

Abstract
Designing and utilization of biomimetic membrane systems generated by bottom-up processes is a rapidly growing scientific and engineering field. Elucidation of the supramolecular construction principle of archaeal cell envelopes composed of S-layer stabilized lipid membranes led to new strategies for generating highly stable functional lipid membranes at meso- and macroscopic scale. In this review, we provide a state-of-the-art survey of how S-layer proteins, lipids and polymers may be used as basic building blocks for the assembly of S-layer-supported lipid membranes. These biomimetic membrane systems are distinguished by a nanopatterned fluidity, enhanced stability and longevity and, thus, provide a dedicated reconstitution matrix for membrane-active peptides and transmembrane proteins. Exciting areas in the (lab-on-a-) biochip technology are combining composite S-layer membrane systems involving specific membrane functions with the silicon world. Thus, it might become possible to create artificial noses or tongues, where many receptor proteins have to be exposed and read out simultaneously. Moreover, S-layer-coated liposomes and emulsomes copying virus envelopes constitute promising nanoformulations for the production of novel targeting, delivery, encapsulation and imaging systems.
Keywords: biomimetics, functionalized interfaces, nanobiotechnology, supported lipid membrane, crystalline cell surface layer, synthetic biology
1. Introduction
Supramolecular architectures building up biological systems offer a dazzling array of ideas and inspiration for nanotechnology. A pivotal role concerning the functionality in biological systems is accorded to (membrane) proteins. For micro- and nano-systems, several key factors such as material interface properties, preserving biological viability, as well as self-assembly and bottom-up strategy as a device-fabrication methodology must be considered [1].
Proteins, as natural building units, have attracted a significant amount of attention owing to their unique features, such as conferring elasticity, promoting cross-linking, facilitating material degradation, selectivity, fostering biomineralization, providing specific binding functions and in particular self-assembly [2]. Integration of these units with non-living systems will promise to bridge the worlds of conventional engineering and biology and could dramatically contribute to the development of both. On the one hand, more powerful tools to study, handle and engineer these natural units could be obtained, while, on the other hand, it may be possible to fabricate novel electronic, optic, high-throughput screening, sequencing, drug targeting and delivery, and sensory nano-systems by enhancing current bottom-up techniques [1].
The importance of cell membranes in biological systems has prompted the development of model membrane platforms that recapitulate fundamental aspects of membrane biology, especially the lipid bilayer environment. Supported lipid mono- and bilayers represent one of the most promising classes of model membranes and are based on the immobilization of a planar lipid membrane on a solid support that enables characterization by a wide range of surface-sensitive analytical techniques. Moreover, as the result of molecular engineering inspired by biology, supported lipid membranes are increasingly able to mimic fundamental properties of natural cell membranes, including fluidity, electrical sealing and hosting integral membrane proteins (MPs). At the same time, new methods have been used to improve the durability of lipid membranes, with shelf-lives now reaching the order of weeks. The capabilities of supported lipid membranes have opened the door to biotechnology applications in medicine, diagnostics, sensor systems, environmental monitoring and energy storage [3–5].
The challenge of integrating non-living systems with biological ones led to problems associated with material interfacing and compatibility, as well as biological issues, such as viability and stability. Furthermore, the integration is also complicated by scaling effects and dynamic interactions between different components. Moreover, water is necessary to maintain biological units, but has adverse effects on engineering components. Although elements used in the biological world are relatively simple, the phenomenon of their assembly is astonishingly diverse. Self-assembly and bottom-up are the most common strategies used for organization of biological units. The extension of this strategy to the creation of hybrid devices is thought to be most promising.
Complex and versatile biological units like cells have extremely elaborate ways to self-assemble functional units like the cell envelope structure. Biomimetic membranes incorporate biological elements or borrow concepts, ideas or inspiration from biological systems [6]. Such membranes can take advantage of the strategies evolved by nature over billions of years. This review is focused on the biomimetic approach of applying the building principle of cell envelope structure for the generation of a versatile biological unit—a protein-supported lipid membrane. The used protein species, termed S-layers (S- is the abbreviation of ‘surface’), are defined as ‘two-dimensional arrays of proteinaceous subunits forming surface layers on prokaryotic cells’ [5,7,8] and are found as the outermost structure in hundreds of different species of almost every taxonomic group of walled bacteria (figure 1) and are an almost universal feature of archaea [4,9–12]. Interestingly, many bacterial and most archaeal S-layer proteins are glycosylated [13–15].
Figure 1.

Transmission electron microscopy image of a freeze-etched and metal shadowed preparation of (a) an archaeal cell (from Methanocorpusculum sinense) and (b) a bacterial cell (from Desulfotomaculum nigrificans). Bars, 200 nm. (Adapted from [5]. Copyright © 2014 with permission from John Wiley & Sons Ltd.)
Nonetheless, not only prokaryotic bacteria and archaea (figure 2) have this kind of oriented self-assembly of proteins at their outermost envelope, but also many viruses do present a regular and well-defined protein layer as the outermost envelope structure.
Figure 2.
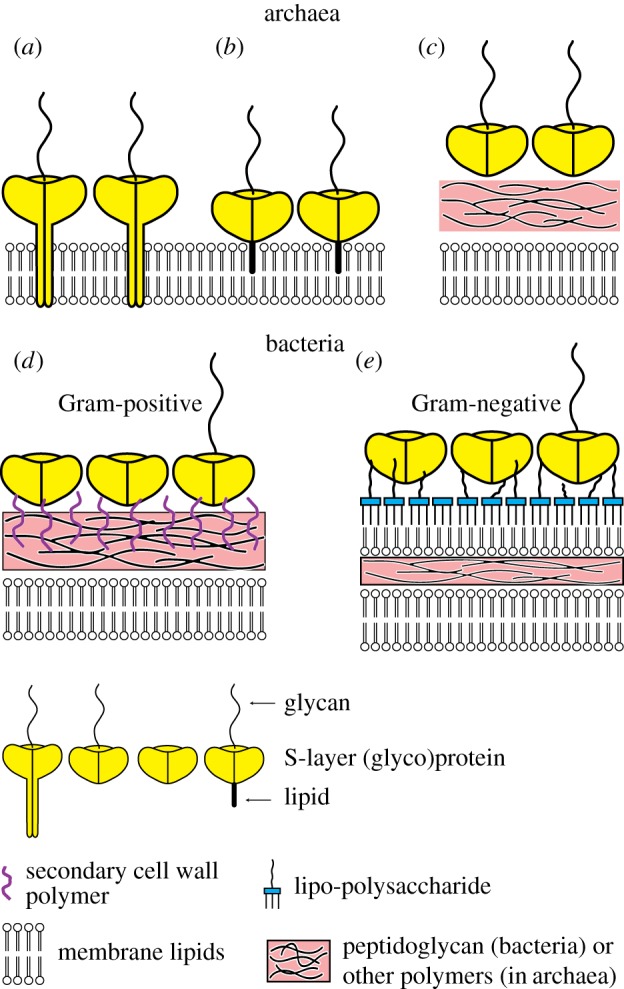
Prokaryotic cell envelopes: schematic of the supramolecular architecture of the major classes of prokaryotic cell envelopes containing surface (S) layers. S-layers in archaea with glycoprotein lattices as an exclusive wall component are composed either of mushroom-like subunits with pillar-like, hydrophobic transmembrane domains (a) or lipid-modified glycoprotein subunits (b). Individual S-layers can be composed of glycoproteins possessing both types of membrane-anchoring mechanisms. Few archaea possess a rigid wall layer (e.g. pseudomurein in methanogenic organisms) as an intermediate layer between the plasmatic membrane and the S-layer (c). In Gram-positive bacteria (d) the S-layer (glyco)proteins are bound to the rigid peptidoglycan-containing layer via secondary cell wall polymers (SCWPs). In Gram-negative bacteria (e) the S-layer is closely associated with the lipopolysaccharide of the outer membrane. (Adapted from [5]. Copyright © 2014 with permission from John Wiley & Sons Ltd.)
In a biomimetic approach copying the supramolecular building principle of archaeal cell envelope structures (figure 3a), two S-layer-supported lipid membranes (SsLMs) differing in their overall shape and hence function have been generated [4,17,18]. First, the planar SsLM comprising a phospho- or tetraether lipid monolayer (figure 3b,c,e) or bilayer (e.g. phospholipid; figure 3d,f) and a closely attached S-layer lattice (figure 3b–e). The S-layer lattice acts as a stabilizing and anchoring scaffold for the lipid membranes. In the case of surface-attached membranes (figure 3e,f), the S-layer also constitutes a biological cushion covering the inorganic support, provides an ionic reservoir and antifouling surface, allows lateral diffusion of membrane lipids and incorporation of integral MPs with domains protruding from the attached lipid membrane.
Figure 3.

Supramolecular structure of an archaeal cell envelope architecture comprising a cytoplasmic membrane, archaeal S-layer proteins incorporated in the lipidic matrix and integral MPs (a). Schematic of various S-layer-supported lipid membranes. (b) Lipid monolayer films at the air/water interphase with an underneath recrystallized S-layer lattice. (1) Tetraether lipid monolayer in the upright conformation. (2) Tetraether lipid monolayer in the U-shaped (bent) conformation. (3) Phospholipid monolayer. (c) A tetraether lipid monolayer membrane is generated across an orifice of a patch-clamp pipette by the tip-dip method. Subsequently a closely attached S-layer lattice is formed by bacterial S-layer proteins on one side of the lipid membrane. In (d), a folded or painted bilayer phospholipid membrane spanning a Teflon aperture is shown. A closed bacterial S-layer lattice can be self-assembled on either one or both (not shown) sides of the lipid membrane. (e) Schematic drawing of a solid support where a closed bacterial S-layer lattice has been assembled. On this biomimetic structure, a tetraether lipid membrane was generated by the modified Langmuir–Blodgett method. Optionally as shown on the left-hand side, a bacterial S-layer lattice can be attached on the external side of the solid-supported lipid membrane. (f) Schematic drawing of a bilayer lipid membrane generated on an S-layer ultrafiltration membrane (SUM). Optionally as shown on the left-hand side, a bacterial S-layer lattice can be attached on the external side of the SUM-supported lipid membrane. In (b–f), the head groups of the lipid molecules interacting with the S-layer protein are shown by a darker colour. As indicated in (c–f), all S-layer-supported model lipid membranes can be functionalized by biomolecules like membrane-active peptides and transmembrane proteins. (Adapted from [16]. Copyright © 2004 with permission from Wiley-VCH.)
The second S-layer/lipid assembly structures concern spherically shaped S-layer-coated liposomes and emulsomes (figure 4). In this manner, a biomimetic archaeal cell envelope or viral envelope composed of an S-layer shell, and some kind of lipidic core (liposome or emulsome), is assembled. These biomimetic nanocarrier systems are mainly designed for drug targeting and delivery and for imaging enhancement purposes [19,21–23]. Moreover, the proteinaceous surface of these nanocarriers not only has a stabilizing effect, but also provides the possibility to endow the liposome or emulsomes with a great variety of functionalities such as packed ligands, antigens (Ag), antibodies, enzymes and stealth molecules (figure 4c). Both types of SsLM may be functionalized with (antimicrobial) membrane-active peptides and peripheral or integral MPs.
Figure 4.

Left: Transmission electron microscopy images of emulsomes coated with the S-layer protein SbsB from G. stearothermophilus PV72/p2 (a) wild-type SbsB and (b) recombinant SbsB. The bars correspond to 100 nm. (Adapted from [19]. Copyright © 2013 with permission from Wiley-VCH.) (c) Schematic drawing of (1) an S-layer-coated emulsome (left) and Iiposome (right) with entrapped functional molecules and (2) functionalized by reconstituted integral proteins. Note, S-layer-coated emulsomes can only transport hydrophobic molecules but with a much higher transport capacity. S-layer-coated emulsomes and liposomes can be used as an immobilization matrix for functional molecules (e.g. human immunoglobulin G) either by direct binding (3) or by immobilization via the Fc-specific ligand protein A (4), or biotinylated ligands can be bound to the S-layer-coated liposome or emulsomes via the biotin–avidin system (5). Alternatively, emulsomes or liposomes can be coated with genetically modified S-layer subunits incorporating functional domains (6). (Adapted from [20]. Copyright © 2002 with permission from Wiley-VCH.)
Although both systems are capable of producing an electrical potential, the charge carriers are different, being ions and electrons, for functionalized lipid membranes and inorganic systems, respectively. These differences result in numerous challenges for their integration. These SsLMs, in particular the planar ones on a solid or porous support (figure 3e,f), are well suited for the development of peptide- or protein-based biosensors. Integrated with inorganic micro- and nano-systems, SsLMs may also constitute a key component for the development of the so-called lipid chips or lab-on-a-chip [24,25].
2. Biological envelope structures
In the following, the envelope structures of bacteria, archaea and viruses will be briefly described to enable the reader to become familiar with the diverse building principles that have evolved in nature. In this context, it is worth mentioning that the so-called Gram stain (the name comes from its inventor, Hans Christian Gram) is an important method of differentiating bacterial species into two large groups: Gram-positive (figure 2d) and Gram-negative (figure 2e). Differentiation is based on the supramolecular structure and consequently the chemical and physical properties of the cell walls by detecting peptidoglycan, which is present in a thick layer in Gram-positive bacteria. One prokaryote domain, the archaea, have such variability of wall structure that the Gram stain is not a useful differentiating tool [26].
2.1. Cell envelope structures of bacteria
Gram-negative bacteria are composed of a cytoplasmic membrane, a thin peptidoglycan layer, and an outer membrane containing lipopolysaccharide (LPS) in its outer leaflet and phospholipids in the inner leaflet (figure 2e). Porins exist in the outer membrane, which act like pores for particular molecules. Gram-positive bacteria, however, are composed of a cytoplasmic membrane and a rigid peptidoglycan-containing layer (murein; teichonic, teichuronic and lipoteichonic acid), which is much thicker than in Gram-negative bacteria (figure 2d) [27,28]. In addition, both types of bacteria may have an S-layer lattice as the outermost structure. The S-layer lattice is directly attached to the outer membrane and the peptidoglycan layer in Gram-negative and Gram-positive bacteria, respectively. In the latter, the rigid cell envelope layer is composed of peptidoglycan and accessory (secondary) cell wall polymers (SCWPs; figure 2d) [29]. The polymer chains are either covalently linked to the peptidoglycan backbone via phosphodiester bonds or tethered to a lipid anchor moiety [30]. By sequence comparison, S-layer-homologous (SLH) motifs [31] have been identified at the N-terminal part of many S-layer proteins [32–34]. It is now evident that SCWPs serve as anchoring structures for many S-layer proteins. In contrast to the SLH motifs of most S-layer proteins, which reveal a positive net charge, those of Bacillus sphaericus strains [34,35] are net negatively charged, which explains why bivalent cations are required for binding of the S-layer subunits to the rigid cell envelope layer [32]. In contrast to most S-layer proteins of Gram-positive bacteria, those of Geobacillus stearothermophilus strains [33,36,37] and lactobacillus [38–41] do not possess SLH motifs. Nevertheless, the N-terminal part of the S-layer proteins of G. stearothermophilus strains is highly conserved and recognizes a net negatively charged SCWP as the binding site [35,37,42].
2.2. Cell envelope structures of archaea
Many archaea dwell in extreme environments, for example at high pressures, salt concentrations or temperatures. Their cell wall differs in structure from that of bacteria and is thought to be more stable in extreme conditions, helping to explain why some archaea can live in many of the most hostile environments on Earth (figure 2a–c).
Studies of the archaeal cell envelope resulted in the recognition of a number of archaea-specific features, including a different lipid composition of the cytoplasmic membrane from bacteria and the lack of a general cell wall polymer, resulting in insensitivity to the most common bacterial cell wall-targeting antibiotics. In most archaea (e.g. Sulfolobus spp.), S-layer proteins are the exclusive cell envelope constituent (figure 2a), whereas in other archaea the cell envelope consists of multiple polymers, including the polysaccharides pseudomurein and methanochondroitin, and can also contain additional S-layer proteins (figure 2c) [9,12]. Archaeal S-layers are mostly composed of a single protein or glycoprotein species, which in many cases is associated with the cytoplasmic membrane. In haloarchaea, methanogens, Staphylothermus spp. and Thermoproteus spp. the main protein constituent of the S-layer is anchored by its carboxy-terminal transmembrane domain to the cytoplasmic membrane (figures 2a and 3a). The transmembrane domain is often preceded by a stretch of serine/threonine residues that are often glycosylated. In some cases, the S-layer is composed of two S-layer proteins; for example, in Sulfolobales spp. the S-layer is composed of the large outer protein which is linked to a small protein called SlaB [12,43]. SlaB, however, is anchored to the cytoplasmic membrane and forms a stalk about 20 nm long [44]. Recently, another type of binding of archaeal S-layer proteins to cytoplasmic membranes has been reported for Haloferax volcanii. It could be demonstrated that a subset of secreted euryarchaeal proteins, including the S-layer glycoprotein, is processed and covalently linked to membrane-embedded lipids involving membrane-spanning enzymes referred to as archaeosortases [45,46]. Because the C-terminal structure recognized by archaeosortases of a large number of euryarchaeal S-layer glycoproteins is highly conserved, it is very likely that this proposed lipid-anchoring mechanism is a broadly conserved surface-anchoring mechanism (figure 2b) [47–49].
2.3. Envelope structures of viruses
Many types of virus particles exist not as naked nucleocapsids but as nucleocapsids surrounded by lipid membranes. These membranes contain various viral-encoded glycoproteins and perform some subset of the functions that are required for successful viral spread. These structures comprise a lipid bilayer and associated proteins are referred to as viral envelopes. The major component of viral envelopes is one or in rare cases more lipid bilayers. The viral lipid bilayers are generally derived from pre-existing membranes of the host cell; therefore, the lipid components are taken from the cellular membrane. In many cases, the acquisition of an envelope occurs as the nucleocapsid buds out from the cytoplasm to the extracellular milieu [50]. The size of many viruses (e.g. influenza and many animal viruses) is 20–400 nm [51], in the same range as handmade lipid structure like liposomes or emulsomes (figure 4a,b).
