

Abstract
Previous studies have demonstrated that cyclin D1, an upstream regulator of the Rb/E2F pathway, is an essential component of the ErbB2/Ras (but not the Wnt/Myc) oncogenic pathway in the mammary epithelium. However, the role of specific E2fs for ErbB2/Ras-mediated mammary tumorigenesis remains unknown. Here we show that in the majority of mouse and human primary mammary carcinomas with ErbB2/HER2 over-expression, E2f3a is up-regulated, raising the possibility that E2F3a is a critical effector of the ErbB2 oncogenic signaling pathway in the mammary gland. We examined the consequence of ablating individual E2fs in mice on ErbB2-triggered mammary tumorigenesis in comparison to a comparable Myc-driven mammary tumor model. We found that loss of E2f1 or E2f3 led to a significant delay on tumor onset in both oncogenic models, whereas loss of E2f2 accelerated mammary tumorigenesis driven by Myc-over-expression. Furthermore, Southern blot analysis of final tumors derived from conditionally deleted E2f3−/loxP mammary glands revealed that there is a selection against E2f3−/− cells from developing mammary carcinomas, and that such selection pressure is higher in the presence of ErbB2 activation than in the presence of Myc activation. Taken together, our data suggest oncogenic activities of E2F1 and E2F3 in ErbB2- or Myc-triggered mammary tumorigenesis, and a tumor suppressor role of E2F2 in Myc-mediated mammary tumorigenesis.
Keywords: Myc, ErbB2, E2F, mammary tumorigenesis, oncogene
Introduction
Uncontrolled cell growth is one of the hallmarks of cancer, and typically involves genetic or epigenetic alterations to genes that directly regulate the cell cycle and cellular proliferation.(1) Previous studies have led to the delineation of a pathway controlling the progression of cells out of quiescence, through G1, and into S phase that involves the activation of cyclin-dependent kinases (CDKs), phosphorylation and inactivation of the retinoblastoma tumor suppressor (Rb), and the subsequent release of E2F transcription factors.(2–4) It is now evident, from studies using both in vitro cell culture systems and in vivo mouse models, that the tumor suppressor function of Rb is largely mediated through its interaction with members of the E2F family and its regulation of E2F-dependent transcriptional activation or repression.(5, 6)
The mammalian E2F family of transcription factors consists of eight known genes (E2F1–8) encoding nine E2F proteins, with the E2f3 locus encoding two distinct isoforms, E2F3a and 3b.(7–10) Based on their structure and function, E2Fs can be divided into two broad groups. The first group, consisting of E2F1, E2F2, and E2F3a, is collectively called activators, since their primary function is to activate genes that are required for entry of cells into S phase. The remaining E2Fs form the repressor group, whose primary function is to repress genes in quiescent or terminally differentiated cells. Early studies using mouse embryonic fibroblasts (MEFs) suggest that the E2F activator subclass is critical for normal cellular proliferation since over-expression of any of the three E2f activators is sufficient to induce quiescent cells to enter the cell cycle. Using MEFs lacking the entire E2F activator subclass, we previously showed that E2F activators are essential for normal cellular proliferation.(11) In addition, we also demonstrated that E2F1, E2F2, and E2F3 are required for aberrant cell growth under oncogenic insults since loss of the three E2Fs prevents Myc and Ras oncogene-induced cellular transformation in primary MEFs,(12) suggesting that E2F1–3 are also required for tumor initiation and/or progression in vivo.
Considering the central role of the Rb/E2F pathway in the control of cell cycle, it is not surprising to find genetic alterations in this pathway in essentially all human malignancies.(13) Moreover, targeted deletion of Rb in mice leads to hyperplasia and carcinomas,(14–18) further supporting an important role of Rb in tumor suppression. Alterations of E2F activators may also contribute to aberrant cell growth and cancer development, through either over-expression/amplification or disruption of their association with Rb. Over-expression of E2F1 is associated with several types of human cancers.(19, 20) More recently, it has been found that E2F3 is up-regulated in 67% of prostate cancers, and patients with E2F3 over-expression have poorer overall survival and reduced cause-specific survival.(21) Consistent with an important role of E2F3 in human cancer development, E2F3 is either up-regulated or amplified in several other cancer types.(22–27) In mice, forced expression of E2f1, E2f2 or E2f3 leads to hyperplasia or neoplasia.(28–32) However, loss-of-function studies in mice do not always support an oncogenic role of E2Fs. For example, deletion of E2f3 in Rb+/− mice reduces the incidence of pituitary tumors but enhances the metastasis of thyroid tumors.(33) In addition, loss of E2f2 accelerates Myc-induced lymphomagenesis while the role of E2f1 in this process remains controversial.(34–36) Furthermore, inactivation of E2f1 or E2f2 enhanced Myc-induced skin tumorigenesis.(37, 38) Nonetheless, collectively these data suggest that E2F activators not only play important roles in regulating normal cellular proliferation, but also contribute to aberrant cell growth and cancer development.
Early studies using MEFs have established a functional link between the Myc or ErbB2/Ras pathway and the Rb/E2F pathway, as Myc or Ras elicits mitogenic signals that activate the cyclin/CDK complexes, leading to the release of E2F activities that promote cell growth.(39) In addition, in MEFs the ability of Myc to induce proliferation or apoptosis is dependent on specific E2F activities.(40) The importance of E2Fs in mediating Myc or Ras signaling is further highlighted by the recent finding that E2f1–3 are essential for Myc/Ras-induced cellular transformation.(12) Finally, recent studies using in vivo mouse models demonstrated that E2f1 and E2f2 mediate Myc-induced lymphomogenesis as loss of E2f1 delays Myc-induced T cell lymphomas,(34) whereas loss of E2f2 accelerates Myc-induced T cell and B cell lymphomas.(35, 36)
To understand the in vivo role of E2F1, E2F2 and E2F3 in regulating oncogene-induced mammary tumorigenesis, we sought to determine whether loss of E2f1, E2f2, or E2f3 in mice impacts on mammary tumorigenesis triggered by the mammary epithelium-specific over-expression of ErbB2/HER2/Neu or Myc, two oncogenes that are over-expressed in up to 30% of human breast cancer patients. Here we show that deletion of E2f1, E2f2, or E2f3 have differential effects on the development of ErbB2- and Myc-induced mammary tumors, raising the possibility that E2Fs, particularly E2F3, are important downstream effectors in Myc- and ErbB2-mediated oncogenic signaling in mammary glands.
Results
E2F activators are up-regulated in ErbB2-overexpressing human primary mammary carcinomas
Considering that ErbB2- or Ras-mediated mammary tumorigenesis in mice is strictly dependent on Cyclin D1,(41) which has been shown to activate E2f activators through phosphorylation and inactivation of Rb, we decided to assess whether E2F activators are relevant factors in human breast cancers. To this end, we collected a set of 12 human primary mammary carcinomas and measured mRNA transcript levels of E2Fs by qPCR. Mammary tissues from six tissue reduction patients with normal breast pathology were used as controls. Among the 12 mammary tumor samples, four had elevated levels (20 to 80 folds) of HER2 transcripts (Figure 1a). Importantly, tumor samples with HER2 over-expression also had much higher levels of E2F activators, E2F1, E2F2, and E2F3a than both normal controls and tumor samples without HER2 over-expression. Furthermore, there were marginal increases in E2F3b transcripts in tumor samples with HER2 over-expression than in the controls, but substantially lower levels of E2F3b transcripts in tumor samples without HER2 over-expression.
Figure 1.
