

Abstract
The study of metabolomics can provide valuable information about biochemical pathways and processes at the molecular level. There have been many reports that have examined the structure, identity and concentrations of metabolites in biological systems. However, the binding of metabolites with proteins is also of growing interest. This review examines past reports that have looked at the binding of various types of metabolites with proteins. An overview of the techniques that have been used to characterize and study metabolite-protein binding is first provided. This is followed by examples of studies that have investigated the binding of hormones, fatty acids, drugs or other xenobiotics, and their metabolites with transport proteins and receptors. These examples include reports that have considered the structure of the resulting solute-protein complexes, the nature of the binding sites, the strength of these interactions, the variations in these interactions with solute structure, and the kinetics of these reactions. The possible effects of metabolic diseases on these processes, including the impact of alterations in the structure and function of proteins, are also considered.
Keywords: Metabolomics, Drug-protein interactions, Hormone-protein interactions, Fatty acid-protein interactions, Xenobiotic-protein interactions, Protein modification
1. Introduction
Metabolomics is a field that involves the study of low mass compounds (i.e., metabolites) that are produced through metabolic processes [1,2]. Metabolites are part of a collection of chemicals known as the “metabolome”, which can include small molecules that are found in cells, tissues, organs, or biological fluids. The area of metabolomics is of interest because the identity and concentration of metabolites can provide information about cellular activity and can be directly related to processes such as protein and gene expression [1–3]. This means that metabolomics can provide information on the phenotypes of individuals at the molecular level [3]. In addition, the characterization and examination of metabolites could lead to new discoveries in biomedical research and personalized medicine [1,3].
Research in metabolomics began in the late 1990s and early 2000s, with the emphasis at that time being on the effects of different metabolites on the gene expression of bacteria and yeast [1]. The first examples of metabolomic studies utilized two-dimensional thin-layer chromatography separations to characterize metabolites in samples. This provided researchers with evidence that variation in the concentrations of metabolites can affect cellular activity [1,4–6]. Further progress in the area of analytical methods such as structural characterization and separation methods has resulted in the development of new instruments and techniques that can be used to provide high resolution information and data from complex samples such as tissues and cells [1,2].
Research in metabolomics can involve either targeted or untargeted approaches [7]. In a targeted approach, researchers use qualitative techniques such as nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS) for the identification, quantification, and structural characterization of specific metabolites. This information can be used to examine specific classes of metabolites and to provide information on the biochemical pathways that are involved in metabolism [2]. In an untargeted approach, scientists use global profiling to analyze the group of chemicals in a metabolome as a whole. This second approach is less specific and sensitive than the targeted approach but allows for the highest possible coverage of the metabolites that may be involved in biochemical pathways [7].
A significant amount of recent research has been devoted to metabolic profiling, or the identification and measurement of the different metabolites that are present and produced in the metabolome [8]. However, it is also important to consider the interactions that occur between metabolites and biological agents, such as the binding of cofactors to enzymes, hormones to receptors, and drugs or their metabolites to proteins [8]. Information on these interactions can be combined with the structural data to provide a better understanding of the regulatory networks and connections in biological pathways. Such information, in turn, could provide a better understanding of how healthy and disease states differ at the molecular level and could provide vital data that can be used for pharmaceutical development [7,9].
This review will look at previous studies that have examined biological interactions as related to metabolites and proteins as binding agents. This will include an overview of the various methods and techniques that have been used in this work to study metabolite-protein interactions. A summary will also be provided of the different types of metabolite-protein binding interactions that have been investigated with these approaches. In addition, the possible effects that metabolic diseases may have on these interactions will be considered.
2. Techniques for examining metabolite-protein interactions
The characterization of metabolite-protein interactions can provide a better understanding in clinical diagnostics of the cellular activity and the biochemical pathways that are present in various medical conditions [1–3,9]. There are many methods that can be used to examine the binding of metabolites with proteins. These methods may involve the direct examination of binding that occurs between proteins and low mass drugs, hormones and their metabolites, or may involve an examination of the free concentrations of these molecules [9–12]. The approaches that are used for this purpose can be divided into three categories: in vitro, in vivo and in silico techniques [9,11–46].
2.1. In vitro methods for studying metabolite-protein interactions
In vitro methods are the most popular techniques used to characterize metabolite-protein interactions. This approach involves the use of standard, well-controlled conditions and reagents that are used in the laboratory to mimic conditions seen in biological systems. To examine metabolite-protein interactions, in vitro methods may use a binding assay (e.g., one based on ultrafiltration or equilibrium dialysis) to examine an interaction or to identify the chemicals that are involved in this process [9]. This approach can provide information such as the strength of the interaction, as well as the thermodynamics and kinetics of binding and possible conformational changes that occur as a result of the interaction [13–15]. Alternatively, an in vitro study may make use of a method that directly examines the structure of a protein and a bound metabolite, such as occurs in X-ray crystallography or NMR spectroscopy [1,16–20]. Other methods may examine the protein-metabolite complex, as demonstrated with mass spectrometry [24–29].
There are many in vitro approaches that can be used to examine the binding of proteins with small molecules and their metabolites. For instance, radiometry and fluorimetry can be used with a binding assay by employing labeled metabolites that contain either a radioisotopic label or fluorophore, respectively [10,21–23]. These labeled metabolites are then incubated with proteins and the signal that is produced from the label is measured, such as through a displacement assay or a proteome microarray [10,23]. Radioisotopic labeling has been applied to enzymes to determine their activity in metabolomic reactions [9]. An example involved the screening of potential inhibitors for an enzyme, in which the substrate was radioactively labeled and the resulting metabolite profiles were analyzed and measured [21]. Fluorescence labeling can provide similar results to radiolabeling; however, this method can also be used to identify and determine the location of a binding site for a metabolite on a protein, such as by observing the displacement of specific probes that are bound to known locations on a protein [10].
Surface plasmon resonance (SPR) and calorimetry are two other methods that can provide information on the strength of protein-metabolite binding and the thermodynamics or kinetics of this interaction [13–15]. Studies based on SPR utilize an immobilized protein on a sensor chip, in which changes in the resonance energy (e.g., from binding of the protein with a target) are detected [9]. The change in this signal is related to the mass of the bound metabolites and can be used to determine the equilibrium constants for this process or, if examined over time, the association and dissociation kinetics that occur between the metabolite and protein during binding [9]. The reaction between a metabolite and protein can result in heat being absorbed or given off [9,13]. Calorimetry can be used to measure the overall enthalpy of the binding reaction between a metabolite and a protein [13].
NMR spectroscopy and X-ray crystallography are two tools that have been used to characterize the structures of metabolite-protein complexes [9,16–20]. NMR spectroscopy has often been used in recent years for characterizing and identifying metabolites in biological samples, but this method can also be used to examine conformational changes that occur during the binding of metabolites with proteins [18–20]. X-Ray crystallography can also give structural information on such interactions by providing detailed information on the binding sites and active sites for hormones, drugs and their metabolites or related compounds on proteins and enzymes [16–17], as is illustrated in Figure 1 [30].
Figure 1.
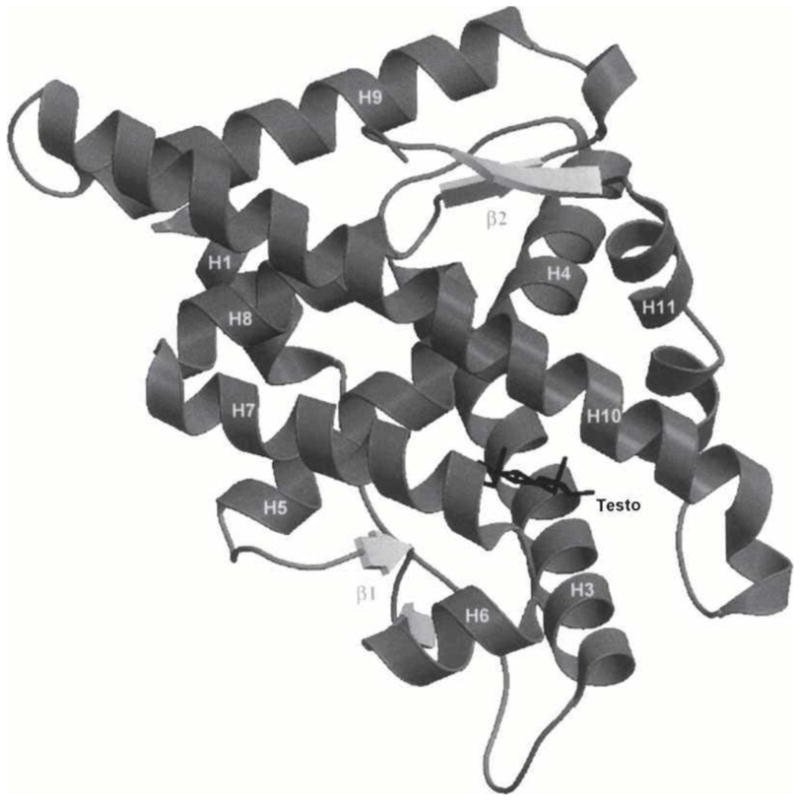
Crystal structure for the complex of human androgen receptor ligand-binding domain with testosterone (Testo). Reproduced with permission from Ref. [30].
Mass spectrometry can not only be used as a tool for analyzing structure and identity of metabolites, but it can be used to analyze metabolite-protein interactions in which information about enzymatic processes or binding by small molecules is generated [9]. Experiments utilizing various types of mass spectrometry, such as quadrupole mass spectrometry or matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS), have allowed for monitoring of the resulting products and analysis of the reaction kinetics of enzyme-substrate reactions [7,24,25]. Further analysis through high resolution mass spectrometers (e.g., an orbitrap or Fourier transform ion cyclotron resonance mass spectrometry) has resulted in accurate analyses of enzymatic activities in which the intermediate steps in enzymatic reactions can be identified [26–29].
Various separation techniques can also be used to examine metabolite-protein interactions. Examples of traditional methods that have often been utilized for this purpose are equilibrium dialysis, ultrafiltration, and ultracentrifugation [9,31–33]. Equilibrium dialysis and ultrafiltration can both be used to separate protein-bound metabolites from free metabolites through the use of a semipermeable membrane. These methods are commonly applied to binding studies to determine the affinity of proteins with drugs and small solutes but can be employed in the same way to examine the interactions of metabolites with proteins [31]. Ultracentrifugation can be used to provide a similar separation of free and protein-bound forms of a metabolite by utilizing a gravitational field in combination with a density gradient to separate these fractions [9,32]. However, each of these methods does have limitations, such as difficulty in detecting small free solute fractions, undesirable adsorption of solutes onto the membrane (e.g., in ultrafiltration or equilibrium dialysis) or overestimation of the free fraction due to release of the bound solute during the separation process [33].
Various chromatographic techniques have also been employed to separate free and protein-bound metabolite fractions [34]. As an example, size exclusion chromatography (SEC) can be applied to this type of analysis when metabolite-protein complexes and free metabolites have a sufficiently large difference in size. In this type of studies, metabolites or small molecules are incubated with a protein, and SEC can be used to remove the small molecules from proteins [34]. Such a method can be used for either the isolation and preparation of metabolite-protein complexes, which can later be analyzed by other methods, or can be used in binding studies to provide information on the association equilibrium constant for a metabolite-protein interaction [8,34,35].
Affinity chromatography and high-performance affinity chromatography (HPAC) have also become popular for analyzing solute-protein interactions [35–38]. These affinity methods have an immobilized biological molecule, such as a protein, as the stationary phase. When used in a low-performance setting, affinity chromatography can be used in a similar way as SEC in that it can be used for preparation and purification. The use of more rigid and efficient supports in HPAC allows this approach to be used as a rapid and relatively high-throughput method for providing information about solute-protein interactions. This information can include data on the affinity, thermodynamics and kinetics of these processes, as well as information on the types of sites that are involved in the interaction (see Figure 2) [35–39].
Figure 2.
