

Abstract
Mammalian mtDNA encodes for 13 core proteins of oxidative phosphorylation. Mitochondrial DNA mutations and deletions cause severe myopathies and neuromuscular diseases. Thus, the integrity of mtDNA is pivotal for cell survival and health of the organism. We here discuss the possible impact of mitochondrial fusion and fission on mtDNA maintenance as well as positive and negative selection processes. Our focus is centred on the important question of how the quality of mtDNA nucleoids can be assured when selection and mitochondrial quality control works on functional and physiological phenotypes constituted by oxidative phosphorylation proteins. The organelle control theory suggests a link between phenotype and nucleoid genotype. This is discussed in the light of new results presented here showing that mitochondrial transcription factor A/nucleoids are restricted in their intramitochondrial mobility and probably have a limited sphere of influence. Together with recent published work on mitochondrial and mtDNA heteroplasmy dynamics, these data suggest first, that single mitochondria might well be internally heterogeneous and second, that nucleoid genotypes might be linked to local phenotypes (although the link might often be leaky). We discuss how random or site-specific mitochondrial fission can isolate dysfunctional parts and enable their elimination by mitophagy, stressing the importance of fission in the process of mtDNA quality control. The role of fusion is more multifaceted and less understood in this context, but the mixing and equilibration of matrix content might be one of its important functions.
Keywords: mitochondrial dynamics, mtDNA, mitophagy, nucleoids, TFAM, OXPHOS
1. Introduction
Mitochondria are remarkable structures inside eukaryotic cells. These organelles emerged roughly two billion years ago from free-living prokaryotic ancestors [1]. This endosymbiotic origin also explains the fact that mitochondria still contain their own genetic material (mitochondrial DNA, mtDNA). During the course of evolution, most mitochondrial genes were transferred to the nucleus, still the genes for 13 proteins are located on the human mtDNA, together with two ribosomal RNA and 22 transfer RNA genes, whose products are essential for mitochondrial protein synthesis [2]. Mitochondria are involved in several essential processes such as apoptosis, calcium homeostasis and fatty acid degradation, but their most important task is the generation of adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS) at the inner mitochondrial membrane. All of the proteins encoded on the mtDNA are subunits of the OXPHOS enzymes.
Thus, damage to the mitochondrial genome has the potential to impact or abolish the main source of cellular energy generation. However, cells contain hundreds of mitochondria, and for a long time, the general opinion was that removal of damaged mitochondria via mitophagy also contributes to a constant removal of mutant mtDNA [3]. This view has now radically changed, because it became clear that mtDNA mutations are at the core of many human diseases such as Leber's hereditary optic neuroretinopathy (LHON), myoclonic epilepsy associated with ragged-red fibres (MERRF), mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), Kearns–Sayre Syndrome (KSS) or chronic progressive external ophthalmoplegia (CPEO) [4,5]. Even though wild-type mtDNA molecules are still present in disease cells (a situation referred to as heteroplasmy1), mutant mtDNA molecules with point mutations, partial deletions or duplications are present at such high heteroplasmy levels per cell that they cause pathological phenotypes. A major question is how these mutations evade mitochondrial quality assurance systems. Obviously, the number of wild-type mtDNA molecules that normally exists per cell does not prevent the spread of disease variants, pointing to some advantage of the latter. This is even more vexing because, with few exceptions, mtDNA disease variants seem to behave as recessive mutations such that they only result in a biochemical or physiological phenotype (e.g. cytochrome c oxidase (COX) deficiency) at high heteroplasmy levels (typically more than 60% mutant mtDNA). This threshold effect is generally considered as an indication of functional complementation, either directly by the co-localization of wild-type (wt) and mutant mtDNA molecules or indirectly by diffusion and mixing with wt gene products.
Interestingly, damage to mtDNA not only causes various diseases, but could also be of relevance in the ageing process. In several species and tissues, it has been observed that mtDNA deletions accumulate with age [6–9]. These early studies were performed on tissue homogenates, and although an accumulation of deletions was found, the overall level was very low. An important insight was gained when single cell studies became possible. They revealed that some cells harbour a large fraction of different deletion mutants, whereas others seemingly contain no mutants at all [10–13]. Furthermore, those cells that do contain deletion mutants are taken over by a single mutant type which is different for different cells. Similarly, clonally expanded deleterious point mutations have been shown to accumulate with age, for example, in colonic crypt stem cells [14–16].
Several mechanisms have been proposed to explain clonal mutation expansion in individual cells with age and that is also the main focus of the article by Holt et al. [17] which is presented elsewhere in this issue. Thus, we give here only a short outline of the various ideas that broadly fall into two categories: those that invoke some sort of selection advantage and those that do not. According to the ‘vicious cycle’ idea, damaged mitochondria generate more radicals and those radicals cause, in turn, more damage to mitochondria [18,19]. This, in principle, should cause various different mtDNA mutants in a cell, which is not supported by observation. ‘Random drift’ is another selection-free hypothesis, which states that chance fluctuations during replication and degradation of mtDNA are sufficient to explain the occurrence of COX-deficient cells in old tissues [20,21]. However, recent computer simulations indicate that such a process can work only in very long-lived species [22]. Very popular among the selection-based theories is the idea that the reduced genome size of a mtDNA deletion mutant confers a replicative advantage [23,24]. Because the replication time of mtDNA is much shorter than the turnover time, it is difficult to see how mtDNA replication could be rate limiting for mitochondrial growth. Recent modelling studies corroborate this problem [25]. The idea of the ‘survival of the slowest’ hypothesis is that damaged mitochondria are degraded less frequently than wild-type organelles [26]. This is, however, contradicted by recent experimental observations showing that damaged organelles are actually preferentially degraded instead of being preferentially kept [27–29]. Finally, it has been suggested that mitochondrial transcription is regulated by a product inhibition feedback that is disrupted in deletion mutants [30]. The tight connection between transcription and priming of replication in metazoan mtDNA can then lead to a selection advantage of mtDNA mutants. This mechanism would work for short- and long-lived animals, is in agreement with the observed clonal expansion and would explain the deletion spectra found in single cell studies.
It is clear from the above introduction that single mutant mtDNA alleles can clonally expand in somatic cells in disease and during ageing to become the predominant genotype. Yet, on the basis of recent insights into mitochondrial fission/fusion and selective mitochondrial degradation via mitophagy, it has been suggested that the combined action of these processes might help to prune the mitochondrial network of less ‘healthy’ branches. This presents a paradox: how do mtDNA mutations accumulate at all if fission and mitophagy would provide an efficient means to clean up those parts of the network where mtDNA mutations result in local energy deficiencies? This apparent paradox is the main topic of this paper in which we discuss mtDNA heteroplasmy dynamics in relation to mitochondrial network dynamics and quality control.
MtDNA is typically present in hundreds to thousands of copies per cell and organized in discrete protein–DNA complexes called nucleoids in an otherwise dynamic mitochondrial network [31]. It has been suggested on the basis of immunofluorescent microscopy [32] and nucleoid mass-spectrometry analysis (see [33,34] for a recent overview) that nucleoids might organize so as to locally facilitate nucleoid maintenance and organize transcription, translation and biogenesis in its immediate surroundings. This also forms the basis for the suggestion that each nucleoid has a limited sphere of influence that could result in a focal mitochondrial OXPHOS deficiency if it contains mutant mtDNA [35]. It has been suggested that nucleoids contain multiple copies of mtDNA [36], but recent work using super-resolution microscopy indicates that each nucleoid contains on average only a single mtDNA copy in various cell types [37]. Furthermore, the confinement of OXPHOS complexes to single cristae owing to their restricted diffusion adds to the focal perspective [38,39]. Together, this would suggest that nucleoids containing a deleterious mutant mtDNA might, indeed, express a focal OXPHOS phenotype allowing it, in principal, to be selectively degraded via fission/mitophagy. However, because many mutations are tolerated in disease and accumulate both in disease and with ageing, this indicates that the link between genotype and phenotype is leaky, and intramitochondrial complementation does occur [35]. One might thus pose the question to what extent functional complementation prevents selective degradation of mutant mtDNA molecules and by what means does this complementation take place? One obvious answer would be that in many tissues the nucleoid copy-number is, in fact, larger than 1. It is therefore of relevance for the further discussion, and for our understanding of functional complementation and the threshold effect in human disease that the nucleoid mtDNA copy-number is also determined in vivo, in human tissues.