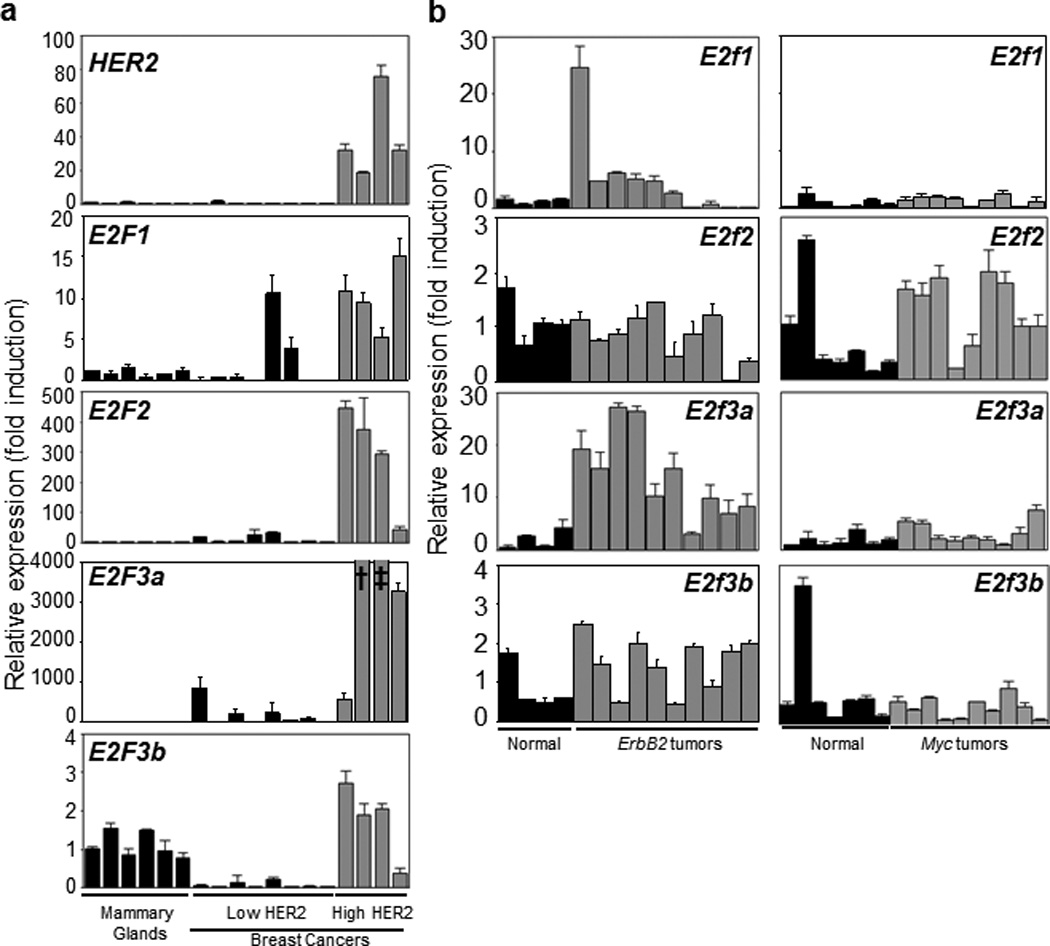
Expression levels of various E2fs in both mouse and human mammary tumors as well as mammary gland controls. (a). Real-time quantitative RT-PCR analysis of E2F1, E2F2, E2F3a, and E2F3b for human breast tumors with high or low levels of HER2 (ErbB2). Normal mammary glands from tissue reduction patients were used as controls. (b). Real-time quantitative RT-PCR analysis of E2f1, E2f2, E2f3a, and E2f3b for mouse mammary tumors with ErbB2 or Myc over-expression, compared to normal mammary gland controls. In all graphs, expression levels of Gapdh were used as a normalization control, and the averaged expression levels of the normal mammary gland control group were normalized to one. Each bar and error bar represent mean ± standard deviation from triplicates except for Figure1a, E2f3a panel, mean ± standard deviation = 92041 ± 24967 for † and = 22879 ± 7986 for ‡.
Up-regulation of E2fs in ErbB2-induced and Myc-induced mouse mammary carcinomas
We next asked whether E2f1, E2f2, and E2f3a are also preferentially up-regulated in mouse mammary carcinomas with ErbB2 over-expression. To this end, we collected a set of 10 primary mammary carcinomas from MMTV-ErbB2 transgenic mice that carry the ErbB2 oncogene driven by the MMTV promoter,(42) and that had gone through two pregnancy/lactation cycles. We then used qPCR to evaluate expression levels of various E2fs compared to those of normal mammary glands from wild-type littermate control mice. In parallel, we also carried out the same analysis with a set of nine primary mammary carcinomas from Wap-Myc mice where the Myc oncogene is driven by the Wap (Whey Acidic Protein) promoter.(43) As shown in Figure 1b, while there was a moderate increase (~2–3 fold) of E2f3a and a marginal increase of E2f2 in Wap-Myc tumors compared to normal controls, there was essentially no change in expression levels of E2f1 or E2f3b. Importantly, in almost all MMTV-ErbB2 tumors, there were substantial increases (~15 fold) in E2f3a expression, and moderate increases (~5 fold) in E2f1 expression.
E2F3 is not required for normal mammary gland development
The data presented above suggest that E2F activators are important mediators of mammary carcinomas with ErbB2 over-expression in both mice and humans. Since E2f3a exhibited the largest up-regulation in those mammary carcinomas, we initially focused on understanding how inactivation of E2f3 impacts on ErbB2- or Myc-induced mammary carcinomas in mice. We first determined whether E2F3 is required for normal mouse mammary gland development by using a Wap-Cre transgene(44) to delete an E2f3 conditional knockout allele (E2f3loxP)(11) specifically in mammary epithelial cells. Consistent with previous report,(44) X-gal staining on whole mount mammary glands from the Rosa26+/LSL-lacZ reporter mice(45) confirmed that Cre expression under the control of the Wap promoter is restricted to the mammary epithelium (Figure 2a). However, Wap-Cre-mediated recombination and subsequent activation of the lacZ gene was incomplete as evidenced by the partial, patchy blue staining in mammary epithelial ducts of day 16.5 pregnant females.
Figure 2.
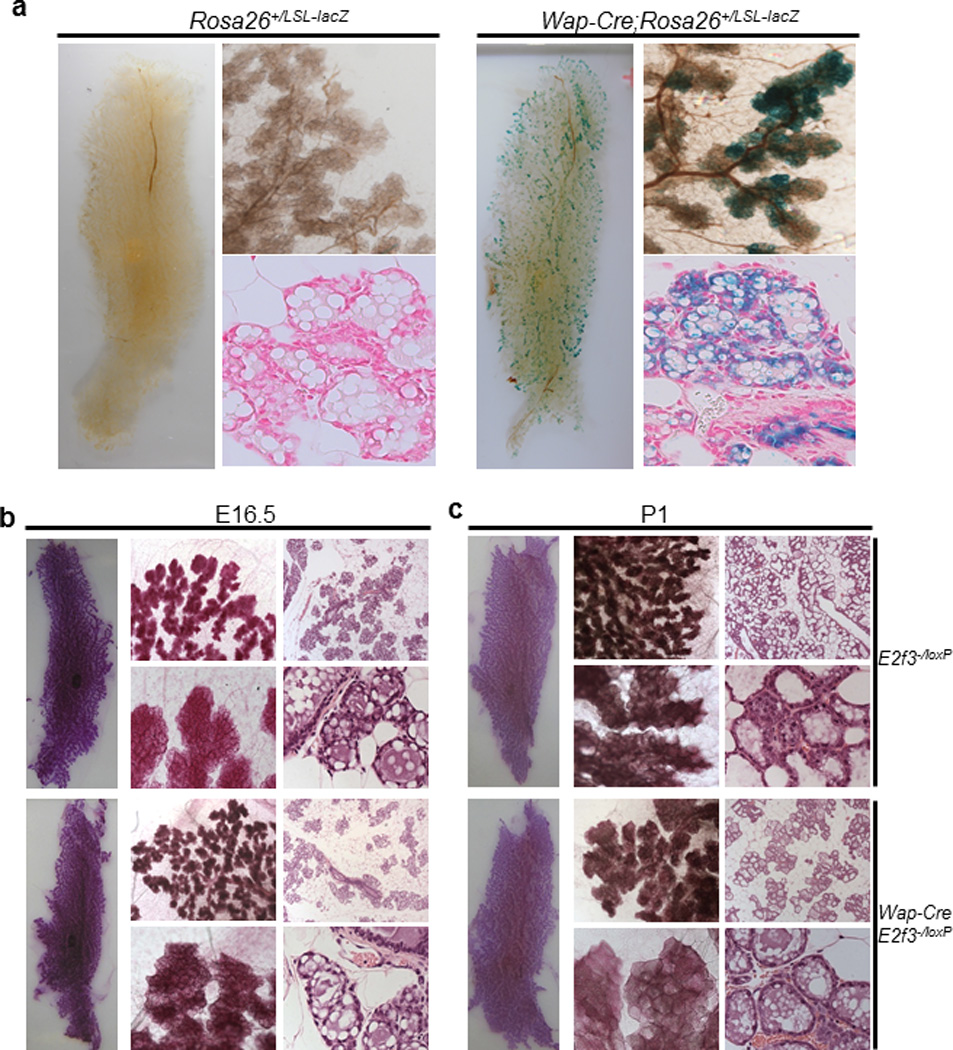
Mammary gland development of wild-type mice and mice deficient for E2f3. (a). X-gal staining of mammary glands from 16.5 days of pregnant mice with the indicated genotypes. For each genotype, left panel: whole mount; top right panel: higher magnification (5×) of the whole mounts; bottom right panel: paraffin-embedded sections counter-stained with eosin (40×). (b & c). Mammary gland morphology of 16.5 days of pregnant (b) or 1 day of lactating (c) mice with indicated genotypes. For each genotype, left panel: whole mount Carmine staining; middle panel: high magnification of whole mounts (top: 2.5×; bottom: 10×); right panel: Hemotoxylin- and eosin-stained paraffin embedded sections (top: 5×; bottom: 40×).
