

Abstract
Cancer is a consequence of mutations in genes that control cell proliferation, differentiation and cellular homeostasis. These genes are classified into two categories: oncogenes and tumor suppressor genes. Together, overexpression of oncogenes and loss of tumor suppressors are the dominant driving forces for tumorigenesis. Hence, targeting oncogenes and tumor suppressors hold tremendous therapeutic potential for cancer treatment. In the last decade, the predominant cancer drug discovery strategy has relied on a traditional reductionist approach of dissecting molecular signaling pathways and designing inhibitors for the selected oncogenic targets. Remarkable therapies have been developed using this approach; however, targeting oncogenes is only part of the picture. Our understanding of the importance of tumor suppressors in preventing tumorigenesis has also advanced significantly and provides a new therapeutic window of opportunity. Given that tumor suppressors are frequently mutated, deleted, or silenced with loss-of-function, restoring their normal functions to treat cancer holds tremendous therapeutic potential. With the rapid expansion in our knowledge on cancer over the last several decades, developing effective anticancer regimens against tumor suppressor pathways has never been more promising. In this article, we will review the concept of tumor suppression, and outline the major therapeutic strategies and challenges of targeting tumor suppressor networks for cancer therapeutics.
Keywords: tumor suppressors, RB, p53, BRCA1, BRCA2, gene therapy, small molecule inhibitors
I. An overview of tumor suppressors
a. A glimpse of history
The genetic basis for cancer was originally proposed by pioneer German pathologist, Theodor Boveri in 1914 (1). Herman J. Muller advanced the notion that cells needed to acquire multiple genetic mutations before becoming cancerous, based on observations of the long latent period between carcinogen exposure and cancer appearance (2). Moreover, epidemiology studies of common cancers revealed that cancer incidence increases with age, which further supported the concept that the initiation of cancer requires multiple mutations (3). Today, it is commonly understood that cancer is a consequence of multiple genetic and epigenetic alterations, which are either inherited or acquired through somatic mutations.
In general, there are two classes of genes that are frequently mutated in cancer: oncogenes and tumor suppressor genes. An oncogene is an oncogenic variant of the normal proto-oncogene that acquired a gain-of-function alteration, resulting from point mutations, chromosome rearrangements, or amplification of the proto-oncogene sequences. In 1911, Peyton Rous discovered the first oncogenic retrovirus, Rous Sarcoma Virus (RSV), and demonstrated that sarcomas could be virally induced by RSV in chickens (4). Sixty years later, in 1970, the first retroviral oncogene, SRC (or v-src), was identified in the oncogenic region of RSV (5). Shortly after the cellular proto-oncogene of SRC (c-src) was isolated, the virally transduced and mutated copy of SRC was demonstrated to be the causative driving force behind RSV induced tumors (6, 7). Since the first oncogene was discovered, finding for additional cancer causing genes has mushroomed.
Despite the significance of oncogenes in the genesis of tumors, many of the altered properties of cancer cells are also attributed to the inactivation or loss of normal cellular regulatory genes, known as tumor suppressor genes. In Boveri’s early works, he predicted the presence of not only oncogenes but also tumor suppressor genes, “…cells of tumor with unlimited growth would arise if those ‘inhibiting chromosomes’ were eliminated.” Tumor suppressor genes play important roles in suppressing uncontrolled proliferation, immortality, and tumorgenicity. Such tumor suppressing properties were first demonstrated in the late 1960s, when Henry Harris reverted highly malignant mouse ascities tumor cells to a nontumorigenic state by fusing the malignant cell with a normal fibroblast. The results from this study indicated that factors present in normal cells could inhibit (or suppress) the tumorigenicity of malignant cells (8). Though controversial at the time, Henry Harris’ observation suggested the existence of certain intrinsic cellular factors that could suppress tumor development in a dominant manner.
b. Discovery of the first tumor suppressor: retinoblastoma susceptibility gene, RB
The very first tumor suppressor gene to be identified and characterized was the Retinoblastoma Susceptibility gene, RB. Retinoblastoma is a rare childhood eye tumor that can be either familial or sporadic (hereditary or non-hereditary). In approximately one-fourth of the retinoblastoma cases, tumors develop in both eyes (bilateral); whereas in the remaining cases, only one eye is affected (unilateral) (9). Using Poisson statistics, Alfred Knudson reasoned that the distribution of the observed bilateral and unilateral retinoblastoma cases could only be caused by two mutational events. This theory became known as the “two-hit” hypothesis, and suggested that tumorigenesis requires two mutational events to inactivate the two functional copies of the “tumor suppressor gene” (10). In familial retinoblastoma, the first inactivating mutation is inherited, while the second mutational event occurs spontaneously in the second allele in the same cell. In the sporadic form, both functional alleles are spontaneously mutated. For children who inherit one mutational “hit” from their parents, the chance of getting another spontaneous mutation in the same retinal cell and developing retinoblastoma is much higher than having two spontaneous mutations on the same susceptibility locus. Consequently, familial retinoblastoma is usually bilateral while sporadic form is unilateral. Later, David Comings unified a general framework for the role of tumor suppressors for all types of cancers. He suggested that dominantly inherited tumors may result from the loss or inactivation of both alleles of suppressor genes that, when active, prevent the expression of transforming genes (possibly oncogenes) normally active only during embryogenesis (11).
Another interesting observation with retinoblastoma is that incipient cancer cells are able to invent ways to eliminate wild-type copies of tumor suppressor genes, a phenomenon now known as “Loss-of-Heterozygosity” (LOH). Using LOH as a guide, subsequent investigations on patients with both a personal and family history of retinoblastoma showed that chromosomal deletion within 13q14, or 13q deletion mosaicism, is associated with an increased risk of developing retinoblastoma (12). The retinoblastoma susceptibility gene, RB, was then identified, which provides a solution to the genetic puzzle of dominantly inherited neoplastic diseases (13, 14). Further analysis showed that RB encodes a nuclear phosphoprotein with molecular weight of about 105kD, RB or pRb, which is found to be absent or present in a defective form in retinoblastoma, osteosarcoma, breast cancer (15), and small-cell lung carcinoma (16). Homozygous knockout mice carrying non-functional RB-1 died before the E. 14 with multiple developmental defects in the hematopoietic and nervous systems. Heterozygous knockout mice developed spontaneous pituitary tumors from 2 to 11 months of age with a nearly 100% incidence (17, 18).
Since its discovery, RB has been the subject of intense study (Figure 1). RB is now known as a universal cell cycle regulator with a central role in governing the passage of cells through the G1 phase, and particularly, the restriction point (R point), control of which is lost in most cancer cells (19). Under normal condition, RB is phosphorylated by cyclin D-CDK4/6 and cyclin E-CDK2 complexes upon mitogenic stimulus (20, 21). The activities of cyclin-CDK complexes are negatively regulated by CDK inhibitors (CKIs), including four INK4 proteins (Inhibitor of CDK4, p16INK4A, p15INK4B, p18INK4C, p19INK4D) that specifically antagonize CDK4/6, and three remaining CDK inhibitors (p21Cip1, p27Kip1, p57Kip2) that have a broader inhibition spectrum (21, 22). The dephosphorylation of RB at the exit of M phase is performed by phosphatase 1α (PP1α), which has been demonstrated to compete with CDKs for a common binding site on RB (23).
Figure 1. RB structure and functions.

(A) A schematic representation of RB protein functional domains and post-translational modification sites, such as phosphorylation (P), acetylation (Ac), and methylation (Me). The central A and B domains mediate interactions with a number of proteins, such as E2Fs and LxCxE motif containing proteins. (B) The cell cycle-dependent phosphorylation status of RB determines the progression of cell cycle progression, chromosome segregation and terminal differentiation.
When RB is unphosphorylated or hypo-phosphorylated, it binds and sequesters the transcriptional activator, E2F, to repress transcription of target genes with the help from chromatin remodeling complexes and Histone Deacetylases (HDACs). However, when hyperphosphorylated, RB dissociates from the E2Fs, allowing E2F/DP to interact with histone acetylase to activate transcription (24). This classic view of RB tumor suppressing activity is focused on this negative regulation of E2F transcriptional activation and cell cycle inhibition. It is surprising when two recent studies demonstrated that the presumably oncogenic E2F family proteins are dispensable for proliferation in vivo (25, 26). Such discoveries, combined with the fact that E2Fs are less frequently mutated in cancer, suggests that RB may have other functions besides E2F-dependent transcriptional regulation. So far, RB has been demonstrated to play an important role in faithful chromosome segregation (27, 28), checkpoint control (29), apoptosis (30), senescence (31), and terminal differentiation (32). Detailed molecular studies revealed that these functions could be mediated by post-translational modifications on the C-terminal domain of RB, such as acetylation (33) and methylation (34), in response to internal or external signals (35). It is undeniable that RB suppresses tumor formation by virtue of its multiple biological activities upon receiving different signals, in addition to its role as a mediator between CDK regulatory pathways and E2F activators.
c. From oncogene to tumor suppressor: p53
Although initially discovered and inaccurately characterized as a weak oncogene, the wild-type p53 was later confirmed to be a bona fide tumor suppressor (36–38). Sequence analysis of the oncogenic version of p53 revealed a single base substitution mutation that led to a “gain of oncogenic function” activity. By the early 1990s, p53 was widely recognized as the most frequently mutated tumor suppressor gene in human cancers. In ovarian cancer, esophageal cancer, colorectal cancer, and head and neck cancer, mutant alleles of p53 or deletion of p53 alleles are found in 40–50% of cases (39). In addition, germline p53 mutations occur in patients with Li-Fraumeni syndrome, a hereditary predisposition to several cancers, especially soft tissue tumors (40, 41). Intriguingly, 75% of the more than 15,000 p53 mutations identified in human tumor cell genomes are missense mutations, quite different from other tumor suppressor genes, such as APC (Adenomatosis Polyposis Coli), where frameshift and nonsense mutations account for more than 60% of mutations identified (42, 43). Biochemical analyses of p53 proteins showed that p53 is capable of forming tetramers, allowing the mutant protein to actively interfere with the function of wild-type protein in a dominant-negative fashion (44, 45). The presence of mutated alleles usually results in an accumulation of a faulty protein in the tumor cells, which also displays loss of heterozygosity (46).