Assuming that the determination of nucleoid copy-number by super-resolution microscopy is a better representation of reality, at least for the cell types studies, the other question arising in the light of the mechanism of complementation is to what extent mitochondrial nucleoids are able to move about in a very confined mitochondrial matrix. We present experimental evidence here that nucleoid movement is equally or even more confined than mitochondrial inner-membrane complexes suggesting that, in principle, there is little room for direct complementation of mutations at the level of mtDNA unless several copies of mtDNA are organized in single nucleoids. Whether nucleoids indeed have a very limited sphere of influence or whether there is room for complementation by more mobile RNA and/or proteins is also discussed in the light of mitochondrial fusion/fission and (the lack of) selective degradation of dysfunctional mitochondria via mitophagy. The questions we are ultimately trying to answer are: (i) can mutant mtDNA alleles result in focal OXPHOS deficiencies in a cellular population of both wt and mutant mtDNA? (ii) under what conditions and which types of mutant alleles are able to clonally expand? and (iii) in relation to the second question, do complementation and/or a replicative advantage play a role in clonal expansion? We propose possible ways in which we can test predictions made here, for example, how we might demonstrate the potential limited spheres of influence of nucleoids.
2. The measurement of mitochondrial dynamics and its application to understand nucleoid mobility
(a). Mitochondrial fusion/fission and nucleoid mobility
Mitochondria are dynamic organelles that fuse and divide frequently and actively move through the cell [40]. This dynamics seems indispensable for proper cellular function as many examples have shown. Reduced dynamics accompanies the process of ageing [41], is associated with the development of neurodegenerative diseases [42–44] and plays an important role in the execution of apoptosis [45,46]. Repetitive fusion and fission of mitochondria during the cell cycle has to be regulated [47–50]; otherwise, the continuity of the mitochondrial genome is not properly guaranteed [51,52], resulting in mitochondrial heterogeneity and malfunction [53]. Obviously, mitochondrial dynamics is important for quality control [27,54]. It has been shown that fused mitochondria exchange their matrix contents including mtDNA [36,55–57], leading to the statement that all mitochondria function as a single dynamic unit [52,58].
Previous studies of mitochondrial dynamics have been primarily performed by two assays: the analysis of the exchange of green- and red fluorescent protein- (GFP- and RFP-) labelled matrix compounds after cell fusion ([57] and as illustrated in figure 1a), and the spreading of photo-activated (pa) or photo-switched matrix-targeted (mt-)fluorescent proteins throughout a single cell owing to mitochondrial dynamics ([59] and as illustrated in figure 1b).
Figure 1.
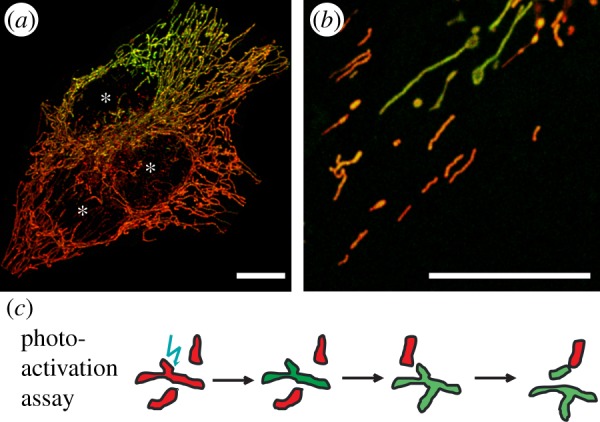
Assays to determine the exchange rates of compounds between mitochondria. (a) Polyethylene glycol- (PEG-)-mediated fusion of cells with differently fluorescent-tagged compounds, here OXPHOS complexes in red (RFP-tagged) and green (GFP-tagged). (b) Use of photo-activatable fluorescent proteins, here OXPHOS complex CI-paGFP to monitor spreading in MitoTracker stained mitochondria. (c) Scheme of photo-activation assay: paGFP is photo-activated in single mitochondria that express paGFP-tagged proteins. Owing to ongoing fusion and fission and mixing of compounds, the fluorescent signal spreads throughout the mitochondrial reticulum. Asterisks indicate nuclei. Scale bars, 10 µm. Images from K.B. (Online version in colour.)
Outer membrane (OM)-targeted GFP and matrix-GFP showed similar dynamics in cell fusion experiments when complete fusion was observed [36]. By contrast, cell fusion and pa-GFP studies indicated that for inner membrane proteins (ABCB10; OXPHOS complexes), diffusion and exchange within a single mitochondrion were significantly slower than for mt-paGFP [60,61]. OXPHOS complexes showed a patchy distribution in cell fusion assays with OXPHOS-GFP and OXPHOS-RFP from different sources [62]. A more detailed study revealed that the reason for the retarded mixing is most likely based on the fact that cristae are preserved during fusion and fission and proteins in the cristae membranes are limited in their diffusion [39,63]. It was similarly shown that cristae architecture limits matrix diffusion [63]. Thus, retarded mixing allows for intramitochondrial heterogeneity at least for some time, but with ongoing fusion and fission eventually results in a homogeneous distribution of compounds (illustrated in figure 2) [39].
Figure 2.

Repetitive fusion and fission cycles are required for good mixing of inner membrane proteins. (a) Dual-colour super-resolution imaging of fused mitochondria with differently labelled OXPHOS complexes (CI-EGFP + CII-mRFP) after few (early state) and frequent (intermediate and late states) fusion and fission events. In short, two stable cell lines expressing OXPHOS complex I fused to monomeric EGFP (CI-EGFP) and complex II fused to monomeric RFP (CII-mRFP), respectively, were co-plated and cell fusion was induced by PEG treatment. Owing to ongoing mitochondrial dynamics, mitochondria in the syncytium fused and divided frequently and mitochondria with a hybrid composition were generated. The yellow arrowheads and colour, respectively, show co-localization of OXPHOS complexes of different origin. (b) Schematic model of the sequence of events eventually generating well-mixed mitochondria. Scale bars, 300 nm (a), 100 nm (b). Adapted with permission from Wilkens et al. [39]. (Online version in colour.)
(b). The spatio-temporal organization of mtDNA in nucleoid microcompartments
It was also shown that mtDNA, such as matrix proteins, in principal, is exchanged between mitochondria [36]. Mitochondrial nucleoids can be visualized in assays by direct staining or using fluorescent mtDNA binding proteins (illustrated in figure 3a,b). Fluorescence images of mtDNA stained with picogreen [64] show an arrangement of distinct foci within mitochondria (figure 3a). Because labelling of mtDNA is potentially harmful owing to intercalation of dyes, the better alternative is the indirect visualization via mtDNA associated proteins. Mitochondrial transcription factor A (TFAM) is an appropriate marker protein because it is not only a mitochondrial transcription factor, but also a packaging factor of mtDNA. It has been shown on several occasions, both for the endogenous protein and overexpressed tagged variants that it constitutively labels all mtDNA molecules [37,65,66]. Indeed, here also the distribution pattern of TFAM (figure 3b) resembles that of mtDNA (figure 3a). Early electron microscopy- (EM-) based research has suggested that mtDNA is associated with the inner mitochondrial membrane [67,68], but recent super-resolution microscopy [65] as well as biochemical analysis [66] also suggest the existence of a dynamic pool of non-membrane associated nucleoids.
Figure 3.