3. Building blocks for biomimetic membranes
Life on Earth is basically composed of the four building blocks: amino acids, nucleic acids, carbohydrates and lipids. The self-assembly and interaction of different polymers of these basic molecules make up the majority of life's structure and function. From a very general point of view, nucleic acids store and transfer genetic information; carbohydrates store energy, reconstitute recognition systems and provide building materials; lipids make membranes and store energy; and proteins are structure elements and perform the chemistry of the cell [52]. Lipids are a loosely defined group of molecules with one main characteristic: they are insoluble in water. Phospholipids are, among lipids, the most important molecules of bacterial and mammalian cells, as they form, with the exception of archaea, the core of all biological membranes. Archaea, in contrast, are composed of ether- or tetraether lipids that show a higher resistance against chemical hydrolysis [53]. Proteins are the most abundant ones of the organic molecules, constituting about 50% of a cell's dry weight. A particular protein's overall conformation can be considered on four levels: primary, secondary, tertiary and quaternary structure. These levels of structure combine to create a complete protein that may serve many different functions within a cell. The building blocks lipids, proteins and carbohydrates (polymers) will be discussed later on the basis of their importance in creating biomimetic surfaces, in particular functional SsLMs.
3.1. Lipids
Natural bacterial cell membranes are complex structures composed of a variety of polymers, lipids and proteins. The weight ratio of protein to lipid varies from 20% to 70%; however, it is the lipid component that gives the membrane the gross morphology of a closed structure [54]. Lipids may not only be regarded as building materials or structural elements, they also have important functional tasks. Phospholipids, in many instances, constitute an important barrier function and are necessary for the stabilization and function of native membrane-bound proteins [55]. The lipidic components of natural cell membranes consist, among other minor components such as sphingolipids, glycol(sphingo)lipids and sterols, mainly of glycerolphospholipids, each differing in charge, acyl chain composition and physical properties [54,56].
However, the most frequently used lipid molecules for membrane formation are synthetic zwitterionic phospholipids carrying isoprene side chains like 1,2-diphytanoyl-sn-glycero-3-phosphatidylcholine (DPhPC; CAS number 207131-40-6) and, thus, forming fluid lamellar structures at biologically relevant ambient temperatures [54,57–59]. Other frequently used phospholipids are lecithin (PC) extracted from egg yolk or soya bean or unsaturated phosphatidylcholine like the zwitterionic palmitoyl-oleoyl-phosphatidylcholine (POPC; CAS number 26853-31-6) doped with small amounts of positively or negatively charged phospholipids such as phosphatidyl-ethanolamine (PE) or phosphatidylglycerol (PG) and -serine (PS), respectively [56,60]. Membrane lipids of archaea, however, are unique and distinct from those found in eukarya and bacteria. The polar lipids consist of isoprenoid chains, 20–40 carbons in length and usually saturated, which are attached via stable ether bonds to the glycerol carbons at the sn-2,3 positions [61]. Polar head groups differ at the genus level of diversity and consist of mixtures of glyco groups (mainly disaccharides), and/or phospho groups, primarily phosphoglycerol, phosphoserine, phosphoethanolamine or phosphoinositol. In this context, it is interesting to note that archaeal lipids comprise isoprene side chains which are bound via an ether linkage to the glycerol moiety of phospholipids. Furthermore, the so-called tetraether lipids, consisting of two different hydrophilic head groups, which are ether-linked by two C40 isoprenoidic chains having up to five cyclopentane rings, have been used to generate a monomolecular membrane (figure 3a–c,e) with the same overall structural features and barrier function as common phospholipid bilayers (figure 3d,f) [62,63]. Glycerol dialkyl nonitol tetraether lipid, extracted and purified from Sulfolobus and Metallosphaera strains and the main phospholipid isolated from Thermoplasma acidophilum, has also frequently been used for membrane formation. The advantage of etherlipids is their pronounced stability towards oxidative degradation, resistance against hydrolysis, even under extreme environmental conditions, and their fluid characteristics over a broad range of temperatures [64–66]. However, a disadvantage is that tetraether lipids may adopt instead an upright conformation (figure 3b(1)), an unstable so-called U-shaped (bent) conformation at the air/water interphase where both hydrophilic head groups interact with the aqueous subphase (figure 3b(2)) [64,67,68]. Thus, films comprising tetraether lipids may not be stable in terms of maintaining the adjusted surface pressure over time as permanent rearrangement of the lipid molecules occurs [62,69,70]. Owing to their amphiphilic nature, ether- and phospholipids can nicely be self-assembled at the air/water interphase with the hydrocarbon chains pointing towards the air and the polar head groups facing the aqueous phase (figure 3b(3)) [71,72].
3.2. S-layer proteins
Despite the diversity of cell envelope structures observed in prokaryotic organisms [28,73], among the most commonly observed cell surface structures are mono- or bimolecular arrays composed of identical species of protein or glycoprotein subunits (figure 1). Because S-layers account for approximately 10% of cellular proteins in bacteria and archaea and because the biomass of prokaryotic organisms surpasses the biomass of eukaryotic organisms [74], S-layers can be considered as one of the most abundant biopolymers on our planet [5,9]. S-layers also represent the simplest biological protein or glycoprotein membranes developed during evolution [75–77].
High-resolution electron and atomic force microscopy studies on the mass distribution of S-layer lattices revealed that the S-layer covers the entire cell surface as a coherent layer [78–81]. Most S-layers are monomolecular assemblies of single subunit species with a molecular weight ranging between 40 and 200 kDa. S-layer lattices generally exhibit oblique (p1, p2), square (p4) or hexagonal (p3, p6) space group symmetry with a centre-to-centre spacing of the morphological units of 3.5–35 nm [12,28,82]. Hexagonal lattice symmetry is predominant among archaea [9,83]. Depending on the lattice type the morphological units consist of one, two, three, four or six protein or glycoprotein monomers, respectively. Bacterial S-layers are generally 5–10 nm thick, whereas archaeal S-layers frequently exhibit a much thicker ‘mushroom-like structure’ with pillar-like domains [12]. Bacterial S-layers reveal a rather smooth outer and a more corrugated inner surface (figure 2d,e). Moreover, S-layers represent highly porous protein lattices (30–70% porosity) with pores of uniform size and morphology in the 2–8 nm range [35,84,85]. Many S-layers possess two or even more distinct classes of pores [12,20,28,82,86].
Biological functions of S-layers vary depending on the different S-layer lattices and organism and may include (i) protection against bacteriophages, bdellovibrios and phagocytosis, (ii) resistance against low pH, (iii) a barrier or protective coat against high- and low-molecular-weight substances (e.g. lytic enzymes), (iv) stabilization of the membrane, (v) provision of adhesion sites for exoproteins, and (vi) provision of a periplasmic compartment in Gram-positive prokaryotes together with the peptidoglycan and the cytoplasmic membranes [5,28,87–90]. Interestingly, in the S-layer of Deinococcus radiodurans ion-gating properties of microbial S-layer protein arrays have also been determined [91]. Ion transport appears to be mainly due to an electrical gradient inside the pores, presumably originating from the negative charges found on this S-layer lattice. The gating characteristics of the nanoporous membranes towards various ionic species were evaluated and revealed that the immobilized S-layers undergo a strong interaction with cations, in particular Ca2+ ions.
Recently, an outstanding antifouling characteristic of the S-layer protein of Lysinibacillus sphaericus CCM 2177 (SbpA) in the presence of high-protein solutions (e.g. 70 mg ml−1 human serum albumin), plasma and whole blood samples was observed. This finding is explained by the inherently (zwitterionic) neutral surface charge of the S-layer protein SbpA [92].
One important feature of S-layer proteins is the capability of isolated native or recombinantly produced subunits to self-assemble into crystalline arrays on many surfaces such as glass, silicon oxide and nitride, mica, noble metals like gold, titanium and platinum, but also on stainless steel or many polymers such as polystyrene, polyester and cellulose, and on technically relevant surfaces like highly oriented pyrolytic graphite or indium tin oxide [92,93]. Transmission electron microscopy (TEM) [94–96] and atomic force microscopy (AFM) [97–101] are the most appropriate techniques to elucidate the recrystallization process of S-layer proteins. Crystal growth at interfaces (e.g. solid supports, air–water interface or lipid membranes) is initiated simultaneously at many randomly distributed nucleation points, and proceeds in plane until the crystalline domains meet, thus leading to a closed, coherent mosaic, of individual, several micrometres large, S-layer domains [97,102–104]. The growth of extended S-layers domains is favoured at low monomer concentrations owing to the corresponding low number of nucleation sites. The individual domains are monocrystalline and separated by grain boundaries.
The formation of coherent crystalline domains depends on the S-layer protein species used, the environmental conditions of the subphase, such as ionic content and strength, the pH value and the surface properties of the interface. Interestingly, it was shown that a self-assembled layer, depending on whether the inner or outer side is exposed to the aqueous environment, can exhibit against cells in tissue cultures either cell adhesive (cytophilic) or cell repulsive (cytophobic) surface properties, respectively. The different orientation and function of the S-layer protein can simply be achieved by altering the recrystallization protocol from a basic (pH 9; resulting in an outer smooth cytophobic side) to an acidic (pH 4; resulting in an inner rough cytophilic surface pattern) condition [105].
While the reassembly of S-layer proteins at the air–water interface and at planar lipid films is well defined [95,96,106–108], the deliberate modification of the surface properties of a solid support allows the reassembly process to be specifically controlled [86,97,103,109]. For example, the S-layer protein SbpA, which is currently one of the most studied S-layer proteins for functionalizing solid supports, forms monolayers with a height of 9 nm on hydrophobic and double layers on hydrophilic silicon supports [97]. The height of the double layer structure is 15 nm and not twice the height of a monolayer, indicating that the two layers are resting on each other like two interdigitated toothed racks. Furthermore, in comparison with hydrophilic surfaces, the layer formation is much faster on hydrophobic supports, starting from many different nucleation sites and thus leading to a mosaic of small crystalline domains (two-dimensional powder) [75]. Along this line, the importance of the interplay between hydrophobic and hydrophilic regions was studied in detail by reassembling the S-layer protein SbpA on self-assembled monolayers (SAMs) on gold composed of dialkyl disulfide derivatives with different end groups (hydroxyl versus methyl) and lengths of the individual methylene bridges [110]. The formation of monolayers was observed when the hydrophobic methyl end groups surmounted the hydrophilic hydroxyl groups. On the contrary, double S-layers were formed when hydrophilic hydroxyl groups superseded the hydrophobic methyl end groups. The threshold for the transition between native and non-native S-layer parameters was four methylene bridges. Finally, it must be noted that different lattice constants were observed.
Self-assembled monolayers were also used to study the influence of the introduced surface chemistry [111]. The SAMs carried methyl, hydroxyl, carboxylic acid or mannose, respectively, as terminating functional groups. It was confirmed that electrostatic interaction (carboxylic acid functional groups) induces a faster adsorption than hydrophobic (methyl groups) or hydrophilic (hydroxyl groups) interaction—as already shown for the reattachment on the bacterial cell [75,112] and at liposomes and polyelectrolyte nanocapsules [113–116].
Although native S-layer proteins have already demonstrated their great potential as patterning elements and nanoscale building blocks, genetic approaches opened up the possibility of modifying and changing the natural properties of S-layer proteins [117,118]. However, although S-layer proteins incorporate insertions or fusions of foreign proteins or domains, the capability to assemble into geometrically well-defined layers must be retained. To date, two different strategies for the production of nanobiotechnologically relevant S-layer fusion proteins are pursued, namely homologous expression and secretion by the cells or production inside a host, mostly Escherichia coli [117]. By these means bio-inspired materials with designed functional properties can be produced. Moreover, the possibility to modify the natural properties of S-layer proteins by genetic manipulations and to incorporate single or multi-functional domains of other proteins has led to a broad spectrum of applications ranging from fluorescent biomarkers, immobilized biocatalysts, vaccine development, diagnostics and sensor development, to biosorption of heavy metals and nanoparticle arrays [5,21,79,119]. In this context, S-layer proteins may also be genetically engineered in order to introduce domains for the covalent binding of lipid molecules and, thus, enhancing the stability of the whole composite SsLM [17,18,120,121]. Table 1 summarizes the possibilities of how S-layer proteins and phospholipid molecules can be interconnected.
Table 1.
Summary of the strategies investigated to date to bind lipid molecules on S-layer protein lattices. SLP, S-layer protein; SMCC, succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate; SPDP, N-succinimidyl 3-(2-pyridyldithio)-propionate; TCEP, Tris (2-carboxyethyl) phosphine hydrochloride; NTA, nitrilotriacetic acid.
| type | reactive group | cross-linker | targeted group | reference |
|---|---|---|---|---|
| natural SLP | ||||
| electrostatic interaction | negatively charges on SLP | zwitterionic or positively charged lipids | [106,107,122] | |
| lectin-type like binding | S-layer-homologous domain on SLP | secondary cell wall polymer coupled to lipids | [123,124] | |
| chemical modification of SLPs | ||||
| covalent bond | carboxyl groups on SLP | carbodiimide analogues | primary amine group from lipids | [125–129] |
| covalent bond | primary amino groups on SLP | SMCC analogues | thiol group from lipids | [130] |
| covalent bond | primary amino groups on SLP | SPDP/TCEP; introduction of thiol group in SLP | maleimide group from lipids | personal communicationa |
| chemical binding of linker on SLPs | ||||
| strong ligation | streptavidin chemically coupled to SLP | biotinylated lipids | [115,131] | |
| genetically engineered SLPs | ||||
| covalent bond | thiol group from introduced cysteine | maleimide group from lipids | [98,132,133] | |
| multiple chelation | multiple histidines (His6-tag) on SLP | nickel(II)-NTA from lipids | [134] | |
| strong ligation | streptavidin fused to SLP | biotinylated lipids | [118] | |
| strong ligation | strep-tag fused to SLP | streptavidin | biotinylated lipids | [99–101] |
aB Schuster et al. 2014, personal communication.
3.3. Cell wall polymers
Studies on a great variety of S-layer proteins from Bacillaceae revealed the existence of specific binding domains on the N-terminal part for sugar polymers, the so-called SCWPs, which are covalently linked to the peptidoglycan of the cell wall [135]. These SCWPs mediate the oriented binding of S-layer proteins to the underlying cell wall.
For Gram-positive bacteria, at least two major types of binding mechanism between S-layer proteins and SCWPs have been described (figure 2d). This specific molecular interaction is often mediated by a recurring structural motif of approximately 55 amino acids, which is mostly found in triplicate at the N-terminus of S-layer proteins. These so-called SLH motifs are involved in cell wall anchoring of S-layer proteins by recognizing a distinct type of SCWP which carries pyruvic acid residues [32,112,136–140]. Moreover, the coexistence of two N-terminally located binding domains for SCWPs and peptidoglycans was also described for the SLH domain carrying S-layer protein of G. stearothermophilus PV72/p2 (SbsB) [29,141].
By contrast, S-layer proteins devoid of SLH motifs are anchored to different types of SCWP via their N- or C-terminal regions. By using affinity studies and surface plasmon resonance spectroscopy, a further main type of binding mechanism was described for G. stearothermophilus which involves a non-pyruvylated SCWP containing 2,3-diacetamido-2,3-dideoxymannuronic acid as the negatively charged component and a highly conserved N-terminal region lacking an SLH domain [36,37,42,142].
In Gram-negative bacteria, no general S-layer anchoring motif has been identified and the S-layer is attached with its N- or C-terminus to the LPS component of the outer membrane (figure 2e) [143–145]. For the Caulobacter crescentus S-layer protein (RsaA), recrystallization on lipid vesicles was obtained only when the vesicles contained the specific species of Caulobacter smooth LPS that previous studies implicated as a requirement for attaching the S-layer to the cell surface [146]. The specific type of phospholipid did not appear critical; phospholipids rather different from those present in Caulobacter membranes or archaeal ether lipids worked equally well. However, the source of LPS was critical. Furthermore, efficient recrystallization and long-range order could not be obtained with pure protein, though it was apparent that calcium was required for crystallization [146].
Few methanogens possess pseudomurein, a polymer with a thickness of approximately 15–20 nm that is similar to bacterial peptidoglycan. In these species (e.g. Methanosphera and Methanothermus), the S-layer proteins interact with the pseudomurein (figure 2c) [12]. However, most archaea, in particular the Sulfolobales and Thermoplasmatales spp., lack a polymer and the S-layer protein is anchored to the cytoplasmic membrane via a protein domain (figure 2a) or a lipid anchor bound to the S-layer protein (figure 2b) [49]. Interestingly, although the members of, for example, Thermoplasmatales do not possess a cell wall, these organisms dwell in a harsh environment such as under extremely low pH values (pH 1–2) and at temperatures of around 60° C [12].
3.4. Membrane-active peptides and integral membrane proteins
MPs as amphiphiles possess complex refolding processes that allow them to attain the three-dimensional configurations only in lipid membranes which are necessary for restoration of functionality. Moreover, these proteins are of major interest in medicine, diagnostics and the pharmaceutical industry. Approximately 60% of the over 430 drug targets that are presently known are MPs [147,148]. The latter are involved with cell sensing, signal transduction, immune recognition, transport of ions and nutrients, and a host of other vital process and, thus, in health and disease [149]. Among this 60%, G-protein-coupled receptors (GPCRs) are the most prevalent (19%), followed by ion channels (17%) and receptors (13%). Membrane-associated enzymes (6%), solute carriers and transporters (together approx. 5%) are also important drug targets [150]. Moreover, the results from the human proteome project suggest that more than 30% of proteins are membrane or membrane-associated proteins like pores, ion channels, membrane-anchored enzymes and most important (G-protein-coupled) receptors [151–154]. However, investigations on these proteins pose significant technical challenges, primarily because of their physico-chemical structure and sensitivity outside their native environment. They may contain large hydrophobic moieties that anchor them in the membrane, but which consequently limit their solubility in most other media, particularly if their structure has to be preserved. For this reason, biomimetic model lipid membranes have attracted lively interest because of their unique feature to provide an amphiphilic matrix for reconstitution of integral MPs [150,155,156]. Hence, MPs as preferred targets for pharmaceuticals received widespread recognition in drug discovery and protein-ligand screening and are of high interest for the development of biosensor platforms [150,155].
Very early after developing techniques for planar lipid bilayer generation membrane-active peptides incorporated in lipid membranes were studied [157]. One very prominent reason is that peptides can more easily be reconstituted in contrast to MPs [156,158,159]. Later on, supported model lipid membrane systems that complemented the existing ones (e.g. Langmuir monolayers, vesicular liposomal dispersions and bimolecular (‘black’) lipid membranes) were introduced [18,160–162]. These planar systems are thought to be promising platforms to study the mode of action of antimicrobial peptides (AMPs) but also for biophysical studies of and with artificial membranes or for sensor development employing, for example, antimicrobial membrane-active peptides and membrane integral proteins. Moreover, it offers the additional advantage of allowing for studies of the influence of membrane structure and order on the function of integral proteins; for example, on how the composition and organization of lipids in a mixed membrane influence the ion translocation activity of integral channel proteins [159,163–165].