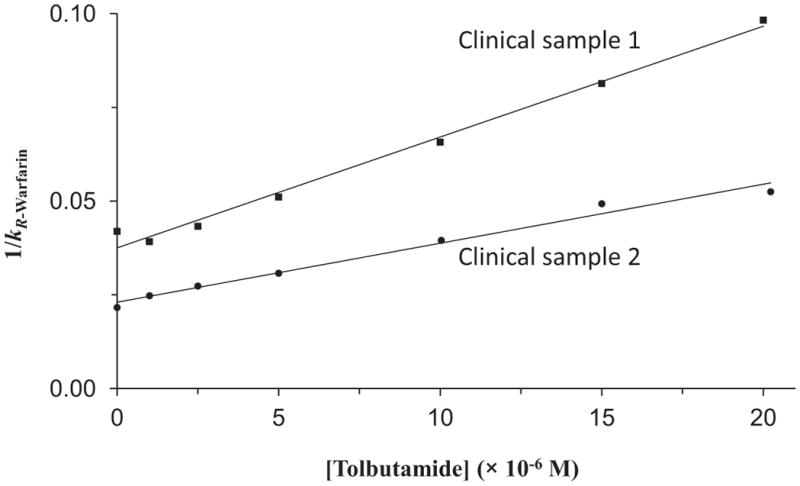
Example of a competition study using high-performance affinity chromatography to examine the interactions of an injected site-selective probe with a solute that is present at a known concentration in the mobile phase. This example shows the change in the retention factor (k) that was measured for R-warfarin as a probe for Sudlow site I of human serum albumin (HSA) in the presence of various concentrations of tolbutamide as a competing agent. These results were obtained for columns that contained two clinical samples of HSA that had different levels of modification due to glycation. Adapted with permission from Ref. [39].
Capillary electrophoresis (CE) is another separation method that can be used to examine metabolite-protein binding [9,37,40,41]. One way this method can be used is to separate the free and bound metabolites through the differences in their size-to-charge ratios. This approach can be utilized alone to determine the affinity of metabolite-protein binding or combined with other methods such as mass spectrometry to examine this interaction [41]. One form of CE is affinity capillary electrophoresis (ACE), in which a biological molecule such as a protein is used as a running buffer additive, thus making it possible to obtain data on the interactions of solute components with this additive [40]. Like HPAC, ACE is a relatively fast method and can be used with small amounts of sample for the screening or analysis of metabolite-protein interactions [38,40].
2.2. In vivo methods for studying metabolite-protein interactions
Although in vitro methods can provide detailed information about metabolite-protein interactions, in vivo analysis can provide a better representation about the metabolite-protein interaction within a biological sample [8,9]. This is particularly true in a situation where a protein may undergo post-translational modifications that result in changes in the protein’s interactions with solutes such as drugs and their metabolites [9]. In vivo methods are often similar to techniques used for in vitro studies but must be able to work with complex samples. In many cases, clinical samples from patients can be obtained and analyzed through approaches such as labeling, NMR or MS structural characterization, and affinity separation methods. By utilizing in vivo studies, researchers are better able to understand the effect of disease states on metabolite-protein interactions, as well as related biochemical pathways and regulatory processes [9,39].
2.3. In silico methods for studying metabolite-protein interactions
Another area of examining metabolite-protein interactions is through in silico tools [9]. These methods utilize computational schemes to determine the docking configurations of a metabolite’s binding sites on proteins or enzymes, as obtained through the use of molecular modeling or quantum mechanics [42,43]. This approach can provide information about the structure of a metabolite-protein complex at a given binding site through an analysis of the most thermodynamically-favorable configurations. These computational methods can result in docking predictions that have a 1.5 to 2 Å accuracy with success rates of 70–80% [43]. If the location of a binding site is not known in advance, a homology method can be used to predict binding sites on a protein through the use of the protein’s amino acid sequence and chemical structures of the metabolites [44]. This method can allow for accurate prediction of ligand-binding proteins and enable the development of a database for these peptide sequences selected for binding to different metabolites [9,45]. These in silico methods can be combined with in vitro analysis to optimize the structural characterization of metabolite-protein interactions, as demonstrated in NMR experiments [46].
3. Interactions of proteins with hormones and related metabolites
Hormones are chemicals that are secreted by endocrine glands. Hormones play a significant role in many regulation pathways, including metabolism, growth and development [47,48]. Examples of low mass hormones include various types of steroids (e.g., estrogens and testosterone) or thyroid hormones (e.g., thyroxine) [49–52]. As these chemicals enter the circulation, they are carried to their target tissue or organ to produce an effect. Many low mass hormones are transported in the bloodstream through their binding to serum proteins [51,52]. These transport proteins may bind to a broad range of hormones and other targets, as occurs for human serum albumin (HSA), or they may be specific for a given hormone or group of hormones, as is the case for thyroxine-binding globulin (TBG) [49]. Once it has been delivered to its target tissue or organ, the hormone can then bind with a receptor to produce an effect. This section will consider interaction studies that have been reported for several types of hormones or their metabolites with serum proteins and hormone receptors.
3.1. Thyroid hormones
Thyroid hormones are a group of iodothyronine compounds that are responsible for metabolism, growth, development, and the regulation of iodine within the body [47]. Many of these hormones are bound in the bloodstream to both HSA through low-to-moderate affinity interactions and to transthyretin or TBG through higher affinity processes [48,49]. An important compound in this group is the hormone L-thyroxine (L-3,5,3′,5′-tetraiodothyronine, or T4), which can be metabolized to form L-3,5,3′-triiodothyronine (T3) [53,54]. Both T4 and T3 are actively involved in regulatory processes and are more than 99% bound to transport proteins in blood [49,53,54].
Several studies have explored the structural differences between thyroid hormones and related compounds as they bind to serum proteins or cell surface receptors [53–55]. One report utilized HPAC to characterize the binding of T4, T3 and related compounds with HSA; the results were used to examine both the affinity constants and thermodynamic properties of these compounds in their interactions with this protein [54]. Some typical results that were obtained in competition studies and through the use of site-specific probes are provided in Table 1. The results indicated that these thyroid hormones were interacting with HSA at both of the major drug-binding sites on this protein for drugs (i.e., Sudlow sites I and II) [53,54]. A comparison of the data obtained for the thyroid hormones and their metabolites indicated that the number and position of iodines, the phenol group, and the thyronine backbone were all important during the binding of these compounds to HSA [54]. Structural studies have also been carried out through the use of modeling and crystallographic data to examine the binding of thyroxine and related compounds to a cell surface receptor for thyroid hormones on αvβ3 integrin [55].
Table 1.
Site-specific association equilibrium constants (Ka) for thyroxine and related compounds with HSA at 37°Ca
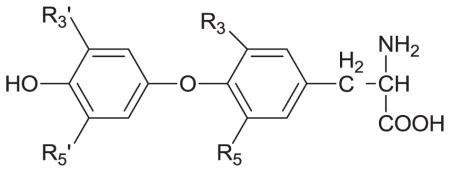
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R3 | R5 | R3′ | R5′ | Chiral form | Ka(M−1 × 105)a | |
| Sudlow site I | Sudlow site II | ||||||
| Thyroxine (T4) | I | I | I | I | L-T4b D-T4 |
1.4 (± 0.1) 5.5 (± 0.9) |
5.7 (± 0.8) 29 (± 2) |
| Triidothyronine (T3) | I | I | I | H | L-T3 D-T3 |
0.170 (± 0.001) 1.45 (± 0.06) |
0.25 (± 0.02) 1.2 (± 0.2) |
| Reverse Triiodothyronine (rT3) | I | H | I | I | L-rT3 | 1.99 (± 0.07) | 3.3 (± 0.6) |
| Diiodothyronine (T2) | I | I | H | H | L-T2 | 0.63 (± 0.03) | 1.16 (± 0.06) |
| Thyronine (T0) | H | H | H | H | L-T0 | 0.18 (± 0.02) | ------- |
3.2. Steroid hormones
The protein binding of steroid hormones and their metabolites has also been characterized through a variety of techniques. As an example, the crystal structure of the serum transport protein sex-hormone binding globulin (SHBG) was determined for a complex of this protein with 5α-dihydrotestosterone [56]. SHBG is an important binding agent in blood for many sex hormones and related compounds, including estradiol, testosterone, androste-5-ene-3β,17β-diol, and 5α-dihydrotestosterone [47,49,56]. The information that was obtained from the crystal structure for the 5α-dihydrotestosterone/SHBG complex was compared with the results of previous binding studies for steroid hormones with SHBG [57,58], and this allowed a model of the binding site for these compounds to be developed. This model gave good agreement with prior data from site-directed mutagenesis [59–61] and photolabeling experiments [62,63] that have been conducted with SHBG [58].
Another structural study looked at the interactions between the human androgen receptor (AR) ligand-binding domain and several androgen-related steroid hormones and metabolites [30]. The compounds that were examined included testosterone, dihydrotestosterone, and tetrahydrogestrinone. An example of some of the results was provided earlier in Figure 1. Both the binding affinity and structural characteristics for the complexes of these agents with AR were explored. Tetrahydrogestrinone was found to have the highest affinity for the AR ligand-binding domain. This strong binding was thought to be due to the presence of greater van der Waals interactions for this compound than for the other steroids that were studied. Dihydrotestosterone had a higher affinity than testosterone, an effect that was proposed to be due to the stronger electrostatic interactions between the structure of dihydrotestosterone and the AR binding domain [30].
4. Interactions of proteins with fatty acids and related metabolites
Fatty acids can also have significant binding to proteins. These compounds are carboxylic acids that contain hydrocarbon chains with lengths of 4 to 36 carbons. In some fatty acids, the hydrocarbon chain is unbranched and fully saturated, such as myristic acid (C14:0). In others, the chain contains one or more double bonds, as is the case of linoleic acid (C18:2) [48]. Long chain fatty acids (i.e., fatty acids with chains containing 16–20 carbons) are particularly critical for a diverse set of cellular and metabolic functions. For instance, long chain fatty acids act as fuel that can be stored as triacylglycerols (or triglycerides) and that can be used to generate ATP through β-oxidation in mitochondria and peroxisomes. In addition, fatty acids are the precursors of phospholipids and glycolipids, which are needed for the construction of membranes [64].
Long chain fatty acids such as oleic (C18:1), palmitic (C16:0), linoleic (C18:2), stearic (C18:0), arachidonic (C20:4) and palmitoleic (C16:1) are crucial intermediates in lipid metabolism [65]. They tend to have low solubility in water and are typically bound in plasma to proteins, with less than 0.1% being present as non-bound, or “free”, fatty acids. Most of the long chain fatty acids in the blood are transported by HSA [65–68]. HSA carries between 0.1 and 2 mol of fatty acids under normal physiological conditions. However, this value can rise to as high as 6 mol fatty acid per mol of HSA in the peripheral vasculature during fasting or exercise or disease states such as diabetes, liver and cardiovascular disease [67].
Many recent studies have attempted to locate fatty acid binding sites on HSA by using X-ray crystallography or NMR spectroscopy [67,69–73]. Some typical results of such studies are provided in Figure 3 [74,75]. In addition, site-directed mutagenesis has been utilized with these methods to see how specific changes in peptide sequence of HSA will affect its binding properties and structure [67,69]. Such studies have revealed that five to seven binding sites on HSA may be occupied by medium and long-chain fatty acids [71]. These binding sites are asymmetrically distributed across the three domains of HSA, with three of these sites overlapping Sudlow sites I and II [70]. All of these sites have similar structural interactions with fatty acids, providing a deep hydrophobic pocket for the methylene tail and containing two or three polar surface residues nearby which provide a binding location for the carboxylic head group of the fatty acid.
Figure 3.