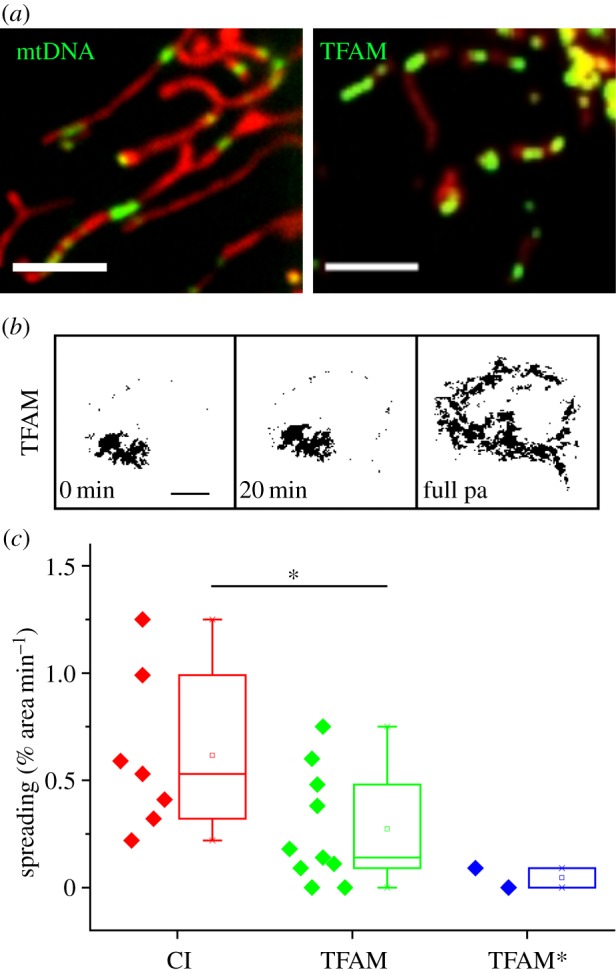
Mitochondrial nucleoids marked by TFAM display remote spreading throughout the chondriom. (a) mtDNA and TFAM localize in similar foci patterns: mtDNA (stained with picogreen) and TFAM-GFP in mitochondria (stained with MitoTracker Red). (b) TFAM/nucleoids from a photo-activated (pa) area show very little dynamics within a single cell during 20 min. (c) Spreading of OXPHOS complex I and TFAM from photo-activated areas in comparison. TFAM*: control measurement in fragmented mitochondria (fragmentation was induced by bacterial contamination). Scale bar, 3 µm (a), 10 µm (b). (Online version in colour.)
Here, we extend the above analyses by the evaluation of nucleoid dynamics monitored by means of a photo-activation assay (figure 3c). This shows that mtDNA nucleoids spread throughout the cell at a rate approximately comparable to complex I of the OXPHOS system (figure 3c). This conforms with an attachment or frequent interaction of nucleoids to the inner mitochondrial membrane, or alternatively with a confinement of nucleoids in a microcompartment [69] rather than with free movement in a continuous matrix lumen. Earlier studies on foci dynamics suggested a restricted nucleoid movement in yeast [48,70] and a very low apparent diffusion constant in human cells in culture [32]. (One has to bear in mind, though, that the monitored dynamics, in general, is an overlay of mitochondrial movement and fusion and fission dynamics).
The size of mtDNA nucleoids roughly ranges from 50 to 300 nm based on EM and super-resolution microscopy [37,65]. This implies that in mitochondria with a regular arrangement of cristae discs with a mean distance of 53 nm, such as observed in HeLa cells [38], most nucleoids would not fit. However, those mitochondria also display disruptions of the regular cristae arrangement resulting in areas with larger free matrix space (illustrated in figure 4a), and it was indeed shown very recently that regions of cristae disruption indicate the presence of nucleoids [71,72]. These matrix sections might constitute special, functional microcompartments, for example, for protein biosynthesis in close proximity to mtDNA [32]. Recently, we established a method to explore and characterize mitochondrial microcompartments by tracking and localization of mobile single molecules within these compartments [38]. To expand this type of analysis to nucleoid dynamics, we used tetramethylrhodamine- (TMR-) labelled TFAM to explore the mtDNA associated microcompartment. We generated trajectory maps of TFAM (figure 4b) and determined the two-dimensional projection area of its accessibility (figure 4b′). The diameter of these areas was comparable to the matrix microcompartments seen in the EM micrographs (figure 4c, marked with asterisks in figure 4a). From the trajectories, it was also possible to determine mean apparent diffusion coefficients for TFAM. The mobile fraction of TFAM had a low diffusion coefficient that was comparable with diverse innermembrane proteins (CIV, Mitofilin-IMMT) but was much slower than, for example, the matrix protein MPP.
Figure 4.
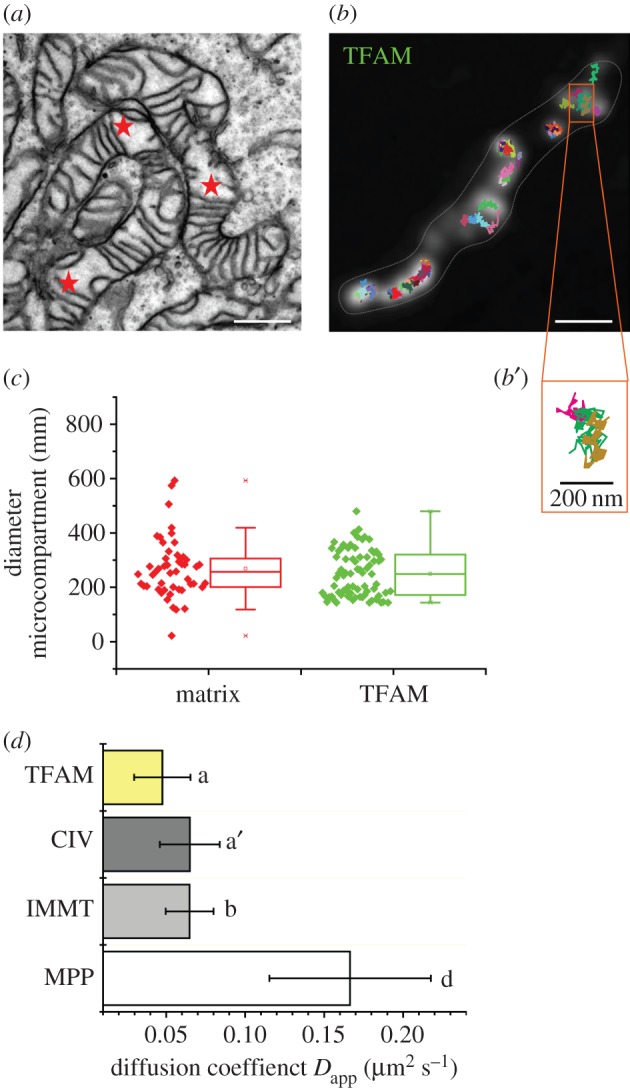
Putative spatio-temporal organization of nucleoids in microcompartments. (a) TEM micrograph of mammalian HeLa mitochondria. (b) Trajectory map of TFAM-Halo/TMR (see electronic supplementary material, Materials and methods) in a single mitochondrion of the same type. The detailed view at the bottom (b′) shows a putative microcompartment explored by three TFAM molecules (magenta, cyan, brown trajectories). (c) Comparison of ultra-structural and dynamics defined microcompartments, respectively, resolved by EM (matrix) and tracking and localization microscopy [38] (TFAM). (d) Apparent diffusion coefficients of the mobile fractions of TFAM, OXPHOS complex IV, mitofilin (IMMT) and mitochondrial matrix processing peptidase (MPP). Mean values from at least two independent measurements, including 12 cells, 50 mitochondria and more than a thousand individual trajectories are depicted (ANOVA significance test; letters indicate significance levels p ≤ 0.05 for a,a′–b; p ≤ 0.001 for a–d). Scale bar, 300 nm (a,b). TEM micrograph courtesy of Verena Wilkens.