By far the most literature can be found for the membrane-active peptides alamethicin, valinomycin and gramicidin [166–172]. The last was also used to determine the fluidity of the lipid membrane and as a molecular force probe [173,174]. In recent years, AMPs gained interest as it turned out that global antibacterial resistance is becoming an increasing public health problem [175]. Indeed, certain naturally derived peptides have been successfully used as antibiotics for many years. Despite the potential obstacles that remain, peptide therapeutics is poised to play a significant role in the treatment of diseases ranging from Alzheimer's disease to cancer [176].
4. The biomimetic approach
It is interesting to note that, in archaea possessing S-layers as the sole cell wall component (figures 2a and 3a), the protein lattices have been identified to be involved in the generation and maintenance of cell shape and in the cell division process [80,177,178]. Moreover, it was postulated by theoretical considerations that S-layers in archaea contribute to osmoprotection [88]. Hence, S-layers must therefore integrate the basic functions of mechanical and osmotic cell stabilization as these organisms dwell under extreme environmental conditions such as temperatures up to 120°C, pH down to zero, high hydrostatic pressure or high salt concentrations [64,179,180]. Since suitable methods for disintegration of archaeal S-layer protein lattices (figure 3a) and their reassembly into monomolecular arrays on lipid films are not yet available, S-layer proteins from Gram-positive bacteria are used to copy the supramolecular building principle of archaeal envelopes for the generation of S-layer-stabilized lipid membranes (figure 3b–f). S-layer proteins can be used as biofunctional surfaces [181] and constitute a fascinating structure for hosting and stabilizing functionalized lipid membranes [4,5,16,18–20,22,76,79,182,183]. These model lipid membranes consist either of a tetraether lipid monolayer (figure 3b,c,e) or of an artificial phospholipid bilayer (figure 3d,f) that replaces the cytoplasmic membrane and isolated bacterial S-layer proteins are assembled as monomolecular lattices onto the lipid membrane (figure 3b–f).
Moreover, a second S-layer acting as a protective molecular sieve and further stabilizing the scaffold can be recrystallized on the top of the previously generated SsLM (figure 3e,f). Hence, S-layer lattices constitute unique supporting architectures resulting in lipid membranes with a highly retained fluidity of the lipid molecules and a considerably extended longevity [17,184–187] (see also §6.1).
Besides archaeal cell envelope structures, many animal and human viruses with viral envelopes covering their protective protein capsids provide the building principle in particular for S-layer-coated liposomes or emulsomes (figure 4). The virus envelopes typically are derived from portions of the host cell membranes (phospholipids and proteins), but include some generally densely packed viral glycoproteins [188,189]. Interestingly, the size of a virus is 20–400 nm [51], i.e. in the same range as the artificial virus envelopes generated by coating liposomes or emulsomes with a monomolecular S-layer lattice (figure 4a,b) [19,22].
The following section intends to give a survey on biomimetic planar and spherical lipid membranes comprising the lipid matrix and a closely associated proteinaceous S-layer lattice. The prerequisite for creating such supramolecular structures is given by the unique non-covalent interaction of S-layer proteins with lipid head groups within planar and spherical lipid mono- and bilayers [17,18,186,187,190].
5. Generation of S-layer-supported lipid membranes
SsLMs have attracted lively interest because of three main reasons. (i) They constitute a versatile biomimetic model to study the characteristics of the archaeal cell envelope by a broad arsenal of surface-sensitive techniques and sophisticated microscopical methods. (ii) Surfaces with new properties such as elevated antifouling characteristics for application in material science and nanomedicine can be generated using SsLMs. And finally, (iii) SsLMs provide an amphiphilic matrix for reconstitution of MPs. This is an important prerequisite to characterize integral MPs in a native environment, to apply the latter in basic research, but also to generate bio-inspired materials like MP-based biosensors and lab-on-a-chip architectures.
Besides the intrinsic interaction of phospholipids with certain domains or amino acid residues of the S-layer protein (which will be discussed in §§5.1 and 5.2), the S-layer protein and lipid molecules can be interconnected by means of (i) chemical modification of the S-layer protein for the subsequent binding of lipid molecules; (ii) chemical binding of molecules (cross-linker) for the indirect coupling of the S-layer protein with lipid molecules; and (iii) introduction of a binding site exposed on the S-layer protein by genetic engineering (table 1). Interestingly, it recently became evident that in nature archaeal proteins are targeted for post-translational modifications such as the addition of a lipid [46]. Hence, our approach to link lipids covalently to S-layer proteins is a biomimetic one occurring in archaea where a portion of membrane-bound proteins are anchored through a covalent association of their carboxy-termini with the lipid bilayer [47].
5.1. Planar lipid membranes
There are two basic methods for the generation of a free-standing bilayer lipid membrane (BLM) across an aperture which links two fluid-filled chambers (figure 3d) [191–194]: first, ‘painted’ bilayers are formed from a dispersion of either one or a mixture of purified phospholipids in a non-polar solvent such as n-decane. Prior to bilayer formation, the hole on which the bilayer is to be formed is ‘primed’ with a small quantity of the phospholipid dispersion. The priming dispersion is allowed to dry, the cup is positioned in the block and both cis and trans chambers are filled with the desired experimental solution. Additional phospholipid dispersion is then drawn across the hole using a ‘stick’. This implement varies considerably from laboratory to laboratory and may be a small brush, a plastic rod or in some cases an air bubble at the end of a glass capillary.
An alternative procedure for the formation of a planar phospholipid bilayer involves the apposition of two phospholipid monolayers formed at the interface of an aqueous solution and the air [192,195]. This so-called ‘folded’ BLM has been adopted by some workers because the resultant bilayer contains somewhat less solvent than painted lipid membranes. However, again prior to bilayer formation, the hole on which the bilayer is to be formed is pretreated with a small quantity of hexadecane dissolved in pentane (1 : 10) [192,196].
Monolayers can be formed by applying phospholipids in a volatile solvent such as pentane, chloroform or hexane to the surface of an aqueous solution; the solvent evaporates in minutes leaving a phospholipid monolayer at the air–solution interface [195,197]. Alternatively, monolayers can be formed by allowing phospholipid liposomes or mixtures of liposomes and native membrane vesicles to equilibrate with a monolayer at an air–water interface [198,199]. The final structure of painted and folded BLMs is schematically depicted in figure 3d.
As implied by its name, this method involves the folding of two monolayers to form a planar bilayer. The apparatus used for bilayer formation via monolayer folding is in many respects similar to that used in the production of painted bilayers, which it involves two chambers separated by a septum containing a hole. A major difference exists in that the septum used for monolayer folding is not of rigid plastic but is composed of a very thin (10–25 µm) polytetrafluorethylene (PTFE; Teflon) membrane. Interestingly, no practical difference in terms of vesicle fusion was observed for painted and folded BLMs, respectively [200].
BLMs may also be formed at the end of conventional patch-clamp pipettes [201–203] with tip diameters in the range 0.5–5 μm either with or without fire polishing [197,204–206]. With this so-called tip-dip technique, the tip of the pipette is immersed in the desired experimental solution in a compartment of a multi-well disposable tray, and a phospholipid monolayer is formed at the air–water interface. Subsequently, a portion of the monolayer is transferred to the pipette tip by raising the pipette into the air. The polar head groups of the phospholipids orientate so that they interact with the aqueous pipette-filling solution and the glass wall of the pipette. The hydrocarbon chains of the molecules face the air. A bilayer is constructed by re-immersion of the pipette in the bath solution. As the tip of the pipette crosses the monolayer at the air–solution interface, a second region of monolayer interacts with the monolayer in the pipette to form a bilayer. The tip-dip technique, however, is also appropriate to generate an electrically tight tetraether lipid monolayer spanning the orifice of the pipette (figure 3c) [207].
Consequently, free-standing SsLMs are generated by two steps. First the lipid membrane is produced by either the painted, folded or tip-dip technique and subsequently an S-layer protein is recrystallized on it (figure 3c,d) [18,120,190]. Although the impact of the attached S-layer lattice on the membrane capacitance and resistance, and the boundary potential on free-standing BLMs, is negligible, the mechanical properties of SsLMs are considerably altered. Hydrostatic pressure applied across painted BLMs caused them to bulge resulting in an area of expansion measured by an increase in membrane capacitance [208]. A significantly higher area of expansion is observed for BLMs than for SsLMs whenever pressure is applied from the S-layer faced side. This experimental result supports the ‘osmoprotecting effect’ of the S-layer lattice, which was proposed 5 years later based on theoretical considerations to be one of the biological functions of S-layer lattices [88].
The membrane tension of BLMs after attachment of S-layer proteins is determined by dynamic light scattering [209]. For plain BLMs, the collective motions of the lipid molecules are dominated by membrane tension rather than by membrane curvature energy. S-layer lattices recrystallized on both sides of the BLM have resulted in a considerable reduction of the membrane tension by a factor of approximately five. However, the membrane bending energy increases by three orders of magnitude, indicating that the attached S-layer lattice facilitates transverse shear motions of lipid molecules [209]. In accordance with voltage pulse experiments [210], a significant increase in the previously negligible surface viscosity of the membrane is observed during S-layer protein attachment [209].
However, although free-standing SsLMs revealed a higher mechanical stability (e.g. against hydrostatic pressure) and longevity, in particular with reconstituted peptides or proteins in comparison with BLMs without an attached S-layer lattice, these membranes are up to now not stable enough for many practical applications [17,18,120]. Hence, a promising strategy is to attach BLMs to porous or solid supports to enhance their practical applicability [25,123,211–214].
Solid-supported lipid membranes involving S-layers as key constituents to provide a stabilizing and defined tethering layer to decouple the BLM from the (inorganic) support, but also to generate an ionic reservoir if desired, have been fabricated in several ways. S-layer proteins have been recrystallized on glass and modified silicon surfaces before generating a tetraether lipid monolayer (figure 3e) or a BLM (figure 3f) by the Langmuir–Blodgett (vertical transfer of a lipid monolayer)/Langmuir–Schaefer (horizontal transfer of a lipid monolayer) (LB/LS) technique [215,216].
SsLMs comprising a tetraether lipid monolayer or a phospholipid bilayer are generated by a modified LB technique on a polymer aperture covering either a porous or solid support [184,217]. SsLMs without the need of an aperture are generated by a combined LB and LS technique and, as recently described, either by the rapid solvent exchange (RSE) technique [125] or by a newly developed vesicle fusion technique [126]. In brief, the latter novel technique is based on the finding that β-diketone-carrying molecules form complexes when europium (Eu3+) ions (Eu) are present. When an amphiphilic β-diketone ligand [218] is incorporated in giant unilamellar vesicles (GUVs) consisting of phospholipids, the addition of Eu induced inter-liposomal complex formation and finally fusion of the GUVs [219]. For this versatile method, small unilamellar vesicles (SUVs) were fabricated comprising, beside, the main membrane forming phospholipid PC, a small amount of β-diketone ligand and linker lipids, capable of binding to chemically activated S-layer proteins. The addition of Eu to SUVs bound on the S-layer lattice resulted in rupture and fusion of the SUVs. By this procedure, a closed planar lipid bilayer is spontaneously formed on the S-layer lattice. This highly reproducible process can exactly be followed by quartz crystal microbalance with dissipation monitoring (QCM-D), but also by combined QCM-D with electrochemical impedance spectroscopy (EIS) measurements [126]. Another approach was to bind disc-shaped micellar lipid structures, called bicelles on the S-layer lattice [127,128,134]. The advantage is that no opening and rearrangement of this structure is necessary as bicelles are bilayered architectures, but, unfortunately, scarce fusion events caused in most cases an incomplete coverage of the S-layer lattice by the lipid membrane.
A further option is to bind thiolated SCWPs on gold surfaces before the S-layer protein is recrystallized on the support. This SCPW layer provides a defined orientation of the S-layer protein on the support and, as determined by AFM measurements, the crystallites of the S-layer lattice are larger and the S-layer lattice appears to be smoother than an S-layer lattice directly recrystallized on a solid support. Moreover, the soft thiolated SCWP layer with a thickness of approximately 6.2 nm [181] decouples the S-layer protein from the gold surface, which might cause denaturation of the protein structure at the area of contact when, for example, a certain electrical potential is applied [220].
5.2. Spherical lipid membranes (liposomes and emulsomes)
Unilamellar liposomes are artificially prepared spherical containers comprising a phospholipid bilayer shell and an aqueous core (figure 4c, right-hand side) [221–223]. Hydrophilic drugs can be stored and transported in the core, whereas the lipidic shell can be loaded with hydrophobic drugs. Emulsomes, however, are spherical systems with an internal solid fat core surrounded by phospholipid mono- and bilayer(s) (figure 4c, left-hand side) [224,225]. Hence, emulsomes show a much higher capacity to transport lipophilic drug molecules. Both nanocarriers can be used for targeted drug delivery for cancer and other diseases [23,225,226]. Furthermore, S-layer lattices as the outermost envelope structure covering the spherical containers constitute biomimetic ‘artificial cell envelopes’ or ‘artificial virus-like particles’, both enabling stabilization of the nanocarriers and presenting addressor molecules in a well-defined orientation and special distribution (figure 4).
Isolated S-layer subunits were recrystallized on positively charged, unilamellar liposomes comprising 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), cholesterol and hexadecylamine [22,114,115]. The S-layer attached to positively charged liposomes by their inner, negatively charged face. This is the orientation identical to the S-layer lattice on intact cells. Coating of the positively charged liposomes with the S-layer protein SbsB from G. stearothermophilus PV72/p2 resulted in inversion of the zeta-potential from an initially positive value to a negative one [114]. A similar behaviour was observed for liposomes coated with S-layer proteins from lactobacilli [227,228].
Emulsomes composed of a solid tripalmitin core and a phospholipid shell were reproducibly generated with an average diameter of approximately 300 nm using temperature-controlled extrusion steps [19]. The advantage of emulsomes is their high capacity to transport lipophilic substances such as curcumin [23]. Both wild-type (wt) and recombinant (r) S-layer protein SbsB formed an S-layer lattice covering the entire surface of emulsomes, as demonstrated by TEM (figure 4a,b). Upon coating with wtSbsB, the positive charge of emulsomes shifts to a highly negative zeta-potential, whereas those coated with rSbsB become charge neutral. This observation is attributed to the presence of the negatively charged SCWP, which is associated only with wtSbsB.
6. Performance of composite S-layer lipid membranes
The most challenging property of model lipid membranes is the feasibility to incorporate membrane-active (antimicrobial) peptides (AMPs) [229,230] and, more important, (complex) integral MPs in a functional state [150].
Up to now, the following membrane-active and/or AMPs have been incorporated in SsLMs: the ion carrier valinomycin, channel-forming alamethicin [184], the pore-forming gramicidin [217] and the AMP analogue peptidyl-glycylleucine-carboxyamide, where all lysine residues were replaced by glutamic acid, termed negatively charged analogue of peptidyl-glycylleucine-carboxyamide (PGLa(–); table 2) [129]. Furthermore, reconstitution of the staphylococcal pore-forming protein α-haemolysin (αHL) [24,231], the M2 segment that forms the ion-conducting channel of the nicotinic acetylcholine receptor (nAChR) [232], the ryanodine receptor/Ca2+ release channel (RyR1) [233] and the voltage-dependent anion channel (VDAC) [234–238] into plain and SsLMs has successfully been performed (table 3).
Table 2.
Summary of membrane-active peptides incorporated in S-layer-supported lipid membranes.
| membrane-active peptide | source | remarks | references |
|---|---|---|---|
| gramicidin A (gA) | Bacillus brevis | linear pentadeca peptide | [217] |
| alamethicin (Ala) | Trichoderma viride | linear, 20 amino acids | [184] |
| valinomycin (Val) | several Streptomyces strains, e.g. S. tsusimaensis and S. fulvissimus | cyclic dodecadepsipeptide, 12 amino acids and esters | [184,207] |
| peptidyl-glycine-leucine-carboxyamide (PGLa) analogue | synthesized via protein chemistry | 20 amino acid; PGLa(–) | [129] |
Table 3.
Summary of transmembrane proteins reconstituted in S-layer-supported lipid membranes.
| transmembrane protein | source | remarks | references |
|---|---|---|---|
| α-HL | exotoxin from Staphylococcus aureus | pore-forming; homo-heptamer | [196,239,240] |
| ryanodine receptor 1 (RyR1) | skeletal muscle cells | Ca2+-release channel; homo-tetramer | [241] |
| nicotinic acetylcholine receptor (nAChR) | plasma membranes of neurons; on postsynaptic side of the neuromuscular junction | ligand-gated ion channel; five subunits |
[130,134] |
| VDAC | located on the outer mitochondrial membrane; also produced by cell-free expression | porin, voltage gated; ion channel monomeric but can cluster | personal communicationa |
| M2 segment from nAChR | segment, forms ion-conducting channel (see nAChR) | ion-conducting channel | [242,243] |
aS Damiati & B Schuster 2013, personal communication.
6.1. Free-standing membranes
SsLMs were first characterized by dual label fluorescence, surface pressure, transmission electron microscopy and AFM, Fourier transform infrared spectroscopy, and X-ray and neutron reflectivity measurements [96,106–108,216,244,245]. Formation of S-layer lattices covering the entire area of lipid films has been observed on zwitterionic phospholipids like PCs and in particular PEs [122]. S-layer proteins did not form crystalline lattices on negatively charged phospholipids. Membranes comprising a small portion of positively charged surfactants [22,210] or lipid derivatives [209] facilitated the crystallization process particularly on PCs. Electrostatic interaction is thought to exist between exposed carboxyl groups on the S-layer lattice and zwitterionic lipid head groups. At least two or three contact points between the S-layer protein and the attached lipid film have been identified [122]. Hence, less than 5% of the lipid molecules of the adjacent monolayer are anchored to these contact points (protein domains) on the S-layer protein whereas the remaining 95% or more of lipid molecules may diffuse freely within the membrane between the pillars consisting of anchored lipid molecules [120,187]. This calculation is based on the lattice constants of SbpA having a square unit cell with a spacing of 13.1 nm [97,116] and an area per lipid molecule of 0.65 nm² [246]. These nanopatterned lipid membranes are also referred to as ‘semifluid lipid membranes’ [95] because of their widely retained fluid behaviour [209,215]. Most important, although peptide side groups of the S-layer protein interpenetrate the phospholipid head group regions almost to their entire depth, no impact on the hydrophobic lipid alkyl chains has been observed [106–108,196,207].
To prove whether a phospholipase is able to hydrolyse a lipid monolayer shielded by an S-layer lattice, porcine pancreatic phospholipase A2 (PLA2) has been injected underneath an S-layer/lipid monolayer membrane. Interestingly, the S-layer lattice has neither constituted a significant barrier for the PLA2 nor induced lipid packing defects, which would have resulted in shorter enzymatic lag periods [247]. Hence, there is significant evidence that the recrystallized S-layer lattice did not modulate a large proportion of the head group region of the phospholipid monolayer to an extent that could seriously impede the recognition of phospholipids by the biological interplay with PLA2 [247].