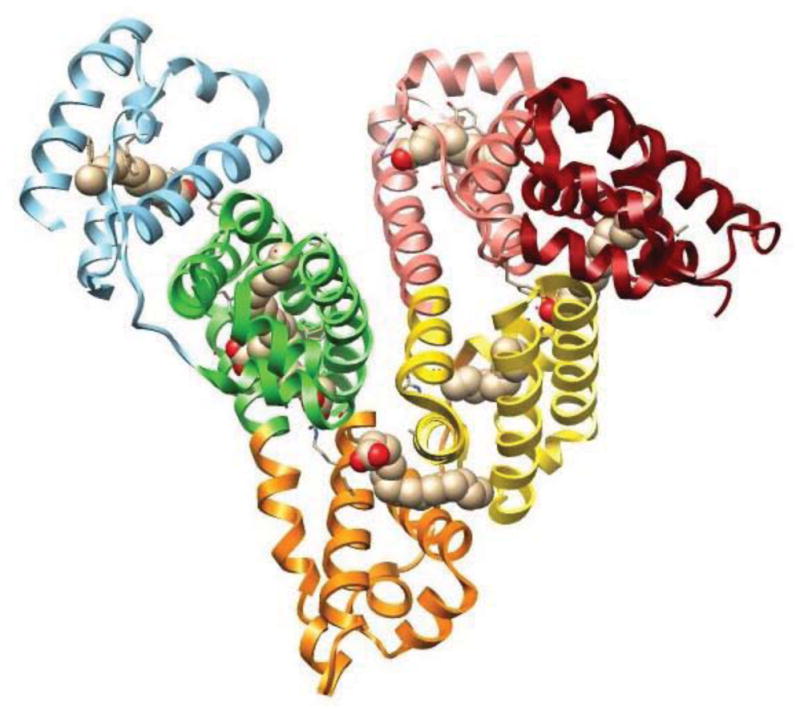
Structure of HSA, showing the regions that bind palmitic acid. This structure was generated using Protein Data Bank (PDB) file ID: 1E7H [75] and is adapted with permission from Ref. [74].
A variety of techniques have also been employed to estimate the binding constants for fatty acids at their sites on HSA. The strongest of these interactions have association equilibrium constants that range from 105 and 108 M−1 [66,74–77]. It has been observed for fatty acids with multiple binding sites on HSA that the value of the individual association constants for each mole of added fatty acid increased as the length of the fatty acid chain was raised [71]. It was later found that the association equilibrium constant for the first bound fatty acid increases with chain length but that this increase does not necessarily occur in a linear fashion; instead, the affinity is generally dependent on the hydrophobic portion of the fatty acid and how it interacts with HSA [69]. It has further been demonstrated that some fatty acids can have direct competition with drugs on HSA or can lead to allosteric effects during these binding processes [72,74,75,78].
5. Interactions of proteins with drugs and related metabolites
Numerous studies have examined the interactions of drugs and their metabolites with proteins. Like low mass hormones, many drugs and their metabolites are transported throughout the body through the use of serum transport proteins. Approximately 43% of the 1500 most commonly used pharmaceutics have at least 90% binding to such binding agents [35,79]. These interactions usually involve proteins that can bind to a broad range of targets, such as HSA and alpha1-acid glycoprotein (AGP), and can play a significant role in determining the activity, distribution, rate of excretion or metabolism, and toxicity of many pharmaceutical agents in the body [80]. In recent years there has also been interest in how the presence of drug metabolites can affect the distribution, apparent activity, and protein interactions of the parent drug [81].
5.1. General effects of metabolites on drug-protein interactions
Many studies have investigated the difference between drugs and their metabolites in their overall binding in serum or to specific serum proteins. For instance, equilibrium dialysis was used to examine the binding of propisomide and its major metabolite to human serum and isolated serum proteins such as AGP [82] and the binding of acetohexamide and its metabolite (−)-hydroxyhexamide to HSA [83]. Another study utilized a similar approach to investigate the binding by tolterodine and its 5-hydroxymethyl or N-dealkylated metabolites to human serum, HSA and AGP [84]. Equilibrium dialysis was further used to measure the binding of tizoxanide, an active metabolite of the drug nitazoxanide, with albumin and AGP [85].
A few studies have been conducted to provide a more detailed comparison of the binding regions and binding constants for drugs and their metabolites on serum proteins. As an example, HPAC and competition studies have been used to compare the binding regions on HSA for the drug phenytoin and its two major metabolites: 5-(3-hydroxyphenyl)-5-phenylhydantoin and 5-(4-hydroxyphenyl)-5-phenylhydantoin (i.e., m-HPPH and p-HPPH, respectively) [81,86]. In an examination of both the major and minor drug binding regions of HSA, phenytoin was found to have direct binding at Sudlow site II and the digitoxin site, with association equilibrium constants at these regions in the range of 0.65–1.04 × 104 M−1 at 37 ºC and pH 7.4 (see Table 2). The same drug had allosteric effects plus possible direct binding at Sudlow site I and the tamoxifen site [86]. However, m-HPPH and p-HPPH only had significant interactions with Sudlow site II, with binding constants of 0.32–0.57 × 103 M−1 for this region [81]. Thus, the parent drug and its metabolites not only had different affinities for HSA but also had differences in the number of their interaction sites with this protein [81,86].
Table 2.
Association equilibrium constants (Ka) and types of binding for phenytoin and its metabolites to various regions on HSAa
| Binding region on HSA | Drug or drug metabolite
|
||
|---|---|---|---|
Phenytoin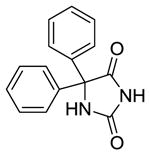
|
m-HPPH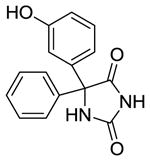
|
p-HPPH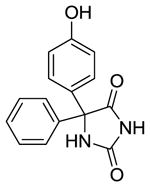
|
|
| Sudlow site Ib | Allosteric effects + possible direct binding | No binding | No binding |
| Sudlow site IIb | Direct binding Ka = 1.04 × 103 M−1 |
Direct binding Ka = 3.2 × 103 M−1 |
Direct binding Ka = 5.7 × 103 M−1 |
| Digitoxin site | Direct binding Ka = 6.5 × 103 M−1 |
No binding | No binding |
| Tamoxifen site | Allosteric effects + possible Direct binding | No binding | No binding |
5.2. Effects of chirality on drug metabolite-protein binding
Another factor to consider for drug- and drug metabolite-protein binding is the effect of chirality on these interactions. Chiral drugs have been estimated to represent 40–50% of all drugs that are currently on the market [87,88]. The separate chiral forms for some drugs can exhibit a wide variation in their toxicology, pharmacokinetics and metabolism. In the extreme case, one enantiomer may produce the desired function in treatment while another may be inactive or even produce undesired or toxic effects. This is because within the body numerous compounds (i.e., enzymes, plasma proteins, and other biomolecules) work as chiral selectors, causing them to sometimes bind or metabolize each chiral form of a drug differently [89–95].
These differences have made it possible in the past to use protein-based HPLC columns, such as those containing serum proteins, for separating the various chiral forms of many drugs [93–95]. The same approach has been utilized to separate and measure chiral drugs and their metabolites in biological samples. For instance, an AGP column was recently used with fluorescence detection to measure the enantiomers of tramadol and its two major metabolites, O-desmethyltramadol and N-desmethyltramadol, in plasma samples (see Figure 4). This method was then used to examine the pharmacokinetics for each of these compounds in the body [96]. A similar approach has been used with LC-MS to examine the chiral forms of methadone and its metabolites 2-ethylidene-1,5-dimethyl-3,3-diphenylpyrrolidine and 2-ethyl-5-methyl-3,3-diphenyl-1-pyrroline in hair samples from patients undergoing methadone maintenance therapy [97].
Figure 4.
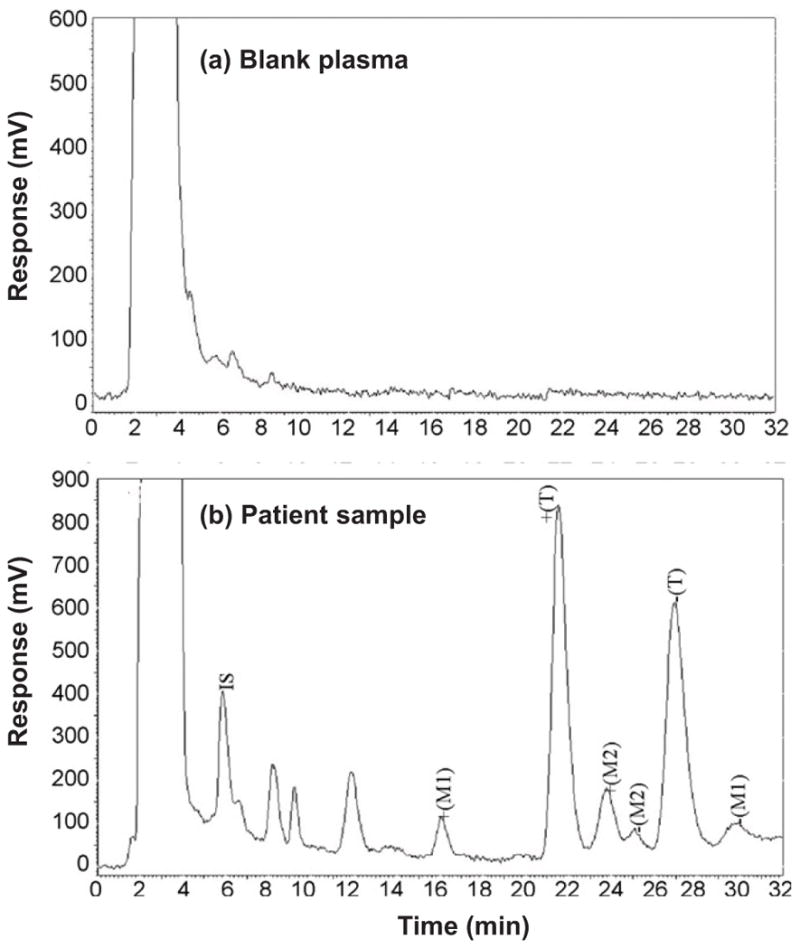
Chiral separation and analysis of tramadol and its major metabolites using HPLC and a column containing immobilized APG as stationary phase. The results in (a) are for a blank human plasma sample. The results in (b) are for a plasma sample taken from a volunteer 2.5 hours after receiving a 100 mg dose of racemic tramadol. Symbols: enantiomers of tramadol, +(T) and −(T); enantiomers of the metabolite O-desmethyltramadol, +(M1) and −(M1); enantiomers of the metabolite N-desmethyltramadol, +(M2) and −(M2); and internal standard (fluconazol), IS. Adapted with permission from Ref. [96].
As has been observed for their parent drugs, the different forms of a chiral metabolite can also differ in how they interact with proteins. One report compared the chiral forms of oxybutynin and its metabolite, N-desethyloxybutynin, in their binding and competition on HSA and AGP. The results showed that the affinity of oxybutynin enantiomers on AGP was much higher than on HSA, and that the enantiomers of N-desethyloxybutynin and oxybutynin were all bound by the same site on AGP [33]. Another study involving the phenytoin metabolites m-HPPH and p-HPPH compared the dissociation rates of the chiral forms of these metabolites from an HPLC column containing immobilized HSA [98]. Dissociation rate constants of 8.2–9.6 s−1 were obtained at pH 7.4 and 37º C for the enantiomers of m-HPPH, while values of 3.2–4.1 s−1 were obtained for the enantiomers of p-HPPH. These results were then used along with separate estimates of the association equilibrium constants to also compare the association rate constants for these metabolites and their enantiomers [98].
5.3. Use of binding data to characterize protein interaction sites for drug metabolites
A number of reports have used binding data for drugs, their metabolites and related analogs to learn about the binding site of these compounds on a protein. Binding and retention data that have been acquired by HPAC have been used to examine the binding of several types of compounds with immobilized serum proteins. This approach has been used to examine the binding of warfarin and coumarin compounds to HSA [99–101], as well as the binding of L-tryptophan and various indole compounds to this protein [102–104]. The same general method has been used to compare the binding of several sulfonylurea drugs with HSA and various preparations of glycated HSA [39,105–110].
If a relatively large group of compounds is considered in a binding study, the results can be used to create a quantitative structure-retention (or reactivity) relationship (QSRR) to describe the site at which these agents are binding to a protein [111–114]. For instance, binding studies based on HPLC or CE using serum proteins can be used to mimic biological systems and to quickly study how changes in the structure of an applied drug or analog will alter these interactions [115,116]. This format has been used to build models that describe the binding of HSA with benzodiazepines [117–119]. Such an approach has also been utilized to examine the binding of AGP with beta-adrenolytic drugs, antihistamines, amino alcohols, cyclic vinca alkaloid analogs, and quinazolone derivatives [116,120–125].