To determine whether Wap-Cre-mediated deletion of E2f3 in mammary epithelial cells affects mammary gland development, we examined the mammary gland structures of monoparous female mice with either 16.5 days of pregnancy or one day of lactation. As shown in Figure 2b and 2c, Carmine-stained whole mount mammary glands of Wap-Cre;E2f3−/loxP mice displayed similar lobuloalveolar structures and ductal branching patterns as those of control E2f3−/loxP mice. Furthermore, analysis of H&E-stained sections of mammary glands did not show obvious differences between Wap-Cre;E2f3−/loxP mice and control mice (Figure 2b and 2c). These data suggest that Wap-Cre-mediated deletion of E2f3 in mammary epithelial cells did not affect the mammary gland development in both pregnant females and lactating females. However, the patchy staining shown in Figure 2a suggests incomplete deletion of E2f3 in mammary epithelium of Wap-Cre;E2f3−/loxP mice. Therefore, our data do not completely rule out the possibility that E2F3 is required for the normal mammary gland development. However, considering that E2f3 germline knockout female mice in a mixed genetic background are fertile and are capable of nursing pups, it is unlikely that E2F3 is required for normal mammary gland development. Lastly, it is important to note that E2f1 or E2f2 knockout female mice are also fertile and can nurse their pups normally. Therefore, neither E2F1 nor E2F2 is likely required for normal mammary gland development.
Inactivation of E2f1, E2f2, or E2f3 alters Myc- and ErbB2-mediated mammary tumorigenesis
To understand the in vivo roles of individual E2Fs in Myc- or ErbB2-mediated mammary tumorigenesis, we determined whether inactivation of E2f1, E2f2, or E2f3 affects Myc- or ErbB2-induced mammary carcinomas. To this end, we interbred E2f1+/−, E2f2+/−, or E2f3−/loxP mice with one of the two parents containing either oncogenic allele to generate cohorts of mice over-expressing either oncogene and with or without E2f1, E2f2, or E2f3. We monitored mammary tumor onset in all cohorts of mice by manual palpation bi-weekly, and evaluated tumor onset by using standard Kaplan-Meier tumor-free curves as well as t50, or the number of days required for 50% of the mice to develop tumors. Tumor-free curves revealed no significant difference in tumor initiation among E2f wild-type (including E2f3+/loxP and E2f3loxP/loxP) mice generated from intercrosses of E2f1+/−, E2f2+/−, or E2f3−/loxP mice over-expressing ErbB2 or Myc (p = 0.598 and 0.117 for ErbB2- and Myc-over-expressing tumor mice, respectively). Therefore, for tumor initiation analyses we pooled all E2f wild-type mice (labeled as +/+ or E2f+/+) over-expressing either oncogene. As shown in Figure 3a and 3b, genetic deletion of individual E2fs had various impacts on mammary carcinogenesis. Specifically, in mice with ErbB2 over-expression, while deletion of E2f2 had no significant impact on tumor initiation (p = 0.261), loss of E2f1 or E2f3 led to significant delays of tumor onsets (p < 0.001 in both cases), with t50 being increased by 34 days and 29 days, respectively. It is interesting to note that MMTV-ErbB2;E2f1+/− mice also had a significant delay of tumor onset compared to MMTV-ErbB2 mice (p < 0.001), with t50 being increased by 14 days. Consistent with important roles of E2F1 and E2F3 in mediating oncogene-triggered mammary tumorigenesis, loss of E2f1 or E2f3 also led to significant delays of tumor onset in mice with Myc over-expression (p = 0.004 and 0.003, respectively), with t50 being increased by 10 days and 21 days, respectively (Figure 3a and 3b). On the other hand, loss of E2f2 in Myc-over-expressing mice led to a significant acceleration of tumor onset (p < 0.001), with t50 being reduced by 9 days. Consistent with specific roles of E2F1–3 on oncogene-triggered mammary tumorigenesis, MMTV-ErbB2;Wap-Cre;E2f3−/loxP mice had a significantly lower average number of tumors compared to MMTV-ErbB2 mice (p < 0.001), and Wap-Myc;E2f2−/− mice had a significantly higher average number of tumors compared to Wap-Myc mice (p < 0.01) (Figure 3c). In addition, MMTV-ErbB2;E2f1−/− mice, MMTV-ErbB2;E2f2−/− mice, and Wap-Myc;Wap-Cre;E2f3−/loxP mice all had moderate but statistically non-significant decreases in average numbers of tumors than control mice (Figure 3c).
Figure 3.
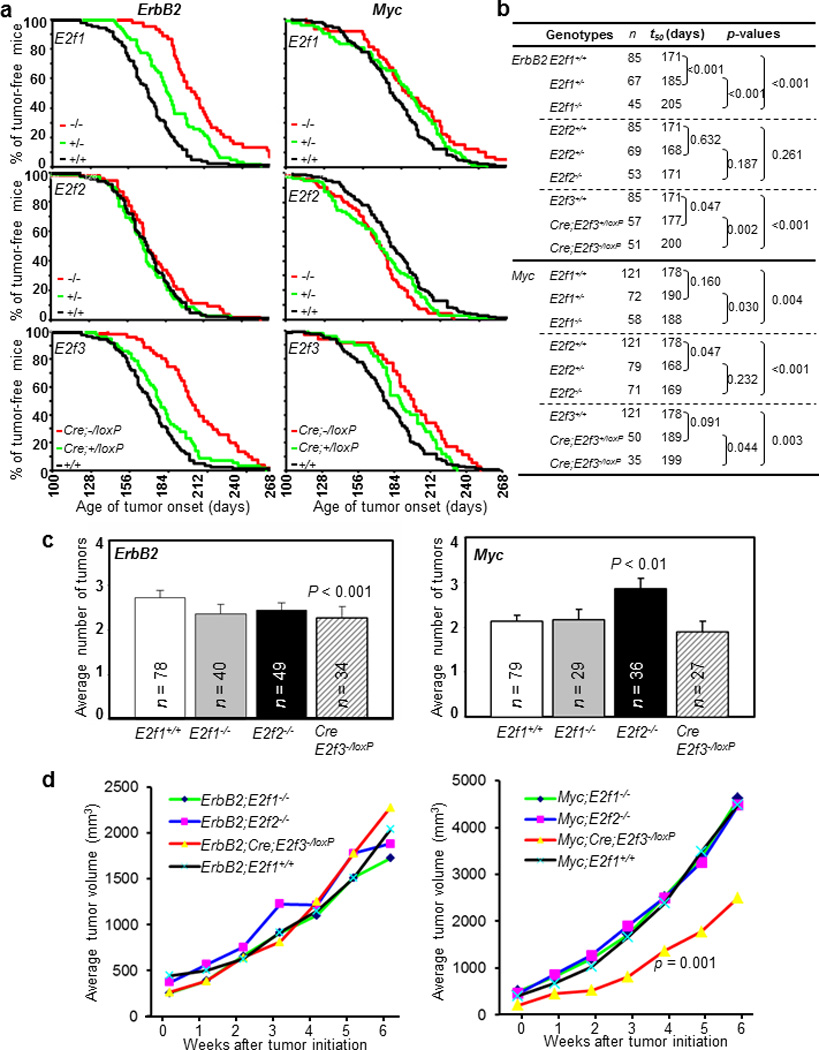
Effects of loss of E2f1, E2f2, or E2f3 on the development of mammary tumors induced by the over-expression of ErbB2 or Myc. (a). Kaplan-Meier tumor-free curves of mice with indicated genotypes. (b). Statistical analysis of tumor incidence among mice with various genotypes. n: number of mice analyzed; t50: the number of days (d) required for 50% of the mice to develop mammary tumors; Cre: Wap-Cre. (c). Average numbers of tumors per mouse at six weeks after the first tumor was detected. n: number of mice analyzed. p: significant p-values between the indicated genotypic group and E2f wild-type group. (d). Average tumor volume per tumor throughout the six weeks after the first tumor was detected. Numbers of mice analyzed are identical to those in Figure 3c. p: significant p-values between the indicated genotypic group and E2f wild-type group.
