leiden, The Netherlands, for hosting the workshop ‘Recent insights in mitochondrial evolution applied to health and ageing’ on which this paper was based, and all the researchers who participated in many healthy discussions. We thank three reviewers who provided constructive comments on a previous version of this manuscript and also thank Ian Holt for his suggestion of the catchy title for this special issue.
Funding statement
We acknowledge the Royal Dutch Academy of Sciences (KNAW) and the IOP Genomics initiative for grants to support this workshop. M.B. is supported by the Australian Research Council (FT120100120 and DP140100560). M.B. also gratefully acknowledges the support provided by an ALW/NWO visitors’ grant (040.11.399). J.N.S. is supported by the Academy of Finland (CoE funding) and The Netherlands Organization for Scientific Research (NWO: VICI grant no. 865.10.004).
References
- 1.Hoekstra RF. 2000. Evolutionary origin and consequences of uniparental mitochondrial inheritance. Hum. Reprod. 15(Suppl. 2), 102–111. ( 10.1093/humrep/15.suppl_2.102) [DOI] [PubMed] [Google Scholar]
- 2.Margulis L. 1970. Origin of eukaryotic cells. New Haven, CT: Yale University Press. [Google Scholar]
- 3.Sagan L. 1967. On origin of mitosing cells. J. Theoret. Biol. 14, 225 ( 10.1016/0022-5193(67)90079-3) [DOI] [PubMed] [Google Scholar]
- 4.Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, Larsson NG. 2008. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 6, e10 ( 10.1371/journal.pbio.0060010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ephrussi B, Slonimski PP. 1955. Subcellular units involved in the synthesis of respiratory enzymes in yeast. Nature 176, 1207–1208. ( 10.1038/1761207b0) [DOI] [PubMed] [Google Scholar]
- 6.Taylor DR, Zeyl C, Cooke E. 2002. Conflicting levels of selection in the accumulation of mitochondrial defects in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 99, 3690–3694. ( 10.1073/pnas.072660299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallace DC. 1999. Mitochondrial diseases in man and mouse. Science 283, 1482–1488. ( 10.1126/science.283.5407.1482) [DOI] [PubMed] [Google Scholar]
- 8.Bandy B, Davison AJ. 1990. Mitochondrial mutations may increase oxidative stress—implications for carcinogenesis and aging. Free Radic. Biol. Med. 8, 523–539. ( 10.1016/0891-5849(90)90152-9) [DOI] [PubMed] [Google Scholar]
- 9.Speijer D, Manjeri GR, Szklarczyk R. 2014. How to deal with oxygen radicals stemming from mitochondrial fatty acid oxidation. Phil. Trans. R. Soc. B 369, 20130446 ( 10.1098/rstb.2013.0446) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szklarczyk R, Nooteboom M, Osiewacz HD. 2014. Control of mitochondrial integrity in ageing and disease. Phil. Trans. R. Soc. B 369, 20130439 ( 10.1098/rstb.2013.0439) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chatterjee A, Mambo E, Sidransky D. 2006. Mitochondrial DNA mutations in human cancer. Oncogene 25, 4663–4674. ( 10.1038/sj.onc.1209604) [DOI] [PubMed] [Google Scholar]
- 12.Legros F, Malka F, Frachon P, Lombes A, Rojo M. 2004. Organization and dynamics of human mitochondrial DNA. J. Cell Sci. 117, 2653–2662. ( 10.1242/jcs.01134) [DOI] [PubMed] [Google Scholar]
- 13.deGrey A. 1997. A proposed refinement of the mitochondrial free radical theory of aging. Bioessays 19, 161–166. ( 10.1002/bies.950190211) [DOI] [PubMed] [Google Scholar]