6. Interactions of proteins with xenobiotics and related metabolites
The term “xenobiotics” refers to chemicals that are produced synthetically and that are not normally found in biological organisms [126]. Drugs represent one type of xenobiotic, but others include environmental pollutants and food additives [126–128]. When they enter the body, xenobiotics can be metabolized through various enzymatic processes. The resulting metabolites, in turn, can sometimes interact with proteins and compete for endogenous compounds for common binding agents [126,129].
Several studies have examined the effects that xenobiotics and their metabolites may have on hormone-protein binding [127,130,131]. For example, the effect of polybrominated diphenyl ethers (PBDEs) on the binding of thyroid hormones to serum proteins has been examined [131]. It has been suggested in several studies that environmental exposure to PBDEs can result in decreased thyroid hormone concentrations in serum, leading to possible neurotoxicity and behavioral effects [131–133]. This effect may be linked to the fact that, when metabolized, PBDEs become hydroxylated and produce a chemical structure similar to that of T4 and its metabolites. It has been further found that PBDE metabolites are able to bind to T4-binding proteins in serum, which could result in the displacement of thyroid hormones. One study examined the binding of transthyretin and TBG with fourteen hydroxylated PBDE compounds through various methods. A fluorescence displacement assay indicated that hydroxylated PBDEs could compete with T4 for binding sites on transthyretin, while work with circular dichroism indicated that hydroxylated PBDEs could bind to the same sites as T4 on TBG and transthyretin [131]. Binding and competition with T4 has also been noted for some polychlorinated biphenyls (PCBs), dichlorodiphenyltrichloroethane (DDT), and related metabolites or compounds with respect to human thyroid receptor, TBG, and transthyretin [130].
Another report examined the effects for a number of xenobiotics and their metabolites on the binding of 17β-estradiol to the estrogen receptor and on the binding of 5α-dihydrotestosterone to the androgen receptor, androgen-binding protein, and SHBG [127]. Compounds that were tested included hexachlorocyclohexane, DDT, methoxychlor, pentachlorophenol, and nonylphenol. It was found that some of these xenobiotics and metabolites could cause a significant decrease in the binding of 5α-hydrotestosterone or 17β-estradiol to their binding proteins. It was further found that binding by these xenobiotic agents could be selective for the steroid receptors and binding proteins that were tested [127].
Polyphenolic compounds are flavonoids that are often used as dietary supplements [128]. Ultrafiltration and CE were used to examine the binding of these compounds to the human serum proteins HSA and AGP. Although similar in structure, these compounds did vary in their affinity towards HSA, with a high level of binding being observed for those compounds with hydrophobic properties and a carbonyl at position C(4) in their structure. It was further noted that these hydrophobic properties did not play a major role in the ability of polyphenolic compounds to bind with AGP [128].
7. Variations in protein structure and binding due to metabolic processes
Another way in which changes in metabolites may affect solute protein interactions is through changes that are created in the structure of the protein. In some cases, these changes may be a direct result of the modification of a protein by a metabolite (e.g., glycation, as discussed in the next section) [39,80,93,134]. In others, this change may be a response to differences in a protein’s environment that are created as the metabolic profile is altered (e.g., as might occur through oxidation) [135,136]. This section will discuss both types of effects using changes that have been observed in serum transport proteins and binding agents as examples.
7.1. Human serum albumin
One protein that has been found to be altered by some metabolic disease is HSA. As has been indicated earlier, HSA is a serum protein that plays a fundamental role in the reversible binding and transport of metabolites, drugs and various endogenous ligands, such as fatty acids [65,137]. HSA is normally found in blood at concentrations ranging from 30–50 g/L and accounts for approximately 60% of the total serum protein content [65]. Binding to HSA is known to greatly influence the pharmacokinetics and activity of many common drugs [49,138–140]. In addition, HSA can increase the solubility of lipophilic drugs, sequester toxins, and act as an important antioxidant in plasma [49,65].
Several past studies have noted that the chemical modification of HSA can alter its binding to drugs, hormones and other solutes. For instance, the reaction of HSA with p-nitropheny acetate, which is thought to mainly modify Tyr-411 at Sudlow site II, can change the binding of various solutes with this protein [141]. The modification of Trp-214 by o-nitrophenylsulphenyl chloride has been demonstrated to change the stereoselectivity and binding affinity of Sudlow site I of HSA [142]. Similar work has been presented that has examined the effects of modifying the lone free cysteine group on HSA by reacting this protein with ethacrynic acid [143,144].
Diabetes is a metabolic disease in which the structure of HSA can be modified. This disease is actually a group of disorders that are characterized by abnormal high levels of blood glucose (i.e., hyperglycemia) that result from insulin deficiency and/or insulin resistance [145]. Many of the long term complications of diabetes, such as heart disease and nerve damage, are associated with the non-enzymatic glycation of proteins [145,146]. Glycation starts with the nucleophilic attack of a reducing sugar (e.g., glucose) onto some of the primary amine groups on proteins to form a reversible Schiff base (see Figure 5). This intermediate can then slowly rearrange to form a more stable Amadori product [145–147]. Oxidation of the Amadori products or free sugars can also generate reactive α-oxaloaldehydes that can react with both lysines and arginines on proteins to form advanced glycation end-products (AGEs) [147].
Figure 5.
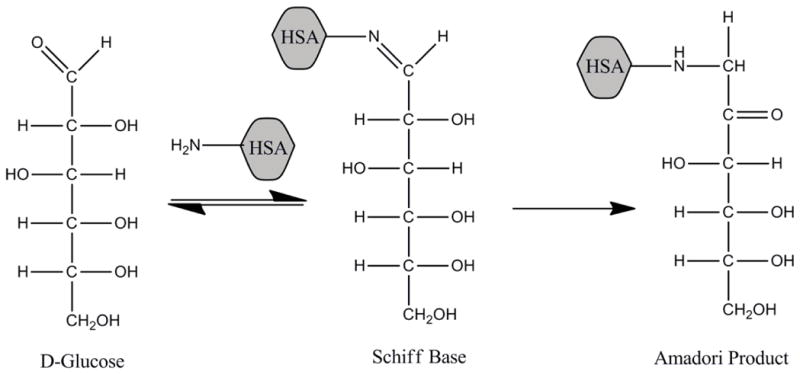
Reactions involved in the early stages of glycation of a protein, using human serum albumin (HSA) as an example [145].
In recent years, it has been found that glycation can affect the binding of several endogenous and exogenous solutes with HSA. For example, L-tryptophan is an essential amino acid [148] and has been extensively used as a site-selective probe for Sudlow site II of glycated HSA and normal HSA [105–108,149]. Recent binding studies using glycated HSA with levels of modification similar to those found in diabetes found an increase of 4.7 to 5.8-fold in the affinity of L-tryptophan for this protein at 37°C [106,149]. Sulfonylurea drugs are a group of anti-diabetic drugs that are used in the management of type 2 diabetes; these drugs are also highly bound to serum proteins such as HSA. Binding studies based on HPAC have found that glycation can affect the binding strength of these drugs to HSA, with both the degree of glycation and the specific type of drug influencing the size of the change [39,105–110].
As indicated in the last section, fatty acids are the major endogenous ligands of HSA and are also known to have many binding sites on this protein [75]. Reports that have examined the combined effect of glycation and the presence of various fatty acids on the binding of sulfonylurea drugs to HSA have found that glycation increases the overall affinity of these drugs to HSA, while the addition of increasing amounts of fatty acids causes a decrease in affinity [74,76]. It has further been noted that glycation could produce changes of at least 3- to 5-fold in the affinities of some fatty acids at their sites of competition with sulfonylurea drugs when comparing the binding of these solutes to normal HSA [76].
Methylglyoxal is a highly reactive metabolite of glucose that has been implicated in several chronic diseases associated with diabetes [150,151]. The elevated concentrations of methylglyoxal in diabetes patients can also lead to protein modification and the formation of AGEs through the reaction of methylglyoxal with arginine or lysine residues. A recent report using quantitative MS and multiple reaction monitoring found that a major site for modification by methylglyoxal on HSA occurs at Arg-257, which is located in Sudlow site I. Molecular modeling conducted in the same study indicated that a decrease in binding by warfarin may occur due to these modifications when comparing glycated HSA and normal HSA [151].
7.2. Alpha1-acid glycoprotein
A second type of serum transport protein that can be affected by metabolic diseases is AGP. AGP is an acute-phase protein that is responsible for binding and delivering numerous basic and neutral drugs in the bloodstream [121]. The concentration of AGP in blood can vary over a wide range and is affected by systemic tissue injury, inflammation and infection. In addition, the glycosylation of AGP can be altered in some disease states, such as rheumatoid arthritis, systemic lupus erythematosus and autoimmune thyroid disease [152]. These changes are important because they can also alter the binding of drugs to AGP. As an example, the affinity of disopyramide for AGP has been found to be affected by the biantennary glycan structures for this protein [153,154]. It has also been reported that genetic variants of AGP can have a significant effect on binding by chiral drugs such as disopyramide and warfarin [153,155].
A number of reports have looked at how changes in AGP binding can affect parent drugs compared to their metabolites. One study evaluated the effect of AGP on lidocaine and its active metabolites monoethylglycinexylidide and glycinexylidide during continuous epidural anesthesia in infants and young children. The results indicated the AGP concentration in plasma could be used as an index to monitor and prevent the toxicity caused by the accumulation of monoethylglycinexylidide during the continuous administration of lidocaine [156]. Another report looked at the concentrations of vecuronium and its metabolite 3-OH desacetylvecuronium in children who were receiving phenytoin or carbamazepine for chronic anticonvulsant therapy [157]. These last two drugs were of interest because many anticonvulsant drugs have been shown to increase the concentration of AGP in plasma, which can then increase protein binding to cationic drugs and alter their distribution. It was found that the increase in AGP concentration associated with the anticonvulsant therapy did not significantly contribute to resistance to vecuronium [158].
7.3. Lipoproteins
Lipoproteins are another set of binding agents in serum that can be affected by metabolic diseases. Lipoproteins are macromolecular complexes of proteins and lipids that transport hydrophobic lipids and related compounds, such as cholesterol and triglycerides, throughout the body [158–161]. Lipoproteins are also known to interact with several basic and neutral hydrophobic drugs in blood [99,162–172]. Examples of drugs that bind to lipoproteins are propranolol and verapamil [37,162–169,173–175].
Lipoprotein concentrations in blood can vary with different disease states. For example, the levels of low-density lipoprotein (LDL) in plasma can increase in diseases such as atherosclerosis and hyperlipidemia [176]. In addition to changes in the levels of lipoproteins in the circulation, metabolic diseases often result in modifications in lipoprotein structures. For instance, increased amounts of LDL that have been modified by AGEs are found in individuals with diabetics and non-diabetics with renal failure. Glycation of LDL may lead to the formation of foam cells and an increase in atherosclerosis. In addition, glycated LDL is more susceptible to further modifications due to oxidation [135].
The oxidation of lipoproteins occurs through free radicals, such as peroxyl radicals, which are released from cells and chemical reactions [136]. These radicals can react with lipoproteins, depleting the particle’s antioxidant defense and initiating oxidation of the lipid core. In the later stages of this process, the surface protein also becomes modified. The oxidation of lipoproteins, specifically LDL, leads to atherosclerosis [136]. In addition to the increased risk of atherosclerosis, oxidized lipoproteins may also impact the ability of the complexes to bind and carry basic and neutral drugs throughout the body [176].
The effects of LDL oxidation on drug binding have been evaluated by using CE and using verapamil and nilvadipine as models for basic and neutral drugs, respectively [176]. It was found that the affinity of these drugs increased with the amount of LDL oxidation. In addition, the binding of verapamil was increased more than it was for nilvadipine, suggesting that basic drugs were more sensitive to oxidation effects. No stereoselective binding was detected between LDL and these model drugs at any oxidation state [176]. However, other studies based on HPAC have noted different binding for the chiral forms of some drugs to LDL [174,175].
8. Conclusion
The field of metabolomics has seen great growth in recent years because of the wealth of information it can provide about biochemical pathways and processes. This review examined previous reports that have looked at the interactions of metabolites with proteins. The first topic discussed was an overview of techniques that have been used to characterize and study metabolite-protein binding. These methods have been used in vitro and in vivo to provide information on the structures of metabolite-protein complexes and to examine the nature of metabolite-protein interactions. Computational studies using in silico tools have been used to provide additional data on metabolite-protein complexes and interactions.