Taken together, the features of mtDNA dynamics at the nucleoid and mitochondrial level reasonably argue for constrained diffusion and internal spreading. This obviously underlines the significance of fusion and fission as a mechanism for remixing.
3. Discussion
(a). An outline of a discussion of mitochondrial network dynamics and the maintenance of mitochondrial genome integrity
We here further discuss the processes of mitochondrial fission/fusion and mitophagy in relation to possible quality control of the mitochondrial genome. Mitochondrial fission combined with selective degradation of parts of the mitochondrial network has been proposed as an efficient means to prune the mitochondrial network of damaged or malfunctioning parts [73], yet there are many examples from mitochondrial disease and in ageing tissues for the selective accumulation of mutant mtDNA with obvious deleterious consequences. Central to the idea that mutant mtDNA alleles might be selected against by means of fission and mitophagy is the idea that nucleoids, often containing only single copies of mtDNA, could have a limited sphere of influence (thus resulting in a direct link between the genotype of individual mtDNA molecules and local phenotype). Limited diffusion of gene products would then result in a local energy deficiency that selection might act upon in a targeted way (figures 5 and 6). Depending on the level of mtDNA mobility/mixing (which, based on the results also presented here, can be assumed to be slow) and the rate of diffusion of mitochondrial gene products away from the nucleoid, we might speak of a more or less ‘leaky link’ [35]. In order to understand how this system might crumble and permit the accumulation of mutant mtDNA alleles, we first discuss if and under what conditions this system might actually work.
Figure 5.

The organelle control theory predicts a ‘leaky link’ between genotype and phenotype [35]. The tight spatial organization of cristae combined with their microcompartment character leads automatically to such a connection, because the diffusion of mRNAs and proteins synthesized by different nucleoids, N, is impeded by these structures.
Figure 6.

Genotype–phenotype linkage as the critical parameter to eliminate dominant negative mtDNA. (a) Mitochondria with wild-type mtDNA can undergo (spontaneous or induced) depolarization, but recovery followed by fusion brings them back into the dynamic mitochondrial network. (b) In a very leaky link, the presence of mutant next to wild-type mtDNA in the same mitochondrion would allow intramitochondrial functional complementation. As long as the decrease of proton motive force (pmf) and membrane potential, respectively, is not too harsh, elimination by mitophagy will not work and mutant mtDNA can escape the quality control. This spares biomaterial on cost on stringency. (c) In a less leaky link, with closer genotype-phenotype coupling, fission can generate fragments with low pmf, unable to fuse again, prone to be eliminated by mitophagosomes.
(b). Mitophagy: a cellular quality assurance system?
The maintenance of mitochondrial function is pivotal for short-term cellular homeostasis and long-term cell and organismal survival. The proton motive force, generated by the proton translocating activity of the respiratory chain complexes I, III and IV, is seen as a general indicator for functionality. By acidification of the intermembrane space, a chemical proton gradient is formed. This gradient—together with the membrane potential ΔΨm—constitutes the proton motive force, pmf = ΔΨm–2,3 RT ΔpH/F [74]. The pmf provides the predominant driving force for ATP synthesis, mitochondrial transport, Ca2+ accumulation and the import of nuclear-encoded, mitochondrially localized proteins. Impaired mitochondrial function caused by different mechanisms habitually results in a decrease in pmf as a common endpoint. Hypopolarized mitochondria are fusion-incompetent, fragment through mitochondrial fission and can be selectively digested by lysosomes [27,54]. Selective mitophagy following fission is part of the quality control system of cells [73,75] to avoid the accumulation of damaged mitochondria and eventually the onset of the intrinsic pathway to cell death (apoptosis). In the process of mitophagy, the PTEN-induced kinase 1 (PINK1) and Parkin, a cytosolic E3 ubiquitin ligase, play a crucial role [76,77]. Mutations in these genes lead to autosomal recessive Parkinsonism. Generally, the maintenance of a healthy mitochondrial population is dependent on balanced organelle fusion and fission [40,78–83]. Although fusion and fission promote the remixing of mitochondrial material, mitochondria display functional heterogeneity as, for example, indicated by variant TMRM+ (a fluorescent indicator of mitochondrial membrane potential) staining [84–86]. One possible explanation for the intramitochondrial heterogeneity is that protein and membranous structures are unevenly distributed and/or unequally functional, which might be the consequence of the focal expression of mutant mtDNA. In combination with selective mitophagy, as outlined above, this could provide a mechanistic basis for a cellular system that preferentially eliminates sequestered mitochondrial fragments and associated nucleoid(s) from the mitochondrial network. On the genetic level, mitochondrial fusion is necessary for the proliferation of mtDNA [87] but, as put forward by Kowald & Kirkwood [35], it also seems that it opens the door for the dissemination of defective mtDNAs which then requires fission and selective degradation to limit this accumulation. One main dilemma probably is that selection works on the phenotype and that the link between genotype and phenotype is not as tight as required, but leaky. This is discussed in the following section.
(c). Restricted nucleoid mobility and the leaky–link model
A mathematical model that formalizes the leaky–link model and the possible accumulation of one type of mtDNA mutation, namely partial deletions, takes into account the level of leakiness on the one hand and a possible replicative advantage of smaller-than-wt mtDNA molecules on the other [35]. Computer simulations showed that these counteracting forces have several possible outcomes for genotype selection depending on the boundary conditions. Assuming a given replicative advantage of mutant over wt mtDNA, at low genotype–phenotype leakiness wt mtDNA becomes the predominant species if a clear selective degradation of mutant mtDNA is invoked. If leakiness and thus complementation would be significant, mutant mtDNA always outcompetes wt mtDNA. The exact level of leakiness above which this takes place depends on the replicative advantage of mutant over wt mtDNA and the rate of selective degradation.
The here demonstrated restricted intramitochondrial nucleoid mobility in an otherwise dynamic mitochondrial network is relevant for ideas that have been proposed to explain the evolution of the mitochondrial fission and fusion cycle. The organelle control theory states that fusion has evolved to equilibrate all matrix proteins across the mitochondrial network and that fission is necessary to counteract the accumulation of defective mtDNA molecules [35]. A central point of this idea is that a physical connection is required between the mtDNA genotype and phenotype for such a cleansing process to work. Only then, the removal of defective proteins also leads to the removal of defective mtDNAs. In its original formulation, it was proposed that nucleoids are attached to their own gene products (OXPHOS complexes) at the inner mitochondrial membrane and also that the different complexes (I–IV) are interconnected. This would establish a link between genotype and phenotype, which in combination with fission and selective mitophagy should be sufficient to allow for the removal of defective mtDNAs. Assuming that some level of complementation between mtDNA molecules could occur, be it directly at the mtDNA level or at the level of RNA and/or protein, the focal genotype–phenotype correlation becomes less clear and thus results in a ‘leaky link’.
The experimental results presented here that demonstrate the restriction of nucleoids and respiratory chain complexes to nearby matrix space or individual mitochondrial cristae [39] renders some of the original assumptions of the organelle control theory unnecessary. The experiments indicate that no direct connection among the OXPHOS complexes and mtDNA molecules is necessary to create a genotype–phenotype link. Instead, the spatial confinement to a cristae microcompartment (if attached to the inner mitochondrial membrane) or the intercristae compartment (if confined by diffusion to specific matrix sections) automatically generates such a link (figure 5). Furthermore, originally, it was proposed that transcription, translation and membrane insertion of proteins are coupled in mitochondria via a process known in bacteria as transertion [88]. The tight spatial arrangement of stacks of cristae together with the compartment character of individual cristae and intercristae space also lifts the requirement for this assumption. Figure 5 shows how this arrangement can quite naturally lead to the link between genotype and phenotype that has been put forward by the organelle control theory. In this model, the link becomes ‘leaky’ mostly by diffusion of RNA and proteins as is also illustrated in figure 5. Mitochondrial fusion and fission as well as cristae dynamics in this model intrinsically affect how tight/leaky the link between genotype–phenotype is, and thus have repercussions for the accumulation of mtDNA mutations. In particular, local remodelling or loss of cristae stacks as a consequence of mtDNA mutations could be an important requirement that would allow intramitochondrial complementation by increased matrix diffusion and mobility of neighbouring nucleoids as well as RNA and proteins, and would also offer an attractive explanation why so many mutations show a biochemical threshold effect. Cristae remodelling would result in a permissive environment for the mutant mtDNA molecules and allow them to go undetected and expand. In this respect, it can be noted that many pathogenic mtDNA mutations do result in ultra-structural mitochondrial abnormalities. In addition, it might provide an alternative explanation why, for example, some optic atrophy 1 (OPA1) and mitofusin (Mfn2) mutations result in the accumulation of multiple mtDNA deletions [89–91].