- 14.Bastiaans E, Aanen DK, Debets AJM, Hoekstra RF, Lestrade B, Maas MFPM. 2014. Regular bottlenecks and restrictions to somatic fusion prevent the accumulation of mitochondrial defects in Neurospora. Phil. Trans. R. Soc. B 369, 20130448 ( 10.1098/rstb.2013.0448) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kowald A, Jendrach M, Pohl S, Bereiter-Hahn J, Hammerstein P. 2005. On the relevance of mitochondrial fusions for the accumulation of mitochondrial deletion mutants: a modelling study. Aging Cell 4, 273–283. ( 10.1111/j.1474-9726.2005.00169.x) [DOI] [PubMed] [Google Scholar]
- 16.Kowald A, Kirkwood TBL. 2011. Evolution of the mitochondrial fusion–fission cycle and its role in aging. Proc. Natl Acad. Sci. USA 108, 10 237–10 242. ( 10.1073/pnas.1101604108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kowald A, Kirkwood TBL. 2013. Mitochondrial mutations and aging: random drift is insufficient to explain the accumulation of mitochondrial deletion mutants in short-lived animals. Aging Cell 12, 728–731. ( 10.1111/acel.12098) [DOI] [PubMed] [Google Scholar]
- 18.Busch KB, Kowald A, Spelbrink JN. 2014. Quality matters: how does mitochondrial network dynamics and quality control impact on mtDNA integrity? Phil. Trans. R. Soc. B 369, 20130442 ( 10.1098/rstb.2013.0442) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frank SA. 2003. Perspective: repression of competition and the evolution of cooperation. Evolution 57, 693–705. [DOI] [PubMed] [Google Scholar]
- 20.Buss LW. 1987. The evolution of individuality. Princeton, NJ: Princeton University Press.
- 21.Frank SA. 1997. Models of symbiosis. Am. Nat. 150, S80–S99. ( 10.1086/286051) [DOI] [PubMed] [Google Scholar]
- 22.Kirkwood TBL. 1977. Evolution of aging. Nature 270, 301–304. ( 10.1038/270301a0) [DOI] [PubMed] [Google Scholar]
- 23.Williams GC. 1957. Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411. ( 10.2307/2406060) [DOI] [Google Scholar]
- 24.Yan MH, Wang X, Zhu X. 2013. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 62, 90–101. ( 10.1016/j.freeradbiomed.2012.11.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallace DC. 2005. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Ann. Rev. Genet. 39, 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wallace DC. 2013. Bioenergetics in human evolution and disease: implications for the origins of biological complexity and the missing genetic variation of common diseases. Phil. Trans. R. Soc. B 368, 20120267 ( 10.1098/rstb.2012.0267) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendel G. 1866. Versuche über Plflanzenhybriden. Verhandlungen des naturforschenden Vereines in Brünn IV, 3–47. [Google Scholar]
- 28.Selosse MA, Albert BR, Godelle B. 2001. Reducing the genome size of organelles favours gene transfer to the nucleus. Trends Ecol. Evol. 16, 135–141. ( 10.1016/s0169-5347(00)02084-x) [DOI] [PubMed] [Google Scholar]
- 29.Bernardi G. 2005. Lessons from a small, dispensable genome: the mitochondrial genome of yeast. Gene 354, 189–200. ( 10.1016/j.gene.2005.03.024) [DOI] [PubMed] [Google Scholar]
- 30.Cosmides LM, Tooby J. 1981. Cytoplasmic inheritance and intragenomic conflict. J. Theor. Biol. 89, 83–129. ( 10.1016/0022-5193(81)90181-8) [DOI] [PubMed] [Google Scholar]
- 31.Frank SA. 1996. Host–symbiont conflict over the mixing of symbiotic lineages. Phil. Trans. R. Soc. Lond. B 263, 339–344. ( 10.1098/rspb.1996.0052) [DOI] [PubMed] [Google Scholar]
- 32.Hoekstra RF. 1987 The evolution of sexes. In Evolution of sex and its consequences, EXS 55 (ed. SC Stearns), pp. 59–91. Basel, Switzerland: Birkhauser. [Google Scholar]