To determine whether loss of various E2fs also affects tumor growth, we measured tumor volumes once a week for six consecutive weeks. As shown in Figure 3d, loss of individual E2fs did not affect the growth of ErbB2-over-expressing mammary tumors as tumors deficient for E2f1, E2f2, or E2f3 grew similarly to those with wild-type E2fs. Interestingly, although loss of E2f1 or E2f2 did not affect tumor growth in mice with Myc-over-expression either, tumors from Wap-Myc;Wap-Cre;E2f3−/loxP mice grew significantly slower than those from Wap-Myc mice (p < 0.001)(Figure 3d).
Most mammary tumors from Wap-Cre;E2f3−/loxP mice retained the E2f3loxP allele
As shown in Figure 3a and 3b, although there was only a marginal difference on tumor initiation between MMTV-ErbB2;Wap-Cre;E2f3+/loxP mice and the control mice (Figure 4A, p = 0.047; t50 of 177 days vs. 171 days), there was a significant delay of tumor onset in MMTV-ErbB2;E2f3−/loxP mice than in the control mice (Figure 4a, p < 0.001; t50 of 190 days vs. 171 days). One potential explanation on the difference of tumor onset between the two types of E2f3 heterozygous mice (MMTV-ErbB2;Wap-Cre;E2f3+/loxP mice vs. MMTV-ErbB2;E2f3−/loxP mice) is that while all cells in MMTV-ErbB2;E2f3−/loxP mice were heterozygous for E2f3, a significant number of mammary epithelial cells in MMTV-ErbB2;Wap-Cre;E2f3+/loxP tumor mice retained the functional E2f3loxP allele due to incomplete deletion of the E2f3loxP allele as indicated in Figure 2a. In this case, we would expect that many tumors derived from MMTV-ErbB2;Wap-Cre;E2f3−/loxP mice and Wap-Myc;Wap-Cre;E2f3−/loxP mice were actually not deleted for E2f3. To test these possibilities, we first assessed the percentage of tumors derived from Wap-Cre mice with Myc or ErbB2 over-expression and with sufficient Wap-Cre-mediated recombination that most likely has no impact on tumorigenesis. To this end, we generated cohorts of MMTV-ErbB2;Wap-Cre;Rosa26+/LSL-lacZ mice and Wap-Myc;Wap-Cre;Rosa26+/LSL-lacZ mice. We allowed these mice to go through two rounds of pregnancies/lactations, and analyzed tumors derived from those mice by whole mount X-gal staining. We quantified the numbers of “blue” tumors and “white” tumors and estimated that the deletion efficiency in MMTV-ErbB2;Wap-Cre;Rosa26+/LSL-lacZ mice and Wap-Myc;Wap-Cre;Rosa26+/LSL-lacZ mice were 42.1% and 44%, respectively (Figure 4d), suggesting that Wap-Cre was sufficiently expressed in about 40% of the mammary epithelial cells with either oncogene over-expression. These data also suggest that the majority of tumors from MMTV-ErbB2;Wap-Cre;E2f3−/loxP mice and Wap-Myc;Wap-Cre;E2f3−/loxP mice did not sufficiently express Cre, thereby retaining the functional E2f3loxP allele.
Figure 4.
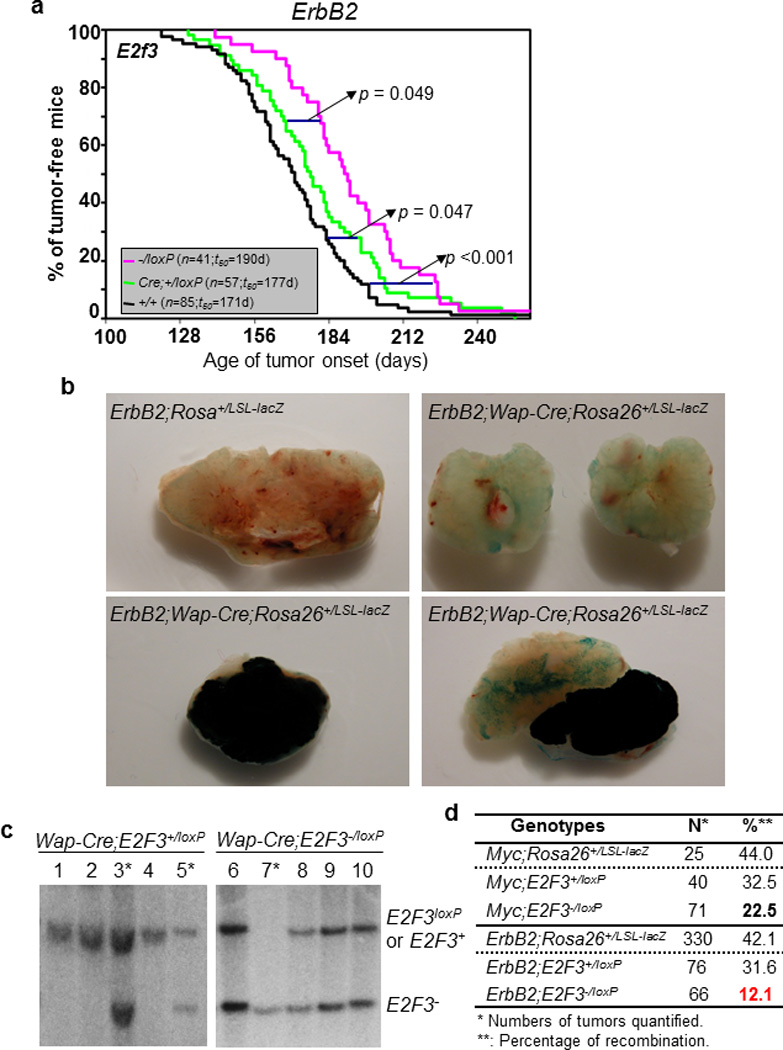
Selection against E2f3−/− cells from developing mammary carcinomas. (a). Kaplan-Meier tumor-free curves of mice with indicated genotypes. n: number of mice analyzed; t50: the number of days (d) required for 50% of the mice to develop mammary tumors; Cre: Wap-Cre; p: p-values between indicated genotypic groups. (b). Whole mount X-gal staining of mammary tumors from mice with indicated genotypes. Under this system, a complete “blue” tumor (bottom left) would indicate that in tumor cells Wap-Cre-mediated deletion of the stop cassette activates the lacZ transgene, a “white” tumor (top right) with similar staining as a control tumor (top left) would indicate that Wap-Cre activity is not sufficient to delete the stop cassette, and a mixed tumor (bottom right) would indicate tumors consisting of both Wap-Cre active cells and Wap-Cre inactive cells. (c). Southern blot analysis of individual mammary tumors derived from mice with indicated genotypes using a probe specific for the E2f3 locus. *: tumors with sufficient Cre expression in mammary epithelial cells to convert the E2f3loxP allele into an E2f3 knockout allele. (d). Percentage (%) of Cre-mediated recombination for Rosa26+/LSL-lacZ allele and E2f3loxP allele estimated by X-gal staining (b) and Southern blot analysis (c), respectively.
To determine the percentage of tumors that retained the E2f3loxP allele, we used Southern blot to analyze tumors from MMTV-ErbB2;Wap-Cre;E2f3−/loxP mice and Wap-Myc;Wap-Cre;E2f3−/loxP mice. As shown in Figure 4c and 4d, in tumors derived from MMTV-ErbB2;Wap-Cre;E2f3+/loxP mice and Wap-Myc;Wap-Cre;E2f3+/loxP mice, the deletion efficiencies of the E2f3loxP allele were 31.6% and 32.5%, respectively. These numbers are about 25% lower than the deletion efficiencies of the stop cassette in the Rosa26LSL-lacZ reporter allele in similar settings (Figure 4d). More importantly, E2f3 deletion efficiencies were further reduced to 22.5% in tumors derived from Wap-Myc;Wap-Cre;E2f3−/loxP mice, and to 12.1% in tumors derived from MMTV-ErbB2;Wap-Cre;E2Ff3−/loxP mice (Figure 4d). Taken together, these data strongly suggest that there is a selection pressure against E2f3−/− mammary epithelial cells to form tumors in both oncogenic models, and such pressure is even higher in tumors with ErbB2 over-expression than in those with Myc over-expression. Considering that the majority of tumors (~88% for MMTV-ErbB2;Wap-Cre;E2f3+/loxP mice and ~77% for Wap-Myc;Wap-Cre;E2f3+/loxP mice) are not deleted for E2f3, the delay of tumor onset in those mice depicted in Figure 3a was most likely substantially under-estimated.