This review next described numerous studies that have investigated the binding of various types of small solutes and their metabolites with proteins. This included work that has been carried out with hormones, fatty acids, drugs or other xenobiotics, and their metabolites with transport proteins and receptors. These examples have considered the structures of the resulting solute-protein complexes, the nature of the binding sites, the strength of these interactions, the variations in these interactions with solute structure, and the kinetics of these reactions. Studies that have examined the effects of various metabolic processes on the structure and activities of proteins, and on the corresponding interactions of solutes with these proteins, were also summarized.
Although most past work in metabolomics has been concerned with the structure and analysis of metabolites, research in metabolite-protein interactions is still a relatively new area. Based on the research that has already been carried out, it is already clear that data on metabolite-protein interactions can provide useful information on biological processes that involve hormones, drugs and other low mass solutes. It is further expected that this type of research will continue to grow in the future as metabolomics becomes more widely used in biomedical research, pharmaceutical science, and personalized medicine.
Highlights.
Interactions involving metabolites and proteins as binding agents are discussed.
An overview is given of previous methods used to study these interactions.
Drug-, hormone-, and fatty acid-protein interactions are considered.
Some effects of metabolic diseases on protein binding are also examined.
Acknowledgments
This research was supported by the National Institutes of Health under grants R01 GM044931 and R01 DK069629 and was conducted in facilities that were renovated under NIH grant RR015468-01. R. Matsuda was supported, in part, under a fellowship through the Molecular Mechanisms of Disease program at the University of Nebraska.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaddurah-Daouk R, Kristal BS, Weishiboum RM. Metabolomics: a global biochemical approach to drug response and disease. Ann Rev Pharmacol Toxicol. 2008;48:653–683. doi: 10.1146/annurev.pharmtox.48.113006.094715. [DOI] [PubMed] [Google Scholar]
- 2.Kuehnbaum NL, Mckibbin PB. New advances in separation science for metabolomics: resolving chemical diversity in post-genomic era. Chem Rev. 2013;113:2437–2468. doi: 10.1021/cr300484s. [DOI] [PubMed] [Google Scholar]
- 3.Patti GJ, Yanes O, Siuzdak G. Metabolomics: the apogee of the omics triology. Nature Rev Mol Cell Biol. 2012;13:263–269. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tweeddale H, Notley-McRobb L, Ferenci T. Effect of slow growth on metabolism of Escherichia coli, as revealed by global metabolite pool (“metabolome”) analysis. J Bacteriol. 1998;180:5109–5116. doi: 10.1128/jb.180.19.5109-5116.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oliver SG, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998;16:373–377. doi: 10.1016/s0167-7799(98)01214-1. [DOI] [PubMed] [Google Scholar]
- 6.Tweeddale H, Notley-McRobb L, Ferenci T. Assessing the effect of reactive oxygen species on Escherichia coli using a metabolome approach. Redox Rep. 1999;4:237–241. doi: 10.1179/135100099101534954. [DOI] [PubMed] [Google Scholar]
- 7.Lee DY, Bowen BP, Northen TR. Mass spectrometry-based metabolomics, analysis of metabolite-protein interactions, and imaging. Biotechniques. 2010;49:557–565. doi: 10.2144/000113451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, Gianoulis TA, Yip KY, Gerstein M, Synder M. Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell. 2010;143:639–650. doi: 10.1016/j.cell.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang GX, Li X, Synder M. Investigating metabolite–protein interactions: an overview of available techniques. Methods. 2012;57:459–466. doi: 10.1016/j.ymeth.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sudlow G, Birkett DJ, Wade DN. Further characterization of specific drug binding sites on human serum albumin. Mol Pharmacol. 1976;12:1052–1061. [PubMed] [Google Scholar]
- 11.Clarke W, Choudhuri AR, Hage DS. Analysis of free drug fractions by ultra-fast immunoaffinity chromatography. Anal Chem. 2001;73:2157–2164. doi: 10.1021/ac0009752. [DOI] [PubMed] [Google Scholar]
- 12.Clarke W, Schiel JE, Moser A, Hage DS. Analysis of free hormone fractions by an ultrafast immunoextraction/displacement immunoassay: studies using free thyroxine as a model system. Anal Chem. 2005;77:1859–1866. doi: 10.1021/ac040127x. [DOI] [PubMed] [Google Scholar]
- 13.Daddaoua A, Krell T, Alfonso C, Morel B, Ramos JL. Compartmentalized glucose metabolism in Pseudomonas putida is controlled by the PtxS repressor. J Bacteriol. 2010;192:4357–4366. doi: 10.1128/JB.00520-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frostell-Karlsson A, Remaeus A, Roos H, Andersson K, Borg P, Hamalainen M, Karlsson R. Biosensor analysis of the interaction between immobilized human serum albumin and drug compounds for prediction of human serum albumin binding levels. J Med Chem. 2000;43:1986–1992. doi: 10.1021/jm991174y. [DOI] [PubMed] [Google Scholar]
- 15.Gestwicki JE, Hsieh HV, Pitner JB. Using receptor conformational change to detect low molecular weight analytes by surface plasmon resonance. Anal Chem. 2001;73:5732–5737. doi: 10.1021/ac0105888. [DOI] [PubMed] [Google Scholar]
- 16.Palmer RA, Niwa H. X-ray crystallographic studies of protein–ligand interactions. Biochem Soc Trans. 2003;31:973–979. doi: 10.1042/bst0310973. [DOI] [PubMed] [Google Scholar]
- 17.Larsen NA, Turner JM, Stevens J, Rosser SJ, Basran A, Lerner RA, Bruce NC, Wilson IA. Crystal structure of a bacterial cocaine esterase. Nature Struct Biol. 2002;9:17–21. doi: 10.1038/nsb742. [DOI] [PubMed] [Google Scholar]
- 18.Vogtherr M, Saxena K, Hoelder S, Grimme S, Betz M, Schieborr U, Pescatore B, Robin M, Delarbre L, Langer T, Wendt KU, Schwalbe H. NMR characterization of kinase p38 dynamics in free and ligand-bound forms. Angew Chem. 2006;45:993–997. doi: 10.1002/anie.200502770. [DOI] [PubMed] [Google Scholar]
- 19.Betz M, Saxena K, Schwalbe H. Biomolecular NMR: a chaperone to drug discovery. Curr Opin Chem Biol. 2006;10:219–225. doi: 10.1016/j.cbpa.2006.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Silva L, Ozdowy P, Krajewski M, Rothweiler U, Singh M, Holak TA. Monitoring the effects of antagonists on protein-protein interactions with NMR spectroscopy. J Am Chem Soc. 2005;127:13220–13226. doi: 10.1021/ja052143x. [DOI] [PubMed] [Google Scholar]
- 21.Schuster I, Egger H, Bikle D, Herzig G, Reddy GS, Stuetz A, Stuetz P, Vorisek G. Selective inhibition of vitamin D hydroxylases in human keratinocytes. Steroids. 2001;66:409–422. doi: 10.1016/s0039-128x(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 22.Goncalves MS. Fluorescent labeling of biomolecules with organic probes. Chem Rev. 2009;109:190–212. doi: 10.1021/cr0783840. [DOI] [PubMed] [Google Scholar]
- 23.Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, Mitchell T, Miller P, Dean RA, Gerstein M, Snyder M. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 24.Liesener A, Karst U. Monitoring enzymatic conversions by mass spectrometry: a critical review. Anal Bioanal Chem. 2005;382:1451–1464. doi: 10.1007/s00216-005-3305-2. [DOI] [PubMed] [Google Scholar]
- 25.Yu Y, Ko KS, Zea CJ, Pohl NL. Discovery of the chemical function of glycosidases: design, synthesis, and evaluation of mass-differentiated carbohydrate libraries. Org Lett. 2004;6:2031–2033. doi: 10.1021/ol049389b. [DOI] [PubMed] [Google Scholar]
- 26.Fischbach MA, Lin H, Liu DR, Walsh CT. In vitro characterization of IroB, a pathogen-associated C-glycosyltransferase. Proc Natl Acad Sci USA. 2005;102:571–576. doi: 10.1073/pnas.0408463102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morozov VN, Morozova TY, Johnson KL, Naylor S. Parallel determination of multiple protein metabolite interactions using cell extract, protein microarrays and mass spectrometric detection. Rapid Comm Mass Spectrom. 2003;17:2430–2438. doi: 10.1002/rcm.1213. [DOI] [PubMed] [Google Scholar]
- 28.Furuya T, Nishi T, Shibata D, Suzuki H, Ohta D, Kino K. Characterization of orphan monooxygenases by rapid substrate screening using FT-ICR mass spectrometry. Chem Biol. 2008;15:563–572. doi: 10.1016/j.chembiol.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 29.Clarke DJ, Stokes AA, Langridge-Smith P, Mackay CL. Online quench-flow electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry for elucidating kinetic and chemical enzymatic reaction mechanisms. Anal Chem. 2010;82:1897–1904. doi: 10.1021/ac9026302. [DOI] [PubMed] [Google Scholar]
- 30.De Jesus-Tran KP, Cote P, Cantin L, Blanchet J, Labrie F, Breton R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonist reveals molecular determinants responsible for binding affinity. Protein Sci. 2006;15:987–999. doi: 10.1110/ps.051905906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vuignier K, Schappler J, Veuthey JL, Carrupt PA, Martel S. Drug-protein binding: a critical review of analytical tools. Anal Bioanal Chem. 2010;398:53–66. doi: 10.1007/s00216-010-3737-1. [DOI] [PubMed] [Google Scholar]
- 32.Comess KM, Schurdak ME, Voorbach MJ, Coen M, Trumbull JD, Yang H, Gao L, Tang H, Cheng X, Lerner CG, McCall O, Burns DJ, Beutel BA. An ultraefficient affinity-based high-throughout screening process: application to bacterial cell wall biosynthesis enzyme MurF. J Biomol Screen. 2006;11:743–754. doi: 10.1177/1087057106289971. [DOI] [PubMed] [Google Scholar]
- 33.Shibukawa A, Yoshikawa Y, Kimura T, Kuroda Y, Nakagawa T, Wainer IW. Binding study of desethyloxybutynin using high-performance frontal analysis method. J Chromatogr B. 2002;768:189–197. doi: 10.1016/s0378-4347(01)00499-6. [DOI] [PubMed] [Google Scholar]
- 34.Muckenschnabel I, Falchetto R, Mayr LM, Filipuzzi I. SpeedScreen: label-free liquid chromatography-mass spectrometry-based high-throughput screening for the discovery of orphan protein ligands. Anal Biochem. 2004;324:241–249. doi: 10.1016/j.ab.2003.09.040. [DOI] [PubMed] [Google Scholar]
- 35.Hage DS, Anguizola JA, Jackson AJ, Matsuda R, Papastavros E, Pfaunmiller E, Tong Z, Vargas-Badilla J, Yoo MJ, Zheng X. Chromatographic analysis of drug interactions in the serum proteome. Anal Methods. 2011;3:1449–1460. doi: 10.1039/C1AY05068K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hage DS. Affinity chromatography: a review of clinical applications. Clin Chem. 1999;45:593–615. [PubMed] [Google Scholar]
- 37.Hage DS. High-performance affinity chromatography: a powerful tool for studying serum protein binding. J Chromatogr B. 2002;768:3–30. doi: 10.1016/s0378-4347(01)00482-0. [DOI] [PubMed] [Google Scholar]
- 38.Hage DS, Tweed SA. Recent advances in chromatographic and electrophoretic methods for the study of drug-protein interactions. J Chromatogr B. 1997;699:499–525. doi: 10.1016/s0378-4347(97)00178-3. [DOI] [PubMed] [Google Scholar]