- 33.Jasmin J-N, Zeyl C. 2014. Rapid evolution of cheating mitochondrial genomes in small yeasts populations. Evolution 68, 269–275. ( 10.1111/evo.12228) [DOI] [PubMed] [Google Scholar]
- 34.Birky CW. 1995. Uniparental inheritance of mitochondrial and chloroplast genes—mechanisms and evolution. Proc. Natl Acad. Sci. USA 92, 11 331–11 338. ( 10.1073/pnas.92.25.11331) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bergstrom CT, Pritchard J. 1998. Germline bottlenecks and the evolutionary maintenance of mitochondrial genomes. Genetics 149, 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hadjivasiliou Z, Lane N, Seymour RM, Pomiankowski A. 2013. Dynamics of mitochondrial inheritance in the evolution of binary mating types and two sexes. Proc. R. Soc. B 280, 20131920 ( 10.1098/rspb.2013.1920) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hastings IM. 1992. Population genetic aspects of deleterious cytoplasmic genomes and their effects on the evolution of sexual reproduction. Genet. Res. 59, 215–225. ( 10.1017/S0016672300030500) [DOI] [PubMed] [Google Scholar]
- 38.Hoekstra RF. 1990. The evolution of male–female dimorphism—older than sex. J. Genet. 69, 11–15. ( 10.1007/BF02931663) [DOI] [Google Scholar]
- 39.Hurst LD, Hamilton WD. 1992. Cytoplasmic fusion and the nature of sexes. Phil. Trans. R. Soc. Lond. B 247, 189–194. ( 10.1098/rspb.1992.0027) [DOI] [Google Scholar]
- 40.Law R, Hutson V. 1992. Intracellular symbionts and the evolution of uniparental cytoplasmic inheritance. Phil. Trans. R. Soc. Lond. B 248, 69–77. ( 10.1098/rspb.1992.0044) [DOI] [PubMed] [Google Scholar]
- 41.Sharpley MS, et al. 2012. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151, 333–343. ( 10.1016/j.cell.2012.09.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rand DM. 2008. Mitigating mutational meltdown in mammalian mitochondria. PLoS Biol. 6, e35 ( 10.1371/journal.pbio.0060035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao L, Shitara H, Horii T, Nagao Y, Imai H, Abe K, Hara T, Hayashi J-I, Yonekawa H. 2007. The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat. Genet. 39, 386–390. ( 10.1038/ng1970) [DOI] [PubMed] [Google Scholar]
- 44.Cree LM, Samuels DC, Lopes S, Rajasimha HK, Wonnapinij P, Mann JR, Dahl HHM, Chinnery PF. 2008. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 40, 249–254. ( 10.1038/ng.2007.63) [DOI] [PubMed] [Google Scholar]
- 45.Hauswirth WW, Laipis PJ. 1982. Mitochondrial DNA polymorphisms in a maternal lineage of Holstein cows. Proc. Natl Acad. Sci. USA 79, 4686–4690. ( 10.1073/pnas.79.15.4686) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moore WS. 1995. Inferring phylogenies from mtDNA variation—mitochondrial gene trees versus nuclear gene trees. Evolution 49, 718–726. ( 10.2307/2410325) [DOI] [PubMed] [Google Scholar]
- 47.Wai T, Teoli D, Shoubridge EA. 2008. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat. Genet. 40, 1484–1488. ( 10.1038/ng.258) [DOI] [PubMed] [Google Scholar]
- 48.Stearns SC. 1987. The selection-arena hypothesis. In Evolution of sex and its consequences, EXS 55 (ed. SC Stearns), pp. 337–349. Basel, Switzerland: Birkhauser. [DOI] [PubMed] [Google Scholar]
- 49.Muller HJ. 1964. The relation of recombination to mutational advance. Mutat. Res. 106, 2–9. ( 10.1016/0027-5107(64)90047-8) [DOI] [PubMed] [Google Scholar]
- 50.Frank SA, Hurst LD. 1996. Mitochondria and male disease. Nature 383, 224 ( 10.1038/383224a0) [DOI] [PubMed] [Google Scholar]