In addition, other hypotheses regarding the evolution of mitochondrial fusion have been proposed, such as the idea that connected mitochondria can serve as power cables transmitting membrane potential throughout the cell [92]. However, they have difficulties explaining the requirement for fission and the observed high fusion and fission rates.
(d). Mitophagy and the removal of deleterious mtDNA mutants
The idea that mitophagy serves as a cellular quality control mechanism to keep damaged mitochondria at bay is an elegant and intriguing hypothesis. It is, however, important to be specific what type of damage should be limited by such a process and on what time-scale.
For short-term quality insurance, it would be sufficient to remove and degrade a dysfunctional membrane patch with impaired OXPHOS complexes. However, the situation is quite different if a respiratory chain deficiency is caused by damage (point mutations or deletions) to mtDNAs. Obviously, in such a case, it is not sufficient to degrade the dysfunctional membrane patch, but it is imperative also to remove the mutant mtDNA that produced the defective respiratory chain components. How a link between genotype and phenotype can be established has already been outlined in the previous section and figure 5. But, as mentioned, such a link might be leaky, and this has important consequences for the efficiency of a mitophagy-based clearance system.
If selective degradation depends on sensing a diminished membrane potential, then it would be optimal if fission would generate mitochondrial fragments that contain single nucleoids (see figure 6 for a proposed mitophagy-based mtDNA clearance system), assuming nucleoids mostly contain single mtDNA molecules as suggested for quite a few different primary and immortal cell lines based on super-resolution microscopy [37]. Fragments containing multiple nucleoids would disguise the presence of mutant mtDNAs, because their combined membrane potential would be difficult to distinguish from a pure wild-type potential. Similarly, in any cell type/tissue in which nucleoids do, in fact, contain multiple mtDNA copies, the local OXPHOS capacity is a sum of the expressed alleles present in that nucleoid. In practice, we can assume that wild-type alleles in mixed wild-type/mutant nucleoids can, at least, to some extent and often fully complement mutant alleles depending on the mutation(s). If future studies in, for example, patient fibroblasts show that single copy mutant mtDNA nucleoids do result in a focal defect and thus in a linked phenotype, it is intriguing to determine if nucleoids in various clinically relevant tissues have single or multiple mtDNA copies.
As can be seen from figure 5, a leakiness of the genotype/phenotype connection would lead to membrane areas that contain OXPHOS complexes which originate from multiple nucleoids. That means even if fission generates single nucleoid fragments, there is always a contamination of OXPHOS complexes originating from other nucleoids. Thus, the cellular mechanism for measuring the membrane potential never results in perfect selectivity. Depending on the degree of leakiness, a certain false-negative rate always remains and fragments containing mutant mtDNAs are not identified as such. The cell can optimize this monitoring system in two ways. First, it can reduce the degree of leakiness. This could be achieved by several mechanisms acting at different levels. At the molecular-ultra-structural level, the diffusibility of RNA, inner membrane proteins and nucleoids could be reduced or limited. At the level of organelle dynamics, fusion/fission rates could be slowed down. Third, the degradation rate of mitochondrial fragments could be increased, which would also limit the diffusion distance and exchange of material. Alternatively, the mechanism sensing the membrane potential could be more stringent, i.e. triggering mitophagy already by a smaller drop of ΔΨm.
However, some routes might be energetically costly, because they involve a higher degradation rate and thus also a higher synthesis rate of mitochondrial components (to replace the degraded ones). Cells might therefore fine tune their rate of mitophagy to control a possible accumulation of mitochondrial damage by just as much as is energetically optimal for a specific species in a specific environment.
(e). Putting the theories to the test
There are many fundamental questions of mitophagy-mediated clearance of mutant mtDNA that researchers only recently have started to address. The first question is whether a bioenergetic deficiency as the consequence of mtDNA mutations is sufficient to elicit a mitophagy response? In a recent study, using 143B osteosarcoma cybrid cell lines carrying various pathogenic mtDNA mutations, it was shown that loss of ΔΨm alone was, in fact, not enough to stimulate mitophagy, but that it could be induced by co-treatment with the mTORC1 inhibitor rapamycin that stimulates macroautophagy [93]. The cybrids in question were all homoplasmic for their respective mutation. Rapamycin only induced mitophagy in cybrids containing a large mtDNA deletion that could be shown to also have resulted in a substantial reduction in ΔΨm. By contrast, in cybrids containing the NARP T8993G or the MELAS A3243G mutation the reduction of ΔΨm was much more moderate and rapamycin could not induce mitophagy. Unfortunately, in this study, it could not be tested whether rapamycin would specifically select against mutant mtDNA. But in a second, very recent study, it was shown that treatment with rapamycin in a neuroblastoma-derived cybrid line (SH-SY5Y) carrying the LHON G11778A mutation, selected against the mutant allele [94]. Based on these findings, it has been suggested that the sensing of deteriorated mitochondrial function is not sufficient to trigger mitophagy but that activation of macroautophagy is also required [95]. Interestingly, in the study by Gilkerson et al. [93], it was shown that loss of ΔΨm did result in recruitment of Parkin but without induction of selective mitophagy. Overall, levels of Parkin in cybrid lines carrying mtDNA mutations or in the mtDNA-less parental cell line however were reduced compared with the wt mtDNA cybrid counterpart. This fits with an earlier study where it was shown that Parkin overexpression in 143B cybrids containing a heteroplasmic mtDNA variant with a pathogenic COXI mutation, resulted in selective elimination of mutant mtDNA [29]. Although, these studies used immortal tumour cell lines and an important question is how these findings apply to primary cells and tissues in the body, they do provide a proof of concept that, once activated, mitophagy can selectively degrade mitochondria or mitochondrial fragments that are deficient owing to a mtDNA mutation, resulting in a reduction of mutant mtDNA levels. This implies that the mitophagy pathway can indeed distinguish between wt and mutant mtDNA alleles, which in the framework of the ‘leaky–link’ hypothesis can be explained by focal OXPHOS deficiency as a consequence of linked nucleoid genotypes and phenotypes. The above studies do raise the question whether in some tissues an additional activation of macroautophagy might not be required and to what extent there is mutation selectivity in activation of mitophagy. In this context, tissue-specific differences in levels of, for example, Parkin expression could also be very relevant.