Under normal conditions, p53 has a very short half-life of 20 minutes and a low steady-state protein level. However, upon receiving signals like UV radiation induced DNA damage, the degradation of p53 is blocked and p53 is stabilized to perform its functions (47). Many of p53’s functions can be traced to its role as a transcription factor. More than 95% of p53 mutations are found in the DNA-binding domain of the p53 protein, compared to other mutations that affect nuclear localization, oligomerization or transactivation. In response to DNA damage, p53 functions as a transcription factor that induces expression of genes such as p21Cip1 to halt cell cycle progression at G1 phase (48). As mentioned above, p21Cip1 inhibits RB activity by preventing the hyperphosphorylation of RB, providing another layer of regulation on cell cycle control. Delaying the cell cycle allows the cell to turn on DNA repair genes to mend the DNA damages. p53 has also been demonstrated to contribute to DNA repair directly by activating genes that facilitate nucleotide excision repair and base excision repair (49, 50). If the DNA damage is too severe to repair, wild-type p53 can redirect the cell into cell cycle arrest, senescence, or even apoptosis, by activating apoptosis-associated genes, such as PUMA (51). Thus, p53 plays essential roles in responding to various cellular stresses signals.
Similar to RB, p53 activities are modulated by a diverse range of post-translational modifications, which includes phosphorylation, acetylation, methylation, ubiquitination, and sumoylation (52). Intriguingly, a recent study investigating the importance of p53 acetylation on tumor suppression revealed that abolishment of p53-dependent apoptosis and/or cell-cycle arrest and senescence in vivo failed to induce early-onset tumorigenesis (53). This finding is consistent with previous studies that demonstrated depletion of p21Cip1 (54) or PUMA (55), the primary mediators of p53-dependent cell-cycle arrest or apoptosis, respectively, does not lead to tumor susceptibility in the same way as loss of p53 does (56). Several other p53 mutants have also been shown to induce apoptosis or cell-cycle arrest independent tumor suppressor activities. This suggested that alternative mechanisms are involved in p53 associated tumorigenesis (57, 58). Indeed, as p53 deficient cancer cells often undergo aerobic glycolysis (also known as the Warburg effect), additional p53 functions in regulating glycolysis and Reactive Oxygen Species (ROS) have been proposed to further contribute to the tumor suppression effect of p53 (59). Understanding the role of p53 in regulating cellular metabolism and protecting cells from oxidative stress may provide new insight into p53 function and window for therapeutic intervention.
d. Breast cancer susceptibility genes: BRCA1 and BRCA2
Breast cancer is one of the most frequent malignancies with a cumulative lifetime risk of nearly 10% in women. Ovarian cancer, on the other hand, has a much lower lifetime risk of 1.8%, but is the most lethal cancers (60). In the 1990s, two Breast Cancer Susceptibility genes, BRCA1 and BRCA2, were identified in patients with hereditary breast and ovarian cancer syndrome (61, 62). Female BRCA1 or BRCA2 mutation carriers have a lifetime breast cancer risk of 50–80% and a lifetime ovarian cancer risk of 10–20%. Studies showed that mutations in BRCA1 account for almost all of the familial breast and ovarian cancer cases and up to 30–50% of families with hereditary breast cancer only. At one point, it was thought that BRCA1 inactivation is not involved in sporadic breast cancers, but now inactive BRCA1 alleles are found in about 10–15% of sporadic breast carcinomas. Mutations in BRCA2 are linked to the other half of inherited breast cancer families and also to male breast cancer (63, 64).
Cellular and molecular studies showed that both BRCA1 and BRCA2 deficient cells tend to accumulate chromosome abnormalities, including chromosomal breaks, aberrant mitotic exchanges and aneuploidy, suggesting their normal function in DNA repair (Figure 2) (65, 66). BRCA1 is a nuclear phosphoprotein that is expressed and phosphorylated by the Ataxia Telangiectasia mutated (ATM) and Checkpoint Kinase 2 (CHK2) proteins in response to DNA damage (67, 68). BRCA1 is an essential component of the RAD50-MRE11-p95 complex, a recombination-mediated DNA double-stranded breaks (DSBs) repair complex, as well as the recombinase, RAD51, located in the nuclear foci of mitotic cells (69, 70). Similar to BRCA1, BRCA2 is also a nuclear phosphoprotein that binds to RAD51 and MRE11 to regulate DSB repair and maintain chromosome integrity (71, 72).
Figure 2. BRCA1 and BRCA2 structure and functions.
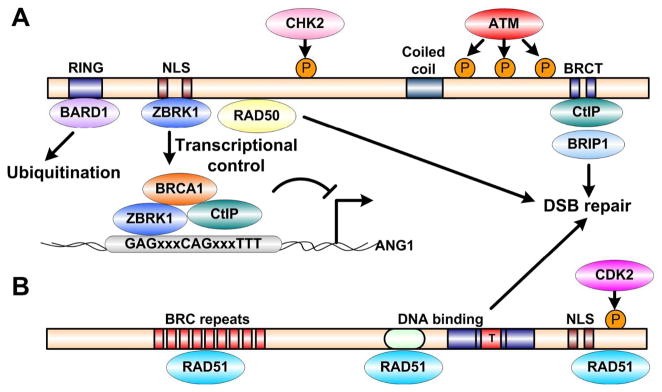
Schematic representations of BRCA1 (A) and BRCA2 (B) structural motifs, interacting proteins, and corresponding kinase phosphorylation sites. Both BRCA1 and BRCA2 regulate DNA double-strand breaks repair; however, BRCA1 is also involved in mediating ubiquitination and transcriptional control.
As aforementioned, DNA damage responses can often activate checkpoints through RB or p53 to arrest the cell cycle either at G1/S transition or G2/M transition (73), which enables the DNA repair pathway to correct and prevent the transmission of genetic errors to daughter cells. A recent study showed that loss of p53-binding protein 1 (53BP1) could abandon the ATM-dependent checkpoint response triggered by the DBSs accumulated in BRCA1-deficient cells (74). Among different types of DNA damages, DSBs are considered to be the most harmful as the integrity of both strands of the DNA duplex are affected simultaneously. There are two major methods to repair DSBs in human, error-free homologous recombination (HR) and error-prone non-homologous end-joining (NHEJ). NHEJ is usually employed during G1 phase due to the absence of a sister chromatid; whereas HR repairs DSBs during the S and G2 phases, when an intact sister chromatid is available as a template for repair (75). Although BRCA1 and BRCA2 share many interacting proteins, their protein sequences are not conserved and they play divergent roles in regulating HR. BRCA1 not only serves as a scaffold protein that integrates different components of the HR machinery (76), but also enhances CtIP-mediated 5′-end resection of DSBs (77). In addition, cells carrying defective BRCA1 do not arrest in G2 phase after DNA damage, which indicates that BRCA1 is involved in mediating DNA-damage checkpoint control (78).
Besides its functions in DNA repair, BRCA1 also has important roles in transcriptional regulation (79, 80), differentiation (81), and ubiquitination as an E3 ligase (82–84). However, a recent study has shown that the E3 ligase activity of BRCA1 seems to be dispensable for tumor suppression (85). BRCA2, on the other side, is primarily involved in recruiting RAD51 to DNA damage sites or stalled replication forks (86). BRCA2 has also been reported to function as a RAD51 loader in facilitating telomere replication (87).
e. New classes of tumor suppressors
Classic tumor suppressors, such as RB and p53, play critical roles in governing the decisions of cells to proliferate, enter senescence or undergo apoptosis; therefore, they are termed as the “gatekeepers” of the cell. BRCA1 and BRCA2, on the other hand, function as “caretakers” to maintain the genome integrity. Loss of genome stability has now been recognized as a new cancer hallmark, which enables generation of random mutations that can potentially provide cancer cells with various growth advantages (88).
Avoiding immune destruction is another new cancer hallmark (89). The immune system can respond to cancer cells by reacting against antigens that are either unique to cancer cells (tumor-specific antigens), or differentially expressed between cancer and normal cells (tumor-associated antigens). Such antigens are often products of oncogenes and can incite strong inflammatory responses. Tumors can suppress the immune system both systemically and locally in the tumor microenvironment by expressing immuno-suppressive cytokines themselves, or activating regulatory T cells that can produce cytokines (90). Novel tumor suppressors have been identified that are capable of provoking immunity to target and destroy tumor cells.
Epigenetics is another exciting area in modern cancer biology research. Epigenetic alterations that can be somatically inherited without changing primary DNA sequences, have been proven to play a key role in tumorigenesis through activation of oncogenes or inactivation of tumor suppressors (91). A number of epigenetic modifiers like DNA methyltransferases, DNA hydroxylases, histone methyltransferases, histone acetyltransferases and chromatin remodeling proteins, are now considered as tumor suppressors since inactivating mutations in these factors can result in decreased expression of classic tumor suppressors (92). Moreover, another group of tumor suppressors have been identified to be genes that are rarely mutated in cancer but frequently silenced by epigenetic changes like DNA methylation (93, 94).
In summary, loss of tumor suppressor genes liberates the cell from its growth-suppressing state or enhances cell survival in various manners. Tumor suppressors play an essential role in the formation of many kinds of human cancers. Therefore, targeting loss of tumor suppressors has tremendous potential as an effective therapeutic strategy against cancer.
f. Conceptual challenges of targeting tumor suppressors for cancer therapeutics
Our knowledge regarding the molecular genetics of tumor suppressors in cancer has grown tremendously over the last several decades. Studies have shown that the inactivation or deletion of certain tumor suppressor genes contribute to tumor development. Conversely, restoring normal expression of the missing tumor suppressor gene in cancer cells can re-establish normal growth suppressive properties and inhibit tumor development. Therefore, the next challenge is to translate this molecular understanding of tumor suppressor genes into viable therapeutic strategies that improve prognostic outcomes for patients.
There are two major strategies for targeting tumor suppressors: (1) reintroduce a functional copy of the tumor suppressor, i.e., gene therapy; (2) develop small molecule inhibitors that reactivate tumor suppressor function. However, both methods have been proven to be technically challenging. First, to re-express a functional tumor suppressor to comparable level as in the normal cells is easily said than done. The major limitations can be attributed to inefficiency in delivering and maintaining the tumor suppressors to all target cells within the bulk of the tumor due to host immune response, insertional mutagenesis, and transduction efficiency. Second, because cancer is a multigenic disorder, where multiple genes contribute to the development of the disease, loss of tumor suppressor function is quite often associated with gain of oncogene activity. Therefore, restoring tumor suppressor activity may not be sufficient to reverse the malignant phenotype.