Deletion of E2f3 leads to a marginal reduction of proliferation in pre-tumor state mammary epithelial cells
The tumor-free curves presented in Figure 3a, together with the qPCR data showing that E2f1 and E2f3a are preferentially up-regulated in mammary carcinomas with ErbB2 or Myc over-expression, raise the possibility that E2F1 and E2F3 are critical downstream effectors of the ErbB2-Ras-Cyclin D1 oncogenic signaling axis in the mammary epithelium. Considering that E2Fs are intimately involved in controlling cellular proliferation,(11, 39, 46) a biological process that often becomes aberrant during tumorigenesis, we investigated whether loss of individual E2fs affects cellular proliferation of mammary epithelium with oncogene-expression. Since loss of E2f1 and E2f3 led to the most significant delays of tumor onset in MMTV-ErbB2 mice, we focused on determining whether loss of E2f1 or E2f3 in pre-tumor state MMTV-ErbB2 mice affects proliferation of mammary epithelial cells from mice that have gone two pregnancy/lactation cycles. As shown in Figure 5a, while the percentages of Ki67-positive cells were very similar between MMTV-ErbB2 mice and MMTV-ErbB2;E2f1−/− mice, there was a marginally significant decrease in percentages of Ki67-positive cells in MMTV-ErbB2;Wap-Cre;E2f3−/loxP mice than in ErbB2 mice (3.6% vs. 6.5%; p = 0.045). Importantly, there was no significant difference in percentages of Ki67-positive cells between tumors with wild-type E2fs and those deficient for E2f1, E2f2, or E2f3 (Figure 5b and 5c). The fact that E2f3−/− tumors had similar numbers of Ki67-positive cells as observed in the control or E2f3 heterozygous tumors suggests that either E2f3 is indispensable for ErbB2- and Myc-induced tumor cells to proliferate, or E2f3−/− tumor cells bypass the requirement of E2f3 for maintaining normal levels of proliferation through compensational mutations.
Figure 5.
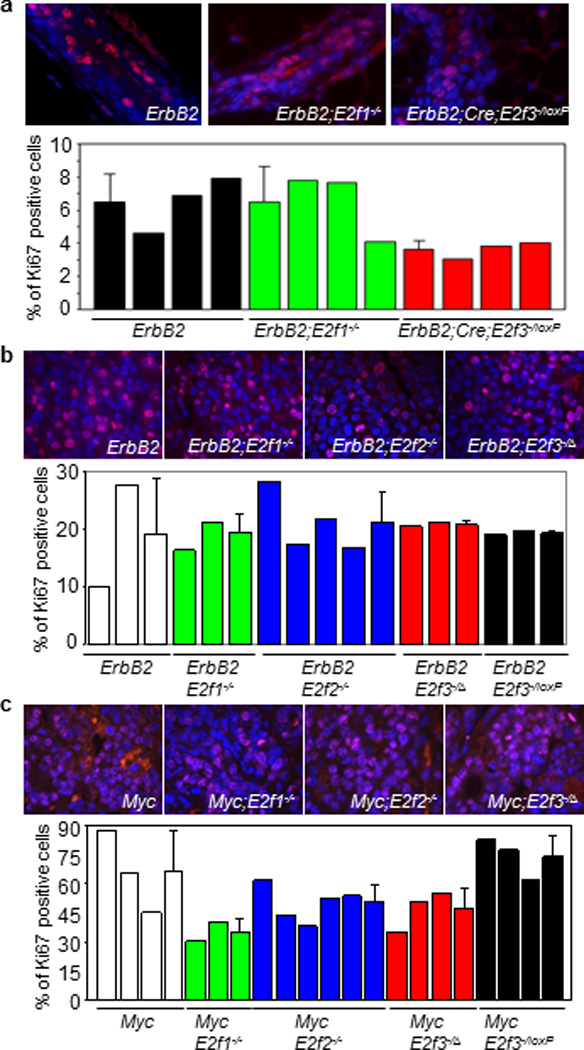
Proliferation status of mammary glands and mammary tumors. Immunofluorescence staining of mammary glands (a) or mammary tumors (b and c) from mice with indicated genotypes with Ki67-specific antibodies (red), counter-stained with DAPI (blue). Graphs with error bars show averaged percentage of Ki67 positive cells for all samples from respective genotypic groups. E2f3−/Δ: tumors from E2f3−/loxP mice with Cre-mediated E2f3 deletion being confirmed by Southern blot analysis as shown in Figure 4c.
Characterization of mammary tumors deficient for various E2fs
To investigate whether deletion of various E2fs affects the biology of mammary tumors with either oncogene over-expression, we first compared the histology of tumors deficient for E2f1, E2f2, or E2f3 to those with wild-type E2fs. As reported before, mouse mammary carcinomas resulted from ErbB2 over-expression were solid tumors, while tumors resulted from Myc over-expression were more glandular (Figure 6a). However, careful examination of H&E-stained sections of primary tumors revealed no major microscopic changes in tumors deficient for various E2fs (Figure 6a, and data not shown), suggesting that deletion of individual E2fs did not change the biological behavior of the mammary tumors.
Figure 6.
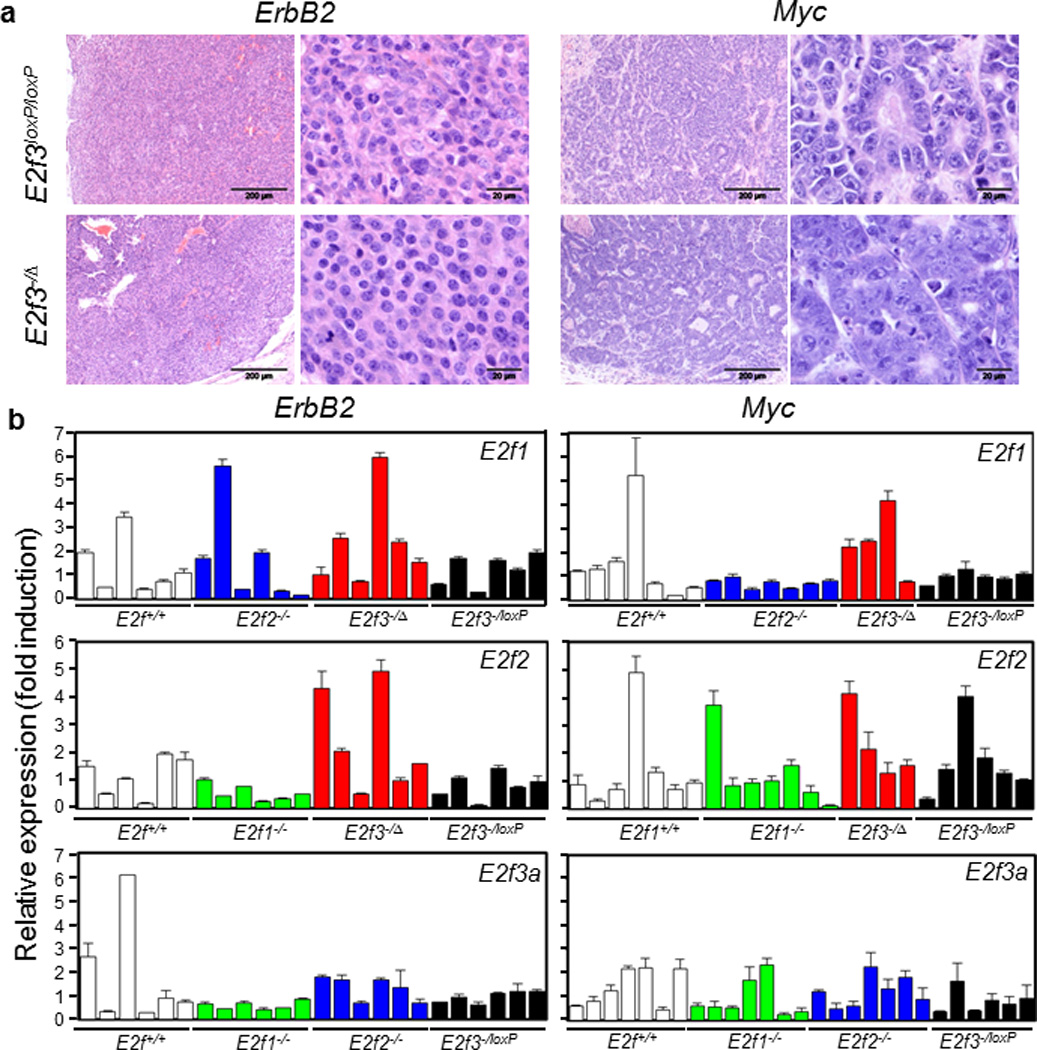
Characterization of final mammary tumors. (a). Hematoxylin- and eosin-staining of paraffin-embedded sections of mammary tumors with indicated genotypes. E2f3−/Δ: tumors from E2f3−/loxP mice with Cre-mediated E2f3 deletion being confirmed by Southern blot analysis as shown in Figure 4c. (b). Real-time quantitative RT-PCR analysis of various E2f genes for mouse mammary tumors with indicated genotypes. In all graphs, expression levels of Gapdh were used as a normalization control, and the averaged expression levels of tumors derived from mice with wild-type E2f were normalized to one. Only statistically significant p-values were shown.