A second question concerns the involvement of the fusion and fission machinery. As outlined above, in order for mitophagy to be selective against mutant mtDNA molecules, the fission machinery would be required to generate fragments with only one or a few mtDNA molecules. To date, there are only a few reports that address the question of whether manipulation of the fusion and fission machinery affects mtDNA integrity and mutation accumulation. Confusingly, it appears that either inhibition of fusion or of fission results in loss of mtDNA integrity and accumulation of mutations, although the mechanisms might be dissimilar. One study showed that in a rhabdomyosarcoma carrying 75–80% of the MELAS A3243G mutations, knockdown of the profission proteins hDrp1 or human fission factor (hFis1) resulted in increased heteroplasmy levels of the mutant allele, which might be attributed to decreased mitophagic clearance of mutant mtDNA containing mitochondrial fragments [96], although this was not formally tested. Alternatively, inhibition of fission by dynamin-related protein-1 (Drp1) knockdown might result in a higher threshold of expression of an OXPHOS phenotype as it also seems to promote nucleoid clustering and thus perhaps functional complementation [96,97],
Conditional knockouts of Mfn1 and 2 in mouse skeletal muscle result in the accumulation of mutations as well as severe mtDNA depletion in double knockout mice [87]. This seems counterintuitive considering that a more fragmented mitochondrial network might stimulate selective mitophagy and thus would help in selecting against mutant mtDNA molecules. However, it seems, in this case, that the lack of fusion capability might result in a primary defect in mtDNA replication [87,98]. One possible reason could be that indeed the mitochondrial proteome is no longer equilibrated over the fragmented mitochondria [87]. This would be in agreement with the proposal that protein mixing and equilibration is the principal evolutionary function of fusion ([35] and above). Increased mutation levels, in this case, would be a direct consequence of this defect rather than be the result of a failure to remove mutant alleles or a selective advantage. Clearly, much more work needs to be done to understand the interplay between the mitochondrial fusion and fission apparatus, nucleoids and the maintenance of mtDNA integrity before we are able to understand how mitochondrial dynamics and mtDNA integrity are balanced.
A fundamental tenet of the ‘leaky–link’ hypothesis is that nucleoids do have a limited sphere of influence. To date, there is no formal proof, although indirect evidence as discussed above suggests that this is indeed the case. The use of primary fibroblasts from patients with heteroplasmic pathogenic mtDNA mutations would allow this to be proved. As nucleoids in primary fibroblasts appear to contain mostly single mtDNA copies and the mitochondrial network is well spread, it should, in principle, be possible using super-resolution fluorescence microscopy or electron microscopy to demonstrate focal deficiencies either of enzyme activities or of concentrations of individual OXPHOS proteins or protein complexes. If we are hoping to fully understand how mitochondrial quality control might deal with mtDNA mutations and to be able to manipulate this quality control system to purge mutant mtDNA molecules, addressing this question should have the highest possible priority.
Funding statement
J.N.S. is supported by the Academy of Finland (CoE funding) and the Netherlands Organization for Scientific Research (NWO: VICI grant no. 865.10.004). K.B. was supported by a DFG grant to Karin Busch (Bu2288/1-1) and the SFB 944.
Endnote
Presence of a mixture of more than one type of an organellar genome (mitochondrial DNA (mtDNA) or plastid DNA) within a cell.
References
- 1.Schwartz RM, Dayhoff MO. 1978. Origins of prokaryotes, eukaryotes, mitochondria, and chloroplasts. Science 199, 395–403. ( 10.1126/science.202030) [DOI] [PubMed] [Google Scholar]
- 2.Anderson S, et al. 1981. Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. ( 10.1038/290457a0) [DOI] [PubMed] [Google Scholar]
- 3.Comfort A. 1974. The position of aging studies. Mech. Ageing Dev. 3, 1–31. ( 10.1016/0047-6374(74)90002-5) [DOI] [PubMed] [Google Scholar]
- 4.Wallace DC. 2010. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 51, 440–450. ( 10.1002/em.20586) [DOI] [PubMed] [Google Scholar]
- 5.Wallace DC. 1999. Mitochondrial diseases in man and mouse. Science 283, 1482–1488. ( 10.1126/science.283.5407.1482) [DOI] [PubMed] [Google Scholar]
- 6.Cortopassi GA, Shibata D, Soong NW, Arnheim N. 1992. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl Acad. Sci. USA 89, 7370–7374. ( 10.1073/pnas.89.16.7370) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee HC, Pang CY, Hsu HS, Wei YH. 1994. Differential accumulations of 4977 bp deletion in mitochondrial DNA of various tissues in human ageing. Biochim. Biophys. Acta 1226, 37–43. [DOI] [PubMed] [Google Scholar]
- 8.Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. 1998. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann. Neurol. 43, 217–223. ( 10.1002/ana.410430212) [DOI] [PubMed] [Google Scholar]
- 9.Wanagat J, Cao Z, Pathare P, Aiken JM. 2001. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 15, 322–332. ( 10.1096/fj.00-0320com) [DOI] [PubMed] [Google Scholar]
- 10.Khrapko K, et al. 1999. Cell by cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 27, 2434–2441. ( 10.1093/nar/27.11.2434) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gokey NG, Cao Z, Pak JW, Lee D, McKiernan SH, McKenzie D, Weindruch R, Aiken JM. 2004. Molecular analyses of mtDNA deletion mutations in microdissected skeletal muscle fibers from aged rhesus monkeys. Aging Cell 3, 319–326. ( 10.1111/j.1474-9728.2004.00122.x) [DOI] [PubMed] [Google Scholar]
- 12.Cao Z, Wanagat J, McKiernan SH, Aiken JM. 2001. Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: analysis by laser-capture microdissection. Nucleic Acids Res. 29, 4502–4508. ( 10.1093/nar/29.21.4502) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herbst A, Pak JW, McKenzie D, Bua E, Bassiouni M, Aiken JM. 2007. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J. Gerontol. A 62, 235–245. ( 10.1093/gerona/62.3.235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDonald SA, Preston SL, Greaves LC, Leedham SJ, Lovell MA, Jankowski JA, Turnbull DM, Wright NA. 2006. Clonal expansion in the human gut: mitochondrial DNA mutations show us the way. Cell Cycle 5, 808–811. ( 10.4161/cc.5.8.2641) [DOI] [PubMed] [Google Scholar]
- 15.Greaves LC, et al. 2006. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc. Natl Acad. Sci. USA 103, 714–719. ( 10.1073/pnas.0505903103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor RW, et al. 2003. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Invest. 112, 1351–1360. ( 10.1172/JCI19435) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holt IJ, Speijer D, Kirkwood TBL. 2014. The road to rack and ruin: selecting deleterious mitochondrial DNA variants. Phil. Trans. R. Soc. B 369, 20130451 ( 10.1098/rstb.2013.0451) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bandy B, Davison AJ. 1990. Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Radic. Biol. Med. 8, 523–539. [DOI] [PubMed] [Google Scholar]
- 19.Arnheim N, Cortopassi G. 1992. Deleterious mitochondrial DNA mutations accumulate in aging human tissues. Mutat. Res. 275, 157–167. ( 10.1016/0921-8734(92)90020-P) [DOI] [PubMed] [Google Scholar]
- 20.Elson JL, Samuels DC, Turnbull DM, Chinnery PF. 2001. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am. J. Hum. Genet. 68, 802–806. ( 10.1086/318801) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chinnery PF, Samuels DC. 1999. Relaxed replication of mtDNA: a model with implications for the expression of disease. Am. J. Hum. Genet. 64, 1158–1165. ( 10.1086/302311) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kowald A, Kirkwood TB. 2013. Mitochondrial mutations and aging: random drift is insufficient to explain the accumulation of mitochondrial deletion mutants in short-lived animals. Aging Cell 12, 728–731. ( 10.1111/acel.12098) [DOI] [PubMed] [Google Scholar]