Regardless the difficulties, numerous studies have shown that restoring or reactivating tumor suppressor function in tumor cells can induce cell-cycle arrest and/or apoptosis in cancer cells. Therefore, there is no doubt that targeting tumor suppressors is a therapeutically viable strategy. Although the challenges are great, there is reason to be optimistic. For example, new technological advancements in vector design have improved the efficiency and efficacy of targeted gene therapy in the last decade. Powerful computational and structural studies have also revealed new “drug-able” protein-protein interaction interfaces for small molecule inhibitors. The current endeavors in developing therapeutics that target the tumor suppressor network will be elaborated and discussed in the following sections.
II. Gene therapy
a. Concept
In the 1960s, scientists proposed to use the exogenous “good” DNA to replace the defective DNA in patients suffering from genetic diseases (95, 96). The very first trial was carried out in 1990 on a 4-year-old girl with ADA-SCID, a severe immune deficient disease (97). Since then, more than 1,900 clinical trials employing gene therapy have been conducted worldwide (For a searchable database on gene therapy clinical trials: J. Gene Med. Gene Therapy Clinical Trials Database. www.wiley.com).
The most common form of gene therapy utilizes DNA that encodes a functional gene to compensate for a dysfunctional gene. Alternatively, synthetic DNA that encodes a therapeutic protein drug or small interference RNA (siRNA) can also be used as treatment. Given that tumor suppressors are often deleted, mutated, or silenced, using gene therapy to restore wild-type tumor suppressor function is a logical approach for treating cancer patients. In theory, such manipulation can be performed on germline cells to correct the mutation or compensate the deletion of tumor suppressor genes; however, most of current investigations are focused on targeting somatic cells, partly due to safety concerns. Currently, there are two major methods of introducing genetic material into the body: viral and non-viral.
b. Viral delivery methods
Harnessing the innate power of viruses to deliver genetic information has been the mainstay strategy since the advent of gene therapy. Among the many different types of viruses, adenoviral vectors and adeno-associated viral vectors are most commonly used as they can be easily manipulated in vitro and are relatively safe in vivo. In the early investigations, a recombinant adenovirus carrying RB cDNA was injected intratumorally into spontaneous pituitary melanotroph tumors that arose in RB heterozygous mice (Figure 3). Such experiments demonstrated the proof of concept with promising efficacy in reducing tumor cell proliferation and prolonging the lifespan of the treated animals (98). In glioblastoma, the most malignant form of glioma, RB is inactivated in at least 30% of tumors and greater than 50% have disrupted RB pathway including homozygous p16INK4 gene deletions (99). Alternative therapies are also highly desirable in glioblastoma as the existence of the blood-brain barrier renders strong resistance to most anticancer therapeutic agents due to low permeability across the barrier. However, such barrier favors viral-mediated gene therapy as the potential anti-adenoviral immune response is limited by the very blood-brain barrier (100). In addition, intratumoral injection of therapeutic adenoviruses is easily achieved using established surgical procedures for glioblastoma.
Figure 3. Representation of tumor suppressor gene therapy model.
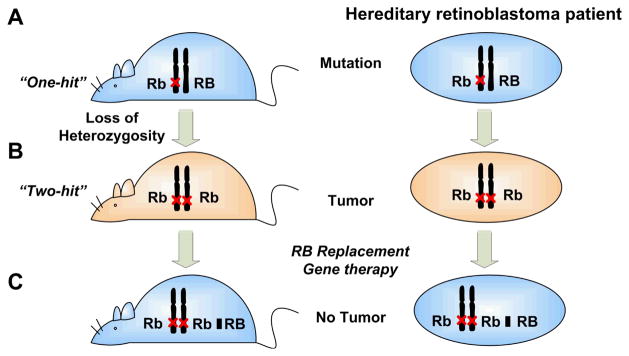
(A) For patients with hereditary retinoblastoma, one copy of the mutated or deleted RB gene is inherited from their parents (the first hit). (B) Acquisition of a second hit, a somatic mutation, causes “loss of heterozygosity” and retinoblastoma. (C) Replacement of the mutated or deleted RB with a normal RB cDNA inhibits tumor growth.
Besides RB, a large body of cancer gene therapy studies has focused on restoring p53 activity. Reactivating p53 has been of particular interest not only because loss of p53 tumor suppressor function is commonly present in most human cancers, but also because continuous p53 inactivation is required for tumor growth maintenance. Gendicine, a recombinant adenovirus carrying p53 cDNA, was the first commercially available, viral-based gene therapy that was approved by regulatory agencies for treating head and neck squamous cell carcinoma (China, 2003) (101). Subsequent trials have also shown Gendicine effective in other types of cancers, including hepatocellular carcinoma and gastric cancer (102, 103).
One major challenge with adenoviruses is their low infection efficiency. As a result, insufficient tumor cells are infected to show a significant therapeutic effect. To overcome this issue, tumor selective oncolytic adenoviruses were developed (104). This new generation of adenoviral vectors can selectively replicate in tumor cells but not normal cells, thereby, amplify the input viral dose and destroy the infected cells by virus-mediated cytolysis, and cause a progressive spread of virus particles in a given tumor. Oncolytic adenoviruses that can specifically replicate in tumor cells with different p53 or RB status, ONYX-015 (105) and AdΔ24 (106), respectively, are now being subjected to various phases of clinical trials in many countries. Although the initial clinical trial results suggest oncolytic adenoviruses have have limited efficacy in the clinic, combination treatment with standard chemotherapies show significantly better responses (107). In addition, great potential for targeting cancer stem cells has been reported in oncolytic adenovirus studies (108). Other types of viruses have also been tested for cancer gene therapy, including retroviruses and lentiviruses (109), as they are advantageous for their stably integration of the transferred gene into the chromosomes and capability for genetic engineering of the glycoprotein to target different cell types (110). However, oncogene activation by the retroviral and lentiviral vectors is a bigger safety concern compared to adenovirus.
Virus targeting, compounded by systemic versus local administration, is another obstacle for cancer gene therapies using viral vector. The virus with the most anticancer therapeutic potential must be capable of infecting cancer cells while sparing normal cells, although the native tropisms of viruses do not match such therapeutic needs. So far, substantial progress has been achieved with improvement in virus targeting in preclinical research (111). Alternatively, the possibility of using neural stem cells or human mesenchymal stem cells to systemically deliver the oncolytic viruses has been tested in glioblastoma. In the preclinical studies, the stem cells, which display a tropism for brain tumor, are loaded with oncolytic viruses before either intravascular or intracranial injections; hence this strategy is also referred to as “Trojan horse” strategy (112, 113). Nevertheless, the clinical efficacy for these novel viral delivery methods awaits further validation.
c. Non-viral delivery methods
Although less pursued compared to virus-mediated gene therapy methods, non-viral delivery methods possess advantages over viral delivery methods in terms of safety, low host immunogenicity, and simplicity for large scale production. Such non-viral approaches can be further divided into two categories: naked plasmids and plasmids encapsulated by liposome or polymer into nanoparticles, i.e., lipoplexes or polyplex, respectively.
Direct injection of naked plasmid DNA into muscle was first tested in vivo as it was simple and inexpensive. Despite a few studies showing that pressurized vascular delivery improves gene transfer in animals with a more systemic effect (114), the overall low expression levels of the transferred genes limited the potential applications of this method in the clinic. With continued improvement in technology, treatments coupling plasmid injection with electroporation (114) or ultrasound (115), have significantly improved the efficacy of gene delivery. Electroporation, in particular, where a pair of electrode needles are inserted into the DNA injection site to deliver electric pulses, has been shown to enhance gene expression in targeted tissues by 2–3 orders of magnitude with minimal tissue damage as compared to the injection of plasmid DNA alone (116). Introducing cytokines into melanomas or head and neck cancer using direct intratumoral injection of DNA plasmids followed by electroporation is now undergoing extensive preclinical and clinical trials at various phases (117, 118). Other assisting methods such as laser and magnetic fields have also been assessed, although the efficiency of gene transfer in vivo needs further improvement (119, 120).
Usage of DNA encapsulating complexes is another method of huge interest. They have tremendous potential for high dosage gene encapsulation, chemical modification and genetic engineering to minimize immune response and improve targeting towards tumor cells. Various compositions have been tested for efficient gene delivery, including cationic polymers such as poly(ethylenimine) and poly(L-lysine), peptides and liposomes, where the positive charges of the capsule favor interactions with negatively charged DNA and cell membrane (121). A recent publication reported the Phase I clinical trial of p53 cDNA administration into patients with solid tumor via a systematic liposomal nanoparticle delivery. Their results are encouraging as p53 protein specifically accumulated in metastatic tumors, but not in normal skin tissue, while the majority of patients demonstrated stable disease with minimal side effects (122).
d. Challenges for cancer gene therapy
The goal for cancer gene therapy is to safely and stably restore normal tumor suppressor functions in cancer cells to inhibit cell growth or eliminate cancer cells. Low and transient expressions of transferred gene often fail to show significant therapeutic effect, while uncontrolled transductions lead to severe side-effect, both compromising the therapeutic outcome. These problems for cancer gene therapy need to be addressed in preclinical investigations before entering human clinical trials. Nevertheless, cancer models, such as glioma and melanoma, which are malignant and lacking of effective treatment, seem to favor cancer gene therapy applications as they are more accessible for intratumoral injections to achieve high dosages of anticancer drugs at the local administration site. Advances in technology may further contribute to the development of successful cancer gene therapy.
III. Inhibitors to restore tumor suppressor functions
Currently, small molecule inhibitors are the most common anticancer therapeutic agents. Following the success of Gleevec for Chronic Myelogenous Leukemia (CML), tremendous effort has been invested in developing inhibitors for various oncogenic kinases. At present, numerous small molecule inhibitors targeting oncogenic kinases are being tested in preclinical and clinical investigations, and about a dozen have been approved by the United State Food and Drug Administration (FDA) for cancer therapy (123). The majority of these inhibitors are specifically directed toward to the ATP binding pocket to inhibit the oncogenic kinase activities. As tumor suppressor gene function is often diminished in cancers, designing small molecule inhibitors to restore tumor suppressor gene will need to take a different approach.
a. Inhibitors targeting RB network
RB’s tumor suppressor function is largely mediated through the transcriptional repression of RB/E2F, which can be alleviated by phosphorylation on RB via cyclin/CDK complexes (124). Cyclin-CDK complexes themselves are subjects of multiple signaling pathways, including Ras, β-catenin and NF-κB (125). Inhibitors for these oncogenic kinases have been shown to indirectly activate RB and restore transcriptional repressions. Such RB-reactivating agents include MEK inhibitor (Trametinib) (126), PPARα agonist (Fenofibrate) (127), and PI3K inhibitor (LY294002) (128).