Previous studies demonstrated substantial functional redundancies among various E2F activators(11) or repressors(47) for cellular proliferation and/or mouse development. To determine whether mammary tumors deficient for individual E2fs experience any compensational up-regulation of other members of E2f activators, we used qPCR to measure expression levels of various E2f genes in tumors deficient for E2f1, E2f2, or E2f3. As shown in Figure 6b, there was no obvious compensational up-regulation of any E2f activators in tumors deficient for E2f1, E2f2, or E2f3 compared to tumors with wild-type E2fs, suggesting that mammary tumors in mice deficient for individual E2fs arose without compensational up-regulation of other E2f activators. Interestingly, de-regulation of some repressor E2fs was observed in tumors lacking certain E2Fs (Supplementary Figure 1).
We previously showed that E2F1–3 control E2F target gene activation and cellular proliferation through a p53-dependent negative feedback loop.(48) To determine whether mammary tumors deficient with various E2fs contain p53 mutations, we sequenced the coding region of the p53 gene from E2f3−/Δ tumors with Myc or ErbB2 over-expression compared to tumors with wild-type or floxed E2f3 alleles. No mutation was found in E2f3−/Δ tumors compared to control tumors, suggesting that the p53 locus is most likely unaltered in mammary tumors deficient for E2f3.
Discussion
Previous studies have demonstrated that cyclin D1 is an essential component of the ErbB2/Ras (but not the Wnt/Myc) oncogenic pathway in the mouse mammary epithelium.(41) However, the role of specific E2Fs in ErbB2/Ras- or Wnt/Myc-mediated mammary tumorigenesis remains largely unknown. In this study, we found that E2f3a is up-regulated in the majority of mouse and human primary mammary carcinomas with ErbB2 or Myc over-expression (Figure 1), raising the possibility that E2F3a is a critical downstream effector of the ErbB2- or Myc-oncogenic pathway. Indeed, loss of E2f3 (and E2f1 to a lesser extent) leads to significant delays of mammary tumor onset in both oncogenic models (Figure 3a and 3b). Importantly, Southern blot analysis of the final tumors revealed a strong selection against E2f3−/− cells from developing mammary tumors as the majority of the tumors (~77% for Myc mice and ~88% for ErbB2 mice) retained a functional E2f3loxP allele (Figure 4); Wap-Cre-mediated deletion efficiency for a Rosa26-LSL-lacZ reporter allele is about 40% in both oncogenic models, whereas the E2f3 deletion efficiency is reduced to ~23% in tumors with Myc-over-expression, and is further reduced to about 12% in tumors with ErbB2 over-expression (Figure 4). Taken together, these data strongly suggest that E2F3 is a critical mediator for the ErbB2- and Myc-triggered mammary carcinogenesis. Since the E2f3 locus encodes E2F3a and E2F3b isoforms and since the E2f3 knockout mouse line used in the present study lacks both isoforms, it remains to be determined whether E2F3a or E2F3b or both play important roles in mediating ErbB2- or Myc-triggered mammary tumorigenesis. Considering that E2F3a is believed to be an activator E2F, whereas E2F3b a repressor E2f, and that E2f3a is up-regulated in the majority of mouse and human primary mammary carcinomas with ErbB2 or Myc over-expression, we postulate that E2F3a is the primary isoform in modulating oncogene-induced mammary tumorigenesis. It is interesting to note that loss of E2f2 in mice with Myc over-expression accelerates mammary tumor development, consistent with its tumor suppressor role that was recently demonstrated in Myc-induced T-cell lymphomagenesis (35) and epithelial tumorigenesis.(37) The fact that deletion of E2f2 in mice accelerates Myc-induced mammary tumorigenesis but does not affect ErbB2-mediated mammary tumorigenesis suggests that the tumor suppression role of the E2f2 locus is most likely dependent on Myc oncogene activation.
It has been well documented that E2F activators are critical for normal cellular proliferation.(11, 46) Several recent studies using in vivo mouse models also implicated E2F activators in mediating oncogene-induced tumorigenesis. For example, while loss of E2f1 impairs the ability of Myc to induce T cell lymphomas,(34) loss of E2f2 accelerates Myc-induced lymphomagenesis.(35, 36) In the present study, we showed that E2F activators are important downstream effectors in mediating ErbB2- and Myc-induced mammary tumorigenesis, since loss of E2f1 or E2f3 led to significant delays of tumor onset in both oncogenic models, and loss of E2f2 accelerated tumor onset in the Wap-Myc model. While our data supports a tumor suppressor role of E2f2 in the context of Myc activation that was recently postulated in several mouse tumor models,(35–37) it is seemingly inconsistent with a recent report suggesting a tumor promoting role of E2f2 in a MMTV-Myc-driven mammary tumor model.(49) In addition, in the MMTV-Myc-driven mammary tumor model, E2F1 seems to play a tumor suppressor role as loss of E2f1 accelerated mammary tumorigenesis.(49) These discrepancies may be explained by different promoters that drive the expression of the Myc oncogene (i.e. MMTV vs. Wap), different physiological states of the mice (i.e. non-parous mice vs. mice undergoing two rounds of pregnancies), and different genetic backgrounds. It is important to note that despite their differences, both the Wap-Myc model and the MMTV-Myc model support a tumor promoting role of E2F3. These data are consistent with recent findings of elevated levels of E2F3 in various human cancers.(21–25) The emerging evidence from both in vivo mouse models and human clinical samples raises the possibility that E2F3 or its downstream targets are potential candidates for targeted therapies of cancer.
Breast cancer represents the highest estimated new cancer cases and the second leading cause of cancer mortality in woman in the United State.(50) Unfortunately, breast cancer therapy has been hampered by limited knowledge on the molecular bases of the disease. Although activation/over-expression of ERBB2 or MYC has been found in up to 30% of breast cancer patients, the critical effector(s) induced by either oncogene remains unknown. In the present studies we identified E2F3 (and E2F1 to a lesser extent) as an important downstream effectors in mediating ErbB2- and Myc-triggered mammary tumorigenesis, and E2F2 as a tumor suppressor in mediating Myc-triggered mammary tumorigenesis. Future studies to delineate the mechanistic link between E2Fs and the Myc and ErbB2 oncogenes in mouse mammary tumor development would allow us to link pathway deregulation with potential therapeutic strategies for breast cancer. Such studies may provide critical insights in developing customized molecular therapies and/or combinatorial therapies of cancers based on individual patients’ defined genetic alterations of particular signaling pathways or molecules.