Free-standing S-layer-supported tetraether lipid monolayers generated on the tip of a glass micropipette (tip-dip technique) and functionalized with valinomycin (figure 3c) revealed a 10-fold higher life time than a membrane without an attached S-layer lattice (table 2) [207]. No reconstitution of αHL could be achieved with tetraether lipid membranes. In membranes mainly composed of the branched phospholipid DPhPC, αHL formed lytic pores when added to the lipid-exposed side of the SsLM (figure 3d). No pore formation was detected upon addition of αHL monomers to the S-layer face of the SsLM. Therefore, one can conclude that the intrinsic molecular sieving properties of the S-layer lattice do not allow passage of αHL monomers through the S-layer pores to the lipid membrane [196]. In addition, these data represent a quality control for the existence of a closed S-layer lattice without any defects and a tight attachment to the BLM. Compared with plain BLMs, SsLMs have a decreased tendency to rupture in the presence of αHL, again indicating an enhanced stability owing to the attached S-layer lattice [196]. Nevertheless, even single pore recordings have been performed with αHL reconstituted in free-standing SsLMs (table 3) [239].
SsLMs formed by the tip-dip technique were functionalized with M2 ion channels [242]. The M2 ion channel characteristics were studied and it turned out that the attached S-layer lattices were non-intrusive to the channel functionality and characteristics. The ability to stabilize BLMs and their non-intrusive character on ion channel activity make S-layer proteins attractive for biosensor applications, especially those that enhance the stability of BLMs beyond the use of tethers or polymer supports [3,242,243]. nAChR is a ligand-triggered ion channel consisting of five channel-forming subunits, which are arranged symmetrically around a central pore. For reconstitution studies in SsLMs, nAChR was isolated from the electric organ of the Pacific electric ray Torpedo californica. Reconstitution of nAChR was followed in liposomes, free-standing membranes with and without an attached S-layer lattice. When activated with carbamoylcholine, the nAChR channels reconstituted in all model lipid membranes were found to be functional and revealed a single channel conductance very close to those reported by others [127,130,206].
6.2. Surface-attached membranes
SsLMs prepared by the LB/LS technique without the need for an aperture have been compared with a silane- and dextran-supported phospholipid bilayer [215]. Most probably owing to the repetitive local interaction of the S-layer lattice with the lipid head groups, the nanopatterned fluidity of lipids was highest in SsLMs compared with the other supported bilayers, as determined by the fluorescence recovery after photobleaching technique. Phospholipid bilayers and tetraether lipid monolayers have also been generated on S-layer-covered gold electrodes. The tetraether lipid monolayer sandwiched by an S-layer lattice on each side (figure 3e) revealed an exceptional long-term robustness of approximately one week [17,18,120,187]. This finding also reflects the optimization of the archaeal cell envelope structure by nature over billions of years of evolution.
Lipid membranes generated on a porous support combine the advantage of easy manual handling, individual access to both membrane surfaces, and an essentially unlimited ionic reservoir on each side of the BLM (figure 3f). This is seen as a basic requirement of experiments copying the in vivo situation (e.g. plasmatic/exoplasmatic side). However, the surface properties of porous supports, such as roughness or great differences in pore size, have significantly impaired the stability of attached BLMs [248]. Hence, a straightforward approach is the use of S-layer ultrafiltration membranes (SUMs) with the S-layer as a stabilizing and smoothening biomimetic layer between the lipid membrane and the porous support [184,217,240].
SUMs were produced by depositing S-layer fragments as a coherent layer on microfiltration membranes [22,249–251]. The mechanical and chemical stability of their composite structure was subsequently obtained by inter- and intramolecular cross-linking [250,252–254]. The uniformity of functional groups on both the surface and within the pore area of the S-layer lattice could be used for very accurate chemical modifications in the sub-nanometre range, allowing the molecular sieving as well as antifouling characteristics of SUMs to be tuned [250,254,255]. Moreover, SUMs can be prepared with different net charges, hydrophilic or hydrophobic surface properties. That is why SUMs have been used as supporting and stabilizing structures for functional lipid membranes [5,18,121,182].
Whereas composite SUM-supported DPhPC bilayers were found to be highly isolating structures with a life time of up to 17 h [184,217,240], BLMs on plain microfiltration membranes revealed a life time of only approximately 3 h. The life time increased significantly to about 1 day by formation of an S-layer–lipid membrane–S-layer sandwich-like structure, i.e. an additional monomolecular S-layer protein lattice recrystallized on the lipid-faced side (figure 3f) [184,240]. An even further increase in the stability of this composite supramolecular structure can be expected upon cross-linking those lipid head groups involved in direct contact with the S-layer proteins. Hence, the nanopatterned anchoring of the membrane is a romising strategy for generating stable and fluid lipid membranes.
The functionality of SsLMs resting on solid supports has been investigated by the incorporation of the membrane-active peptides valinomycin, alamethicin, gramicidin D and the AMP analogue PGLa(–) (table 2) [184,256,257]. SsLMs with incorporated valinomycin, a potassium-selective ion carrier, revealed a remarkable high resistance bathed in sodium buffer. However, bathed in potassium buffer a decrease in resistance by a factor of 500 was observed for the same membrane owing to valinomycin-mediated ion transport [184].
SsLMs generated by the RSE technique are used to perform combined surface-sensitive QCM-D and EIS measurements. This study evidenced not only the attachment and/or insertion of PGLa(–) in the supported lipid membrane but also indicated toroidal pore formation in a concentration-dependent fashion [129]. Hence, SsLMs constitute a promising platform for studying the interaction and insertion of membrane-active (antimicrobial) peptides [229].
Incorporation of the membrane-active peptide gramicidin D could be demonstrated by measurements on single gramicidin D pores in all the above-mentioned SsLMs [217].
Finally, alamethicin channels could not only be incorporated in SsLMs on solid supports, the channels could even be specifically blocked as increasing amounts of inhibitor (amiloride) gave rise to a significant increase in membrane resistance (table 2) [184]. Thus, proof of concept for the applicability of these composite S-layer/lipid structures for biosensing purposes has been demonstrated. In future, the ability to reconstitute integral MPs in defined structures on, for example, sensor surfaces is one of the most important concerns in designing biomimetic sensing devices [3,18,258].
MPs have also been successfully reconstituted in SsLMs (table 3). Reconstitution of αHL, moreover even single pore recordings, could be achieved with SUM-supported DPhPC bilayers but no pore formation was observed with BLMs generated on the pure micro-filtration membranes [240].
RyR1, isolated from rabbit muscle cells, was successfully reconstituted in SsLMs [241]. For this purpose, the supported membranes were formed by the β-diketone ligand-triggered vesicle fusion technique either on glass for fluorescence experiments or on gold for QCM-D measurements. Preliminary measurements clearly indicated that incorporation of RyR1 occurred, which was verified by control experiments to exclude misinterpretation due to unspecific adsorption to the bilayer or the S-layer lattice [241]. Nevertheless, further experiments, for example combined QCM-D with EIS studies or patch-clamp measurements on a chip, have to be performed. Finally, SsLMs may constitute a versatile and stabilizing scaffold allowing the detailed investigation of different drugs on isolated RyR1 in high-throughput screening-like devices.
SsLMs made by the newly developed europium-induced vesicle fusion technique were incubated with VDAC (table 3). A significant decrease in membrane resistance could be observed but the membrane capacitance did not vary significantly. Moreover, increasing VDAC concentration decreased membrane resistance, which indicates an increasing number of channels reconstituted spontaneously into the SsLM (S Damiati & B Schuster 2013, personal communication). It is well known that VDAC reconstituted in artificial membranes forms a voltage-gated channel. At low membrane potentials (less than 10 mV), VDAC is in the open state and switches to the closed state at high membrane potentials [235,236]. Indeed, this behaviour could also be clearly observed for the SsLMs with incorporated VDAC channels. Furthermore, the membrane resistance decreased again after reducing the voltage from 10 mV back to zero but the resistance was higher than the first measurement. This may be explained by the re-opening of some channels while others remain closed. In addition, it is conceivable that keeping the channels in the closed state for a long period of time during the measurements may reduce the rate of re-opening of VDAC and cause some structural rearrangements in order to achieve a more stable closed conformation [259]. Moreover, it has been shown that the presence of the nucleotides nicotinamide adenine dinucleotide hydride (NADH) or nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) induce channel closure and, thus, the conductance of the VDAC channels is significantly reduced [260–262]. Indeed, addition of NADH to the SsLM with reconstituted VDAC caused a significant increase in the membrane resistance; this is strong evidence for the blocking of VDAC channels by NADH molecules (S Damiati & B Schuster 2013, personal communication).
6.3. Interphases in solution (liposomes and emulsomes)
The influence of an S-layer coating on the stability of liposomes was studied by the release of the encapsulated hydrophilic marker carboxyfluorescein (CF) during the application of mechanical and thermal challenges. S-layer-coated liposomes (figure 4c(1)) released only half the amount of enclosed CF compared with plain liposomes upon exposure to shear forces or ultrasonication as mechanical stress [114]. Furthermore, temperature shifts from 25°C to 55°C and vice versa induced considerably less CF release from S-layer-coated than from plain liposomes. Moreover, the S-layer protein on the liposome can be cross-linked with glutaraldehyde or bis(sulfosuccinimidyl)suberate [257,263,264]. In addition, cross-linking can also be used for covalent attachment of macromolecules (figure 4c(3,4)). In turn, a layer of intact liposomes can also be reversibly tethered via the specific nickel–His-tag linkage on an S-layer lattice [130]. Moreover, liposomes functionalized with reconstituted integral proteins may also be stabilized by a coat comprising an S-layer lattice (figure 4c(2)).
The thermotropic phase behaviour of liposomes with (figure 4c(1)) or without an S-layer coating was characterized by differential scanning microcalorimetry. The data indicated for both preparations a broad phase transition around 50°C due to the chain melting from a liquid-ordered gel-like to a liquid-ordered fluid phase, similar to that described for DPPC/cholesterol mixtures. The slightly higher phase transition temperature for the S-layer-coated liposomes was explained by increased intermolecular order [265].
S-layer-coated liposomes have been investigated for their ability to act as a versatile system for entrapping and binding target molecules (figure 4). Indeed, S-layer-coated liposomes constitute a proper matrix for the covalent attachment of macromolecules such as ferritin [22]. Another approach was the biotinylation of S-layer-coated liposomes which resulted in two accessible biotin residues for subsequent avidin binding per S-layer subunit (figure 4c(5)) [115]. An ordered monomolecular layer of streptavidin was formed on the surface of the S-layer-coated liposomes as visualized by labelling with biotinylated ferritin. Furthermore, biotinylated anti-human immunoglobulin G (IgG) was attached via streptavidin to the biotinylated S-layer-coated liposomes. The biological activity of the bound anti-human IgG was confirmed by enzyme-linked immunosorbent assay [115].
S-layer proteins recrystallized on liposomes were exploited as the immobilization matrix for anti-human IgG (figure 4c(3)). The interaction of rabbit or swine anti-human IgG as Ag was studied by measuring changes in ultrasound velocity [131]. The ultrasound velocity decreased linearly following an increase in Ag concentration, presumably caused by changes in hydration of the membrane due to the binding process. Finally, no substantial differences in the behaviour of ultrasound velocity were observed for interaction of human IgG with rabbit or swine anti-human IgG [131].
Moreover, S-layer/streptavidin fusion proteins have been constructed (figure 4c(6)), and hence the biotinylated binding partner can be bound in a much more defined orientation and position. By this method, three biotin residues accessible for subsequent avidin binding were introduced per S-layer subunit [118].
An interesting approach is the recrystallization of functional chimeric S-layer fusion proteins carrying the sequence of the enhanced green fluorescent protein (EGFP) on liposomes (figure 4c(6)) [21]. The uptake of S-layer/EGFP fusion protein-coated liposomes into eukaryotic cells, for example a cell type in an immortal human cell line (HeLa cell) could be nicely visualized by the intrinsic EGFP fluorescence. The major part of the coated liposomes was internalized by HeLa cells within 2 h of incubation by endocytosis [21]. With regard to further experiments, the most interesting advantage can be seen in co-recrystallization of, for example, S-layer/EGFP fusion protein and the S-layer/streptavidin fusion protein [118] on the same liposomal surface. The uptake of these specially coated liposomes by target cells and the functionality of transported drugs could be investigated simultaneously without the need for any additional labels.
In vitro cell culture studies revealed that S-layer-coated emulsomes (figure 4) can be taken up by human liver carcinoma cells (HepG2) without showing any significant cytotoxicity [19]. Moreover, in a very recent study the low-water-soluble polyphenolic compound curcumin was encapsulated inside the solid lipid core of emulsomes. These emulsomes were termed CurcuEmulsomes [23]. Not only the particular characteristics of CurcuEmulsomes were investigated but also the delivery of curcumin via CurcuEmulsomes into HepG2 cells as the first cellular model system. Hence, this type of nanocarrier provides an interesting platform for the delivery of hydrophobic bioactive agents whose medical use is otherwise limited. The utilization of S-layer fusion proteins equipped in a nanopatterned fashion by identical or diverse functional domains or biomolecules may lead to attractive nanobiotechnological applications, particularly as carrier and/or drug targeting and delivery systems, as artificial virus envelopes in, for example, medicinal applications, as diagnostic tools, for molecular imaging and in gene therapy [18,115,121,263,266].
7. Conclusion and perspectives
Considerable knowledge has accumulated over past years concerning the isolation and purification of S-layer proteins [257], the experimental conditions required for obtaining coherent extended S-layer lattices on solid or porous supports, but also on the generation and intrinsic properties of S-layer-supported lipid films, and application of various microscopic and biophysical techniques. The study and use of biomimetic membrane systems generated by bottom-up processes is a rapidly growing scientific and engineering field. Elucidation of the supramolecular construction principle of archaeal cell envelopes composed of S-layer-stabilized lipid membranes (figure 2a–c), as well as envelopes of a great variety of animal and human viruses, led to new strategies for generating more stable functional lipid membranes with nanopatterned fluidity at the meso- and macroscopic scale. In this biomimetic architecture, artificial or isolated lipids replace the cytoplasmic membrane and isolated native or recombinant S-layer proteins derived from Bacillaceae are attached on either one or both sides of the lipid membrane (figure 3).
An important strategy to design stable membranes on surfaces and interfaces concerns the application of SCWPs as constituents of the molecular construction kit. The use of SCWPs enables reassembly of S-layer lattices on surfaces and lipid films in a predetermined orientation (figure 5a) and is, thus, essential for many nanobiotechnological applications. Moreover, SCWPs [29,267], chemically modified SCWPs (e.g. pyridyl disulfide-activated ones) [112] or SCWPs linked to lipid molecules [123] provide useful anchors or spacers between S-layer lattices and inorganic supports (figure 5 and table 1) [124]. The distance between the lipid membrane and the electrode, and, thus, the ionic reservoir, may be modulated in the nanometre range by this intermediate polymer. SCWP-linked phospholipids incorporated in the outer leaflet of the lipid membrane will allow the recrystallization of a second S-layer lattice on the top of the composite architecture at a certain distance so that previously incorporated integral MPs are shielded from the external environment (figure 5a). Moreover, the crystalline top layer composed of glycoproteins serves as a nanoporous filter with antifouling characteristics and as a protecting layer for, for example, mechanical challenges (figure 5b). This approach mimics the functional aspects of many bacterial and archaeal S-layer lattices [5,28].
Figure 5.
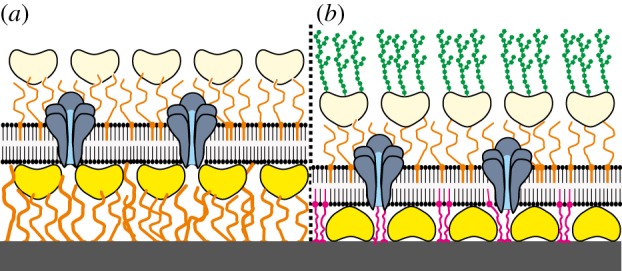
Schematic drawing of a lipid membrane on an S-layer-covered porous or solid support. (a) A thiolated SCWP is chemisorbed on the surface of a gold sensor and an S-layer lattice is recrystallized on it. The orientation of the S-layer lattice is determined by the SCWP layer. Subsequently, the lipid membrane is generated on the S-layer lattice and may be functionalized by integral MPs. Finally, a further proteinaceous lattice composed of S-layer proteins can be recrystallized on the top via SCWP-linked phospholipids which are anchored in the outer leaflet of the lipid membrane. (b) The lipid membrane is bound to the surface by membrane-anchored tethered lipid molecules reaching from the support through the pores of the S-layer lattice. Again, in this lipid membrane, transmembrane proteins can be reconstituted. Finally, a further proteinaceous lattice composed of S-layer glycoproteins (with carbohydrate moieties pointing to the external side) can be recrystallized on the top via SCWP-linked phospholipids which again are anchored in the outer leaflet of the lipid membrane. (Adapted from [18]. Copyright © 2009 with permission from Elsevier Inc.)
Chemical binding of the head groups of lipids on the S-layer protein was revealed to be an appropriate approach to generate stable and functional SsLMs. In addition, chimeric S-layer fusion proteins containing streptavidin can be recrystallized on their corresponding SCWPs or directly on solid supports [118]. As a result biotinylated lipids mixed with synthetic phospho- or isolated tetraether lipids of the fluid lipid membrane can be linked to this S-layer fusion protein via the biotin/streptavidin bridge to obtain an enhanced stability (table 1).
Another opportunity is the application of biotinylated integral MPs which may also be linked to the streptavidin S-layer fusion protein. The concept of these functional SsLMs is new and might have considerable impact on the development of solid-supported nanopatterned biomimetic membranes with functional proteins distributed in regular arrays following the lattice constant of the S-layer.
Finally, as most of the native S-layer proteins do not contain cysteine residues, it is possible to introduce cysteine residues on surface-exposed positions [132,133] that will be used for binding lipid molecules carrying a maleimide residue on their head group.
The very accurate nanopatterned fluidity of the lipid molecules and maintenance of lateral mobility of membrane-active peptides and MPs [18,120,217,240] in these biomimetic membranes provides significant advantage compared with other supported membrane systems [268]. Moreover, the combination of S-layer proteins and the specific interacting SCWPs enables a defined variation of the distance between the S-layer lattice and the solid support [76,77,124], which is seen as a basic requirement for the incorporation of MPs with domains exposed on the inner and/or outer membrane surface and for the assembly of multi-component membrane functions such as GPCRs, kinases, etc.