Alternatively, downstream effectors of RB could also be potential targets. Hec1 (Highly Expressed in Cancer 1) was originally identified as an RB-interacting protein in a yeast two-hybrid screen. As shown in Figure 1, interaction with Hec1 has linked RB to the regulation of mitotic chromosome segregation, during which RB provides a chaperone-like activity to aid in accurate distribution of genetic material (28). Hec1 overexpression has been observed in a variety of human cancers and one of the eleven cancer gene signatures (129–132). In addition, phosphorylation of Hec1 by the mitotic kinase Nek2 is critical for the Hec1 function in cells (133). Thus, the specific interaction between Hec1 and Nek2 represents an ideal anticancer target.
Traditionally, using small molecules to interrupt protein-protein interactions have been shunned by many small molecule developers (134). Unlike the enzymatic active sites of kinases, which are often represented by well-defined pockets, protein-protein interacting interface can involve large surface areas that are shallow and lack of well-defined features (135, 136). Initially, most of researches focused on developing endogenous or engineered peptide inhibitors that mimic the target protein. However, most of the peptide inhibitors were quickly proved to be unsuccessful due to the limitation of oral bioavailability. Small molecule inhibitors provide more opportunities than peptide inhibitors, though the binding affinity, specificity and cellular permeability are still the essential criteria for evaluating small molecule inhibitors.
To find a small molecule inhibitor, we have adapted a unique reverse yeast two-hybrid system, allowing high-throughput screening for compounds that can interrupt specific protein-protein interaction (Figure 4). In this system, we genetically fused TetR with the C-terminal binding region of Hec1 (TetR-Hec1), which was then constitutively expressed in yeast. On the other hand, activation domain of GAL1 fused with Nek2 (AD-Nek2) was under the control of GAL1-inducible promoter. Without the disruption of Hec1/Nek2 interaction, the 5-FOA gene will be activated and hydrolyzed 5-FOA into toxic metabolites, inhibiting yeast growth. Conversely, if an inhibitor is able to disrupt the Hec1/Nek2 interaction, 5-FOA will not be metabolized, thus permitting yeast growth. Using this approach, we have discovered a novel compound, INH1, which can specifically disrupt the Hec1/Nek2 interaction via direct Hec1 binding. The antitumor cytotoxicity was validated both in cultured tumor cells and xenograft models (137). INH1 was further modified via organic chemistry synthesis, though the efficacies for its analogs were not significantly improved (138). Nevertheless, this is a direct proof of concept that it is possible to target tumor suppressor gene network by targeted disruption of protein-protein interaction.
Figure 4. Small molecule inhibitors, INHs, inhibit tumor growth by disrupting interaction between RB-interacting protein, Hec1, and Nek2.

(A) A schematic representation of a reverse yeast two-hybrid system for high-throughput screening for Hec1/Nek2 inhibitors. TetR fused with the C-terminal binding region of Hec1 (TetR-Hec1) was constitutively expressed; activation domain of GAL1 fused with Nek2 (AD-Nek2) was under the control of GAL1-inducible promoter. If an inhibitor abolishes the Hec1/Nek2 interaction, 5-FOA will not be metabolized, thus permitting yeast growth; otherwise, 5-FOA will be hydrolyzed into toxic metabolites and inhibit yeast growth. (B) Structures of two candidate INH compounds that promote yeast growth, i.e., disrupt Hec1/Nek2 interaction. (Figure adapted from (137))
b. p53/MDM2 disruptors
Although about 50% of human tumors lack both p53 alleles or carry a mutated form of p53, in many tumors, a wild-type form of p53 is present, offering an opportunity to boost its tumor suppressor functions to eradicate the tumors. As aforementioned, p53 is a short-lived protein with a low ‘steady-state’ level. Activated p53 will transcriptionally activate MDM2 (also known as HDM2), an oncogene that is usually amplified in cancers, to carry out p53 degradation by the ubiquitin-proteasome pathways (139). Binding of MDM2 to p53 transactivation domain inhibits p53 mediated transcriptional activation (140), and promotes nuclear export of p53 to cytoplasm. In the cytoplasm, MDM2 functions as an E3 ligase to induce attachment of ubiquitin moieties onto p53 for degradation (141). This auto-regulatory loop can be modulated by several signaling pathways via regulating p53/MDM2 interaction (51). For example, the p53/MDM2 interaction can be disrupted by phosphorylations of p53, especially on amino acids in its N-terminal domain. These phosphorylations, which in turn stabilize p53, can be achieved by kinases activated in response to DNA damage in normal cells (142, 143). Therefore, inhibiting p53/MDM2 interaction is a promising because it not only abolishes MDM2-mediated p53 degradation, but also restores p53 transcriptional activities.
The crystal structure of the p53/MDM2 complex revealed that the interacting interface relies on a relative deep hydrophobic cleft of MDM2 that accommodates three amino acid side chains in the helical region of the p53 transactivation domain (144). This discovery led to the development of p53 mimicking peptide inhibitors as therapeutic agents to disrupt p53/MDM2 interaction in tumor cells (145–147). A number of more effective small molecule inhibitors have been identified through high-throughput screening of large chemical libraries, including Nutlins (148) and MI-219 (149), which structurally mimic the p53 peptide and bind to MDM2 pocket. On the other hand, small molecules mimicking MDM2, such as RITA (150), have also been identified to bind p53, although the cellular activities for RITA seemed to be p53-independent (151). In addition, a recent study using computational methods has revealed a novel pocket in the p53 core domain, mutation of which can abolish the reactivation of p53 by known reactivating drugs. This pocket can be a potential target for direct pharmaceutical reactivation of p53 (152).
Taken together, these inhibitors disrupt p53/MDM2 interaction and induce accumulation of wild-type p53 in cells. However, their cellular activities are quite diverse in terms p53 accumulation, and cell cycle arrest versus apoptosis. The effects on tumor cells and normal cells are not homogenous as well (153). Moreover, the presence of the other MDM2 family member, MDMX (also known as HDMX or MDM4), has been reported to reduce the efficacy of these inhibitors, probably due to redundant activities between MDM2 and MDMX (154). Some of the p53/MDM2 disruptors, such as MI-219 and Nutlin-3, which demonstrated high binding affinity and specificity to MDM2 and potent induction of cellular p53 activity, have entered advanced preclinical development or early clinical trials (155). The Phase I clinical trial for RG7112, a much more potent Nutlin analog with better pharmacological properties, showed promising results with increased p53 activation and decreased cell proliferation in liposarcoma patients (156). Further investigations on RG7112 and other p53/MDM2 disruptors are under way.
c. PARP1 inhibitors for BRCA1/BRCA2 deficient tumors
DNA is damaged thousands of times during each cell cycle, and these genetic insults must be repaired. As aforementioned, BRCA1/2 play essential roles in DNA DSBs repair using the error-free HR mechanism. On the other hand, Poly(ADP-ribose) Polymerase 1 (PARP1) is a protein required for repairing single-strand DNA breaks (157, 158). If these “nicks” are not repaired, they can lead to double-strand breaks during replication. Since tumor cells undergo rapid cell division rate, it is obvious that tumor cells will be more susceptible for DNA breaks, making PARP1 a good therapeutic target (159). As human PARP1 catalyzes the transfer of ADP-ribose moiety from nicotinamide adenine dinucleotide (NAD+) donor to a recipient protein, preferentially to glutamate or lysine residues, most PAPR1 inhibitors are designed to compete with NAD+ at the enzymatic site. Currently, there are more than 40 clinical trials in development or ongoing for PARP1 inhibitors as anticancer agents, and major pharmaceutical companies are heavily investing in these PAPR1 inhibitors (160).
Treating cells with PARP1 inhibitors abolishes the cell’s ability to repair of single-strand breaks and generates multiple double strand breaks. This approach exploits a synthetic lethal strategy in cancers that harbor deficient BRCA1/2. Hence, PARP1 inhibitors have been tested in BRCA1/2-associated breast and ovarian cancers (161). A Phase I clinical trial on Olaparib (AZD2281) showed that BRCA1/2 deficient tumors are sensitive to PARP inhibitors with only mild side effects, which can be due to the selectively targeting of PARP inhibitors in BRCA1/2 defective cells, but not the normal cells with intact HR (162). This is consistent with the results that PARP knockout mice are viable and healthy in general, suggesting depletion of only PARP function is tolerable under normal condition (163, 164).
However, the clinical study by Fong et al. showed that not all BRCA1 or BRCA2 mutation carriers respond to the treatment (162). Similarly, some BRCA1-deficient cancers are found with null or low PARP1 expression, potentially limiting the application of PARP1 inhibitors (165). Additionally, an intragenic deletion in BRCA2 was later reported to render resistance to PARP1 inhibitors (166). These studies suggested that only in a small subset of the patient population benefitted from synthetic lethal therapies using PARP1 inhibitors. Nevertheless, in some preclinical studies, inhibiting CDK1 or PI3K can sensitize BRCA-proficient cancers to PARP inhibition (167, 168). Therefore, understanding the unique characteristics of PARP inhibition within the patient specific context is important for improving the efficacy.
d. BRCA2/RAD51 disruptor
Cancers are often under DNA replication stress and high levels of DNA damage, which require functional DNA repair pathways including HR. Elevated recombinase RAD51 expression and enhanced HR rates have been observed in many types of cancers, including CML, and often contributes to drug-resistance (169, 170). To perform HR, RAD51 needs to multimerize and form nucleo-filaments on ssDNA, critical steps which are facilitated by many accessory factors including the breast cancer susceptibility gene product BRCA2 (71). Thus, RAD51 is an attractive target molecule for developing tumor-selective inhibitors.