Materials and Methods
Mice and PCR-based genotyping
MMTV-Neu mice(42) and Wap-Myc mice(43) were obtained on a FVB background, whereas Wap-Cre, Rosa26-LSL-lacZ, E2f1−/−, E2f2−/−, and E2f3−/loxP mice were backcrossed to a FVB background for five or six generations to minimize the heterogeneity arose from mixed genetic backgrounds. All mice for tumor studies had gone through two pregnancy/lactation cycles to maximize the Wap-Cre expression. Genotypes of mice were determined by standard PCR of genomic DNA isolated from mouse tails. All mice were maintained in a barrier facility in accordance with standards established by the Institutional Animal Care and Use Committee at the Ohio State University. Mice at end points were euthanized by CO2 asphyxiation. Mammary glands or mammary tumors were either snap-frozen for molecular analysis, or fixed in 10% formalin for histological and immunocytochemical analyses.
Whole mounts, histology and immunofluorescence staining
Mammary glands number 4 or number 9 were dissected and mounted onto glass slides. After being fixed at 4°C overnight in a Carnoy’s fixative, the whole mount glands were stained overnight with carmine following a standard protocol. For histological analysis, 5 um paraffin-embedded sections were stained with hematoxylin and eosin (H&E). To assess lacZ expression patterns, we stained whole mount glands/tumors or 5 um paraffin-embedded sections with X-gal using standard X-gal staining protocols. For immunofluorescence staining, 5 um sections were de-paraffinized in xylene, sequentially rehydrated, and rinsed in de-ionized water. Antigen retrieval was performed using a DAKO Target Retrieval solution. Slides were incubated with primary antibodies against Ki67 (BD Biosciences, 1:100 dilution) and a fluorescence-conjugated secondary antibody (Alexa 488 from Molecular Probes, 1:500 dilution), and counterstained with DAPI.
Southern blot analysis
Genomic DNA isolated from mammary tumors was digested with EcoRV. Southern blot was carried out as previously described.(11)
Real-time quantitative RT-PCR (qPCR)
Frozen mammary tissues were used for total RNA isolation using TRIzol (Life Technologies). Human patient samples were obtained and processed in accordance with the Institutional Review Board approval at the Ohio State University. Reverse transcription of 2–5ug of total RNA was performed to generate cDNA using Superscript III reverse transcriptase (Invitrogen). qPCR was carried out in triplicates using a SYBR-Green-based system (BioRad) and a BioRad iCycler. Data were analyzed using the ΔCt method, and were normalized by the expression levels of Gapdh.
Statistical analysis
Pairwise comparisons on Kaplan-Meier tumor-free curves were performed using the Log-rank test. For each comparison, we used a Bonferonni adjusted alpha level to determine statistical significance. To assess tumor growth, we used an electronic caliper to measure palpable tumors once a week for six weeks after tumor onset. Tumor volumes were estimated by calculating 0.4 × L × S2 where L was the longest dimension and S was the shortest dimension. Tumor growth was analyzed by two way analysis of variance (ANOVA) for repeated measures over the course of the 6-week measurements and then by unpaired t-test to compare values at six weeks. Average number of palpable tumors was determined at six weeks following the onset of the first palpable tumor of each mouse and was tested for statistical significance by unpaired t-test. All other pairwise statistical analyses were done using unpaired T-test.
Supplementary Material
Acknowledgements
We thank L. Rawahneh, J. Moffitt, and N. Lovett for technical assistance with histology. We also thank OSUCCC Shared Resources for Microarray and Nucleic Acid. This work was funded by NIH grants to G.L. (R01CA85619, R01CA82259, R01HD047470, R01 CA121275; P01CA097189). G.L. is a Pew Charitable Trust Scholar and a Leukemia and Lymphoma Society Scholar.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
The authors declare no conflict of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. Epub 2011/03/08. [DOI] [PubMed] [Google Scholar]
- 2.DeGregori J. The Rb network. Journal of cell science. 2004;117(Pt 16):3411–3413. doi: 10.1242/jcs.01189. Epub 2004/07/15. [DOI] [PubMed] [Google Scholar]
- 3.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12(15):2245–2262. doi: 10.1101/gad.12.15.2245. Epub 1998/08/08. [DOI] [PubMed] [Google Scholar]
- 4.Nevins JR. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 1998;9(8):585–593. Epub 1998/08/26. [PubMed] [Google Scholar]
- 5.Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24(17):2810–2826. doi: 10.1038/sj.onc.1208612. Epub 2005/04/20. [DOI] [PubMed] [Google Scholar]
- 6.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3(1):11–20. doi: 10.1038/nrm714. Epub 2002/02/02. [DOI] [PubMed] [Google Scholar]
- 7.Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem Sci. 2004;29(8):409–417. doi: 10.1016/j.tibs.2004.06.006. Epub 2004/09/15. [DOI] [PubMed] [Google Scholar]
- 8.DeGregori J, Johnson DG. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Curr Mol Med. 2006;6(7):739–748. doi: 10.2174/1566524010606070739. Epub 2006/11/15. [DOI] [PubMed] [Google Scholar]
- 9.He Y, Cress WD. E2F-3B is a physiological target of cyclin A. J Biol Chem. 2002;277(26):23493–23499. doi: 10.1074/jbc.M202629200. Epub 2002/05/01. [DOI] [PubMed] [Google Scholar]
- 10.Leone G, Nuckolls F, Ishida S, Adams M, Sears R, Jakoi L, et al. Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol Cell Biol. 2000;20(10):3626–3632. doi: 10.1128/mcb.20.10.3626-3632.2000. Epub 2000/04/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, et al. The E2F1–3 transcription factors are essential for cellular proliferation. Nature. 2001;414(6862):457–462. doi: 10.1038/35106593. Epub 2001/11/24. [DOI] [PubMed] [Google Scholar]
- 12.Sharma N, Timmers C, Trikha P, Saavedra HI, Obery A, Leone G. Control of the p53-p21CIP1 Axis by E2f1, E2f2, and E2f3 is essential for G1/S progression and cellular transformation. J Biol Chem. 2006;281(47):36124–36131. doi: 10.1074/jbc.M604152200. Epub 2006/09/30. [DOI] [PubMed] [Google Scholar]
- 13.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10(7):699–703. doi: 10.1093/hmg/10.7.699. Epub 2001/03/21. [DOI] [PubMed] [Google Scholar]
- 14.Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev. 2000;14(19):2410–2434. doi: 10.1101/gad.819500. Epub 2000/10/06. [DOI] [PubMed] [Google Scholar]
- 15.Maddison LA, Sutherland BW, Barrios RJ, Greenberg NM. Conditional deletion of Rb causes early stage prostate cancer. Cancer Res. 2004;64(17):6018–6025. doi: 10.1158/0008-5472.CAN-03-2509. Epub 2004/09/03. [DOI] [PubMed] [Google Scholar]
- 16.Simin K, Wu H, Lu L, Pinkel D, Albertson D, Cardiff RD, et al. pRb inactivation in mammary cells reveals common mechanisms for tumor initiation and progression in divergent epithelia. PLoS biology. 2004;2(2):E22. doi: 10.1371/journal.pbio.0020022. Epub 2004/02/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006;66(16):7889–7898. doi: 10.1158/0008-5472.CAN-06-0486. Epub 2006/08/17. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Z, Flesken-Nikitin A, Nikitin AY. Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts. Cancer Res. 2007;67(12):5683–5690. doi: 10.1158/0008-5472.CAN-07-0768. Epub 2007/06/08. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki T, Yasui W, Yokozaki H, Naka K, Ishikawa T, Tahara E. Expression of the E2F family in human gastrointestinal carcinomas. International journal of cancer Journal international du cancer. 1999;81(4):535–538. doi: 10.1002/(sici)1097-0215(19990517)81:4<535::aid-ijc5>3.0.co;2-4. Epub 1999/05/04. [DOI] [PubMed] [Google Scholar]