Copying nature's solution for the assembly of lipid membranes dwelling under most extreme environmental conditions is seen as one of the most promising strategies for various applications. Exciting areas for application of composite S-layer membrane systems concern mainly sensor systems involving specific membrane functions. Up to date, the use of such devices is primarily hampered by the instability and short longevity of membranes and, in particular, problems emerging upon drying biomimetic membranes. Native S-layer glycoproteins or S-layer proteins with carbohydrate moieties attached by chemical procedures should significantly help to overcome this problem (figure 5b). Moreover, previous studies have demonstrated that chemical cross-linking of S-layer glycoproteins through their glycan chains is feasible [269].
This approach is inspired by nature both in the nanobiotechnological design of supported lipid membranes and in the use of MPs [270] as they have attracted interest in their potential with respect to possible applications such as MP-based biosensors for detecting small hydrophobic molecules, ligand fishing, DNA sequencing, high-throughput screening and in (lab-on-a-) biochip technology. The same development is needed for biosensors such as artificial noses or tongues, in which many receptor proteins have to be exposed and read out simultaneously to create the response.
Another area of future development concerns copying virus envelopes by reassembly of S-layer (fusion) proteins on liposomes and emulsomes (figure 4) for the production of new targeting, delivery, encapsulation, and imaging systems, and vaccines.
The understanding of the mechanisms involved in the interaction of biological systems with inorganic materials is of interest in both fundamental and applied disciplines. A deep knowledge of the surface/biological fluid interface processes is needed for the design of new biocompatible materials for implants, drug targeting and delivery and nanodevices for diagnosis and therapy.
Funding statement
Financial support provided from the Austrian Science Fund (FWF), project P 20256-B11, is gratefully acknowledged.
References
- 1.Xi J, Ho D, Chu B, Montemagno CD. 2005. Lessons learned from engineering biologically active hybrid nano/micro devices. Adv. Funct. Mater. 15, 1233–1240. ( 10.1002/adfm.200400619) [DOI] [Google Scholar]
- 2.Srinivasan N, Kumar S. 2012. Ordered and disordered proteins as nanomaterial building blocks. WIREs Nanomed. Nanobiotechnol. 4, 204–218. ( 10.1002/wnan.1160) [DOI] [PubMed] [Google Scholar]
- 3.Jackman JA, Knoll W, Cho N-J. 2012. Biotechnology applications of tethered lipid bilayer membranes. Materials 5, 2637–2657. ( 10.3390/ma5122637) [DOI] [Google Scholar]
- 4.Sleytr UB, Pum D, Egelseer EM, Ilk N, Schuster B. 2013. S-layer proteins. In Handbook of biofunctional surfaces (ed. Knoll W.), pp. 507–568. Singapore: Pan Stanford Publishing. [Google Scholar]
- 5.Sleytr UB, Schuster B, Egelseer EM, Pum D. In press S-layers: principles and applications. FEMS Microbiol. Rev. ( 10.1111/1574-6976.12063) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen Y-X, Saboe PO, Sines IT, Erbakan M, Kumar M. 2014. Biomimetic membranes: a review. J. Memb. Sci. 454, 359–381. ( 10.1016/j.memsci.2013.12.019) [DOI] [Google Scholar]
- 7.Sleytr UB. 1976. Self-assembly of the hexagonally and tetragonally arranged subunits of bacterial surface layers and their reattachment to cell walls. J. Ultrastruct. Res. 55, 360–377. ( 10.1016/S0022-5320(76)80093-7) [DOI] [PubMed] [Google Scholar]
- 8.Sleytr UB, Messner P, Pum D, Sára M. 1988. Crystalline bacterial cell surface layers. Berlin, Germany: Springer. [DOI] [PubMed] [Google Scholar]
- 9.Messner P, Schäffer C, Egelseer EM, Sleytr UB. 2010. Occurrence, structure, chemistry, genetics, morphogenesis, and function of S-layers. In Prokaryotic cell wall compounds—structure and biochemistry (eds König H, Claus H, Varma A.), pp. 53–109. Heidelberg, Germany: Springer. [Google Scholar]
- 10.Sleytr UB, Messner P, Pum D, Sára M. 1996. Crystalline surface layers on eubacteria and archaeobacteria. In Crystalline bacterial cell surface proteins (eds Sleytr UB, Messner P, Pum D, Sára M.). Austin, TX: R. G. Landes Company and Academic Press, Inc. [Google Scholar]
- 11.Sleytr UB, Messner P, Pum D, Sára M. 1999. Crystalline bacterial cell surface layers (S layers): from supramolecular cell structure to biomimetics and nanotechnology. Angew. Chem. Int. Ed. 38, 1035–1054. ( 10.1002/(SICI)1521-3773(19990419)38:8) [DOI] [PubMed] [Google Scholar]
- 12.Albers SV, Meyer BH. 2011. The archaeal cell envelope. Nat. Rev. Microbiol. 9, 414–426. ( 10.1038/nrmicro2576) [DOI] [PubMed] [Google Scholar]
- 13.Jarrell KF, Jones GM, Kandiba L, Nair DB, Eichler J. 2010. S-layer glycoproteins and flagellins: reporters of archaeal posttranslational modifications. Archaea 2010, 1–13. ( 10.1155/2010/612948) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Messner P, Steiner K, Zarschler K, Schäffer C. 2008. S-layer nanoglycobiology of bacteria. Carbohydr. Res. 343, 1934–1951. ( 10.1016/j.carres.2007.12.025). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schäffer C, Graninger M, Messner P. 2001. Prokaryotic glycosylation. Proteomics 1, 248–261. () [DOI] [PubMed] [Google Scholar]
- 16.Sleytr UB, Egelseer EM, Pum D, Schuster B. 2004. S-Layers. In NanoBiotechnology: concepts, methods and perspectives (eds Niemeyer CM, Mirkin CA.), pp. 77–92. Weinheim, Germany: Wiley-VCH. [Google Scholar]
- 17.Schuster B, Sleytr UB. 2005. 2D-protein crystals (S-layers) as support for lipid membranes. In Advances in planar lipid bilayers and liposomes (eds Tien TH, Ottova A.), pp. 247–293. Amsterdam, The Netherlands: Elsevier Science. [Google Scholar]
- 18.Schuster B, Sleytr UB. 2009. Composite S-layer lipid structures. J. Struct. Biol. 168, 207–216. ( 10.1016/j.jsb.2009.03.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ücisik MH, Küpcü S, Debreczeny M, Schuster B, Sleytr UB. 2013. S-layer coated emulsomes as potential nanocarriers. Small 9, 2895–2904. ( 10.1002/smll.201203116) [DOI] [PubMed] [Google Scholar]
- 20.Sleytr UB, Sára M, Pum D, Schuster B, Messner P, Schäffer C. 2002. Self-assembly protein systems: microbial S-layers. In Biopolymers: polyamides and complex proteinaceous materials I (eds Steinbüchel A, Fahnestock SR.), pp. 285–338. Weinheim, Germany: Wiley-VCH. [Google Scholar]
- 21.Ilk N, Küpcü S, Moncayo G, Klimt S, Ecker RC, Hofer-Warbinek R, Egelseer EM, Sleytr UB, Sára M. 2004. A functional chimaeric S-layer-enhanced green fluorescent protein to follow the uptake of S-layer-coated liposomes into eukaryotic cells. Biochem. J. 379, 441–448. ( 10.1042/BJ20031900) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Küpcü S, Sára M, Sleytr UB. 1995. Liposomes coated with crystalline bacterial cell surface protein (S-layers) as immobilization structures for macromolecules. Biochim. Biophys. Acta 1235, 263–269. ( 10.1016/0005-2736(95)80013-6) [DOI] [PubMed] [Google Scholar]
- 23.Ucisik MH, Küpcü S, Schuster B, Sleytr UB. 2013. Characterization of CurcuEmulsomes: nanoformulation for enhanced solubility and delivery of curcumin. J. Nanobiotechnol. 11, 37 ( 10.1186/1477-3155-11-37) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bayley H, Cremer PS. 2001. Stochastic sensors inspired by biology. Nature 413, 226–230. ( 10.1038/35093038) [DOI] [PubMed] [Google Scholar]
- 25.Castellana ET, Cremer PS. 2006. Solid supported lipid bilayers: from biophysical studies to sensor design. Surf. Sci. Rep. 61, 429–444. ( 10.1016/j.surfrep.2006.06.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beveridge TJ. 2001. Use of the Gram stain in microbiology. Biotech. Histochem. 76, 111–118. ( 10.1080/714028139) [DOI] [PubMed] [Google Scholar]
- 27.Beveridge TJ. 1994. Bacterial S-layers. Curr. Opin. Struct. Biol. 4, 204–212. ( 10.1016/S0959-440X(94)90309-3) [DOI] [Google Scholar]
- 28.Sleytr UB, Beveridge TJ. 1999. Bacterial S-layers. Trends Microbiol. 7, 253–260. ( 10.1016/S0966-842X(99)01513-9) [DOI] [PubMed] [Google Scholar]
- 29.Sára M. 2001. Conserved anchoring mechanisms between crystalline cell surface S-layer proteins and secondary cell wall polymers in Gram-positive bacteria? Trends Microbiol. 9, 47–49. ( 10.1016/S0966-842X(00)01905-3) [DOI] [PubMed] [Google Scholar]
- 30.Archibald AR, Hancock IC, Harwood CR. 1993. Cell wall structure, synthesis, and turnover. In Bacillus subtilis and other Gram-positive bacteria (eds Sonenshein AL, Hoch JA, Losick R.), pp. 381–410. Washington, DC: American Society for Microbiology. [Google Scholar]
- 31.Lupas A, Engelhardt H, Peters J, Santarius U, Volker S, Baumeister W. 1994. Domain structure of the Acetogenium kivui surface layer revealed by electron crystallography and sequence analysis. J. Bacteriol. 176, 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ilk N, Kosma P, Puchberger M, Egelseer EM, Mayer HF, Sleytr UB, Sára M. 1999. Structural and functional analyses of the secondary cell wall polymer of Bacillus sphaericus CCM 2177 that serves as an S-layer-specific anchor. J. Bacteriol. 181, 7643–7646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuen B, Koch A, Asenbauer E, Sára M, Lubitz W. 1997. Molecular characterization of the Bacillus stearothermophilus PV72 S-layer gene sbsB induced by oxidative stress. J. Bacteriol. 179, 1664–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowditch RD, Baumann P, Yousten AA. 1989. Cloning and sequencing of the gene encoding a 125-kilodalton surface-layer protein from Bacillus sphaericus 2362 and of a related cryptic gene. J. Bacteriol. 171, 4178–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sára M, Sleytr UB. 2000. S-layer proteins. J. Bacteriol. 182, 859–868. ( 10.1128/JB.182.4.859-868.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jarosch M, Egelseer EM, Huber C, Moll D, Mattanovich D, Sleytr UB, Sára M. 2001. Analysis of the structure-function relationship of the S-layer protein SbsC of Bacillus stearothermophilus ATCC 12980 by producing truncated forms. Microbiology 147, 1353–1363. [DOI] [PubMed] [Google Scholar]
- 37.Jarosch M, Egelseer EM, Mattanovich D, Sleytr UB, Sára M. 2000. S-layer gene sbsC of Bacillus stearothermophilus ATCC 12980: molecular characterization and heterologous expression in Escherichia coli. Microbiology 146, 273–281. [DOI] [PubMed] [Google Scholar]
- 38.Avall-Jääskeläinen S, Kylä-Nikkilä K, Kahala M, Miikkulainen-Lahti T, Palva A. 2002. Surface display of foreign epitopes on the Lactobacillus brevis S-layer. Appl. Environ. Microbiol. 68, 5943–5951. ( 10.1128/AEM.68.12.5943-5951) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Avall-Jaaskelainen S, Palva A. 2005. Lactobacillus surface layers and their applications. FEMS Microbiol. Rev. 29, 511–529. ( 10.1016/j.femsre.2005.04) [DOI] [PubMed] [Google Scholar]
- 40.Hynonen U, Palva A. 2013. Lactobacillus surface layer proteins: structure, function and applications. Appl. Microbiol. Biotechnol. 97, 5225–5243. ( 10.1016/j.femsre.2005.04.0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vilen H, Hynonen U, Badelt-Lichtblau H, Ilk N, Jaaskelainen P, Torkkeli M, Palva A. 2009. Surface location of individual residues of SlpA provides insight into the Lactobacillus brevis S-layer. J. Bacteriol. 191, 3339–3349. ( 10.1128/JB.01782-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egelseer EM, Leitner K, Jarosch M, Hotzy C, Zayni S, Sleytr UB, Sára M. 1998. The S-layer proteins of two Bacillus stearothermophilus wild-type strains are bound via their N-terminal region to a secondary cell wall polymer of identical chemical composition. J. Bacteriol. 180, 1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veith A, Klingl A, Zolghadr B, Lauber K, Mentele R, Lottspeich F, Rachel R, Albers SV, Kletzin A. 2009. Acidianus, Sulfolobus and Metallosphaera surface layers: structure, composition and gene expression. Mol. Microbiol. 73, 58–72. ( 10.1111/j.1365-2958.2009.06746.x) [DOI] [PubMed] [Google Scholar]
- 44.Baumeister W, Lembcke G. 1992. Structural features of archaebacterial cell envelopes. J. Bioenerg. Biomembr. 24, 567–575. ( 10.1007/BF00762349) [DOI] [PubMed] [Google Scholar]
- 45.Eichler J, Maupin-Furlow J. 2013. Post-translation modification in Archaea: lessons from Haloferax volcanii and other haloarchaea. FEMS Microbiol. Rev. 37, 583–606. ( 10.1111/1574-6976.12012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szabo Z, Pohlschroder M. 2012. Diversity and subcellular distribution of archaeal secreted proteins. Front. Microbiol. 3, 207 ( 10.3389/fmicb.2012.00207) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abdul Halim MF, et al. 2013. Haloferax volcanii archaeosortase is required for motility, mating, and C-terminal processing of the S-layer glycoprotein. Mol. Microbiol. 88, 1164–1175. ( 10.1111/mmi.12248) [DOI] [PubMed] [Google Scholar]
- 48.Haft DH, Payne SH, Selengut JD. 2012. Archaeosortases and exosortases are widely distributed systems linking membrane transit with posttranslational modification. J. Bacteriol. 194, 36–48. ( 10.1128/JB.06026-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kandiba L, Guan Z, Eichler J. 2013. Lipid modification gives rise to two distinct Haloferax volcanii S-layer glycoprotein populations. Biochim. Biophys. Acta Biomembr. 1828, 938–943. ( 10.1016/j.bbamem.2012.11.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lucas W. 2010. Viral capsids and envelopes: structure and function. eLS. ( 10.1002/9780470015902.a0001091.pub2) [DOI] [Google Scholar]
- 51.Marx JL. 1991. Virus. In The encyclopedia Americana, International Edition (ed. Grolier), pp. 172 New York, NY: Scholastic. [Google Scholar]
- 52.Cambell N, Reece J. 2008. The structure and function of large biological molecules. In Biology (eds Campbell NA, Reece JB, Urry LA, Cain ML, Wasserman SA, Minorsky PV, Jackson RB.), pp. 68–91. San Francisco, CA: Pearson Benjamin Cummings. [Google Scholar]
- 53.Paltauf F. 1994. Ether lipids in biomembranes. Chem. Phys. Lipids 74, 101–139. ( 10.1016/0009-3084(94)90054-X) [DOI] [PubMed] [Google Scholar]
- 54.van Meer G, Voelker DR, Feigenson GW. 2008. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124. ( 10.1038/nrm2330) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sandermann H., Jr 1978. Regulation of membrane enzymes by lipids. Biochim. Biophys. Acta 513, 209–237. ( 10.1016/0304-4157(78)90015-1) [DOI] [PubMed] [Google Scholar]
- 56.Seddon AM, Casey D, Law RV, Gee A, Templer RH, Ces O. 2009. Drug interactions with lipid membranes. Chem. Soc. Rev. 38, 2509–2519. ( 10.1039/b813853m) [DOI] [PubMed] [Google Scholar]
- 57.Hanke W, Schlue WR. 1993. Physical properties of biological membranes and planar lipid bilayers. London, UK: Academic Press. [Google Scholar]
- 58.Marsh D. 1996. Lateral pressure in membranes. Biochim. Biophys. Acta Rev. Biomembr. 1286, 183–223. ( 10.1016/S0304-4157(96)00009-3) [DOI] [PubMed] [Google Scholar]
- 59.Marsh D. 1996. Membrane assembly studied by spin-label electron spin resonance. Braz. J. Med. Biol. Res. 29, 863–871. [PubMed] [Google Scholar]
- 60.Sengupta P, Baird B, Holowka D. 2007. Lipid rafts, fluid/fluid phase separation, and their relevance to plasma membrane structure and function. Semin. Cell Dev. Biol. 18, 583–590. ( 10.1016/j.semcdb.2007.07.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sprott GD. 2011. Archaeal membrane lipids and applications. eLS. ( 10.1002/9780470015902.a0000385.pub3) [DOI] [Google Scholar]