Structural studies demonstrated that RAD51 directly binds to the six conserved BRC repeats of BRCA2 (171, 172). As shown in Figure 5, using the same strategy as for Hec1/Nek2, we identified a novel small molecule inhibitor, IBR2, which is capable of disrupting RAD51 multimerization, reducing ionizing radiation-induced RAD51 foci formation, impairing HR, inhibiting cancer cell growth and inducing apoptosis. In a murine CML model bearing the T315I BCR-ABL mutation that is resistant to Gleevec (173), IBR2 significantly prolonged animal survival. Consistently, IBR2 effectively inhibits the proliferation of CD34+ progenitor cells derived from CML patients that are resistant to known BCR-ABL inhibitor (174). The efficacy of such small molecule inhibitor of RAD51 on other difficult-to-treat cancers warrants further investigation both preclinically and clinically.
Figure 5. IBR2 inhibits tumor growth by disrupting interaction between RAD51 and BRC repeats.

(A) Structures of IBR compounds (B6 is the negative compound) that disrupt RAD51/BRC repeats interaction. (B) IBR2-RAD51 docking model. IBR2 is shown in ball-and-stick model and colored by hydrophobicity. (Figure adapted from (194))
e. Inhibitors of epigenetic modulators
Over the last decade, our understanding on how epigenetic modifiers can alter the expression of cancer-associated genes provides another opportunity for therapeutic intervention. The most extensively studied epigenetic changes that can silence tumor suppressors are DNA methylation and histone acetylation.
DNA methylation is primarily thought to play a repressive role via CpG island methylation at promoter sequences (175). It is not surprising that many have made the observation of hypermethylation phenotypes in numerous cancers (176). One of the first chemotherapeutic drugs found to modify the epigenome was a cytotoxic nucleoside analog derived in the 1960s called 5-azacytidine, or azacytidine (177). At the time, it was thought that azacytidine exerted its cytotoxic effects solely through random incorporation into the genome and RNA, leaving cells in S-phase and susceptible to cytotoxic stress. It was later discovered that this nucleoside analog could also inhibit DNA methyltransferases by covalently binding to the enzyme (178). Mechanistically, azacytidine was demonstrated to non-selectively reduce methylation levels at promoters of tumor suppressor genes and restore their function via increased gene expression. A number of DNA methyltransferase inhibitors, such as the azacytidine derivate, 5-aza-20-deoxycytidine (Decitabine), have been FDA approved and are used in chemotherapy regimens.
Mutations in pathways regulating the demethylation of DNA have also been implicated in CpG Island Hypermethylator Phenotypes (CIMP) in cancer and are currently the focus of cancer therapeutics development (179). Tet Methylcytosine Dioxygenases (TETs) and Isocitrate Dehydrogenases (IDHs), are another group of interesting epigenetic modulators that regulate DNA methylation and are found to be mutated in numerous hematological cancers and malignancies (180–182). TET family proteins are known to initiate the demethylation cascade that begins after the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), which is eventually converted back to cytosine (183, 184). The activation of TET enzymes require α-ketoglutarate as a cofactor, while mutant IDHs have gain of function that use α-ketoglutarate to generate 2-hydroxyglutarate, which stereochemically resembles α-ketoglutarate and inhibits TETs (181). Such complicated cross-talk between TET and IDH may in part explain the observation of mutually exclusive presence of mutant TET and IDH in glioma and leukemia (185, 186). Currently, clinical trials are underway to test the efficacy of using small molecules to inhibit 2-hydroxyglutarate production by IDH (187, 188). Such inhibition may effectively reverse the inhibition of chromatin demethylation to restore tumor suppressor expression, and demonstrate great pharmacological potential as cancer remedy.
Besides DNA modifications, histone modifications have been proven to be powerful and essential epigenetic regulation on gene expression. Acetylation of histones, for example, is required to maintain chromatin in an open state for active transcription, while HDACs remove acetyl groups to transcriptionally silence gene expression. Therefore, HDACs have become promising therapeutic targets that are under extensive study. Vorinostat and Romideosin were the first FDA approved HDAC inhibitors that came out in 2006 and 2009, respectively (189, 190). The cellular effects of these inhibitors have been shown to include the induction of apoptosis, cell cycle arrest, differentiation, inhibition of angiogenesis and alteration of gene expression patterns (191, 192). Unfortunately, these inhibitors do not spare non-transformed cells, highlighting the need for additional HDAC inhibitors that are specific to HDAC isoforms or specific to mutations found only in cancerous cells.
f. Challenges for developing inhibitors targeting tumor suppressors
Because tumor suppressors are usually mutated, deleted, or silenced in cancers, the first challenge for developing small-molecule inhibitors is to define a specific target. Therefore, possible targets may not necessarily target the tumor suppressor itself, but can target proteins that regulate the tumor suppressor activity. Hence, identification of specific structural interfaces on tumor suppressors or its regulatory proteins is crucial for the synthesis and design of novel compounds. Selecting a target with well-defined structural characteristic, such as a pocket, has proven to be beneficial as in the cases of p53/MDM2 and BRCA2/RAD50 disruptors.
Once the target is identified, multiple approaches have been developed to screen for such small molecule inhibitors, including small focused library screening, high-throughput screening of large chemical libraries, computational 3D database screening and structure-based de novo design. The advancement in computational modeling software has been demonstrated extremely useful in providing information in prediction and subsequent optimization in preclinical research. However, low efficacy and lack of specificity observed in the clinical trials have remained hurdles yet to be overcome.
IV. Concluding remarks and perspectives
A tremendous amount of financial resources and manpower have been invested to understand the malignant nature of cancer in hopes of finding a cure. For the past 40 years, our knowledge of cancer has advanced dramatically. Identifications of oncogenes and tumor suppressors have played a pivotal role in enhancing our understanding of the unique biology of cancer, as well as aiding in the development of new cancer therapies. Discovery of anticancer drug target like the oncogenic BCR-ABL has led to the development of Gleevec, one of the most successful therapeutic drugs for leukemia. On the other hand, the development of therapies that target the tumor suppressor network has failed to translate into viable treatments options for cancer patients. Because tumor suppressor genes are often mutated, deleted or epigenetically silenced in cancer cells, effective anticancer therapies that target tumor suppressor genes must restore the normal functions of tumor suppressor genes. Therefore, gene therapies have been developed for cancers, where the loss of tumor suppressor functions is compensated via ectopic expression of wild-type genes. Various virus-mediated and non-virus-mediated delivery methods have been developed over the past 25 years. However, many experiments and clinical trials to date are hindered by efficacy and safety concerns. Alternatively, numerous inhibitors have been identified to either disrupt interactions between tumor suppressor genes and their negative regulators, or induce synthetic lethality, although the effectiveness of these inhibitors needs to be validated with solid clinical data.
One problem encountered in clinical trials for anticancer therapeutic agents is the small percentage of patients that respond to the treatment, as observed in the case of PARP1 inhibitors for BRCA mutation carriers (162). However, this is a common issue for clinical trials in general, including those for oncogene inhibitors. To overcome this obstacle, identification and incorporation of more reliable biomarkers will be beneficial or necessary to discriminate patient populations with the best responses to a particular treatment (192, 193). In addition, despite the fact that mouse models have been used as a standard system for preclinical testing, the obvious difference between mouse and human genetics cannot be ignored, which can undermine the validity of such testing. More reliable and faithful models are highly desired for testing anticancer therapeutic drugs.
Nevertheless, the ultimate judgment of any anticancer therapy targeting tumor suppressor genes will be based on the potential risk and benefit for the cancer patient. With the tremendous efforts to overcome the difficulties and discoveries of novel biotechnologies, we should expect breakthroughs in the near future towards finding the cure of cancer.
Acknowledgments
This work was supported by NIH grants (R01-CA107568) and (R01-CA94170) to W.-H. L.