- 39.Anguizola J, Joseph KS, Barnaby OS, Matsuda R, Alvarado G, Clarke W, Cerny RL, Hage DS. Development of affinity microcolumns for drug-protein binding studies in personalized medicine: interactions of sulfonylurea drugs with in vivo glycated human serum albumin. Anal Chem. 2013;85:4453–4460. doi: 10.1021/ac303734c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heegard NHH, Schou C. In: Handbook of Affinity Chromatography. 2. Hage DS, editor. Chap. 26 CRC Press; Boca Raton: 2006. [Google Scholar]
- 41.Hoffmann T, Martin MM. CE-ESI-MS/MS as a rapid screening tool for the comparison of protein-ligand interactions. Electrophoresis. 2010;31:1248–1255. doi: 10.1002/elps.200900585. [DOI] [PubMed] [Google Scholar]
- 42.Sun H, Scott DO. Structure-based drug metabolism predictions for drug design. Chem Biol Drug Des. 2010;75:3–17. doi: 10.1111/j.1747-0285.2009.00899.x. [DOI] [PubMed] [Google Scholar]
- 43.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 44.de Groot MJ, Wakenhut F, Whitlock G, Hyland R. Understanding CYP2D6 interactions. Drug Discovery Today. 2009;14:964–972. doi: 10.1016/j.drudis.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 45.Mandava S, Makowski L, Devarapalli S, Uzubell J, Rodi DJ. RELIC - a bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics. 2004;4:1439–1460. doi: 10.1002/pmic.200300680. [DOI] [PubMed] [Google Scholar]
- 46.Bertini I, Fragai M, Giachetti A, Luchinat C, Maletta M, Parigi G, Yeo KJ. Combining in silico tools and NMR data to validate protein-ligand structural models: application to matrix metalloproteinases. J Med Chem. 2005;48:7544–7559. doi: 10.1021/jm050574k. [DOI] [PubMed] [Google Scholar]
- 47.Lemke TL, Williams DA, Roche VF, Zito SW. Medicinal Chemistry. 6. Lippincott Williams and Wilkins; Philadelphia: 2008. [Google Scholar]
- 48.Nelson DL, Cox MM, editors. Lehninger Principles of Biochemistry. 6. W.H. Freeman Publishers; New York: 2005. [Google Scholar]
- 49.Tietz NW, editor. Textbook of Clinical Chemistry. Saunders; Philadelphia: 1986. [Google Scholar]
- 50.Clarke W. Contemporary Practice in Clinical Chemistry. 2. AACC Press; Washington, DC: 2011. [Google Scholar]
- 51.Gornall AG, editor. Applied Biochemistry of Clinical Disorders. 2. Lippincott; Philadelphia: 1986. [Google Scholar]
- 52.Anderson DC. Sex-hormone-binding globulin. Clin Endocrinol. 1974;3:69–96. doi: 10.1111/j.1365-2265.1974.tb03298.x. [DOI] [PubMed] [Google Scholar]
- 53.Loun B, Hage DS. Characterization of thyroxine-albumin binding using high-performance affinity chromatography I. Interactions at the warfarin and indole sites of albumin. J Chromatogr. 1992;579:22S–235. [PubMed] [Google Scholar]
- 54.Loun B, Hage DS. Characterization of thyroxine-albumin binding using high-performance-affinity chromatography II. Comparison of the binding of thyroxine, triiodothyronines and related compounds at the warfarin and indole sites of human serum albumin. J Chromatogr B. 1995;665:303–311. doi: 10.1016/0378-4347(94)00547-i. [DOI] [PubMed] [Google Scholar]
- 55.Cody V, Davis PJ, Davis FB. Molecular modeling of the thyroid hormone interaction with αvβ3 integrin. Steroids. 2007;72:165–170. doi: 10.1016/j.steroids.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 56.Grishkovskaya I, Avvakumov GV, Sklenar G, Dales D, Hammond GL, Muller YA. Crystal structure of human sex hormone-binding globulin: steroid transport by laminin G-like domain. EMBO J. 2000;19:504–512. doi: 10.1093/emboj/19.4.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Renoir JM, Mercier-Bodard C, Baulieu EE. Hormonal and immunological aspects of the phylogeny of sex steroid binding plasma protein. Proc Natl Acad Sci USA. 1980;77:4578–4582. doi: 10.1073/pnas.77.8.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Westphal AJC. Steroid-Protein Interactions II. Springer-Verlag; Berlin, Germany: 1986. [Google Scholar]
- 59.Bocchinfuso WP, Hammond GL. Steroid-binding and dimerization domains of human sex hormone-binding globulin partially overlap: steroids and Ca2+ stabilize dimer formation. Biochemistry. 1994;33:10622–10629. doi: 10.1021/bi00201a008. [DOI] [PubMed] [Google Scholar]
- 60.Bocchinfuso WP, Warmels Rodenhiser S, Hammond GL. Structure/function analyses of human sex hormone-binding globulin by site-directed mutagenesis. FEBS Lett. 1992;301:227–230. doi: 10.1016/0014-5793(92)81253-i. [DOI] [PubMed] [Google Scholar]
- 61.Sui LM, Cheung AW, Namkung PC, Petra PH. Localization of the steroid-binding site of the human sex steroid-binding protein of plasma (SBP or SHBG) by site-directed mutagenesis. FEBS Lett. 1992;310:115–118. doi: 10.1016/0014-5793(92)81309-a. [DOI] [PubMed] [Google Scholar]
- 62.Grenot C, de Montard A, Blachere T, de Ravel MR, Mappus E, Cuilleron CY. Characterization of Met-139 as the photolabeled amino acid residue in the steroid binding site of sex hormone binding globulin using delta 6 derivatives of either testosterone or estradiol as unsubstituted photoaffinity labeling reagents. Biochemistry. 1992;31:7609–7621. doi: 10.1021/bi00148a024. [DOI] [PubMed] [Google Scholar]
- 63.Kassab D, Pichat S, Chambon C, Blachere T, Rolland de Ravel M, Mappus E, Grenot C, Cuilleron CY. Photoaffinity labeling of homologous Met-133 and Met-139 amino acids of rabbit and sheep sex hormone-binding globulins with the unsubstituted Delta 6-testosterone photoreagent. Biochemistry. 1998;37:14088–14097. doi: 10.1021/bi9806347. [DOI] [PubMed] [Google Scholar]
- 64.Schaffer JE. Fatty acid transport: the roads taken. Am J Physiol Endocrinol Metab. 2002;282:E239–E246. doi: 10.1152/ajpendo.00462.2001. [DOI] [PubMed] [Google Scholar]
- 65.Peters T., Jr . All About Albumin: Biochemistry, Genetics, and Medical Applications. Academic Press; San Diego: 1996. [Google Scholar]
- 66.Richieri GV, Kleinfeld AM. Unbound free fatty acid levels in human serum. J Lipid Res. 1995;36:229–240. [PubMed] [Google Scholar]
- 67.Simard JR, Zunszain PA, Ha CE, Hang JS, Bhagavan NV, Petitpas I, Curry S, Hamilton JA. Locating high-affinity fatty acid-binding sites on albumin by X-ray crystallography and NMR spectroscopy. Proc Natl Acad Sci USA. 2005;102:17958–17963. doi: 10.1073/pnas.0506440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chaung VT, Otagiri M. How do fatty acids cause allosteric binding of drugs to human serum albumin? Pharm Res. 2002;19:1458–1464. doi: 10.1023/a:1020496314081. [DOI] [PubMed] [Google Scholar]
- 69.Kragh-Hansen U, Watanabe H, Nakajou K, Iwao Y, Otagiri M. Chain length-dependent binding of fatty acid anions to human serum albumin studied by site-directed mutagenesis. J Mol Biol. 2006;363:702–712. doi: 10.1016/j.jmb.2006.08.056. [DOI] [PubMed] [Google Scholar]
- 70.Petitpas I, Grune T, Bhattacharya AA, Curry S. Crystal structures of human serum albumin complexed with monounsaturated and polyunsaturated fatty acids. J Mol Biol. 2001;314:S955–960. doi: 10.1006/jmbi.2000.5208. [DOI] [PubMed] [Google Scholar]
- 71.Bhattacharya AA, Grüne T, Curry S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J Mol Biol. 2000;303:721–732. doi: 10.1006/jmbi.2000.4158. [DOI] [PubMed] [Google Scholar]
- 72.Simard JR, Zunszain PA, Hamilton JA, Curry S. Location of high and low affinity fatty acid binding sites on human serum albumin revealed by NMR drug-competition analysis. J Mol Biol. 2006;361:336–351. doi: 10.1016/j.jmb.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 73.Curry S, Mandelkow H, Brick P, Franks N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nature Struct Biol. 1998;5:827–835. doi: 10.1038/1869. [DOI] [PubMed] [Google Scholar]
- 74.Anguizola JA, Basiaga SBG, Hage DS. Effects of fatty acids and glycation on drug interactions with human serum albumin. Curr Metabolomics. 2013;1:239–250. doi: 10.2174/2213235x1130100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Spector AA. Fatty acid binding to plasma albumin. J Lipids Res. 1976;16:165–179. [PubMed] [Google Scholar]
- 76.Basiaga SB, Hage DS. Chromatographic studies of changes in binding of sulfonylurea drugs to human serum albumin due to glycation and fatty acids. J Chromatogr B. 2010;878:3193–3197. doi: 10.1016/j.jchromb.2010.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Demant EJ, Richieri GV, Kleinfeld AM. Stopped-flow kinetic analysis of long-chain fatty acid dissociation from bovine serum albumin. Biochem J. 2002;363:809–815. doi: 10.1042/0264-6021:3630809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Noctor TAG, Wainer IW, Hage DS. Allosteric and competitive displacement of drugs from human serum albumin by octanoic acid, as revealed by high-performance liquid affinity chromatography, on a human serum albumin-based stationary phase. J Chromatogr. 1992;577:305–315. doi: 10.1016/0378-4347(92)80252-l. [DOI] [PubMed] [Google Scholar]
- 79.Kratochowil NA, Huber W, Muller F, Kansy M, Gerber PR. Predicting plasma protein binding of drugs: a new approach. Biochem Pharmacol. 2002;64:1355–1374. doi: 10.1016/s0006-2952(02)01074-2. [DOI] [PubMed] [Google Scholar]
- 80.Hage DS, Anguizola J, Barnaby O, Jackson A, Yoo MJ, Papastavros E, Pfaunmiller E, Sobansky M, Tong Z. Characterization of drug interactions with serum proteins by using high-performance affinity chromatography. Curr Drug Metab. 2011;12:313–328. doi: 10.2174/138920011795202938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ohnmacht CM, Chen S, Tong Z, Hage DS. Studies by biointeraction chromatography of binding by phenytoin metabolites to human serum albumin. J Chromatogr B. 2006;836:83–91. doi: 10.1016/j.jchromb.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 82.Zini R, Barre J, Defer G, Jeanniot JP, Houin G, Tillement JP. Protein binding of propisomide. J Pharm Sci. 1985;74:530–533. doi: 10.1002/jps.2600740507. [DOI] [PubMed] [Google Scholar]
- 83.Imamura Y, Kojima Y, Ichibagase H. Effect of simultaneous administration of drugs on absorption and excretion. XIX. Binding of acetohexamide and its major metabolite, (−)-hydroxyhexamide, to human serum albumin. Chem Pharm Bull. 1985;33:1281–1284. doi: 10.1248/cpb.33.1281. [DOI] [PubMed] [Google Scholar]
- 84.Påhlman I, Gozzi P. Serum protein binding of tolterodine and its major metabolites in humans and several animal species. Biopharm Drug Dispos. 1999;20:91–99. doi: 10.1002/(sici)1099-081x(199903)20:2<91::aid-bdd162>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 85.Zhao Z, Xue F, Zhang L, Zhang K, Fei C, Zheng W, Wang X, Wang M, Zhao Z, Meng X. The pharmacokinetics of nitazoxanide active metabolite (tizoxanide) in goats and its protein binding ability in vitro. J Vet Pharmacol Ther. 2009;33:147–153. doi: 10.1111/j.1365-2885.2009.01119.x. [DOI] [PubMed] [Google Scholar]
- 86.Chen J, Ohnmacht C, Hage DS. Studies of phenytoin binding to human serum albumin by high-performance affinity chromatography. J Chromatogr B. 2004;809:137–145. doi: 10.1016/j.jchromb.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 87.Shen Q, Wang L, Zhou H, Jiang H, Yu L, Zheng S. Stereoselective binding of chiral drugs to plasma proteins. Acta Pharmacologica Sinica. 2013;34:998–1006. doi: 10.1038/aps.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sekhon BS. Exploiting the power of stereochemistry in drugs: an overview of racemic and enantiopure drugs. J Mod Med Chem. 2013;1:10–36. [Google Scholar]