- 23.Wallace DC. 1992. Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science 256, 628–632. ( 10.1126/science.1533953) [DOI] [PubMed] [Google Scholar]
- 24.Lee CM, Lopez ME, Weindruch R, Aiken JM. 1998. Association of age-related mitochondrial abnormalities with skeletal muscle fiber atrophy. Free Radic. Biol. Med. 25, 964–972. ( 10.1016/S0891-5849(98)00185-3) [DOI] [PubMed] [Google Scholar]
- 25.Kowald A, Dawson M, Kirkwood TB. 2014. Mitochondrial mutations and ageing: can mitochondrial deletion mutants accumulate via a size based replication advantage? J. Theor. Biol. 340, 111–118. ( 10.1016/j.jtbi.2013.09.009) [DOI] [PubMed] [Google Scholar]
- 26.de Grey ADNJ. 1997. A proposed refinement of the mitochondrial free radical theory of aging. Bioessays 19, 161–166. ( 10.1002/bies.950190211) [DOI] [PubMed] [Google Scholar]
- 27.Twig G, et al. 2008. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. ( 10.1038/sj.emboj.7601963) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim I, Lemasters JJ. 2011. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid. Redox Signal. 14, 1919–1928. ( 10.1089/ars.2010.3768) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. 2010. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc. Natl Acad. Sci. USA 107, 11 835–11 840. ( 10.1073/pnas.0914569107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kowald A, Kirkwood TB. 2014. Transcription could be the key to the selection advantage of mitochondrial deletion mutants in aging. Proc. Natl Acad. Sci. USA 111, 2972–2977. ( 10.1073/pnas.1314970111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spelbrink JN. 2010. Functional organization of mammalian mitochondrial DNA in nucleoids: history, recent developments, and future challenges. IUBMB Life 62, 19–32. ( 10.1002/iub.282) [DOI] [PubMed] [Google Scholar]
- 32.Iborra FJ, Kimura H, Cook PR. 2004. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2, 9 ( 10.1186/1741-7007-2-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bogenhagen DF, Rousseau D, Burke S. 2008. The layered structure of human mitochondrial DNA nucleoids. J. Biol. Chem. 283, 3665–3675. ( 10.1074/jbc.M708444200) [DOI] [PubMed] [Google Scholar]
- 34.Hensen F, Cansiz S, Gerhold JM, Spelbrink JN. 2013. To be or not to be a nucleoid protein: a comparison of mass-spectrometry based approaches in the identification of potential mtDNA-nucleoid associated proteins. Biochimie 100, 219 226–10 242 ( 10.1016/j.biochi.2013.09.017) [DOI] [PubMed] [Google Scholar]
- 35.Kowald A, Kirkwood TB. 2011. Evolution of the mitochondrial fusion-fission cycle and its role in aging. Proc. Natl Acad. Sci. USA 108, 10 237–10 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Legros F, Malka F, Frachon P, Lombes A, Rojo M. 2004. Organization and dynamics of human mitochondrial DNA. J. Cell Sci. 117, 2653–2662. ( 10.1242/jcs.01134) [DOI] [PubMed] [Google Scholar]
- 37.Kukat C, Wurm CA, Spåhr H, Falkenberg M, Larsson N-G, Jakobs S. 2011. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl Acad. Sci. USA 108, 13 534–13 539. ( 10.1073/pnas.1109263108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Appelhans T, Richter CP, Wilkens V, Hess ST, Piehler J, Busch KB. 2012. Nanoscale organization of mitochondrial microcompartments revealed by combining tracking and localization microscopy. Nano Lett. 12, 610–616. ( 10.1021/nl203343a) [DOI] [PubMed] [Google Scholar]
- 39.Wilkens V, Kohl W, Busch K. 2013. Restricted diffusion of OXPHOS complexes in dynamic mitochondria delays their exchange between cristae and engenders a transitory mosaic distribution. J. Cell Sci. 126, 103–116. ( 10.1242/jcs.108852) [DOI] [PubMed] [Google Scholar]
- 40.Bereiter-Hahn J, Voth M. 1994. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc. Res. Tech. 27, 198–219. ( 10.1002/jemt.1070270303) [DOI] [PubMed] [Google Scholar]
- 41.Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, Jendrach M. 2010. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 123, 917–926. ( 10.1242/jcs.059246) [DOI] [PubMed] [Google Scholar]
- 42.Lin MT, Beal MF. 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. ( 10.1038/nature05292) [DOI] [PubMed] [Google Scholar]
- 43.Knott AB, Bossy-Wetzel E. 2008. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann. N.Y. Acad. Sci. 1147, 283–292. ( 10.1196/annals.1427.030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oettinghaus B, Licci M, Scorrano L, Frank S. 2012. Less than perfect divorces: dysregulated mitochondrial fission and neurodegeneration. Acta Neuropathol. 123, 189–203. ( 10.1007/s00401-011-0930-z) [DOI] [PubMed] [Google Scholar]
- 45.Scorrano L. 2003. Divide et impera: Ca2+ signals, mitochondrial fission and sensitization to apoptosis. Cell Death Differ. 10, 1287–1289. ( 10.1038/sj.cdd.4401310) [DOI] [PubMed] [Google Scholar]
- 46.Desagher S, Martinou JC. 2000. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 10, 369–377. ( 10.1016/S0962-8924(00)01803-1) [DOI] [PubMed] [Google Scholar]
- 47.Margineantu DH, Gregory Cox W, Sundell L, Sherwood SW, Beechem JM, Capaldi RA. 2002. Cell cycle dependent morphology changes and associated mitochondrial DNA redistribution in mitochondria of human cell lines. Mitochondrion 1, 425–435. ( 10.1016/S1567-7249(02)00006-5) [DOI] [PubMed] [Google Scholar]
- 48.Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW, Walter P. 1997. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell 8, 1233–1242. ( 10.1091/mbc.8.7.1233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zunino R, Braschi E, Xu L, McBride HM. 2009. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J. Biol. Chem. 284, 17 783–17 795. ( 10.1074/jbc.M901902200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arakaki N, et al. 2006. Dynamics of mitochondria during the cell cycle. Biol. Pharm. Bull. 29, 1962–1965. ( 10.1248/bpb.29.1962) [DOI] [PubMed] [Google Scholar]
- 51.Arimura S, Yamamoto J, Aida GP, Nakazono M, Tsutsumi N. 2004. Frequent fusion and fission of plant mitochondria with unequal nucleoid distribution. Proc. Natl Acad. Sci. USA 101, 7805–7808. ( 10.1073/pnas.0401077101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheahan MB, McCurdy DW, Rose RJ. 2005. Mitochondria as a connected population: ensuring continuity of the mitochondrial genome during plant cell dedifferentiation through massive mitochondrial fusion. Plant J. 44, 744–755. ( 10.1111/j.1365-313X.2005.02561.x) [DOI] [PubMed] [Google Scholar]
- 53.Chen H, Chomyn A, Chan DC. 2005. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 280, 26 185–26 192. ( 10.1074/jbc.M503062200) [DOI] [PubMed] [Google Scholar]
- 54.Tatsuta T, Langer T. 2008. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 27, 306–314. ( 10.1038/sj.emboj.7601972) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meeusen S, McCaffery JM, Nunnari J. 2004. Mitochondrial fusion intermediates revealed in vitro. Science 305, 1747–1752. ( 10.1126/science.1100612) [DOI] [PubMed] [Google Scholar]
- 56.Malka F, Guillery O, Cifuentes-Diaz C, Guillou E, Belenguer P, Lombes A, Rojo M. 2005. Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 6, 853–859. ( 10.1038/sj.embor.7400488) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishihara N, Jofuku A, Eura Y, Mihara K. 2003. Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochem. Biophys. Res. Commun. 301, 891–898. ( 10.1016/S0006-291X(03)00050-0) [DOI] [PubMed] [Google Scholar]
- 58.Hayashi J, Takemitsu M, Goto Y, Nonaka I. 1994. Human mitochondria and mitochondrial genome function as a single dynamic cellular unit. J. Cell Biol. 125, 43–50. ( 10.1083/jcb.125.1.43) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Molina AJ, Shirihai OS. 2009. Monitoring mitochondrial dynamics with photoactivatable green fluorescent protein. Methods Enzymol. 457, 289–304. ( 10.1016/S0076-6879(09)05016-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Twig G, et al. 2006. Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am. J. Phys. Cell Phys. 291, C176–C184. [DOI] [PubMed] [Google Scholar]