- 62.Bakowsky U, Rothe U, Antonopoulos E, Martini T, Henkel L, Freisleben HJ. 2000. Monomolecular organization of the main tetraether lipid from Thermoplasma acidophilum at the water-air interface. Chem. Phys. Lipids 105, 31–42. ( 10.1016/S0009-3084(99)00131-0) [DOI] [PubMed] [Google Scholar]
- 63.Strobl C, Six L, Heckmann K, Henkel B, Ring K. 1984. Physicochemical characterization of the tetraether lipids from Thermoplasma acidophilum. II. Film balance studies on the monomolecular organization of the main glycophospholipid in monofilms. Z. Naturforsch. 40c, 219–222. [Google Scholar]
- 64.De Rosa M. 1996. Archaeal lipids: structural features and supramolecular organization. Thin Solid Films 284–285, 13–17. ( 10.1016/S0040-6090(96)08832-3) [DOI] [Google Scholar]
- 65.Gliozzi A, Relini A, Chong PLG. 2002. Structure and permeability properties of biomimetic membranes of bolaform archaeal tetraether lipids. J. Membr. Sci. 206, 131–147. ( 10.1016/S0376-7388(01)00771-2) [DOI] [Google Scholar]
- 66.Nicolini C. 1996. Supramolecular architecture and molecular bioelectronics. Thin Solid Films 284–285, 1–5. ( 10.1016/S0040-6090(96)08834-7) [DOI] [Google Scholar]
- 67.Jacquemet A, Terme N, Benvegnu T, Vié V, Lemiègre L. 2013. Collapsed bipolar glycolipids at the air/water interface: effect of the stereochemistry on the stretched/bent conformations. J. Colloid Interface Sci. 412, 72–81. ( 10.1016/j.jcis.2013.09.010) [DOI] [PubMed] [Google Scholar]
- 68.Jeworrek C, Evers F, Erlkamp M, Grobelny S, Tolan M, Chong PLG, Winter R. 2011. Structure and phase behavior of archaeal lipid monolayers. Langmuir 27, 13 113–13 121. ( 10.1021/la202027s) [DOI] [PubMed] [Google Scholar]
- 69.Dote JL, Barger WR, Behroozi F, Chang EL, Lo SL, Montague CE, Nagumo M. 1990. Monomolecular film behavior of tetraether lipids from a thermoacidophilic archaebacterium at the air/water interface. Langmuir 6, 1017–1023. ( 10.1021/la00095a023) [DOI] [Google Scholar]
- 70.Gliozzi A, Relini A, Rolandi R, Dante S, Gambacorta A. 1994. Organization of bipolar lipids in monolayers at the air-water interface. Thin Solid Films 242, 208–212. ( 10.1016/0040-6090(94)90531-2) [DOI] [Google Scholar]
- 71.Pichot R, Watson RL, Norton IT. 2013. Phospholipids at the interface: current trends and challenges. Int. J. Mol. Sci. 14, 11 767–11 794. ( 10.3390/ijms130611767) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang DN, Stieglitz H, Marden J, Tamm LK. 2013. Benjamin Franklin, Philadelphia's favorite son, was a membrane biophysicist. Biophys. J. 104, 287–291. ( 10.1016/j.bpj.2012.12.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beveridge TJ, Graham LL. 1991. Surface layers of bacteria. Microbiol. Rev. 55, 684–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whitman WB, Coleman DC, Wiebe WJ. 1998. Prokaryotes: the unseen majority. Proc. Natl Acad. Sci. USA 95, 6578–6583. ( 10.1073/pnas.95.12.6578) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sleytr UB. 1975. Heterologous reattachment of regular arrays of glycoproteins on bacterial surfaces. Nature 257, 400–402. ( 10.1038/257400a0) [DOI] [PubMed] [Google Scholar]
- 76.Sleytr UB, Egelseer EM, Ilk N, Pum D, Schuster B. 2007. S-layers as a basic building block in a molecular construction kit. FEBS J. 274, 323–334. ( 10.1111/j.1742-4658.2006.05606.x) [DOI] [PubMed] [Google Scholar]
- 77.Sleytr UB, Huber C, Ilk N, Pum D, Schuster B, Egelseer EM. 2007. S-layers as a tool kit for nanobiotechnological applications. FEMS Microbiol. Lett. 267, 131–144. ( 10.1111/j.1574-6968.2006.00573) [DOI] [PubMed] [Google Scholar]
- 78.Sleytr UB. 1978. Regular arrays of macromolecules on bacterial cell walls: structure, chemistry, assembly, and function. Int. Rev. Cytol. 53, 1–62. ( 10.1016/S0074-7696(08)62240-8) [DOI] [PubMed] [Google Scholar]
- 79.Sleytr UB, Egelseer EM, Ilk N, Messner P, Schäffer C, Pum D, Schuster B. 2010. Nanobiotechnological applications of S-layers. In Prokaryotic cell wall compounds—structure and biochemistry (eds König H, Claus H, Varma A.), pp. 459–481. Heidelberg, Germany: Springer. [Google Scholar]
- 80.Sleytr UB, Messner P. 1989. Self-assembly of crystalline bacterial cell surface layers (S-layers). In Electron microscopy of subcellular dynamics (ed. Plattner H.), pp. 13–31. Boca Raton, FL: CRC Press. [Google Scholar]
- 81.Sleytr UB, Schuster B, Egelseer EM, Pum D, Horejs CM, Tscheliessnig R, Ilk N. 2011. Nanobiotechnology with S-layer proteins as building blocks. In Progress in molecular biology and translational science, molecular assembly in natural and engineered systems (ed. Howorka S.), pp. 277–352. Burlington, VT: Elsevier Academic Press Inc. [DOI] [PubMed] [Google Scholar]
- 82.Pavkov-Keller T, Howorka S, Keller W. 2011. The structure of bacterial S-layer proteins. In Molecular assembly in natural and engineered systems (ed. Howorka S.), pp. 73–130. Burlington, VT: Elsevier Academic Press Inc. [DOI] [PubMed] [Google Scholar]
- 83.Messner P, Sleytr UB. 1992. Crystalline bacterial cell-surface layers. Adv. Microb. Physiol. 33, 213–275. ( 10.1016/S0065-2911(08)60218-0) [DOI] [PubMed] [Google Scholar]
- 84.Sára M, Pum D, Sleytr UB. 1992. Permeability and charge-dependent adsorption properties of the S-layer lattice from Bacillus coagulans E38–E66. J. Bacteriol. 174, 3487–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sleytr UB, Messner P. 2009. Crystalline bacterial cell surface layers (S-layers). In Encyclopedia of microbiology (ed. Schaechter M.), pp. 89–98, 3rd edn San Diego, CA: Academic Press/Elsevier Science. [Google Scholar]
- 86.Sleytr UB, Messner P, Pum D, Sára M. 1999. Kristalline Zelloberflächen-Schichten prokaryotischer Organismen (S Schichten): von der supramolekularen Zellstruktur zur Biomimetik und Nanotechnologie. Angew. Chem. 111, 1098–1120. () [DOI] [Google Scholar]
- 87.Engelhardt H. 2007. Are S-layers exoskeletons? The basic function of protein surface layers revisited. J. Struct. Biol. 160, 115–124. ( 10.1016/j.jsb.2007.08.003) [DOI] [PubMed] [Google Scholar]
- 88.Engelhardt H. 2007. Mechanism of osmoprotection by archaeal S-layers: a theoretical study. J. Struct. Biol. 160, 190–199. ( 10.1016/j.jsb.2007.08.004) [DOI] [PubMed] [Google Scholar]
- 89.de la Fuente-Nunez C, Mertens J, Smit J, Hancock REW. 2012. The bacterial surface layer provides protection against antimicrobial peptides. Appl. Environ. Microbiol. 78, 5452–5456. ( 10.1128/AEM.01493-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fagan RP, Fairweather NF. 2014. Biogenesis and functions of bacterial S-layers. Nat. Rev. Microbiol 12, 211–222. ( 10.1038/nrmicro3213) [DOI] [PubMed] [Google Scholar]
- 91.Sotiropoulou S, Mark SS, Angert ER, Batt CA. 2007. Nanoporous S-layer protein lattices. A biological ion gate with calcium selectivity. J. Phys. Chem. C 111, 13 232–13 237. ( 10.1021/jp072132l) [DOI] [Google Scholar]
- 92.Picher MM, Kupcu S, Huang CJ, Dostalek J, Pum D, Sleytr UB, Ertl P. 2013. Nanobiotechnology advanced antifouling surfaces for the continuous electrochemical monitoring of glucose in whole blood using a lab-on-a-chip. Lab Chip 13, 1780–1789. ( 10.1039/c3lc41308j) [DOI] [PubMed] [Google Scholar]
- 93.Sleytr UB, Györvary E, Pum D. 2003. Crystallization of S-layer protein lattices on surfaces and interfaces. Prog. Organ Coat 47, 279–287. ( 10.1016/S0300-9440(03)00143-7) [DOI] [Google Scholar]
- 94.Pum D, Sára M, Sleytr UB. 1989. Structure, surface charge, and self-assembly of the S-layer lattice from Bacillus coagulans E38–66. J. Bacteriol. 171, 5296–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pum D, Sleytr UB. 1994. Large-scale reconstruction of crystalline bacterial surface layer proteins at the air-water interface and on lipids. Thin Solid Films 244, 882–886. ( 10.1016/0040-6090(94)90592-4) [DOI] [Google Scholar]
- 96.Pum D, Weinhandl M, Hödl C, Sleytr UB. 1993. Large-scale recrystallization of the S-layer of Bacillus coagulans E38–66 at the air/water interface and on lipid films. J. Bacteriol. 175, 2762–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Györvary ES, Stein O, Pum D, Sleytr UB. 2003. Self-assembly and recrystallization of bacterial S-layer proteins at silicon supports imaged in real time by atomic force microscopy. J. Microsc. 212, 300–306. ( 10.1111/j.1365-2818.2003.01270.x) [DOI] [PubMed] [Google Scholar]
- 98.Tang J, Badelt-Lichtblau H, Ebner A, Preiner J, Kraxberger B, Gruber HJ, Sleytr UB, Ilk N, Hinterdorfer P. 2008. Fabrication of highly ordered gold nanoparticle arrays templated by crystalline lattices of bacterial S-layer protein. Chem. Phys. Chem. 9, 2317–2320. ( 10.1002/cphc.200800507) [DOI] [PubMed] [Google Scholar]
- 99.Tang J, et al. 2008. Recognition imaging and highly ordered molecular templating of bacterial S-layer nanoarrays containing affinity-tags. Nano Lett. 8, 4312–4319. ( 10.1021/nl802092c) [DOI] [PubMed] [Google Scholar]
- 100.Tang J, et al. 2008. High-affinity tags fused to S-layer proteins probed by atomic force microscopy. Langmuir 24, 1324–1329. ( 10.1021/la702276k) [DOI] [PubMed] [Google Scholar]
- 101.Tang J, Ebner A, Kraxberger B, Badelt-Lichtblau H, Gruber HJ, Sleytr UB, Ilk N, Hinterdorfer P. 2010. Mapping short affinity tags on bacterial S-layer with an antibody. Chem. Phys. Chem. 11, 2323–2326. ( 10.1002/cphc.201000295) [DOI] [PubMed] [Google Scholar]
- 102.Sleytr UB, Sára M, Pum D, Schuster B. 2005. Crystalline bacterial cell surface layers (S-layers): a versatile self-assembly system. In Supramolecular polymers (ed. Ciferri A.), pp. 583–612, 2nd edn Boca Raton, FL: Taylor & Francis Group. [Google Scholar]
- 103.Pum D, Sleytr UB. 1995. Monomolecular reassembly of a crystalline bacterial cell surface layer (S layer) on untreated and modified silicon surfaces. Supramol. Sci. 2, 193–197. ( 10.1016/0968-5677(96)89675-1) [DOI] [Google Scholar]
- 104.Pum D, Sleytr UB. 1995. Anisotropic crystal growth of the S-layer of Bacillus sphaericus CCM 2177 at the air/water interface. Colloids Surf. A: Physicochem. Eng. Aspects 102, 99–104. ( 10.1016/0927-7757(95)03190-O) [DOI] [Google Scholar]
- 105.Rothbauer M, Küpcü S, Sticker D, Sleytr UB, Ertl P. 2013. Exploitation of S-layer anisotropy: pH-dependent nanolayer orientation for cellular micropatterning. ACS Nano 7, 8020–8030. ( 10.1021/nn403198a) [DOI] [PubMed] [Google Scholar]
- 106.Weygand M, Wetzer B, Pum D, Sleytr UB, Cuvillier N, Kjaer K, Howes PB, Losche M. 1999. Bacterial S-layer protein coupling to lipids: X-ray reflectivity and grazing incidence diffraction studies. Biophys. J. 76, 458–468. ( 10.1016/S0006-3495(99)77213-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weygand M, Schalke M, Howes PB, Kjaer K, Friedmann J, Wetzer B, Pum D, Sleytr UB, Lösche M. 2000. Coupling of protein sheet crystals (S-layers) to phospholipid monolayers. J. Mat. Chem. 10, 141–148. ( 10.1039/A905196A) [DOI] [Google Scholar]
- 108.Weygand M, Kjaer K, Howes PB, Wetzer B, Pum D, Sleytr UB, Lösche M. 2002. Structural reorganization of phospholipid headgroups upon recrystallization of an S-layer lattice. J. Phys. Chem. B 106, 5793–5799. ( 10.1021/jp014641) [DOI] [Google Scholar]
- 109.Comolli LR. 2013. Conformational transitions driving lattice growth at an S-layer boundary resolved by cryo-TEM. Biophys. J. 104, 353A ( 10.1016/j.bpj.2012.11.1958) [DOI] [PubMed] [Google Scholar]
- 110.Moreno-Flores S, Kasry A, Butt HJ, Vavilala C, Schmittel M, Pum D, Sleytr UB, Toca-Herrera JL. 2008. From native to non-native two-dimensional protein lattices through underlying hydrophilic/hydrophobic nanoprotrusions. Angew. Chem. Int. Ed. 47, 4707–4710. ( 10.1002/ange.200800151) [DOI] [PubMed] [Google Scholar]
- 111.López AE, Pum D, Sleytr UB, Toca-Herrera JL. 2011. Influence of surface chemistry and protein concentration on the adsorption rate and S-layer crystal formation. Phys. Chem. Chem. Phys. 13, 11 905–11 913. ( 10.1039/C1CP00052G) [DOI] [PubMed] [Google Scholar]
- 112.Mader C, Huber C, Moll D, Sleytr UB, Sára M. 2004. Interaction of the crystalline bacterial cell surface layer protein SbsB and the secondary cell wall polymer of Geobacillus stearothermophilus PV72 assessed by real-time surface plasmon resonance biosensor technology. J. Bacteriol. 186, 1758–1768. ( 10.1128/JB.186.6.1758-1768.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Küpcü S, Mader C, Sára M. 1995. The crystalline cell surface layer of Thermoanaerobacter thermohydrosulfuricus L111–69 as an immobilization matrix: influence of the morphological properties and the pore size of the matrix on activity loss of covalently bound enzymes. Biotechnol. Appl. Biochem. 21, 275–286. [Google Scholar]
- 114.Mader C, Küpcü S, Sára M, Sleytr UB. 1999. Stabilizing effect of an S-layer on liposomes towards thermal or mechanical stress. Biochim. Biophys. Acta 1418, 106–116. ( 10.1016/S0005-2736(99)00030-9) [DOI] [PubMed] [Google Scholar]
- 115.Mader C, Küpcü S, Sleytr UB, Sára M. 2000. S-layer-coated liposomes as a versatile system for entrapping and binding target molecules. Biochim. Biophys. Acta 1463, 142–150. ( 10.1016/S0005-2736(99)00190-X) [DOI] [PubMed] [Google Scholar]
- 116.Toca-Herrera JL, Krastev R, Bosio V, Küpcü S, Pum D, Fery A, Sára M, Sleytr UB. 2005. Recrystallization of bacterial S-layers on flat polyelectrolyte surfaces and hollow polyelectrolyte capsules. Small 1, 339–348. ( 10.1002/smll.200400035) [DOI] [PubMed] [Google Scholar]
- 117.Ilk N, Egelseer EM, Sleytr UB. 2011. S-layer fusion proteins: construction principles and applications. Curr. Opin. Biotechnol. 22, 824–831. ( 10.1016/j.copbio.2011.05.510) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moll D, Huber C, Schlegel B, Pum D, Sleytr UB, Sára M. 2002. S-layer-streptavidin fusion proteins as template for nanopatterned molecular arrays. Proc. Natl Acad. Sci. USA 99, 14 646–14 651. ( 10.1073/pnas.232299399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sára M, Egelseer EM, Huber C, Ilk N, Pleschberger M, Pum D, Sleytr UB. 2006. S-layer proteins: potential application in nano(bio)technology. In Microbial bionanotechnology: biological self-assembly systems and biopolymer-based nanostructures (ed. Rehm B.), pp. 307–338. Wymondham, UK: Horizon Bioscience. [Google Scholar]
- 120.Schuster B. 2005. Biomimetic design of nano-patterned membranes. Nanobiotechnology 1, 153–164. ( 10.1385/Nano:1:2:153) [DOI] [Google Scholar]
- 121.Schuster B, Kepplinger C, Sleytr UB. 2010. Biomimetic S-layer stabilized lipid membranes. In Biomimetics in biophysics: model systems, experimental techniques and computation (ed. Toca-Herrera JL.), pp. 1–12. Kerala, India: Research Signpost. [Google Scholar]
- 122.Wetzer B, Pfandler A, Györvary E, Pum D, Lösche M, Sleytr UB. 1998. S-layer reconstitution at phospholipid monolayers. Langmuir 14, 6899–6906. ( 10.1021/la980547f) [DOI] [Google Scholar]
- 123.Knoll W, et al. 2008. Solid supported lipid membranes: new concepts for the biomimetic functionalization of solid surfaces. Biointerphases 3, FA125–FA135. ( 10.1116/1.2913612) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sleytr UB, Sara M, Mader C, Schuster B, Unger FM. 2006. Use of secondary cell wall polymer of procaryotic microorganisms. US Patent Number 7,125,707 B2.