- 89.Landoni MF, Soraci A. Pharmacology of chiral compounds: 2-arylpropionic acid derivatives. Curr Drug Metab. 2001;2:37–51. doi: 10.2174/1389200013338810. [DOI] [PubMed] [Google Scholar]
- 90.Drayer DE. Clinical pharmacology through the looking glass: reflections on the racemate vs enantiomer debate. Clin Pharmacol Ther. 1986;40:125–133. [Google Scholar]
- 91.Patocka J, Dvorak A. Biomedical aspects of chiral molecules. J Appl Med. 2004;2:95–100. [Google Scholar]
- 92.Jamali F, Mehvar R, Pasutto FM. Enantioselective aspects of drug action and disposition: therapeutic pitfalls. J Pharm Sci. 1989;78:695–715. doi: 10.1002/jps.2600780902. [DOI] [PubMed] [Google Scholar]
- 93.Patel S, Wainer IW, Lough WJ. In: Handbook of Affinity Chromatography. 2. Hage DS, editor. Vol. 21 CRC Press; Boca Raton: 2006. [Google Scholar]
- 94.Patel S, Wainer IW, Lough WJ. In: Handbook of Affinity Chromatography. 2. Hage DS, editor. Vol. 24 CRC Press; Boca Raton: 2006. [Google Scholar]
- 95.Hage DS. Chromatographic and electrophoretic studies of protein binding to chiral solutes. J Chromatogr A. 2001;906:459–481. doi: 10.1016/s0021-9673(00)00957-2. [DOI] [PubMed] [Google Scholar]
- 96.Ardakani YH, Mehvar R, Foroumadi A, Rouini MR. Development and validation of a rapid HPLC method for simultaneous determination of tramadol, and its two main metabolites in human plasma. J Chromatogr B. 2008;864:109–115. doi: 10.1016/j.jchromb.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 97.Kelly T, Doble P, Dawson M. Chiral analysis of methadone and its major metabolites (EDDP and EMDP) by liquid chromatography–mass spectrometry. J Chromatogr B. 2005;814:315–323. doi: 10.1016/j.jchromb.2004.10.053. [DOI] [PubMed] [Google Scholar]
- 98.Tong Z, Hage DS. Characterization of interaction kinetics between chiral solutes and human serum albumin by using high-performance affinity chromatography and peak profiling. J Chromatogr A. 2011;1218:6892–6897. doi: 10.1016/j.chroma.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Loun B, Hage DS. Chiral separation mechanisms in protein-based HPLC columns. I. Thermodynamic studies of (R)- and (S)-warfarin binding to immobilized human serum albumin. Anal Chem. 1994;66:3814–3822. doi: 10.1021/ac00093a043. [DOI] [PubMed] [Google Scholar]
- 100.Loun B, Hage DS. Chiral separation mechanisms in protein-based HPLC columns. II. Kinetic studies of R- and S-warfarin binding to immobilized human serum albumin. Anal Chem. 1996;68:1218–1225. doi: 10.1021/ac950827p. [DOI] [PubMed] [Google Scholar]
- 101.Joseph KS, Moser AC, Basiaga S, Schiel JE, Hage DS. Evaluation of alternatives to warfarin as probes for Sudlow site I of human serum albumin: characterization by high performance affinity chromatography. J Chromatogr A. 2009;1216:3492–3500. doi: 10.1016/j.chroma.2008.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang J, Hage DS. Characterization of the binding and chiral separation of D- and L-tryptophan on a high-performance immobilized human serum albumin column. J Chromatogr. 1993;645:241–250. doi: 10.1016/0021-9673(93)83383-4. [DOI] [PubMed] [Google Scholar]
- 103.Yang J, Hage DS. Effect of mobile phase composition on the binding kinetics of chiral solutes on a protein-based HPLC column: interactions of D- and L-tryptophan with immobilized human serum albumin. J Chromatogr A. 1997;766:15–25. doi: 10.1016/s0021-9673(96)01040-0. [DOI] [PubMed] [Google Scholar]
- 104.Conrad ML, Moser AC, Hage DS. Evaluation of indole-based probes for studying drug binding to human serum albumin in high-performance affinity separations. J Sep Sci. 2009;32:1145–1155. doi: 10.1002/jssc.200800567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Matsuda R, Anguizola J, Joseph KS, Hage DS. High-performance affinity chromatography and the analysis of drug interactions with modified proteins: binding of gliclazide with glycated human serum albumin. Anal Bioanal Chem. 2011;401:2811–2819. doi: 10.1007/s00216-011-5382-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jackson AJ, Anguizola J, Pfaunmiller EL, Hage DS. Use of entrapment and high-performance affinity chromatography to compare the binding of drugs and site-specific probes with normal and glycated human serum albumin. Anal Bioanal Chem. 2013;405:5833–5841. doi: 10.1007/s00216-013-6981-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Joseph KS, Hage DS. Characterization of the binding of sulfonylurea drugs to HSA by high-performance affinity chromatography. J Chromatogr B. 2010;878:1590–1598. doi: 10.1016/j.jchromb.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Matsuda R, Anguizola J, Joseph KS, Hage DS. Analysis of drug interactions with modified proteins by high-performance affinity chromatography: Binding of glibenclamide to normal and glycated human serum albumin. J Chromatogr A. 2012;1265:114–122. doi: 10.1016/j.chroma.2012.09.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Joseph KS, Anguizola J, Jackson AJ, Hage DS. Chromatographic analysis of acetohexamide binding to glycated human serum albumin. J Chromatogr B. 2010;878:2775–2781. doi: 10.1016/j.jchromb.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Joseph KS, Anguizola J, Hage DS. Binding of tolbutamide to glycated human serum albumin. J Pharm Biomed Anal. 2011;54:426–432. doi: 10.1016/j.jpba.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kaliszan R. Retention data from affinity high-performance liquid chromatography in view of chemometrics. J Chromatogr B. 1998;715:229–244. doi: 10.1016/s0378-4347(98)00175-3. [DOI] [PubMed] [Google Scholar]
- 112.Markuszewski M, Kaliszan R. Quantitative structure-retention relationships in affinity high-performance liquid chromatography. J Chromatogr B. 2002;768:55–56. doi: 10.1016/s0378-4347(01)00485-6. [DOI] [PubMed] [Google Scholar]
- 113.Wainer IW. Enantioselective high-performance liquid affinity chromatography as a probe of ligand-biopolymer interactions: an overview of a different use for high-performance liquid chromatographic chiral stationary phases. J Chromatogr A. 1994;666:221–234. [Google Scholar]
- 114.Kaliszan R. Quantitative structure-retention relationships. Anal Chem. 1994;64:A619–A627. doi: 10.1021/ac9901586. [DOI] [PubMed] [Google Scholar]
- 115.Kaliszan R. Chromatography and capillary electrophoresis in modelling the basic processes of drug action. Trends Anal Chem. 1999;18:400–410. [Google Scholar]
- 116.Kaliszan R. Chemometric analysis of biochromatographic data - implications for molecular pharmacology. Chemometr Intell Lab Systems. 1994;24:89–97. [Google Scholar]
- 117.Kaliszan R, Kaliszan A, Noctor TAG, Purcell WP, Wainer IW. Mechanism of retention of benzodiazepines in affinity, reversed phase and adsorption high performance liquid chromatography in view of quantitative structure retention relationships. J Chromatogr. 1992;609:69–81. [Google Scholar]
- 118.Kaliszan R, Noctor TAG, Wainer IW. Stereochemical aspects of benzodiazepine binding to human serum albumin. 2. Quantitative relationships between structure and enantioselective retention in high-performance liquid affinity-chromatography. Mol Pharmacol. 1992;42:512–517. [PubMed] [Google Scholar]
- 119.Kaliszan R, Noctor TAG, Wainer IW. Quantitative structure-enantioselective retention relationships for the chromatography of 1,4-benzodiazepines on a human serum-albumin based HPLC chiral stationary phase - an approach to the computational prediction of retention and enantioselectivity. Chromatographia. 1992;33:546–550. [Google Scholar]
- 120.Nasal A, Radwanska A, Osmialowsk K, Bucinski A, Kaliszan R, Barker GE, Sun P, Hartwick RA. Quantitative relationships between structure of β-adrenolytic and antihistamine drugs and their retention on an α1-acid glycoprotein HPLC column. Biomed Chromatogr. 1994;8:125–129. doi: 10.1002/bmc.1130080306. [DOI] [PubMed] [Google Scholar]
- 121.Kaliszan R, Nasal A, Turowski M. Quantitative structure-retention relationships in the examination of the topography of the binding site of antihistamine drugs on alpha(1)-acid glycoprotein. J Chromatogr A. 1996;722:25–32. doi: 10.1016/0021-9673(95)00523-4. [DOI] [PubMed] [Google Scholar]
- 122.Karlsson A, Aspegren A. Enantiomeric separation of amino alcohols on protein phases using statistical experimental design. A comparative study. J Chromatogr A. 2000;866:15–23. doi: 10.1016/s0021-9673(99)01040-7. [DOI] [PubMed] [Google Scholar]
- 123.Kaliszan R, Nasal A, Turowski M. Binding site for basic drugs on alpha 1-acid glycoprotein as revealed by chemometric analysis of biochromatographic data. Biomed Chromatogr. 1995;707:211–215. doi: 10.1002/bmc.1130090504. [DOI] [PubMed] [Google Scholar]
- 124.Fitos I, Visy J, Simonyi M, Hermansson J. Chiral high-performance liquid chromatographic separations of vinca alkaloid analogues on alpha 1-acid glycoprotein and human serum albumin columns. J Chromatogr A. 1992;609:163–171. doi: 10.1016/0021-9673(92)80159-r. [DOI] [PubMed] [Google Scholar]
- 125.Gyimesi-Forras K, Szasz G, Gergely A, Szabo M, Kokosi J. Study on the sorption properties of alpha1-acid glycoprotein (AGP)-based stationary phase modified by organic solvents. J Chromatogr Sci. 2000;38:430–434. [Google Scholar]
- 126.Johnson CH, Patterson AD, Idle JI, Gonzalez FL. Xenobiotic metabolomics: major impact on the metabolome. Annu Rev Pharmacol Toxicol. 2012;52:37–56. doi: 10.1146/annurev-pharmtox-010611-134748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Danzo BJ. Environmental xenobiotics may disrupt normal endocrine function by interfering with the binding of physiological ligands to steroid receptors and binding proteins. Environ Health Perspect. 1997;105:294–301. doi: 10.1289/ehp.97105294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Diniz A, Escuder-Gilabert L, Lopes NP, Villanueva-Camanas RM, Sagrado S, Medina-Hernandez MJ. Characterization of interactions between polyphenolic compounds and human serum proteins by capillary electrophoresis. Anal Bioanal Chem. 2008;391:625–632. doi: 10.1007/s00216-008-2046-4. [DOI] [PubMed] [Google Scholar]
- 129.Omiecinski CJ, Vauden Huevel JP, Perdew GH, Peters JH. Xenobiotic metabolism, disposition, and regulation by receptors: from biochemical phenomenon to predictors of major toxicities. Toxicol Sci. 2011;120:S49–S75. doi: 10.1093/toxsci/kfq338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cheek AO, Kow K, Chen J, McLachlan JA. Potential mechanisms of thyroid disruption in humans: interaction of organochlorine compounds with thyroid receptor, transthyretin, and thyroid-binding globulin. Environ Health Perspect. 1999;107:273–278. doi: 10.1289/ehp.99107273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cao J, Lin Y, Guo L, Zhang A, Wei Y, Yang Y. Structure-based investigation on the binding interaction of hydroxylated polybrominated diphenyl ethers with thyroxine transport proteins. Toxicol. 2010;277:20–28. doi: 10.1016/j.tox.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 132.Fowles JR, Fairbrother A, Baecher-Steppan L, Kerkvliet NI. Immunologic and endocrine effects of the flame-retardant pentabromodiphenyl ether (DE-71) in C57BL/6J mice. Toxicol. 1994;86:49–61. doi: 10.1016/0300-483x(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 133.Hallgren S, Sinjari T, Hakansson H, Darnerud PO. Effects of polybrominated diphenyl ethers (PBDEs) and polychlorinated biphenyls (PCBs) on thyroid hormone and vitamin A levels in rats and mice. Arch Toxicol. 2001;75:200–208. doi: 10.1007/s002040000208. [DOI] [PubMed] [Google Scholar]