- 20.Iwamoto M, Banerjee D, Menon LG, Jurkiewicz A, Rao PH, Kemeny NE, et al. Overexpression of E2F-1 in lung and liver metastases of human colon cancer is associated with gene amplification. Cancer biology & therapy. 2004;3(4):395–399. Epub 2004/01/17. [PubMed] [Google Scholar]
- 21.Foster CS, Falconer A, Dodson AR, Norman AR, Dennis N, Fletcher A, et al. Transcription factor E2F3 overexpressed in prostate cancer independently predicts clinical outcome. Oncogene. 2004;23(35):5871–5879. doi: 10.1038/sj.onc.1207800. Epub 2004/06/09. [DOI] [PubMed] [Google Scholar]
- 22.Cooper CS, Nicholson AG, Foster C, Dodson A, Edwards S, Fletcher A, et al. Nuclear overexpression of the E2F3 transcription factor in human lung cancer. Lung Cancer. 2006;54(2):155–162. doi: 10.1016/j.lungcan.2006.07.005. Epub 2006/08/30. [DOI] [PubMed] [Google Scholar]
- 23.Feber A, Clark J, Goodwin G, Dodson AR, Smith PH, Fletcher A, et al. Amplification and overexpression of E2F3 in human bladder cancer. Oncogene. 2004;23(8):1627–1630. doi: 10.1038/sj.onc.1207274. Epub 2004/01/13. [DOI] [PubMed] [Google Scholar]
- 24.Grasemann C, Gratias S, Stephan H, Schuler A, Schramm A, Klein-Hitpass L, et al. Gains and overexpression identify DEK and E2F3 as targets of chromosome 6p gains in retinoblastoma. Oncogene. 2005;24(42):6441–6449. doi: 10.1038/sj.onc.1208792. Epub 2005/07/12. [DOI] [PubMed] [Google Scholar]
- 25.Oeggerli M, Tomovska S, Schraml P, Calvano-Forte D, Schafroth S, Simon R, et al. E2F3 amplification and overexpression is associated with invasive tumor growth and rapid tumor cell proliferation in urinary bladder cancer. Oncogene. 2004;23(33):5616–5623. doi: 10.1038/sj.onc.1207749. Epub 2004/05/04. [DOI] [PubMed] [Google Scholar]
- 26.Orlic M, Spencer CE, Wang L, Gallie BL. Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosomes Cancer. 2006;45(1):72–82. doi: 10.1002/gcc.20263. Epub 2005/09/24. [DOI] [PubMed] [Google Scholar]
- 27.Hurst CD, Tomlinson DC, Williams SV, Platt FM, Knowles MA. Inactivation of the Rb pathway and overexpression of both isoforms of E2F3 are obligate events in bladder tumours with 6p22 amplification. Oncogene. 2008;27(19):2716–2727. doi: 10.1038/sj.onc.1210934. Epub 2007/11/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agger K, Santoni-Rugiu E, Holmberg C, Karlstrom O, Helin K. Conditional E2F1 activation in transgenic mice causes testicular atrophy and dysplasia mimicking human CIS. Oncogene. 2005;24(5):780–789. doi: 10.1038/sj.onc.1208248. Epub 2004/11/09. [DOI] [PubMed] [Google Scholar]
- 29.Conner EA, Lemmer ER, Omori M, Wirth PJ, Factor VM, Thorgeirsson SS. Dual functions of E2F-1 in a transgenic mouse model of liver carcinogenesis. Oncogene. 2000;19(44):5054–5062. doi: 10.1038/sj.onc.1203885. Epub 2000/10/24. [DOI] [PubMed] [Google Scholar]
- 30.Lazzerini Denchi E, Attwooll C, Pasini D, Helin K. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol. 2005;25(7):2660–2672. doi: 10.1128/MCB.25.7.2660-2672.2005. Epub 2005/03/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulson QX, McArthur MJ, Johnson DG. E2F3a stimulates proliferation, p53-independent apoptosis and carcinogenesis in a transgenic mouse model. Cell Cycle. 2006;5(2):184–190. doi: 10.4161/cc.5.2.2307. Epub 2005/12/13. [DOI] [PubMed] [Google Scholar]
- 32.Scheijen B, Bronk M, van der Meer T, De Jong D, Bernards R. High incidence of thymic epithelial tumors in E2F2 transgenic mice. J Biol Chem. 2004;279(11):10476–10483. doi: 10.1074/jbc.M313682200. Epub 2003/12/20. [DOI] [PubMed] [Google Scholar]
- 33.Ziebold U, Lee EY, Bronson RT, Lees JA. E2F3 loss has opposing effects on different pRB-deficient tumors, resulting in suppression of pituitary tumors but metastasis of medullary thyroid carcinomas. Mol Cell Biol. 2003;23(18):6542–6552. doi: 10.1128/MCB.23.18.6542-6552.2003. Epub 2003/08/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baudino TA, Maclean KH, Brennan J, Parganas E, Yang C, Aslanian A, et al. Myc-mediated proliferation and lymphomagenesis, but not apoptosis, are compromised by E2f1 loss. Mol Cell. 2003;11(4):905–914. doi: 10.1016/s1097-2765(03)00102-3. Epub 2003/04/30. [DOI] [PubMed] [Google Scholar]
- 35.Opavsky R, Tsai SY, Guimond M, Arora A, Opavska J, Becknell B, et al. Specific tumor suppressor function for E2F2 in Myc-induced T cell lymphomagenesis. Proc Natl Acad Sci U S A. 2007;104(39):15400–15405. doi: 10.1073/pnas.0706307104. Epub 2007/09/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rempel RE, Mori S, Gasparetto M, Glozak MA, Andrechek ER, Adler SB, et al. A role for E2F activities in determining the fate of Myc-induced lymphomagenesis. PLoS Genet. 2009;5(9):e1000640. doi: 10.1371/journal.pgen.1000640. Epub 2009/09/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pusapati RV, Weaks RL, Rounbehler RJ, McArthur MJ, Johnson DG. E2F2 suppresses Myc-induced proliferation and tumorigenesis. Molecular carcinogenesis. 2010;49(2):152–156. doi: 10.1002/mc.20584. Epub 2009/10/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rounbehler RJ, Rogers PM, Conti CJ, Johnson DG. Inactivation of E2f1 enhances tumorigenesis in a Myc transgenic model. Cancer Res. 2002;62(11):3276–3281. Epub 2002/05/31. [PubMed] [Google Scholar]
- 39.Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature. 1997;387(6631):422–426. doi: 10.1038/387422a0. Epub 1997/05/22. [DOI] [PubMed] [Google Scholar]
- 40.Leone G, Sears R, Huang E, Rempel R, Nuckolls F, Park CH, et al. Myc requires distinct E2F activities to induce S phase and apoptosis. Mol Cell. 2001;8(1):105–113. doi: 10.1016/s1097-2765(01)00275-1. Epub 2001/08/21. [DOI] [PubMed] [Google Scholar]
- 41.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411(6841):1017–1021. doi: 10.1038/35082500. Epub 2001/06/29. [DOI] [PubMed] [Google Scholar]
- 42.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54(1):105–115. doi: 10.1016/0092-8674(88)90184-5. Epub 1988/07/01. [DOI] [PubMed] [Google Scholar]
- 43.Sandgren EP, Schroeder JA, Qui TH, Palmiter RD, Brinster RL, Lee DC. Inhibition of mammary gland involution is associated with transforming growth factor alpha but not c-myc-induced tumorigenesis in transgenic mice. Cancer Res. 1995;55(17):3915–3927. Epub 1995/09/01. [PubMed] [Google Scholar]
- 44.Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, et al. Cre-mediated gene deletion in the mammary gland. Nucleic acids research. 1997;25(21):4323–4330. doi: 10.1093/nar/25.21.4323. Epub 1997/10/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature genetics. 1999;21(1):70–71. doi: 10.1038/5007. Epub 1999/01/23. [DOI] [PubMed] [Google Scholar]
- 46.Humbert PO, Verona R, Trimarchi JM, Rogers C, Dandapani S, Lees JA. E2f3 is critical for normal cellular proliferation. Genes Dev. 2000;14(6):690–703. Epub 2000/03/25. [PMC free article] [PubMed] [Google Scholar]
- 47.Gaubatz S, Lindeman GJ, Ishida S, Jakoi L, Nevins JR, Livingston DM, et al. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol Cell. 2000;6(3):729–735. doi: 10.1016/s1097-2765(00)00071-x. Epub 2000/10/13. [DOI] [PubMed] [Google Scholar]
- 48.Timmers C, Sharma N, Opavsky R, Maiti B, Wu L, Wu J, et al. E2f1, E2f2, and E2f3 control E2F target expression and cellular proliferation via a p53-dependent negative feedback loop. Mol Cell Biol. 2007;27(1):65–78. doi: 10.1128/MCB.02147-05. Epub 2006/12/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujiwara K, Yuwanita I, Hollern DP, Andrechek ER. Prediction and genetic demonstration of a role for activator E2Fs in Myc-induced tumors. Cancer Res. 2011;71(5):1924–1932. doi: 10.1158/0008-5472.CAN-10-2386. Epub 2011/01/20. [DOI] [PubMed] [Google Scholar]
- 50.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. Epub 2013/01/22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.