List of Abbreviations
- RSV
Rous Sarcoma Virus
- SRC
Sarcoma
- RB
Retinoblastoma Susceptibility gene
- LOH
Loss of Heterozygosity
- R Point
Restriction Point
- CDK
Cyclin Dependent Kinase
- CKI
Cdk Inhibitors
- INK4
Inhibitor of CDK4
- PP1α
Phosphatase 1α
- HDAC
Histone Deacetylase
- APC
Adenomatosis Polyposis Coli
- ROS
Reactive Oxygen Species
- PUMA
P53 Up-Regulated Modulator of Apoptosis
- BRCA1/2
Breast Cancer Susceptibility gene 1/2
- ATM
Ataxia Telangiectasia
- CHK2
Checkpoint Kinase 2
- DSB
Double-Strand Break
- 53BP1
p53-binding protein 1
- MRE11
Meiotic Recombination 11
- HR
Homologous Recombination
- NHEJ
Non-Homologous End-Joining
- ADA-SCID
Adenosine Deaminase Deficiency caused Severe Combined Immunodeficiency
- siRNA
Small Interference RNA
- CML
Chronic Myelogeneous Leukemia
- FDA
The United States Food and Drug Administration
- MEK
MAP Kinase Kinase
- PPARα
Peroxisome Proliferator-Activated Receptor A
- PI3K
Phosphoinositide 3-Kinase
- Hec1
Highly Expressed in Cancer 1
- Nek2
NIMA-Related Kinase 2
- 5-FOA
5-Fluoroorotic Acid
- MDM2
Mouse Double Minute 2
- PARP1
Poly(ADP-Ribose) Polymerase 1
- NAD
Nicotinamide Adenine Dinucleotide
- ssDNA
Single-Strand DNA
- CIMP
CpG Island Hypermethylator Phenotype
- TET
Tet Methylcytosin Dioxygenase
- IDH
Isocitrate Dehydrogenase 1
- 5mc
5-methylcytosine
- 5hmC
5-hydroxymethylcytosine
- 2-HG
2-hydroxyglutarate
- BCR-ABL
Breakpoint Cluster Region-Ableson
References
- 1.Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121 (Suppl 1):1–84. doi: 10.1242/jcs.025742. [DOI] [PubMed] [Google Scholar]
- 2.Muller HJ. Genetic Damage Produced by Radiation. Science. 1955;122(3173):759. doi: 10.1126/science.122.3173.759. [DOI] [PubMed] [Google Scholar]
- 3.Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8(1):1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen L, Issa JP. Epigenetics in colorectal cancer. Current opinion in gastroenterology. 2002;18(1):68–73. doi: 10.1097/00001574-200201000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Duesberg PH, Vogt PK. Differences between the ribonucleic acids of transforming and nontransforming avian tumor viruses. Proceedings of the National Academy of Sciences of the United States of America. 1970;67(4):1673–80. doi: 10.1073/pnas.67.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stehelin D, Fujita DJ, Padgett T, Varmus HE, Bishop JM. Detection and enumeration of transformation-defective strains of avian sarcoma virus with molecular hybridization. Virology. 1977;76(2):675–84. doi: 10.1016/0042-6822(77)90250-1. [DOI] [PubMed] [Google Scholar]
- 7.Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260(5547):170–3. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- 8.Harris H, Miller OJ, Klein G, Worst P, Tachibana T. Suppression of malignancy by cell fusion. Nature. 1969;223(5204):363–8. doi: 10.1038/223363a0. [DOI] [PubMed] [Google Scholar]
- 9.Broaddus E, Topham A, Singh AD. Incidence of retinoblastoma in the USA: 1975–2004. Br J Ophthalmol. 2009;93(1):21–3. doi: 10.1136/bjo.2008.138750. [DOI] [PubMed] [Google Scholar]
- 10.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1971;68(4):820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comings DE. A general theory of carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 1973 Dec;70(12):3324–8. doi: 10.1073/pnas.70.12.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benedict WF, Murphree AL, Banerjee A, Spina CA, Sparkes MC, Sparkes RS. Patient with 13 chromosome deletion: evidence that the retinoblastoma gene is a recessive cancer gene. Science. 1983;219(4587):973–5. doi: 10.1126/science.6336308. [DOI] [PubMed] [Google Scholar]
- 13.Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323(6089):643–6. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 14.Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY. Human retinoblastoma susceptibility gene: cloning, identification, and sequence. Science. 1987;235(4794):1394–9. doi: 10.1126/science.3823889. [DOI] [PubMed] [Google Scholar]
- 15.Lee EY, To H, Shew JY, Bookstein R, Scully P, Lee WH. Inactivation of the retinoblastoma susceptibility gene in human breast cancers. Science. 1988;241(4862):218–21. doi: 10.1126/science.3388033. [DOI] [PubMed] [Google Scholar]
- 16.Harbour JW, Lai SL, Whang-Peng J, Gazdar AF, Minna JD, Kaye FJ. Abnormalities in structure and expression of the human retinoblastoma gene in SCLC. Science. 1988;241(4863):353–7. doi: 10.1126/science.2838909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359(6393):288–94. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 18.Hu N, Gutsmann A, Herbert DC, Bradley A, Lee WH, Lee EY. Heterozygous Rb-1 delta 20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene. 1994;9(4):1021–7. [PubMed] [Google Scholar]
- 19.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nature reviews Cancer. 2002;2(12):910–7. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 20.Dyson N, Buchkovich K, Whyte P, Harlow E. Cellular proteins that are targetted by DNA tumor viruses for transformation. Princess Takamatsu symposia. 1989;20:191–8. [PubMed] [Google Scholar]
- 21.Lee WH, Chen PL, Riley DJ. Regulatory networks of the retinoblastoma protein. Annals of the New York Academy of Sciences. 1995;752:432–45. doi: 10.1111/j.1749-6632.1995.tb17453.x. [DOI] [PubMed] [Google Scholar]
- 22.Cobrinik D, Dowdy SF, Hinds PW, Mittnacht S, Weinberg RA. The retinoblastoma protein and the regulation of cell cycling. Trends Biochem Sci. 1992;17(8):312–5. doi: 10.1016/0968-0004(92)90443-d. [DOI] [PubMed] [Google Scholar]
- 23.Hirschi A, Cecchini M, Steinhardt RC, Schamber MR, Dick FA, Rubin SM. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nature structural & molecular biology. 2010;17(9):1051–7. doi: 10.1038/nsmb.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harbour JW, Dean DC. Chromatin remodeling and Rb activity. Current opinion in cell biology. 2000;12(6):685–9. doi: 10.1016/s0955-0674(00)00152-6. [DOI] [PubMed] [Google Scholar]
- 25.Chen D, Pacal M, Wenzel P, Knoepfler PS, Leone G, Bremner R. Division and apoptosis of E2f-deficient retinal progenitors. Nature. 2009;462(7275):925–9. doi: 10.1038/nature08544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chong JL, Wenzel PL, Saenz-Robles MT, Nair V, Ferrey A, Hagan JP, et al. E2f1-3 switch from activators in progenitor cells to repressors in differentiating cells. Nature. 2009;462(7275):930–4. doi: 10.1038/nature08677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manning AL, Longworth MS, Dyson NJ. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010;24(13):1364–76. doi: 10.1101/gad.1917310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng L, Chen Y, Lee WH. Hec1p, an evolutionarily conserved coiled-coil protein, modulates chromosome segregation through interaction with SMC proteins. Mol Cell Biol. 1999;19(8):5417–28. doi: 10.1128/mcb.19.8.5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Talluri S, Isaac CE, Ahmad M, Henley SA, Francis SM, Martens AL, et al. A G1 checkpoint mediated by the retinoblastoma protein that is dispensable in terminal differentiation but essential for senescence. Mol Cell Biol. 2010;30(4):948–60. doi: 10.1128/MCB.01168-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ianari A, Natale T, Calo E, Ferretti E, Alesse E, Screpanti I, et al. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer cell. 2009;15(3):184–94. doi: 10.1016/j.ccr.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer cell. 2010;17(4):376–87. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen PL, Riley DJ, Chen Y, Lee WH. Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes & Development. 1996;10(21):2794–804. doi: 10.1101/gad.10.21.2794. [DOI] [PubMed] [Google Scholar]
- 33.Chan HM, Krstic-Demonacos M, Smith L, Demonacos C, La Thangue NB. Acetylation control of the retinoblastoma tumour-suppressor protein. Nature cell biology. 2001;3(7):667–74. doi: 10.1038/35083062. [DOI] [PubMed] [Google Scholar]
- 34.Carr SM, Munro S, Kessler B, Oppermann U, La Thangue NB. Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. The EMBO journal. 2011;30(2):317–27. doi: 10.1038/emboj.2010.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Markham D, Munro S, Soloway J, O’Connor DP, La Thangue NB. DNA-damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO reports. 2006;7(2):192–8. doi: 10.1038/sj.embor.7400591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278(5701):261–3. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 37.Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17(1):43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 38.Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57(7):1083–93. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 39.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253(5015):49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 40.Malkin D. p53 and the Li-Fraumeni syndrome. Cancer Genet Cytogenet. 1993;66(2):83–92. doi: 10.1016/0165-4608(93)90233-c. [DOI] [PubMed] [Google Scholar]
- 41.Li FP, Fraumeni JF., Jr Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71(4):747–52. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 42.Wu G, Wu W, Hegde M, Fawkner M, Chong B, Love D, et al. Detection of sequence variations in the adenomatous polyposis coli (APC) gene using denaturing high-performance liquid chromatography. Genetic testing. 2001 Winter;5(4):281–90. doi: 10.1089/109065701753617408. [DOI] [PubMed] [Google Scholar]
- 43.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer research. 1994;54(18):4855–78. [PubMed] [Google Scholar]
- 44.Pavletich NP, Chambers KA, Pabo CO. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 1993;7(12B):2556–64. doi: 10.1101/gad.7.12b.2556. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Reed M, Wang P, Stenger JE, Mayr G, Anderson ME, et al. p53 domains: identification and characterization of two autonomous DNA-binding regions. Genes Dev. 1993;7(12B):2575–86. doi: 10.1101/gad.7.12b.2575. [DOI] [PubMed] [Google Scholar]
- 46.Bartek J, Bartkova J, Vojtesek B, Staskova Z, Lukas J, Rejthar A, et al. Aberrant expression of the p53 oncoprotein is a common feature of a wide spectrum of human malignancies. Oncogene. 1991;6(9):1699–703. [PubMed] [Google Scholar]
- 47.Hofseth LJ, Hussain SP, Harris CC. p53: 25 years after its discovery. Trends Pharmacol Sci. 2004;25(4):177–81. doi: 10.1016/j.tips.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 48.Lane DP. p53, guardian of the genome. Nature. 1992;358(6381):15–6. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 49.Zhou J, Ahn J, Wilson SH, Prives C. A role for p53 in base excision repair. The EMBO journal. 2001;20(4):914–23. doi: 10.1093/emboj/20.4.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adimoolam S, Ford JM. p53 and regulation of DNA damage recognition during nucleotide excision repair. DNA repair. 2003;2(9):947–54. doi: 10.1016/s1568-7864(03)00087-9. [DOI] [PubMed] [Google Scholar]
- 51.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 52.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137(4):609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149(6):1269–83. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer research. 2001;61(16):6234–8. [PubMed] [Google Scholar]
- 55.Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer cell. 2003;4(4):321–8. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 56.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 57.Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nature genetics. 2004;36(1):63–8. doi: 10.1038/ng1282. [DOI] [PubMed] [Google Scholar]