- 134.Hage DS, Austin J. High-performance affinity chromatography and immobilized serum albumin as probes for drug- and hormone-protein binding. J Chromatogr B. 2000;739:39–54. doi: 10.1016/s0378-4347(99)00445-4. [DOI] [PubMed] [Google Scholar]
- 135.Tabas I. Nonoxidative modifications of lipoproteins in atherogenesis. Annu Rev Nutr. 1999;19:123–139. doi: 10.1146/annurev.nutr.19.1.123. [DOI] [PubMed] [Google Scholar]
- 136.Cobbold CA, Sherratt JA, Maxwell SRJ. Lipoprotein oxidation and its significance for atherosclerosis: a mathematical approach. Bull Math Biol. 2002;64:65–95. doi: 10.1006/bulm.2001.0267. [DOI] [PubMed] [Google Scholar]
- 137.Bertucci C, Domenici E. Reversible and covalent binding of drugs to human serum albumin: methodological approaches and physiological relevance. Curr Med Chem. 2002;9:1463–1481. doi: 10.2174/0929867023369673. [DOI] [PubMed] [Google Scholar]
- 138.Ascoli GA, Domenici E, Bertucci C. Drug binding to human serum albumin: abridged review of results obtained with high-performance liquid chromatography and circular dichroism. Chirality. 2006;18:667–679. doi: 10.1002/chir.20301. [DOI] [PubMed] [Google Scholar]
- 139.He XM, Carter DC. Atomic structure and chemistry of human serum albumin. Nature. 1992;358:209–15. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 140.Fanali G, di Masi A, Trezza V, Marino M, Fasano M, Ascenzi P. Human serum albumin: from bench to bedside. Mol Aspects Med. 2012;33:209–290. doi: 10.1016/j.mam.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 141.Noctor TAG, Wainer IW. The in situ acetylation of an immobilized human serum albumin chiral stationary phase for high-performance liquid chromatography in the examination of drug-protein binding phenomena. Pharmaceut Res. 1992;9:480–484. doi: 10.1023/a:1015884112039. [DOI] [PubMed] [Google Scholar]
- 142.Chattopadhyay A, Tian T, Kortum L, Hage DS. Development of tryptophan-modified human serum albumin columns for site-specific studies of drug-protein interactions by high-performance affinity chromatography. J Chromatogr B. 1998;715:183–190. doi: 10.1016/s0378-4347(98)00140-6. [DOI] [PubMed] [Google Scholar]
- 143.Hage DS, Chen J. In: Handbook of Affinity Chromatography. 2. Hage DS, editor. Vol. 22 CRC Press; Boca Raton: 2006. [Google Scholar]
- 144.Bertucci C, Nanni B, Raffaelli A, Salvadori P. Chemical modification of human albumin at Cys34 by ethacrynic acid: structural characterization and binding properties. J Pharm Biomed Anal. 1998;18:127–136. doi: 10.1016/s0731-7085(98)00163-0. [DOI] [PubMed] [Google Scholar]
- 145.Anguizola J, Matsuda R, Barnaby OS, Hoy KS, Wa C, DeBolt E, Koke M, Hage DS. Review: glycation of human serum albumin. Clin Chim Acta. 2013;425:64–76. doi: 10.1016/j.cca.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hartog JWL, Voors AA, Bakker SJL, Smit AJ, Veldhuisen DJV. Advanced glycation end-products (AGEs) and heart failure: pathophysiology and clinical implications. Eur J Heart Fail. 2007;9:1146–1155. doi: 10.1016/j.ejheart.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 147.Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J. 1999;344:109–116. [PMC free article] [PubMed] [Google Scholar]
- 148.Richard DM, Dawes MA, Mathias CW, Acheson A, Hill-Kapturczak N, Dougherty DM. L-Tryptophan: basic metabolic functions, behavioral research and therapeutic indications. Int J Tryptophan Res. 2009;2:45–60. doi: 10.4137/ijtr.s2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Joseph KS, Hage DS. The effects of glycation on the binding of human serum albumin to warfarin and L-tryptophan. J Pharm Biomed Anal. 2010;53:811–818. doi: 10.1016/j.jpba.2010.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kimzey MJ, Yassine HN, Riepel BM, Tsaprailis G, Monks TJ, Lau SS. New site(s) of methylglyoxal-modified human serum albumin, identified by multiple reaction monitoring, alter warfarin binding and prostaglandin metabolism. Chem Biol Int. 2011;192:122–128. doi: 10.1016/j.cbi.2010.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tang D, Zhu JX, Wu AG, Xu YH, Duan TT, Zheng ZG, Wang RS, Li D, Zhu Q. Pre-column incubation followed by fast liquid chromatography analysis for rapid screening of natural methylglyoxal scavengers directly from herbal medicines: case study of Polygonum cuspidatum. J Chromatogr A. 2013;1286:102–110. doi: 10.1016/j.chroma.2013.02.058. [DOI] [PubMed] [Google Scholar]
- 152.Ceciliani F, Pocacqua V. The acute phase protein α1-acid glycoprotein: a model for altered glycosylation during diseases. Curr Protein Pept Sci. 2007;8:91–108. doi: 10.2174/138920307779941497. [DOI] [PubMed] [Google Scholar]
- 153.Kuroda Y, Kita Y, Shibukawa A, Nakagawa T. Role of biantennary glycans and genetic variants of human alpha1-acid glycoprotein in enantioselective binding of basic drugs as studied by high performance frontal analysis/capillary electrophoresis. Pharm Res. 2001;18:389–393. doi: 10.1023/a:1011023518144. [DOI] [PubMed] [Google Scholar]
- 154.Kishino S, Nomura A, Itoh S, Nakagawa T, Takekuma Y, Sugawara M, Furukawa H, Todo S, Miyazaki K. Age- and gender-related differences in carbohydrate concentrations of alpha1-acid glycoprotein variants and the effects of glycoforms on their drug-binding capacities. Eur J Clin Pharmacol. 2002;58:621–628. doi: 10.1007/s00228-002-0530-x. [DOI] [PubMed] [Google Scholar]
- 155.Nakagawa T, Kishino S, Itoh S, Sugawara M, Miyazaki K. Differential binding of disopyramide and warfarin enantiomers to human alpha(1)-acid glycoprotein variants. Br J Clin Pharmacol. 2003;56:664–669. doi: 10.1046/j.1365-2125.2003.01909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kakiuchi Y, Kohda Y, Miyabe M, Momose Y. Effect of plasma alpha1-acid glycoprotein concentration on the accumulation of lidocaine metabolites during continuous epidural anesthesia in infants and children. Int J Clin Pharmacol Ther. 1999;37:493–498. [PubMed] [Google Scholar]
- 157.Soriano SG, Sullivan LJ, Venkatakrishnan K, Greenblatt DJ, Martyn JA. Pharmacokinetics and pharmacodynamics of vecuronium in children receiving phenytoin or carbamazepine for chronic anticonvulsant therapy. Br J Anaesth. 2001;86:223–229. doi: 10.1093/bja/86.2.223. [DOI] [PubMed] [Google Scholar]
- 158.McNamara JR, Warnick GR, Cooper GR. A brief history of lipid and lipoprotein measurements and their contribution to clinical chemistry. Clin Chim Acta. 2006;369:158–167. doi: 10.1016/j.cca.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 159.Jonas A. In: Biochemistry of Lipids, Lipoproteins, and Membranes. Vance DE, Vance JE, editors. Elsevier-Science Publishers; Amsterdam: 2002. p. 483. [Google Scholar]
- 160.Barklay M. In: Blood Lipids and Lipoproteins Quantitation, Composition, and Metabolism. Nelson GJ, editor. Wiley-Interscience; New York: 1972. p. 587. [Google Scholar]
- 161.Wasan KM, Cassidy SM. Role of plasma lipoproteins in modifying the biological activity of hydrophobic drugs. J Pharm Sci. 1998;87:411–424. doi: 10.1021/js970407a. [DOI] [PubMed] [Google Scholar]
- 162.Durrington PN. In: Lipoproteins and Lipids. Durrington PN, editor. Wright; London: 1989. p. 255. [Google Scholar]
- 163.Havel RJ, Kane JP. In: The Metabolic and Molecular Basis of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. McGraw-Hill Professional; New York: 1995. p. 1129. [Google Scholar]
- 164.Skipski VR. In: Blood Lipids and Lipoproteins Quantitation, Composition, and Metabolism. Nelson GJ, editor. Wiley-Interscience; New York: 1972. p. 587. [Google Scholar]
- 165.Kwong TC. Free drug measurements: methodology and clinical significance. Clin Chim Acta. 1985;151:193–216. doi: 10.1016/0009-8981(85)90082-8. [DOI] [PubMed] [Google Scholar]
- 166.Glasson S. The distribution of bound propranolol between the different human serum proteins. Mol Pharmacol. 1980;17:187–191. [PubMed] [Google Scholar]
- 167.Ohnishi T, Mohamed NAL, Shibukawa A, Kuroda Y, Nakagawa T, El Gizawy S, Askal HF, El Kommos ME. Frontal analysis of drug–plasma lipoprotein binding using capillary electrophoresis. J Pharm Biomed Anal. 2002;27:607–614. doi: 10.1016/s0731-7085(01)00569-6. [DOI] [PubMed] [Google Scholar]
- 168.Mohamed NAL, Kuroda Y, Shibukawa A, Nakagawa T, El Gizawy S, Askal HF, El Kommos ME. Enantioselective binding analysis of verapamil to plasma lipoproteins by capillary electrophoresis-frontal analysis. J Chromatogr A. 2000;875:447–453. doi: 10.1016/s0021-9673(99)01288-1. [DOI] [PubMed] [Google Scholar]
- 169.Mohamed NAL, Kuroda Y, Shibukawa A, Nakagawa T, El Gizawy S, Askal HF, El Kommos ME. Binding analysis of nilvadipine to plasma lipoproteins by capillary electrophoresis-frontal analysis. J Pharm Biomed Anal. 1999;21:1037–1043. doi: 10.1016/s0731-7085(99)00197-1. [DOI] [PubMed] [Google Scholar]
- 170.Kim HS, Wainer IW. Rapid analysis of the interactions between drugs and human serum albumin (HSA) using high-performance affinity chromatography (HPAC) J Chromatogr B. 2008;870:22–26. doi: 10.1016/j.jchromb.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hollósy F, Valkó K, Hersey A, Nunhuck S, Kéri G, Bevan CJ. Estimation of volume of distribution in humans from high throughput HPLC-based measurements of human serum albumin binding and immobilized artificial membrane partitioning. Med Chem. 2006;49:6958–6971. doi: 10.1021/jm050957i. [DOI] [PubMed] [Google Scholar]
- 172.Buchholz L, Cai CH, Andress L, Cleton A, Brodfuehrer J, Cohen L. Evaluation of the human serum albumin column as a discovery screening tool for plasma protein binding. Eur J Pharm Sci. 2002;15:209–215. doi: 10.1016/s0928-0987(01)00219-6. [DOI] [PubMed] [Google Scholar]
- 173.Chen S, Sobansky MR, Hage DS. Analysis of drug interactions with high density lipoproteins by high-performance affinity chromatography. Anal Biochem. 2010;397:107–114. doi: 10.1016/j.ab.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sobansky MR, Hage DS. Identification and analysis of stereoselective drug interactions with low density lipoprotein by high-performance affinity chromatography. Anal Bioanal Chem. 2012;403:563–571. doi: 10.1007/s00216-012-5816-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sobansky MR, Hage DS. In: Advances in Medicine and Biology. Berhardt LV, editor. Vol. 53. Vol. 9 Nova Science; Hauppage: 2012. [Google Scholar]
- 176.Kuroda Y, Cao B, Shibukawa A, Nakagawa T. Effect of oxidation of low-density lipoprotein on drug binding affinity studied by high performance frontal analysis-capillary electrophoresis. Electrophoresis. 2001;22:3401–3407. doi: 10.1002/1522-2683(200109)22:16<3401::AID-ELPS3401>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]