- 125.Schrems A, Kibrom A, Küpcü S, Kiene E, Sleytr UB, Schuster B. 2011. Bilayer lipid membrane formation on a chemically modified S-layer lattice. Langmuir 27, 3731–3738. ( 10.1021/la104238e) [DOI] [PubMed] [Google Scholar]
- 126.Schrems A, Larisch VD, Stanetty C, Dutter K, Damiati S, Sleytr UB, Schuster B. 2011. Liposome fusion on proteinaceous S-layer lattices triggered via β-diketone ligand–europium(III) complex formation. Soft Matter 7, 5514–5518. ( 10.1039/C1SM05468F) [DOI] [Google Scholar]
- 127.Pisecker M. 2005. Biophysical investigations on S-layer supported lipid membranes. Master's thesis, University of Natural Resources and Life Sciences, Vienna, Austria. [Google Scholar]
- 128.Schuster D. 2005. Fabrication and characterisation of nanostructures as anchoring matrix for lipid assemblies. Master's thesis, University of Natural Resources and Life Sciences, Vienna, Austria. [Google Scholar]
- 129.Schrems A, Larisch VD, Sleytr UB, Hohenegger M, Lohner K, Schuster B. 2013. Insertion of an anionic analogue of the antimicrobial peptide PGLa in lipid architectures including S-layer supported lipid bilayers. Curr. Nanosci. 9, 262–270. ( 10.2174/1573413711309020016) [DOI] [Google Scholar]
- 130.Kepplinger C. 2007. Anchoring of lipid assemblies to native and modified S-layer lattices—a nanobiotechnological study. PhD thesis, University of Natural Resources and Life Sciences, Vienna, Austria, Vienna, Austria. [Google Scholar]
- 131.Krivanek R, Rybar P, Küpcü S, Sleytr UB, Hianik T. 2002. Affinity interactions on a liposome surface detected by ultrasound velocimetry. Bioelectrochemistry 55, 57–59. ( 10.1016/S1567-5394(01)00161-X) [DOI] [PubMed] [Google Scholar]
- 132.Badelt-Lichtblau H, Kainz B, Völlenkle C, Egelseer EM, Sleytr UB, Pum D, Ilk N. 2009. Genetic engineering of the S-layer protein SbpA of Lysinibacillus sphaericus CCM 2177 for the generation of functionalized nanoarrays. Bioconjug. Chem. 20, 895–903. ( 10.1021/bc800445r) [DOI] [PubMed] [Google Scholar]
- 133.Howorka S, Sára M, Wang Y, Kuen B, Sleytr UB, Lubitz W, Bayley H. 2000. Surface-accessible residues in the monomeric and assembled forms of a bacterial surface layer protein. J. Biol. Chem. 275, 37 876–37 886. ( 10.1074/jbc.M003838200) [DOI] [PubMed] [Google Scholar]
- 134.Kiene E. 2011. Potential and limitations of S-layers as support for planar lipid bilayers. PhD thesis, University of Natural Resources and Life Sciences, Vienna, Austria. [Google Scholar]
- 135.Egelseer EM, Ilk N, Pum D, Messner P, Schäffer C, Schuster B, Sleytr UB. 2010. S-layers, microbial, biotechnological applications. In The encyclopedia of industrial biotechnology: bioprocess, bioseparation, and cell technology (ed. Flickinger MC.), pp. 4424–4448. Hoboken, NJ: John Wiley & Sons, Inc. [Google Scholar]
- 136.Huber C, Ilk N, Rünzler D, Egelseer EM, Weigert S, Sleytr UB, Sára M. 2005. The three S-layer-like homology motifs of the S-layer protein SbpA of Bacillus sphaericus CCM 2177 are not sufficient for binding to the pyruvylated secondary cell wall polymer. Mol. Microbiol. 55, 197–205. ( 10.1111/j.1365-2958.2004.04351.x) [DOI] [PubMed] [Google Scholar]
- 137.Ries W, Hotzy C, Schocher I, Sleytr UB, Sára M. 1997. Evidence that the N-terminal part of the S-layer protein from Bacillus stearothermophilus PV72/p2 recognizes a secondary cell wall polymer. J. Bacteriol. 179, 3892–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rünzler D, Huber C, Moll D, Köhler G, Sára M. 2004. Biophysical characterization of the entire bacterial surface layer protein SbsB and its two distinct functional domains. J. Biol. Chem. 279, 5207–5215. ( 10.1074/jbc.M308819200) [DOI] [PubMed] [Google Scholar]
- 139.Mesnage S, Fontaine T, Mignot T, Delepierre M, Mock M, Fouet A. 2000. Bacterial SLH domain proteins are non-covalently anchored to the cell surface via a conserved mechanism involving wall polysaccharide pyruvylation. Embo J. 19, 4473–4484. ( 10.1093/emboj/19.17.4473) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mesnage S, Tosi-Couture E, Mock M, Fouet A. 1999. The S-layer homology domain as a means for anchoring heterologous proteins on the cell surface of Bacillus anthracis. J. Appl. Microbiol. 87, 256–260. ( 10.1046/j.1365-2672.1999.00880.x) [DOI] [PubMed] [Google Scholar]
- 141.Sára M, Egelseer EM, Dekitsch C, Sleytr UB. 1998. Identification of two binding domains, one for peptidoglycan and another for a secondary cell wall polymer, on the N-terminal part of the S-layer protein SbsB from Bacillus stearothermophilus PV72/p2. J. Bacteriol. 180, 6780–6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schäffer C, Wugeditsch T, Kählig H, Scheberl A, Zayni S, Messner P. 2002. The surface layer (S-layer) glycoprotein of Geobacillus stearothermophilus NRS 2004/3a. Analysis of its glycosylation. J. Biol. Chem. 277, 6230–6239. ( 10.1074/jbc.M108873200) [DOI] [PubMed] [Google Scholar]
- 143.Bingle WH, Nomellini JF, Smit J. 1997. Linker mutagenesis of the Caulobacter crescentus S-layer protein: toward a definition of an N-terminal anchoring region and a C-terminal secretion signal and the potential for heterologous protein secretion. J. Bacteriol. 179, 601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Doig P, McCubbin WD, Kay CM, Trust TJ. 1993. Distribution of surface-exposed and non-accessible amino acid sequences among the two major structural domains of the S-layer protein of Aeromonas salmonicida. J. Mol. Biol. 233, 753–765. ( 10.1006/jmbi.1993.1550) [DOI] [PubMed] [Google Scholar]
- 145.Thomas S, Austin JW, McCubbin WD, Kay CM, Trust TJ. 1992. Roles of structural domains in the morphology and surface anchoring of the tetragonal paracrystalline array of Aeromonas hydrophila. Biochemical characterization of the major structural domain. J. Mol. Biol. 228, 652–661. ( 10.1016/0022-2836(92)90847-D) [DOI] [PubMed] [Google Scholar]
- 146.Nomellini JF, Küpcü S, Sleytr UB, Smit J. 1997. Factors controlling in vitro recrystallization of the Caulobacter crescentus paracrystalline S-layer. J. Bacteriol. 179, 6349–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bleicher KH, Böhm HJ, Müller K, Alanine AI. 2003. Hit and lead generation: beyond high-throughput screening. Nat. Rev. Drug Discov. 2, 369–378. ( 10.1038/nrd1086) [DOI] [PubMed] [Google Scholar]
- 148.Ellis C, Smith A. 2004. Highlighting the pitfalls and possibilities of drug research. Nat. Rev. Drug Discov. 3, 238–278. ( 10.1038/nrd1332) [DOI] [PubMed] [Google Scholar]
- 149.Viviani B, Gardoni F, Marinovich M. 2007. Cytokines and neuronal ion channels in health and disease. Int. Rev. Neurobiol. 82, 247–263. ( 10.1016/S0074-7742(07)82013-7) [DOI] [PubMed] [Google Scholar]
- 150.Tiefenauer L, Demarche S. 2012. Challenges in the development of functional assays of membrane proteins. Materials 5, 2205–2242. ( 10.3390/ma5112205) [DOI] [Google Scholar]
- 151.Galdiero S, Falanga A, Vitiello M, Raiola L, Russo L, Pedone C, Isernia C, Galdiero M. 2010. The presence of a single N-terminal histidine residue enhances the fusogenic properties of a membranotropic peptide derived from herpes simplex virus type 1 glycoprotein H. J. Biol. Chem. 285, 17 123–17 136. ( 10.1074/jbc.M110.114819) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Galdiero S, Galdiero M, Pedone C. 2007. Beta-barrel membrane bacterial proteins: structure, function, assembly and interaction with lipids. Curr. Protein Pept. Sci. 8, 63–82. ( 10.2174/138920307779941541) [DOI] [PubMed] [Google Scholar]
- 153.Gerstein M, Hegyi H. 1998. Comparing genomes in terms of protein structure: surveys of a finite parts list. FEMS Microbiol. Rev. 22, 277–304. ( 10.1111/j.1574-6976.1998.tb00371.x) [DOI] [PubMed] [Google Scholar]
- 154.Marassi FM, Ramamoorthy A, Opella SJ. 1997. Complete resolution of the solid-state NMR spectrum of a uniformly 15N-labeled membrane protein in phospholipid bilayers. Proc. Natl Acad. Sci. USA 94, 8551–8556. ( 10.1073/pnas.94.16.8551) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Demarche S, Sugihara K, Zambelli T, Tiefenauer L, Vörös J. 2011. Techniques for recording reconstituted ion channels. Analyst 136, 1077–1089. ( 10.1039/c0an00828a) [DOI] [PubMed] [Google Scholar]
- 156.Shen H-H, Lithgow T, Martin LL. 2013. Reconstitution of membrane proteins into model membranes: seeking better ways to retain protein activities. Int. J. Mol. Sci. 14, 1589–1607. ( 10.3390/ijms14011589) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mueller P, Rudin DO. 1968. Action potentials induced in biomolecular lipid membranes. Nature 217, 713–719. ( 10.1038/217713a0) [DOI] [PubMed] [Google Scholar]
- 158.Morera FJ, Vargas G, Gonzalez C, Rosenmann E, Latorre R. 2007. Ion-channel reconstitution. Methods Mol. Biol. 400, 571–585. ( 10.1007/978-1-59745-519-0_38) [DOI] [PubMed] [Google Scholar]
- 159.Wiese A, Gutsmann T, Seydel U. 2003. Towards antibacterial strategies: studies on the mechanisms of interaction between antibacterial peptides and model membranes. J. Endotoxin Res. 9, 67–84. ( 10.1179/096805103125001441) [DOI] [PubMed] [Google Scholar]
- 160.Knoll W, Köper I, Naumann R, Sinner EK. 2008. Tethered bimolecular lipid membranes. A novel model membrane platform. Electrochim. Acta 53, 6680–6689. ( 10.1016/j.electacta.2008.02.121) [DOI] [Google Scholar]
- 161.Sinner EK, Ritz S, Naumann R, Schiller S, Knoll W. 2009. Self-assembled tethered bimolecular lipid membranes. Adv. Clin. Chem. 49, 159–179. ( 10.1016/S0065-2423(09)49007-3) [DOI] [PubMed] [Google Scholar]
- 162.Knoll W, Park H, Sinner EK, Yao D, Yu F. 2004. Supramolecular interfacial architectures for optical biosensing with surface plasmons. Surf. Sci. 1–2, 30–42. ( 10.1016/j.susc.2004.06.192) [DOI] [Google Scholar]
- 163.Chen X, Chen Z. 2006. SFG studies on interactions between antimicrobial peptides and supported lipid bilayers. Biochim. Biophys. Acta Biomembr. 1758, 1257–1273. ( 10.1016/j.bbamem.2006.01.017) [DOI] [PubMed] [Google Scholar]
- 164.Duclohier H. 2010. Antimicrobial peptides and peptaibols, substitutes for conventional antibiotics. Curr. Pharmac. Des. 16, 3212–3223. ( 10.2174/138161210793292500) [DOI] [PubMed] [Google Scholar]
- 165.Jelinek R, Kolusheva S. 2005. Membrane interactions of host-defense peptides studied in model systems. Curr. Protein Pept. Sci. 6, 103–114. ( 10.2174/1389203053027511) [DOI] [PubMed] [Google Scholar]
- 166.Volinsky R, Kolusheva S, Berman A, Jelinek R. 2006. Investigations of antimicrobial peptides in planar film systems. Biochim. Biophys. Acta Biomembr. 1758, 1393–1407. ( 10.1016/j.bbamem.2006.03.002) [DOI] [PubMed] [Google Scholar]
- 167.Anrather D, Smetazko M, Saba M, Alguel Y, Schalkhammer T. 2004. Supported membrane nanodevices. J. Nanosci. Nanotechnol. 4, 1–22. ( 10.1166/jnn.2004.226) [DOI] [PubMed] [Google Scholar]
- 168.Cornell BA, Braach-Maksvytis VLB, King LG, Osman PDJ, Raguse B, Wieczorek L, Pace RJ. 1997. A biosensor that uses ion-channel switches. Nature 387, 580–583. ( 10.1038/42432) [DOI] [PubMed] [Google Scholar]
- 169.Cornell BA, Braach-Maksvytis VLB, King LG, Osman PDJ, Raguse B, Wieczorek L, Pace RJ. 1999. The gramicidin-based biosensor: a functioning nano-machine. Novartis Found. Symp. 225, 231–254. [DOI] [PubMed] [Google Scholar]
- 170.Valina-Saba M, Weiss-Wichert C, Smetazko MM, Anrather D, Schmutzer SE, Bauer GD, Schalkhammer TG. 1997. A new highly sensitive gated ion channel biosensor for antibody detection—setup, ion channel design, supported membranes . Proc. SPIE 3199, 24–34. ( 10.1117/12.301103) [DOI] [Google Scholar]
- 171.Woodhouse G, King L, Wieczorek L, Osman P, Cornell B. 1999. The ion channel switch biosensor. J. Mol. Recogn. 12, 328–334. () [DOI] [PubMed] [Google Scholar]
- 172.Zagnoni M. 2012. Miniaturised technologies for the development of artificial lipid bilayer systems. Lab Chip Miniaturisation Chem. Biol. 12, 1026–1039. ( 10.1039/c2lc20991h) [DOI] [PubMed] [Google Scholar]
- 173.Knoll W, Schmidt G, Rotzer H, Henkel T, Pfeiffer W, Sackmann E, Mittler-Neher S, Spinke J. 1991. Lateral order in binary lipid alloys and its coupling to membrane functions. Chem. Phys. Lipids 57, 363–374. ( 10.1016/0009-3084(91)90086-Q) [DOI] [PubMed] [Google Scholar]
- 174.Lundbæk JA, Collingwood SA, Ingólfsson HI, Kapoor R, Andersen OS. 2010. Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes. J. R. Soc. Interface 7, 373–395. ( 10.1098/rsif.2009.0443) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bax R, Mullan N, Verhoef J. 2000. The millennium bugs—the need for and development of new antibacterials. Int. J. Antimicrob. Agents 16, 51–59. ( 10.1016/S0924-8579(00)00189-8) [DOI] [PubMed] [Google Scholar]
- 176.Lien S, Lowman HB. 2003. Therapeutic peptides. Trends Biotechnol. 21, 556–562. ( 10.1016/j.tibtech.2003.10.005) [DOI] [PubMed] [Google Scholar]
- 177.Messner P, Pum D, Sára M, Stetter KO, Sleytr UB. 1986. Ultrastructure of the cell envelope of the archaebacteria Thermoproteus tenax and Thermoproteus neutrophilus. J. Bacteriol. 166, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pum D, Messner P, Sleytr UB. 1991. Role of the S layer in morphogenesis and cell division of the archaebacterium Methanocorpusculum sinense. J. Bacteriol. 173, 6865–6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hanford MJ, Peeples TL. 2002. Archaeal tetraether lipids. Appl. Biochem. Biotechnol. 97, 45–62. ( 10.1385/ABAB:97:1:45) [DOI] [PubMed] [Google Scholar]
- 180.Stetter KO. 1999. Extremophiles and their adaptation to hot environments. FEBS Lett. 452, 22–25. ( 10.1016/S0014-5793(99)00663-8) [DOI] [PubMed] [Google Scholar]
- 181.Schuster B, Sleytr UB. 2009. Tailor-made crystalline structures of truncated S-layer proteins on heteropolysaccharides. Soft Matter 5, 334–341. ( 10.1039/b810211b) [DOI] [Google Scholar]
- 182.Schuster B, Sleytr UB. 2008. Fabrication and characterization of functionalized S-layer supported lipid membranes. In Bioelectrochemistry research developments (ed. Bernstein EM.), pp. 105–124. Hauppauge, NY: Nova Science Publishers, Inc. [Google Scholar]
- 183.Sleytr UB, Pum D, Sára M, Schuster B. 2004. Molecular nanotechnology and nanobiotechnology with 2-D protein crystals. In Encylopedia of nanoscience and nanotechnology (ed. Nalwa HS.), pp. 693–702. San Diego, CA: Academic Press. [Google Scholar]
- 184.Gufler PC, Pum D, Sleytr UB, Schuster B. 2004. Highly robust lipid membranes on crystalline S-layer supports investigated by electrochemical impedance spectroscopy. Biochim. Biophys. Acta 1661, 154–165. ( 10.1016/j.bbamem.2003.12.009) [DOI] [PubMed] [Google Scholar]
- 185.Schuster B, Gufler PC, Pum D, Sleytr UB. 2004. S-layer proteins as supporting scaffoldings for functional lipid membranes. IEEE Trans. Nanobiosci. 3, 16–21. ( 10.1109/TNB.2004.824267) [DOI] [PubMed] [Google Scholar]
- 186.Schuster B, Pum D, Sleytr UB. 2008. S-layer stabilized lipid membranes. Biointerphases 3, FA3–FA11. ( 10.1116/1.2889067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Schuster B, Sleytr UB. 2006. Biomimetic S-layer supported lipid membranes. Curr. Nanosci. 2, 143–152. ( 10.2174/157341306776875749) [DOI] [Google Scholar]
- 188.Schambach A, Zychlinski D, Ehrnstroem B, Baum C. 2013. Biosafety features of lentiviral vectors. Hum. Gene Therapy 24, 132–142. ( 10.1089/hum.2012.229) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tong S, Li J, Wands JR, Wen Y-m. 2013. Hepatitis B virus genetic variants: biological properties and clinical implications. Emerg. Microb. Infect. 2, e10 ( 10.1038/emi.2013.10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Schuster B, Sleytr UB. 2000. S-layer-supported lipid membranes. J. Biotechnol. 74, 233–254. [DOI] [PubMed] [Google Scholar]
- 191.Bartsch P, Walter C, Selenschik P, Honigmann A, Wagner R. 2012. Horizontal bilayer for electrical and optical recordings. Materials 5, 2705–2730. ( 10.3390/ma5122705) [DOI] [Google Scholar]
- 192.Hanke W, Schlue W-R. 1993. Planar lipid bilayer. Methods and applications. San Diego, CA: Academic PressLimited. [Google Scholar]
- 193.Mueller P, Rudin DO, Ti Tien H, Wescott WC. 1962. Reconstitution of cell membrane structure in vitro and its transformation into an excitable system. Nature 194, 979–980. ( 10.1038/194979a0) [DOI] [PubMed] [Google Scholar]
- 194.Mueller P, Rudin DO, Tien HT, Wescott WC. 1963. Methods for the formation of single bimolecular lipid membranes in aqueous solution. J. Phys. Chem. 67, 534–535. ( 10.1021/j100796a529) [DOI] [Google Scholar]