- 58.Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145(4):571–83. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Puzio-Kuter AM. The Role of p53 in Metabolic Regulation. Genes & cancer. 2011;2(4):385–91. doi: 10.1177/1947601911409738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Claus EB, Risch N, Thompson WD. Genetic analysis of breast cancer in the cancer and steroid hormone study. American journal of human genetics. 1991;48(2):232–42. [PMC free article] [PubMed] [Google Scholar]
- 61.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 62.Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265(5181):2088–90. doi: 10.1126/science.8091231. [DOI] [PubMed] [Google Scholar]
- 63.Nathanson KL, Wooster R, Weber BL. Breast cancer genetics: what we know and what we need. Nat Med. 2001;7(5):552–6. doi: 10.1038/87876. [DOI] [PubMed] [Google Scholar]
- 64.Wooster R, Weber BL. Breast and ovarian cancer. N Engl J Med. 2003 Jun 5;348(23):2339–47. doi: 10.1056/NEJMra012284. [DOI] [PubMed] [Google Scholar]
- 65.Zheng L, Li S, Boyer TG, Lee WH. Lessons learned from BRCA1 and BRCA2. Oncogene. 2000;19(53):6159–75. doi: 10.1038/sj.onc.1203968. [DOI] [PubMed] [Google Scholar]
- 66.Yun J, Zhong Q, Kwak JY, Lee WH. Hypersensitivity of Brca1-deficient MEF to the DNA interstrand crosslinking agent mitomycin C is associated with defect in homologous recombination repair and aberrant S-phase arrest. Oncogene. 2005;24(25):4009–16. doi: 10.1038/sj.onc.1208575. [DOI] [PubMed] [Google Scholar]
- 67.Chen Y, Farmer AA, Chen CF, Jones DC, Chen PL, Lee WH. BRCA1 is a 220-kDa nuclear phosphoprotein that is expressed and phosphorylated in a cell cycle-dependent manner. Cancer research. 1996;56(14):3168–72. [PubMed] [Google Scholar]
- 68.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286(5442):1162–6. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 69.Scully RCJ, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997 doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 70.Zhong Q, Chen CF, Li S, Chen Y, Wang CC, Xiao J, et al. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science. 1999;285(5428):747–50. doi: 10.1126/science.285.5428.747. [DOI] [PubMed] [Google Scholar]
- 71.Chen JJ, Silver D, Cantor S, Livingston DM, Scully R. BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer research. 1999 Apr 1;59(7 Suppl):1752s–6s. [PubMed] [Google Scholar]
- 72.Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386(6627):804–10. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- 73.Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer cell. 2002 Aug;2(2):103–12. doi: 10.1016/s1535-6108(02)00102-2. [DOI] [PubMed] [Google Scholar]
- 74.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature structural & molecular biology. 2010;17(6):688–95. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nature reviews Cancer. 2012;12(1):68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316(5828):1194–8. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459(7245):460–3. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gorski JJ, Savage KI, Mulligan JM, McDade SS, Blayney JK, Ge Z, et al. Profiling of the BRCA1 transcriptome through microarray and ChIP-chip analysis. Nucleic acids research. 2011;39(22):9536–48. doi: 10.1093/nar/gkr679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ahmed KM, Tsai CY, Lee WH. Derepression of HMGA2 via removal of ZBRK1/BRCA1/CtIP complex enhances mammary tumorigenesis. The Journal of biological chemistry. 2010;285(7):4464–71. doi: 10.1074/jbc.M109.062265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Furuta S, Wang JM, Wei S, Jeng YM, Jiang X, Gu B, et al. Removal of BRCA1/CtIP/ZBRK1 repressor complex on ANG1 promoter leads to accelerated mammary tumor growth contributed by prominent vasculature. Cancer cell. 2006;10(1):13–24. doi: 10.1016/j.ccr.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 81.Furuta S, Jiang X, Gu B, Cheng E, Chen PL, Lee WH. Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(26):9176–81. doi: 10.1073/pnas.0503793102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baer R, Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Current opinion in genetics & development. 2002;12(1):86–91. doi: 10.1016/s0959-437x(01)00269-6. [DOI] [PubMed] [Google Scholar]
- 83.Mallery DL, Vandenberg CJ, Hiom K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. The EMBO journal. 2002;21(24):6755–62. doi: 10.1093/emboj/cdf691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Poole AJ, Li Y, Kim Y, Lin SC, Lee WH, Lee EY. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314(5804):1467–70. doi: 10.1126/science.1130471. [DOI] [PubMed] [Google Scholar]
- 85.Reid LJ, Shakya R, Modi AP, Lokshin M, Cheng JT, Jasin M, et al. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proceedings of the National Academy of Sciences of the United States of America. 2008 Dec 30;105(52):20876–81. doi: 10.1073/pnas.0811203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Molecular cell. 2001 Feb;7(2):263–72. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 87.Badie S, Escandell JM, Bouwman P, Carlos AR, Thanasoula M, Gallardo MM, et al. BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nature structural & molecular biology. 2010;17(12):1461–9. doi: 10.1038/nsmb.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 89.Old LJ. Cancer immunology: the search for specificity--G. H. A. Clowes Memorial lecture. Cancer research. 1981;41(2):361–75. [PubMed] [Google Scholar]
- 90.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nature reviews Immunology. 2006;6(4):295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 91.Hughes J, Alusi G, Wang Y. Gene therapy and nasopharyngeal carcinoma. Rhinology. 2012;50(2):115–21. doi: 10.4193/Rhino11.239. [DOI] [PubMed] [Google Scholar]
- 92.INGN 201: Ad-p53, Ad5CMV-p53 adenoviral p53, p53 gene therapy--introgen, RPR/INGN 201. Drugs in R&D. 2007;8(3):176–87. doi: 10.2165/00126839-200708030-00005. [DOI] [PubMed] [Google Scholar]
- 93.Fathi AT, Grant S, Karp JE. Exploiting cellular pathways to develop new treatment strategies for AML. Cancer treatment reviews. 2010;36(2):142–50. doi: 10.1016/j.ctrv.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Daskalakis M, Blagitko-Dorfs N, Hackanson B. Decitabine. Recent results in cancer research. Fortschritte der Krebsforschung Progres dans les recherches sur le cancer. 2010;184:131–57. doi: 10.1007/978-3-642-01222-8_10. [DOI] [PubMed] [Google Scholar]
- 95.Osterman JV, Waddell A, Aposhian HV. Gene therapy systems: the need, experimental approach, and implications. Annals of the New York Academy of Sciences. 1971;179:514–9. doi: 10.1111/j.1749-6632.1971.tb46929.x. [DOI] [PubMed] [Google Scholar]
- 96.Rogers S. Gene therapy: a potentially invaluable aid to medicine and mankind. Research communications in chemical pathology and pharmacology. 1971;2(4):587–600. [PubMed] [Google Scholar]
- 97.Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. 1995;270(5235):475–80. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- 98.Riley DJ, Nikitin AY, Lee WH. Adenovirus-mediated retinoblastoma gene therapy suppresses spontaneous pituitary melanotroph tumors in Rb+/− mice. Nat Med. 1996;2(12):1316–21. doi: 10.1038/nm1296-1316. [DOI] [PubMed] [Google Scholar]
- 99.Lee SH, Kim MS, Kwon HC, Park IC, Park MJ, Lee CT, et al. Growth inhibitory effect on glioma cells of adenovirus-mediated p16/INK4a gene transfer in vitro and in vivo. Int J Mol Med. 2000 Nov;6(5):559–63. [PubMed] [Google Scholar]
- 100.Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends in immunology. 2007;28(1):12–8. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 101.Zhang SW, Xiao SW, Liu CQ, Sun Y, Su X, Li DM, et al. Treatment of head and neck squamous cell carcinoma by recombinant adenovirus-p53 combined with radiotherapy: a phase II clinical trial of 42 cases. Zhonghua yi xue za zhi. 2003;83(23):2023–8. [PubMed] [Google Scholar]
- 102.Guan YS, Liu Y, Zhou XP, Li X, He Q, Sun L. p53 gene (Gendicine) and embolisation overcame recurrent hepatocellular carcinoma. Gut. 2005 Sep;54(9):1318–9. doi: 10.1136/gut.2005.069237. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 103.Xie YS, Zhang YH, Liu SP, Liu SQ, Peng CW, Wu L, et al. Synergistic gastric cancer inhibition by chemogenetherapy with recombinant human adenovirus p53 and epirubicin: an in vitro and in vivo study. Oncology reports. 2010;24(6):1613–20. [PubMed] [Google Scholar]
- 104.Alemany R, Balague C, Curiel DT. Replicative adenoviruses for cancer therapy. Nature biotechnology. 2000;18(7):723–7. doi: 10.1038/77283. [DOI] [PubMed] [Google Scholar]
- 105.Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3(6):639–45. doi: 10.1038/nm0697-639. [DOI] [PubMed] [Google Scholar]
- 106.Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19(1):2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 107.Rogulski KR, Freytag SO, Zhang K, Gilbert JD, Paielli DL, Kim JH, et al. In vivo antitumor activity of ONYX-015 is influenced by p53 status and is augmented by radiotherapy. Cancer research. 2000;60(5):1193–6. [PubMed] [Google Scholar]
- 108.Short JJ, Curiel DT. Oncolytic adenoviruses targeted to cancer stem cells. Molecular cancer therapeutics. 2009;8(8):2096–102. doi: 10.1158/1535-7163.MCT-09-0367. [DOI] [PubMed] [Google Scholar]
- 109.Huber BE, Richards CA, Krenitsky TA. Retroviral-mediated gene therapy for the treatment of hepatocellular carcinoma: an innovative approach for cancer therapy. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(18):8039–43. doi: 10.1073/pnas.88.18.8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Anliker B, Abel T, Kneissl S, Hlavaty J, Caputi A, Brynza J, et al. Specific gene transfer to neurons, endothelial cells and hematopoietic progenitors with lentiviral vectors. Nature methods. 2010;7(11):929–35. doi: 10.1038/nmeth.1514. [DOI] [PubMed] [Google Scholar]
- 111.Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nature reviews Genetics. 2007;8(8):573–87. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W, et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(23):12846–51. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sonabend AM, Ulasov IV, Tyler MA, Rivera AA, Mathis JM, Lesniak MS. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem cells. 2008;26(3):831–41. doi: 10.1634/stemcells.2007-0758. [DOI] [PubMed] [Google Scholar]
- 114.Wells DJ. Gene therapy progress and prospects: electroporation and other physical methods. Gene therapy. 2004;11(18):1363–9. doi: 10.1038/sj.gt.3302337. [DOI] [PubMed] [Google Scholar]
- 115.Taniyama Y, Tachibana K, Hiraoka K, Aoki M, Yamamoto S, Matsumoto K, et al. Development of safe and efficient novel nonviral gene transfer using ultrasound: enhancement of transfection efficiency of naked plasmid DNA in skeletal muscle. Gene therapy. 2002;9(6):372–80. doi: 10.1038/sj.gt.3301678. [DOI] [PubMed] [Google Scholar]
- 116.Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo. Nature biotechnology. 1998;16(9):867–70. doi: 10.1038/nbt0998-867. [DOI] [PubMed] [Google Scholar]
- 117.O’Malley BW, Jr, Li D, McQuone SJ, Ralston R. Combination nonviral interleukin-2 gene immunotherapy for head and neck cancer: from bench top to bedside. The Laryngoscope. 2005;115(3):391–404. doi: 10.1097/00005537-200503000-00002. [DOI] [PubMed] [Google Scholar]
- 118.Tamura T, Sakata T. Application of in vivo electroporation to cancer gene therapy. Current gene therapy. 2003;3(1):59–64. doi: 10.2174/1566523033347462. [DOI] [PubMed] [Google Scholar]
- 119.Zeira E, Manevitch A, Khatchatouriants A, Pappo O, Hyam E, Darash-Yahana M, et al. Femtosecond infrared laser-an efficient and safe in vivo gene delivery system for prolonged expression. Molecular therapy: the journal of the American Society of Gene Therapy. 2003;8(2):342–50. doi: 10.1016/s1525-0016(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 120.Scherer F, Anton M, Schillinger U, Henke J, Bergemann C, Kruger A, et al. Magnetofection: enhancing and targeting gene delivery by magnetic force in vitro and in vivo. Gene therapy. 2002;9(2):102–9. doi: 10.1038/sj.gt.3301624. [DOI] [PubMed] [Google Scholar]