- 51.Beekman M, Dowling DK, Aanen DK. 2014. The costs of being male: are there sex-specific effects of uniparental mitochondrial inheritance? Phil. Trans. R. Soc. B 369, 20130440 ( 10.1098/rstb.2013.0440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Innocenti P, Morrow EH, Dowling DK. 2011. Experimental evidence supports a sex-specific selective sieve in mitochondrial genome evolution. Science 332, 845–848. ( 10.1126/science.1201157) [DOI] [PubMed] [Google Scholar]
- 53.Camus MF, Clancy DJ, Dowling DK. 2012. Mitochondria, maternal inheritance, and male aging. Curr. Biol. 22, 1717–1721. ( 10.1016/j.cub.2012.07.018) [DOI] [PubMed] [Google Scholar]
- 54.Ruiz-Pesini E, et al. 2000. Human mtDNA haplogroups associated with high or reduced spermatozoa motility. Am. J. Hum. Genet. 67, 682–696. ( 10.1086/303040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Budar F, Touzet P, De Paepe R. 2003. The nucleo-mitochondrial conflict in cytoplasmic male sterilities revisited. Genetica 117, 3–16. ( 10.1023/a:1022381016145) [DOI] [PubMed] [Google Scholar]
- 56.Aanen DK, Kuyper TW, Debets AJM, Hoekstra RF. 2004. The evolution of non-reciprocal nuclear exchange in mushrooms as a consequence of genomic conflict. Phil. Trans. R. Soc. Lond. B 271, 1235–1241. ( 10.1098/rspb.2004.2693) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dowling DK, Friberg U, Lindell J. 2008. Evolutionary implications of non-neutral mitochondrial genetic variation. Trends Ecol. Evol. 23, 546–554. ( 10.1016/j.tree.2008.05.011) [DOI] [PubMed] [Google Scholar]
- 58.Rand DM, Haney RA, Fry AJ. 2004. Cytonuclear coevolution: the genomics of cooperation. Trends Ecol. Evol. 19, 645–653. ( 10.1016/j.tree.2004.10.003) [DOI] [PubMed] [Google Scholar]
- 59.Yee KM, et al. 2012. Neuron-specific enolase is elevated in asymptomatic carriers of Leber's hereditary optic neuropathy. Invest. Ophthalmol. Vis. Sci. 53, 6389–6392. ( 10.1167/iovs.12-9677) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hadjivasiliou Z, Pomiankowski A, Seymour RM, Lane N. 2012. Selection for mitonuclear co-adaptation could favour the evolution of two sexes. Proc. R. Soc. B 279, 1865–1872. ( 10.1098/rspb.2011.1871) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wolff JN, Ladoukakis ED, Enríquez JA, Dowling DK. 2014. Mitonuclear interactions: evolutionary consequences over multiple biological scales. Phil. Trans. R. Soc. B 369, 20130443 ( 10.1098/rstb.2013.0443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Holt IJ, Harding AE, Morgan-Hughes JA. 1988. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 331, 717–719. ( 10.1038/331717a0) [DOI] [PubMed] [Google Scholar]
- 63.Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AMS, Elsas LJ, Nikoskelainen EK. 1988. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 242, 1427–1430. ( 10.1126/science.3201231) [DOI] [PubMed] [Google Scholar]
- 64.Holt IJ, Speijer D, Kirkwood TBL. 2014. The road to rack and ruin: selecting deleterious mitochondrial DNA variants. Phil. Trans. R. Soc. B 369, 20130451 ( 10.1098/rstb.2013.0451) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goto Y-I, Nonaka I, Horai S. 1990. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 348, 651–653. ( 10.1038/348651a0) [DOI] [PubMed] [Google Scholar]
- 66.Ciafaloni E, et al. 1992. MELAS: clinical features, biochemistry, and molecular genetics. Ann. Neurol. 31, 391–398. ( 10.1002/ana.410310408) [DOI] [PubMed] [Google Scholar]