- 61.Busch KB, Bereiter-Hahn J, Wittig I, Schagger H, Jendrach M. 2006. Mitochondrial dynamics generate equal distribution but patchwork localization of respiratory complex I. Mol. Membr. Biol. 23, 509–520. ( 10.1080/09687860600877292) [DOI] [PubMed] [Google Scholar]
- 62.Muster B, Kohl W, Wittig I, Strecker V, Joos F, Haase W, Bereiter-Hahn J, Busch K. 2010. Respiratory chain complexes in dynamic mitochondria display a patchy distribution in life cells. PLoS ONE 5, e11910 ( 10.1371/journal.pone.0011910) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dieteren CEJ, Gielen SCAM, Nijtmans LGJ, Smeitink JAM, Swarts HG, Brock R, Willems PHGM, Koopman WJH. 2011. Solute diffusion is hindered in the mitochondrial matrix. Proc. Natl Acad. Sci. USA 108, 8657–8662. ( 10.1073/pnas.1017581108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bereiter-Hahn J, Vöth M. 1997. Distribution and dynamics of mitochondrial nucleoids in animal cells in culture. Exp. Biol. 1, 1–17. [Google Scholar]
- 65.Brown TA, Tkachuk AN, Shtengel G, Kopek BG, Bogenhagen DF, Hess HF, Clayton DA. 2011. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol. Cell Biol. 31, 4994–5010. ( 10.1128/mcb.05694-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rajala N, Gerhold JM, Martinsson P, Klymov A, Spelbrink JN. 2013. Replication factors transiently associate with mtDNA at the mitochondrial inner membrane to facilitate replication. Nucleic Acids Res. 42, 952–967. ( 10.1093/nar/gkt988) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Albring M, Griffith J, Attardi G. 1977. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc. Natl Acad. Sci. USA 74, 1348–1352. ( 10.1073/pnas.74.4.1348) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nass MM. 1969. Mitochondrial DNA. I. Intramitochondrial distribution and structural relations of single- and double-length circular DNA. J. Mol. Biol. 42, 521–528. [DOI] [PubMed] [Google Scholar]
- 69.Busch KB, Deckers-Hebestreit G, Hanke GT, Mulkidjanian AY. 2013. Dynamics of bioenergetic microcompartments. Biol. Chem. 394, 163–188. ( 10.1515/hsz-2012-0254) [DOI] [PubMed] [Google Scholar]
- 70.Okamoto K, Perlman PS, Butow RA. 1998. The sorting of mitochondrial DNA and mitochondrial proteins in zygotes: preferential transmission of mitochondrial DNA to the medial bud. J. Cell Biol. 142, 613–623. ( 10.1083/jcb.142.3.613) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kopek BG, Shtengel G, Xu CS, Clayton DA, Hess HF. 2012. Correlative 3D superresolution fluorescence and electron microscopy reveal the relationship of mitochondrial nucleoids to membranes. Proc. Natl Acad. Sci. USA 109, 6136–6141. ( 10.1073/pnas.1121558109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kopek BG, Shtengel G, Grimm JB, Clayton DA, Hess HF. 2013. Correlative photoactivated localization and scanning electron microscopy. PLoS ONE 8, e77209 ( 10.1371/journal.pone.0077209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Youle RJ, van der Bliek AM. 2012. Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. ( 10.1126/science.1219855) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mitchell P. 1966. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. Camb. Philos. Soc. 41, 445–502. ( 10.1111/j.1469-185X.1966.tb01501.x) [DOI] [PubMed] [Google Scholar]
- 75.Rugarli EI, Langer T. 2012. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 31, 1336–1349. ( 10.1038/emboj.2012.38) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Narendra DP, Youle RJ. 2011. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 14, 1929–1938. ( 10.1089/ars.2010.3799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jin SM, Youle RJ. 2012. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 125, 795–799. ( 10.1242/jcs.093849) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yaffe MP. 1999. Dynamic mitochondria. Nat. Cell Biol. 1, E149–E150. ( 10.1038/14101) [DOI] [PubMed] [Google Scholar]
- 79.Jensen RE, Hobbs AE, Cerveny KL, Sesaki H. 2000. Yeast mitochondrial dynamics: fusion, division, segregation, and shape. Microsc. Res. Tech. 51, 573–583. ( 10.1002/1097-0029(20001215)51:6%3C573::AID-JEMT7%3E3.0.CO;2-2) [DOI] [PubMed] [Google Scholar]
- 80.Shaw JM, Nunnari J. 2002. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 12, 178–184. ( 10.1016/S0962-8924(01)02246-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karbowski M, Youle RJ. 2003. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 10, 870–880. ( 10.1038/sj.cdd.4401260) [DOI] [PubMed] [Google Scholar]
- 82.Perfettini JL, Roumier T, Kroemer G. 2005. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol. 15, 179–183. ( 10.1016/j.tcb.2005.02.005) [DOI] [PubMed] [Google Scholar]
- 83.Rube DA, van der Bliek AM. 2004. Mitochondrial morphology is dynamic and varied. Mol. Cell. Biochem. 256-257, 331–339. [DOI] [PubMed] [Google Scholar]
- 84.Collins TJ, Berridge MJ, Lipp P, Bootman MD. 2002. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 21, 1616–1627. ( 10.1093/emboj/21.7.1616) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Distelmaier F, Koopman WJ, Testa ER, de Jong AS, Swarts HG, Mayatepek E, Smeitink JA, Willems PH. 2008. Life cell quantification of mitochondrial membrane potential at the single organelle level. Cytometry A 73, 129–138. ( 10.1002/cyto.a.20503) [DOI] [PubMed] [Google Scholar]
- 86.Bereiter-Hahn J, Vöth M. 1998. Do single mitochondria contain zones with different membrane potential? Exp. Biol. 3, 12. [Google Scholar]
- 87.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. 2010. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141, 280–289. ( 10.1016/j.cell.2010.02.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Woldringh CL. 2002. The role of co-transcriptional translation and protein translocation (transertion) in bacterial chromosome segregation. Mol. Microbiol. 45, 17–29. ( 10.1046/j.1365-2958.2002.02993.x) [DOI] [PubMed] [Google Scholar]
- 89.Rouzier C, et al. 2012. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain 135, 23–34. ( 10.1093/brain/awr323) [DOI] [PubMed] [Google Scholar]
- 90.Hudson G, et al. 2008. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain 131, 329–337. ( 10.1093/brain/awm272) [DOI] [PubMed] [Google Scholar]
- 91.Amati-Bonneau P, et al. 2008. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 131, 338–351. ( 10.1093/brain/awm298) [DOI] [PubMed] [Google Scholar]
- 92.Skulachev VP. 2001. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem. Sci. 26, 23–29. ( 10.1016/S0968-0004(00)01735-7) [DOI] [PubMed] [Google Scholar]
- 93.Gilkerson RW, De Vries RLA, Lebot P, Wikstrom JD, Torgyekes E, Shirihai OS, Przedborski S, Schon EA. 2012. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 21, 978–990. ( 10.1093/hmg/ddr529) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dai Y, Zheng K, Clark J, Swerdlow RH, Pulst SM, Sutton JP, Shinobu LA, Simon DK. 2013. Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum. Mol. Genet. 23, 637–647 ( 10.1093/hmg/ddt450) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Vries RL, Gilkerson RW, Przedborski S, Schon EA. 2012. Mitophagy in cells with mtDNA mutations: being sick is not enough. Autophagy 8, 699–700. ( 10.4161/auto.19470) [DOI] [PubMed] [Google Scholar]
- 96.Malena A, Loro E, Di Re M, Holt IJ, Vergani L. 2009. Inhibition of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum. Mol. Genet. 18, 3407–3416. ( 10.1093/hmg/ddp281) [DOI] [PubMed] [Google Scholar]
- 97.Ban-Ishihara R, Ishihara T, Sasaki N, Mihara K, Ishihara N. 2013. Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc. Natl Acad. Sci. USA 110, 11 863–11 868. ( 10.1073/pnas.1301951110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vielhaber S, et al. 2013. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol. 125, 245–256. ( 10.1007/s00401-012-1036-y) [DOI] [PubMed] [Google Scholar]