- 195.Montal M, Mueller P. 1972. Formation of bimolecular membranes from lipid monolayers and a study of their electrical properties. Proc. Natl Acad. Sci. USA 69, 3561–3566. ( 10.1073/pnas.69.12.3561) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schuster B, Pum D, Braha O, Bayley H, Sleytr UB. 1998. Self-assembled alpha-hemolysin pores in an S-layer-supported lipid bilayer. Biochim. Biophys. Acta 1370, 280–288. ( 10.1016/S0005-2736(97)00274-5) [DOI] [PubMed] [Google Scholar]
- 197.Coronado R, Latorre R. 1983. Phospholipid bilayers made from monolayers on patch-clamp pipettes. Biophys. J. 43, 231–236. ( 10.1016/S0006-3495(83)84343-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Schindler H. 1980. Formation of planar bilayers from artificial or native membrane vesicles. FEBS Lett. 122, 77–79. ( 10.1016/0014-5793(80)80405-4) [DOI] [PubMed] [Google Scholar]
- 199.Schindler H, Quast U. 1980. Formation of planar membranes from natural liposomes. Application to acetylcholine receptor from Torpedo. Ann. N. Y. Acad. Sci. 358, 361 ( 10.1111/j.1749-6632.1980.tb15421.x) [DOI] [PubMed] [Google Scholar]
- 200.Anzai K, Ogawa K, Ozawa T, Yamamoto H. 2001. Quantitative comparison of two types of planar lipid bilayers—folded and painted—with respect to fusion with vesicles. J. Biochem. Biophys. Methods 48, 283–291. ( 10.1016/S0165-022X(01)00160-9) [DOI] [PubMed] [Google Scholar]
- 201.Sakmann B. 2006. Patch pipettes are more useful than initially thought: simultaneous pre- and postsynaptic recording from mammalian CNS synapses in vitro and in vivo. Pflügers Arch. 453, 249–259. ( 10.1007/s00424-006-0172-4) [DOI] [PubMed] [Google Scholar]
- 202.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391, 85–100. ( 10.1007/BF00656997) [DOI] [PubMed] [Google Scholar]
- 203.Neher E, Sakmann B, Steinbach JH. 1978. The extracellular patch clamp: a method for resolving currents through individual open channels in biological membranes. Pflugers Arch. 375, 219–228. ( 10.1007/BF00584247) [DOI] [PubMed] [Google Scholar]
- 204.Detwiler PB, Conner JD, Bodoia RD. 1982. Gigaseal patch clamp recordings from outer segments of intact retinal rods. Nature 300, 59–61. ( 10.1038/300059a0) [DOI] [PubMed] [Google Scholar]
- 205.Montal M, Darszon A, Schindler H. 1981. Functional reassembly of membrane proteins in planar lipid bilayers. Q. Rev. Biophys. 14, 1–79. ( 10.1017/S0033583500002079) [DOI] [PubMed] [Google Scholar]
- 206.Suarez-Isla BA, Wan K, Lindstrom J, Montal M. 1983. Single-channel recordings from purified acetylcholine receptors reconstituted in bilayers formed at the tip of patch pipets. Biochemistry 22, 2319–2323. ( 10.1021/bi00279a003) [DOI] [PubMed] [Google Scholar]
- 207.Schuster B, Pum D, Sleytr UB. 1998. Voltage clamp studies on S-layer-supported tetraether lipid membranes. Biochim. Biophys. Acta 1369, 51–60. ( 10.1016/S0005-2736(97)00206-X) [DOI] [PubMed] [Google Scholar]
- 208.Schuster B, Sleytr UB. 2002. The effect of hydrostatic pressure on S-layer-supported lipid membranes. Biochim. Biophys. Acta 1563, 29–34. ( 10.1016/S0005-2736(02)00370-X) [DOI] [PubMed] [Google Scholar]
- 209.Hirn R, Schuster B, Sleytr UB, Bayerl TM. 1999. The effect of S-layer protein adsorption and crystallization on the collective motion of a planar lipid bilayer studied by dynamic light scattering. Biophys. J. 77, 2066–2074. ( 10.1016/S0006-3495(99)77048-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Schuster B, Sleytr UB, Diederich A, Bähr G, Winterhalter M. 1999. Probing the stability of S-layer-supported planar lipid membranes. Eur. Biophys. J. 28, 583–590. ( 10.1007/s002490050240) [DOI] [PubMed] [Google Scholar]
- 211.Chan YHM, Boxer SG. 2007. Model membrane systems and their applications. Curr. Opin. Chem. Biol. 11, 581–587. ( 10.1016/j.cbpa.2007.09.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Katsaras J, Kučerka N, Nieh MP. 2008. Structure from substrate supported lipid bilayers. Biointerphases 3, FB55–FB63. ( 10.1116/1.2992133) [DOI] [PubMed] [Google Scholar]
- 213.Reimhult E, Kumar K. 2008. Membrane biosensor platforms using nano- and microporous supports. Trends Biotechnol. 26, 82–89. ( 10.1016/j.tibtech.2007.11.004) [DOI] [PubMed] [Google Scholar]
- 214.Steinem C, Janshoff A. 2010. Multicomponent membranes on solid substrates: interfaces for protein binding. Curr. Opin. Colloid Interface Sci. 15, 479–488. ( 10.1016/j.cocis.2010.06.004) [DOI] [Google Scholar]
- 215.Györvary E, Wetzer B, Sleytr UB, Sinner A, Offenhäuser A, Knoll W. 1999. Lateral diffusion of lipids in silane-, dextrane- and S-layer protein-supported mono- and bilayers. Langmuir 15, 1337–1347. ( 10.1021/la980827v) [DOI] [Google Scholar]
- 216.Wetzer B, Pum D, Sleytr UB. 1997. S-layer stabilized solid supported lipid bilayers. J. Struct. Biol. 119, 123–128. ( 10.1006/jsbi.1997.3867) [DOI] [PubMed] [Google Scholar]
- 217.Schuster B, Weigert S, Pum D, Sára M, Sleytr UB. 2003. New method for generating tetraether lipid membranes on porous supports. Langmuir 19, 2392–2397. ( 10.1021/La026691p) [DOI] [Google Scholar]
- 218.Marchi-Artzner V, Brienne MJ, Gulik-Krzywicki T, Dedieu J-C, Lehn J-M. 2004. Selective complexation and transport of europium ions at the interface of vesicles. Chem. Eur. J. 10, 2342–2350. ( 10.1002/chem.200305423) [DOI] [PubMed] [Google Scholar]
- 219.Haluska CK, Riske KA, Marchi-Artzner V, Lehn J-M, Lipowsky R, Dimova R. 2006. Time scales of membrane fusion revealed by direct imaging of vesicle fusion with high temporal resolution. Proc. Natl Acad. Sci. USA 103, 15 841–15 846. ( 10.1073/pnas.0602766103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zafiu C, Trettenhahn G, Pum D, Sleytr UB, Kautek W. 2011. Structural control of surface layer proteins at electrified interfaces investigated by in situ Fourier transform infrared spectroscopy. Phys. Chem. Chem. Phys. 13, 13 232–13 237. ( 10.1039/c0cp02127j) [DOI] [PubMed] [Google Scholar]
- 221.Bangham AD, Standish MM, Watkins JC. 1965. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 13, 238–252. ( 10.1016/S0022-2836(65)80093-6) [DOI] [PubMed] [Google Scholar]
- 222.Cui HF, Ye JS, Leitmannova Lui A, Tien HT. 2006. Lipid microvesicles: on the four decades of liposome research. In Advances in planar lipid bilayers and liposomes (eds Leitmannova Lui A, Tien HT.), pp. 1–48. Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- 223.Tien HT, Ottova-Leitmannova A. 2000. Membrane biophysics: as viewed from experimental bilayer lipid membranes. Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- 224.Amselem AS, Yogev A, Zawoznik E, Friedman D. 1994. Emulsomes, a novel drug delivery technology. Proc. Int. Symp. Control Release Bioactive Materials 21, 1369. [Google Scholar]
- 225.Vyas SP, Subhedar R, Jain S. 2006. Development and characterization of emulsomes for sustained and targeted delivery of an antiviral agent to liver. J. Pharm. Pharmacol. 58, 321–326. ( 10.1211/jpp.58.3.0005) [DOI] [PubMed] [Google Scholar]
- 226.Andresen TL, Jensen SS, Jorgensen K. 2005. Advanced strategies in liposomal cancer therapy: problems and prospects of active and tumor specific drug release. Progr. Lipid Res. 44, 68–97. ( 10.1016/j.plipres.2004.12.001) [DOI] [PubMed] [Google Scholar]
- 227.Hollmann A, Delfederico L, Glikmann G, De Antoni G, Semorile L, Disalvo EA. 2007. Characterization of liposomes coated with S-layer proteins from lactobacilli. Biochim. Biophys. Acta 1768, 393–400. ( 10.1016/j.bbamem.2006.09.009) [DOI] [PubMed] [Google Scholar]
- 228.Hollmann A, Delfederico L, De Antoni G, Semorile L, Disalvo EA. 2010. Interaction of bacterial surface layer proteins with lipid membranes: synergysm between surface charge density and chain packing. Colloids Surf. B: Biointerfaces 79, 191–197. ( 10.1016/j.colsurfb.2010.03.046) [DOI] [PubMed] [Google Scholar]
- 229.Wimley WC, Hristova K. 2011. Antimicrobial peptides; successes, challenges and unanswered questions. J. Membr. Biol. 239, 27–34. ( 10.1007/s00232-011-9343-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hancock REW, Chapple DS. 1999. Peptide antibiotics. Antimicrob. Agents Chemother. 43, 1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Bhakdi S, Tranum-Jensen J. 1991. Alpha-toxin of Staphylococcus aureus. Microbiol. Microbiol. Rev. 55, 733–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP. 1991. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 353, 846–849. ( 10.1038/353846a0) [DOI] [PubMed] [Google Scholar]
- 233.Marx SO, Ondrias K, Marks AR. 1998. Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science 281, 818–821. ( 10.1126/science.281.5378.818) [DOI] [PubMed] [Google Scholar]
- 234.Colombini M. 1980. Structure and mode of action of a voltage dependent anion-selective channel (VDAC) located in the outer mitochondrial membrane. Ann. N. Y. Acad. Sci. 341, 552–563. ( 10.1111/j.1749-6632.1980.tb47198.x) [DOI] [PubMed] [Google Scholar]
- 235.Colombini M. 1989. Voltage gating in the mitochondrial channel, VDAC. J. Membr. Biol. 111, 103–111. ( 10.1007/BF01871775) [DOI] [PubMed] [Google Scholar]
- 236.Colombini M. 2004. VDAC: the channel at the interface between mitochondria and the cytosol. Mol. Cell. Biochem. 256–257, 107–115. ( 10.1023/B:Mcbi.0000009862.17396.8d) [DOI] [PubMed] [Google Scholar]
- 237.Colombini M. 2012. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta Biomembr. 1818, 1457–1465. ( 10.1016/j.bbamem.2011.12.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Colombini M, Mannella CA. 2012. VDAC, the early days. Biochim. Biophys. Acta Biomembr. 1818, 1438–1443. ( 10.1016/j.bbamem.2011.11.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Schuster B, Sleytr UB. 2002. Single channel recordings of alpha-hemolysin reconstituted in S-layer-supported lipid bilayers. Bioelectrochemistry 55, 5–7. ( 10.1016/S1567-5394(01)00148-7) [DOI] [PubMed] [Google Scholar]
- 240.Schuster B, Pum D, Sára M, Braha O, Bayley H, Sleytr UB. 2001. S-layer ultrafiltration membranes: a new support for stabilizing functionalized lipid membranes. Langmuir 17, 499–503. ( 10.1021/la0008784) [DOI] [Google Scholar]
- 241.Larisch V-D. 2012. Characterization of the ryanodine receptor 1 in model lipid membranes. Master's thesis, University of Natural Resources and Life Sciences, Vienna, A; ustria. [Google Scholar]
- 242.Keizer HM, Andersson M, Chase C, Laratta WP, Proemsey JB, Tabb J, Long JR, Duran RS. 2008. Prolonged stochastic single ion channel recordings in S-layer protein stabilized lipid bilayer membranes. Colloids Surf. B Biointerfaces 65, 178–185. ( 10.1016/j.colsurfb.2008.04.015) [DOI] [PubMed] [Google Scholar]
- 243.Keizer HM, et al. 2007. Functional ion channels in tethered bilayer membranes—implications for biosensors. Chembiochem 8, 1246–1250. ( 10.1002/cbic.200700094) [DOI] [PubMed] [Google Scholar]
- 244.Diederich A, Sponer C, Pum D, Sleytr UB, Lösche M. 1996. Reciprocal influence between the protein and lipid components of a lipid-protein membrane model. Colloids Surf. B 6, 335–346. ( 10.1016/0927-7765(96)01267-2) [DOI] [Google Scholar]
- 245.Hollmann A, Delfederico L, De Antoni G, Semorile L, Disalvo EA. 2010. Relaxation processes in the adsorption of surface layer proteins to lipid membranes. J. Phys. Chem. B 114, 16 618–16 624. ( 10.1021/jp107062e) [DOI] [PubMed] [Google Scholar]
- 246.Lee BW, Faller R, Sum AK, Vattulainen I, Patra M, Karttunen M. 2004. Structural effects of small molecules on phospholipid bilayers investigated. Fluid Phase Equilib. 225, 63–68. ( 10.1016/j.fluid.2004.07.008) [DOI] [Google Scholar]
- 247.Schuster B, Gufler PC, Pum D, Sleytr UB. 2003. Interplay of phospholipase A2 with S-layer-supported lipid monolayers. Langmuir 19, 3393–3397. ( 10.1021/la026771t) [DOI] [Google Scholar]
- 248.Nikolelis DP, Hianik T, Krull UJ. 1999. Biosensors based on thin lipid films and liposomes. Electroanalysis 11, 7–15. () [DOI] [Google Scholar]
- 249.Sára M, Manigley C, Wolf G, Sleytr UB. 1988. Isoporous ultrafiltration membranes from bacterial cell envelope layers. J. Membr. Sci. 36, 179–186. ( 10.1016/0376-7388(88)80015-2) [DOI] [Google Scholar]
- 250.Sára M, Sleytr UB. 1988. Membrane biotechnology: two-dimensional protein crystals for ultrafiltration purposes. In Biotechnology (ed. Rehm HJ.), pp. 615–636. Weinheim, Germany: VCH. [Google Scholar]
- 251.Sleytr UB, Sara M. 1986. Ultrafiltration membranes with uniform pores from crystalline bacterial cell envelope layers. Appl. Microbiol. Biotechnol. 25, 83–90. ( 10.1007/BF00938929) [DOI] [Google Scholar]
- 252.Sára M, Küpcü S, Weiner C, Weigert S, Sleytr UB. 1993. Crystalline protein layers as isoporous molecular sieves and immobilisation and affinity matrices. In Immobilised macromolecules: application potentials (eds Sleytr UB, Messner P, Pum D, Sára M.), pp. 71–86. London, UK: Springer. [Google Scholar]
- 253.Weigert S, Sára M. 1996. Ultrafiltration membranes prepared from crystalline bacterial cell surface layers as model systems for studying the influence of surface properties on protein adsorption. J. Membr. Sci. 121, 185–196. ( 10.1016/S0376-7388(96)00176-7) [DOI] [Google Scholar]
- 254.Küpcü S, Sára M, Sleytr UB. 1993. Influence of covalent attachment of low molecular weight substances on the rejection and adsorption properties of crystalline proteinaceous ultrafiltration membranes. Desalination 90, 65–76. ( 10.1016/0011-9164(93)80165-J) [DOI] [Google Scholar]
- 255.Weigert S, Sára M. 1995. Surface modification of an ultrafiltration membrane with crystalline structure and studies on interactions with selected protein molecules. J. Membr. Sci. 106, 147–159. ( 10.1016/0376-7388(95)00085-Q) [DOI] [Google Scholar]
- 256.Lohner K, Prossnigg F. 2009. Biological activity and structural aspects of PGLa interaction with membrane mimetic systems. Biochim. Biophys. Acta 1788, 1656–1666. ( 10.1016/j.bbamem.2009.05.012) [DOI] [PubMed] [Google Scholar]
- 257.Schuster B, Sleytr UB. 2013. Nanotechnology with S-layer proteins. In Protein nanotechnology: protocols, instrumentation and applications (ed. Gerrard JA.), pp. 153–175, 2nd edn New York, NY: Humana Press. [Google Scholar]
- 258.Trojanowicz M. 2001. Miniaturized biochemical sensing devices based on planar lipid membranes. Fresenius J. Anal. Chem. 372, 246–260. ( 10.1007/s002160101005) [DOI] [PubMed] [Google Scholar]
- 259.Colombini M, Blachly-Dyson E, Forte M. 1996. VDAC, a channel in the outer mitochondrial membrane. Ion Channels 4, 169–202. ( 10.1007/978-1-4899-1775-1_5) [DOI] [PubMed] [Google Scholar]
- 260.Komarov AG, Deng D, Craigen WJ, Colombini M. 2005. New insights into the mechanism of permeation through large channels. Biophys. J. 89, 3950–3959. ( 10.1529/biophysj.105.070037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Lee AC, Xu X, Colombini M. 1996. The role of pyridine dinucleotides in regulating the permeability of the mitochondrial outer membrane. J. Biol. Chem. 271, 26 724–26 731. ( 10.1074/jbc.271.43.26724) [DOI] [PubMed] [Google Scholar]
- 262.Zizi M, Forte M, Blachly-Dyson E, Colombini M. 1994. NADH regulates the gating of VDAC, the mitochondrial outer membrane channel. J. Biol. Chem. 269, 1614–1616. [PubMed] [Google Scholar]
- 263.Schuster B, Pum D, Sára M, Sleytr UB. 2006. S-layer proteins as key components of a versatile molecular construction kit for biomedical nanotechnology. Mini-Rev. Med. Chem. 6, 909–920. ( 10.2174/138955706777935026) [DOI] [PubMed] [Google Scholar]
- 264.Schuster B, Györvary E, Pum D, Sleytr UB. 2005. Nanotechnology with S-layer proteins. Methods Mol. Biol. 300, 101–123. [DOI] [PubMed] [Google Scholar]
- 265.Küpcü S, Lohner K, Mader C, Sleytr UB. 1998. Microcalorimetric study on the phase behaviour of S-layer coated liposomes. Mol. Membr. Biol. 15, 69–74. ( 10.3109/09687689809027520) [DOI] [PubMed] [Google Scholar]
- 266.Pum D, Sára M, Schuster B, Sleytr UB. 2006. Bacterial surface layer proteins: a simple but versatile biological self-assembly system in nature. In Nanotechnology: science and computation (eds Chen J, Jonoska N, Rozenberg G.), pp. 277–292. Berlin, Germany: Springer. [Google Scholar]
- 267.Schäffer C, Messner P. 2005. The structure of secondary cell wall polymers: how Gram-positive bacteria stick their cell walls together. Microbiology 151, 643–651. ( 10.1099/mic.0.27749-0) [DOI] [PubMed] [Google Scholar]
- 268.Sinner EK, Knoll W. 2001. Functional tethered membranes. Curr. Opin. Chem. Biol. 5, 705–711. ( 10.1016/S1367-5931(01)00269-1) [DOI] [PubMed] [Google Scholar]
- 269.Messner P, Wellan M, Kubelka W, Sleytr UB. 1993. Reversible cross-linking of crystalline bacterial surface layer glycoproteins through their glycan chains. Appl. Microbiol. Biotechnol. 40, 7–11. ( 10.1007/BF00170420) [DOI] [Google Scholar]
- 270.Schrems A, et al. In press. The grab-and-drop protocol: a novel strategy for membrane protein isolation and reconstitution from single cells. Analyst. ( 10.1039/c4an00059e) [DOI] [PubMed] [Google Scholar]