- 67.Rowland LP, Blake DM, Hirano M, Di Mauro S, Schon EA, Hays AP, Devivo DC. 1991. Clinical syndromes associated with ragged red fibers. Rev. Neurol. 147, 467–473. [PubMed] [Google Scholar]
- 68.Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC. 1990. Myoclonic Epilepsy and Ragged-Red Fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell 61, 931–937. ( 10.1016/0092-8674(90)90059-N) [DOI] [PubMed] [Google Scholar]
- 69.Wallace DC, Zheng X, Lott MT, Shoffner JM, Hodge JA, Kelley RI, Epstein CM, Hopkins LC. 1988. Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell 55, 601–610. ( 10.1016/0092-8674(88)90218-8) [DOI] [PubMed] [Google Scholar]
- 70.Zeviani M, Gellera C, Pannacci M, Uziel G, Prelle A, Servidei S, DiDonato S. 1990. Tissue distribution and transmission of mitochondrial DNA deletions in mitochondrial myopathies. Ann. Neurol. 28, 94–97. ( 10.1002/ana.410280118) [DOI] [PubMed] [Google Scholar]
- 71.McShane MA, Hammans SR, Sweeney MG, Holt IJ, Beattie TJ, Brett EM, Harding AE. 1991. Pearson syndrome and mitochondrial encephalomyopathy in a patient with a deletion of mtDNA. Am. J. Hum. Genet. 48, 39–42. [PMC free article] [PubMed] [Google Scholar]
- 72.Fan W, et al. 2008. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319, 958–962. ( 10.1126/science.1147786) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He Y, et al. 2010. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 464, 610–614. ( 10.1038/nature08802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goto H, Dickins B, Afgan E, Paul I, Taylor J, Makova K, Nekrutenko A. 2011. Dynamics of mitochondrial heteroplasmy in three families investigated via a repeatable re-sequencing study. Genome Biol. 12, R59 ( 10.1186/gb-2011-12-6-r59) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Payne BAI, et al. 2013. Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 22, 384–390. ( 10.1093/hmg/dds435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guo Y, Li C-I, Sheng Q, Winther JF, Cai Q, Boice JD, Shyr Y. 2013. Very low-level heteroplasmy mtDNA variations are inherited in humans. J. Genet. Genom. 40, 607–615. ( 10.1016/j.jgg.2013.10.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aanen DK, Maas MFPM. 2012. Recruitment of healthy mitochondria fuels transmissible cancers. Trends Genet. 28, 1–6. ( 10.1016/j.tig.2011.10.001) [DOI] [PubMed] [Google Scholar]
- 78.Chinnery PF, Samuels DC, Elson J, Turnbull DM. 2002. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet 360, 1323–1325. ( 10.1016/S0140-6736(02)11310-9) [DOI] [PubMed] [Google Scholar]
- 79.Krishnan KJ, Greaves LC, Reeve AK, Turnbull D. 2007. The ageing mitochondrial genome. Nucleic Acids Res. 35, 7399–7405. ( 10.1093/nar/gkm635) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cairns J. 1975. Mutation selection and natural history of cancer. Nature 255, 197–200. ( 10.1038/255197a0) [DOI] [PubMed] [Google Scholar]
- 81.Burger G, Gray MW, Lang BF. 2003. Mitochondrial genomes: anything goes. Trends Genet. 19, 709–716. ( 10.1016/j.tig.2003.10.012) [DOI] [PubMed] [Google Scholar]
- 82.Lynch M. 2006. The origins of eukaryotic gene structure. Mol. Biol. Evol. 23, 450–468. ( 10.1093/molbev/msj050) [DOI] [PubMed] [Google Scholar]
- 83.van Diepeningen AD, Engelmoer DJP, Sellem CH, Huberts DHEW, Slakhorst SM, Sainsard-Chanet A, Zwaan BJ, Hoekstra RF, Debets AJM. 2014. Does autophagy mediate age-dependent effect of dietary restriction responses in the filamentous fungus Podospora anserina? Phil. Trans. R. Soc. B 369, 20130447 ( 10.1098/rstb.2013.0447) [DOI] [PMC free article] [PubMed] [Google Scholar]