- 121.Morille M, Passirani C, Vonarbourg A, Clavreul A, Benoit JP. Progress in developing cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials. 2008;29(24–25):3477–96. doi: 10.1016/j.biomaterials.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 122.Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M, et al. Phase I Study of a Systemically Delivered p53 Nanoparticle in Advanced Solid Tumors. Molecular therapy: the journal of the American Society of Gene Therapy. 2013;21(5):1096–103. doi: 10.1038/mt.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature reviews Cancer. 2009 Jan;9(1):28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zheng L, Lee WH. The retinoblastoma gene: a prototypic and multifunctional tumor suppressor. Experimental cell research. 2001 Mar 10;264(1):2–18. doi: 10.1006/excr.2000.5129. [DOI] [PubMed] [Google Scholar]
- 125.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266(5192):1821–8. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 126.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 127.Saidi SA, Holland CM, Charnock-Jones DS, Smith SK. In vitro and in vivo effects of the PPAR-alpha agonists fenofibrate and retinoic acid in endometrial cancer. Molecular cancer. 2006;5:13. doi: 10.1186/1476-4598-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yuan J, Mehta PP, Yin MJ, Sun S, Zou A, Chen J, et al. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Molecular cancer therapeutics. 2011;10(11):2189–99. doi: 10.1158/1535-7163.MCT-11-0185. [DOI] [PubMed] [Google Scholar]
- 129.Zhong Q, Chen CF, Chen Y, Chen PL, Lee WH. Identification of cellular TSG101 protein in multiple human breast cancer cell lines. Cancer research. 1997;57(19):4225–8. [PubMed] [Google Scholar]
- 130.van’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 131.Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. The Journal of clinical investigation. 2005;115(6):1503–21. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wu G, Wei R, Cheng E, Ngo B, Lee WH. Hec1 contributes to mitotic centrosomal microtubule growth for proper spindle assembly through interaction with Hice1. Molecular biology of the cell. 2009;20(22):4686–95. doi: 10.1091/mbc.E08-11-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen Y, Riley DJ, Zheng L, Chen PL, Lee WH. Phosphorylation of the mitotic regulator protein Hec1 by Nek2 kinase is essential for faithful chromosome segregation. The Journal of biological chemistry. 2002;277(51):49408–16. doi: 10.1074/jbc.M207069200. [DOI] [PubMed] [Google Scholar]
- 134.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nature reviews Drug discovery. 2004;3(4):301–17. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 135.Carry JC, Garcia-Echeverria C. Inhibitors of the p53/hdm2 protein-protein interaction-path to the clinic. Bioorganic & medicinal chemistry letters. 2013;23(9):2480–5. doi: 10.1016/j.bmcl.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 136.Jones S, Thornton JM. Principles of protein-protein interactions. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(1):13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wu G, Qiu XL, Zhou L, Zhu J, Chamberlin R, Lau J, et al. Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer research. 2008;68(20):8393–9. doi: 10.1158/0008-5472.CAN-08-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Qiu XL, Li G, Wu G, Zhu J, Zhou L, Chen PL, et al. Synthesis and biological evaluation of a series of novel inhibitor of Nek2/Hec1 analogues. Journal of medicinal chemistry. 2009;52(6):1757–67. doi: 10.1021/jm8015969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fakharzadeh SS, Trusko SP, George DL. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. The EMBO journal. 1991;10(6):1565–9. doi: 10.1002/j.1460-2075.1991.tb07676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69(7):1237–45. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 141.Tao W, Levine AJ. Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(6):3077–80. doi: 10.1073/pnas.96.6.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, et al. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287(5459):1824–7. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 143.Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(24):13777–82. doi: 10.1073/pnas.96.24.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274(5289):948–53. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 145.Bottger V, Bottger A, Howard SF, Picksley SM, Chene P, Garcia-Echeverria C, et al. Identification of novel mdm2 binding peptides by phage display. Oncogene. 1996;13(10):2141–7. [PubMed] [Google Scholar]
- 146.Garcia-Echeverria C, Chene P, Blommers MJ, Furet P. Discovery of potent antagonists of the interaction between human double minute 2 and tumor suppressor p53. Journal of medicinal chemistry. 2000;43(17):3205–8. doi: 10.1021/jm990966p. [DOI] [PubMed] [Google Scholar]
- 147.Picksley SM, Vojtesek B, Sparks A, Lane DP. Immunochemical analysis of the interaction of p53 with MDM2;--fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene. 1994;9(9):2523–9. [PubMed] [Google Scholar]
- 148.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 149.Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(10):3933–8. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10(12):1321–8. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 151.Di Conza G, Buttarelli M, Monti O, Pellegrino M, Mancini F, Pontecorvi A, et al. IGF-1R/MDM2 relationship confers enhanced sensitivity to RITA in Ewing sarcoma cells. Molecular cancer therapeutics. 2012;11(6):1247–56. doi: 10.1158/1535-7163.MCT-11-0913. [DOI] [PubMed] [Google Scholar]
- 152.Wassman CD, Baronio R, Demir O, Wallentine BD, Chen CK, Hall LV, et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nature communications. 2013;4:1407. doi: 10.1038/ncomms2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(6):1888–93. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nature reviews Cancer. 2013;13(2):83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annual review of pharmacology and toxicology. 2009;49:223–41. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. The lancet oncology. 2012;13(11):1133–40. doi: 10.1016/S1470-2045(12)70474-6. [DOI] [PubMed] [Google Scholar]
- 157.Juarez-Salinas H, Sims JL, Jacobson MK. Poly(ADP-ribose) levels in carcinogen-treated cells. Nature. 1979;282(5740):740–1. doi: 10.1038/282740a0. [DOI] [PubMed] [Google Scholar]
- 158.Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980;283(5747):593–6. doi: 10.1038/283593a0. [DOI] [PubMed] [Google Scholar]
- 159.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nature reviews Cancer. 2010;10(4):293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Banerjee S, Kaye S. PARP inhibitors in BRCA gene-mutated ovarian cancer and beyond. Curr Oncol Rep. Dec;13(6):442–9. doi: 10.1007/s11912-011-0193-9. [DOI] [PubMed] [Google Scholar]
- 162.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 163.de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(14):7303–7. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang ZQ, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, et al. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997;11(18):2347–58. doi: 10.1101/gad.11.18.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Domagala P, Huzarski T, Lubinski J, Gugala K, Domagala W. PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: possible implications for PARP-1 inhibitor therapy. Breast cancer research and treatment. 2011;127(3):861–9. doi: 10.1007/s10549-011-1441-2. [DOI] [PubMed] [Google Scholar]
- 166.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 167.Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. 2011;17(7):875–82. doi: 10.1038/nm.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer discovery. 2012;2(11):1036–47. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Xia SJ, Shammas MA, Shmookler Reis RJ. Elevated recombination in immortal human cells is mediated by HsRAD51 recombinase. Mol Cell Biol. 1997;17(12):7151–8. doi: 10.1128/mcb.17.12.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vispe S, Cazaux C, Lesca C, Defais M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic acids research. 1998;26(12):2859–64. doi: 10.1093/nar/26.12.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chen CF, Chen PL, Zhong Q, Sharp ZD, Lee WH. Expression of BRC repeats in breast cancer cells disrupts the BRCA2-Rad51 complex and leads to radiation hypersensitivity and loss of G(2)/M checkpoint control. The Journal of biological chemistry. 1999;274(46):32931–5. doi: 10.1074/jbc.274.46.32931. [DOI] [PubMed] [Google Scholar]
- 172.Chen PL, Chen CF, Chen Y, Xiao J, Sharp ZD, Lee WH. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(9):5287–92. doi: 10.1073/pnas.95.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 174.Zhu J, Zhou L, Wu G, Konig H, Lin X, Li G, et al. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO molecular medicine. 2012;5(3):353–65. doi: 10.1002/emmm.201201760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Herranz M, Esteller M. DNA methylation and histone modifications in patients with cancer: potential prognostic and therapeutic targets. Methods in molecular biology. 2007;361:25–62. doi: 10.1385/1-59745-208-4:25. [DOI] [PubMed] [Google Scholar]
- 176.Das PM, Singal R. DNA methylation and cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22(22):4632–42. doi: 10.1200/JCO.2004.07.151. [DOI] [PubMed] [Google Scholar]
- 177.Sorm F, Piskala A, Cihak A, Vesely J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia. 1964;20(4):202–3. doi: 10.1007/BF02135399. [DOI] [PubMed] [Google Scholar]
- 178.Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2′-deoxycytidine. The Journal of biological chemistry. 1982;257(4):2041–8. [PubMed] [Google Scholar]
- 179.Issa JP. CpG island methylator phenotype in cancer. Nature reviews Cancer. 2004;4(12):988–93. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 180.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 181.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell. 2010;18(6):553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell. 2010;17(5):510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324(5929):929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–5. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010;28(22):3636–43. doi: 10.1200/JCO.2010.28.3762. [DOI] [PubMed] [Google Scholar]
- 186.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–30. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–6. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 189.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature reviews Drug discovery. 2006;5(9):769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 190.Wagner JM, Hackanson B, Lubbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clinical epigenetics. 2010;1(3–4):117–36. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ma X, Ezzeldin HH, Diasio RB. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs. 2009;69(14):1911–34. doi: 10.2165/11315680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 192.Wang X, Weaver DT. The ups and downs of DNA repair biomarkers for PARP inhibitor therapies. American journal of cancer research. 2011;1(3):301–27. [PMC free article] [PubMed] [Google Scholar]
- 193.Sawyers CL. The cancer biomarker problem. Nature. 2008;452(7187):548–52. doi: 10.1038/nature06913. [DOI] [PubMed] [Google Scholar]
- 194.Zhu J, Zhou L, Wu G, Konig H, Lin X, Li G, et al. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO molecular medicine. 2013;5(3):353–65. doi: 10.1002/emmm.201201760. [DOI] [PMC free article] [PubMed] [Google Scholar]