

Abstract
RNA-guided engineered nucleases (RGENs) derived from the prokaryotic adaptive immune system known as CRISPR (clustered, regularly interspaced, short palindromic repeat)/Cas (CRISPR-associated) enable genome editing in human cell lines, animals, and plants, but are limited by off-target effects and unwanted integration of DNA segments derived from plasmids encoding Cas9 and guide RNA at both on-target and off-target sites in the genome. Here, we deliver purified recombinant Cas9 protein and guide RNA into cultured human cells including hard-to-transfect fibroblasts and pluripotent stem cells. RGEN ribonucleoproteins (RNPs) induce site-specific mutations at frequencies of up to 79%, while reducing off-target mutations associated with plasmid transfection at off-target sites that differ by one or two nucleotides from on-target sites. RGEN RNPs cleave chromosomal DNA almost immediately after delivery and are degraded rapidly in cells, reducing off-target effects. Furthermore, RNP delivery is less stressful to human embryonic stem cells, producing at least twofold more colonies than does plasmid transfection.
The type II CRISPR (clustered, regularly interspaced, short palindromic repeat)/Cas (CRISPR-associated) system is an adaptive immune response in bacteria and archaea, which functions by recognizing and cleaving foreign DNA from phages and plasmids via Cas9 protein and guide RNAs, whose sequences are partially derived from the invaders (Horvath and Barrangou 2010; Wiedenheft et al. 2012). Recently, we and others exploited this system to develop RNA-guided endonucleases or engineered nucleases (RGENs) that enable targeted genome editing in cultured human cells (Cho et al. 2013a; Cong et al. 2013; Jinek et al. 2013; Mali et al. 2013b), zebrafish embryos (Hwang et al. 2013), and bacteria (Jiang et al. 2013). Since then, RGENs have been successfully used to modify genomes in various species including model organisms (Cho et al. 2013b; Dickinson et al. 2013; Friedland et al. 2013; Gratz et al. 2013; Li et al. 2013a,c; Wang et al. 2013; Sung et al. 2014) and plants (Li et al. 2013b; Nekrasov et al. 2013; Shan et al. 2013), rapidly catching up with their precursors, namely, zinc finger nucleases (ZFNs) (Bibikova et al. 2003; Porteus and Baltimore 2003) and transcription activator-like effector nucleases (TALENs) (Miller et al. 2011).
Thus, RGENs are now a new member in the growing family of engineered nucleases (Kim and Kim 2014). These enzymes cleave chromosomal DNA in cells, producing site-specific double-strand breaks (DSBs), the repair of which via endogenous homologous recombination (HR) or nonhomologous end joining (NHEJ) gives rise to targeted mutagenesis and chromosomal rearrangements. We have developed and improved all of these types of programmable nucleases over the last several years (Kim et al. 2009, 2010, 2011; Cho et al. 2013a; Kim et al. 2013a; Sung et al. 2013) and reported that the specificity and activity of RGENs are at least on par with those of their precursors. Unlike ZFNs and TALENs, whose DNA-targeting specificities are altered by protein engineering, new RGENs with desired specificities can be prepared simply by replacing guide RNAs. Furthermore, use of in vitro transcribed guide RNAs rather than plasmids that encode them makes this system cloning-free (Cho et al. 2013a).
For efficient genome editing via RGENs, the successful delivery of guide RNA and Cas9 into cells is essential. In animal experiments, in vitro transcribed Cas9-encoding mRNA or recombinant Cas9 protein can be directly injected into one-cell stage embryos using glass needles to obtain genome-edited animals. To express Cas9 and guide RNA in cultured cells in vitro, typically, plasmids that encode them are transfected via lipofection or electroporation. Unfortunately, use of plasmids is often limited by random integration of all or part of the plasmid DNA into the host genome, a process known as stable transfection. Plasmid DNA can also be inserted at RGEN on-target and off-target sites (Gabriel et al. 2011). Indeed, we found that at least one out of three large insertions and six out of 26 (23%) small insertions at off-target sites, reported in two recent papers (Cradick et al. 2013; Fu et al. 2013), were derived from the Cas9- or sgRNA-encoding plasmid (Supplemental Table 1). Unwanted insertions of plasmid DNA sequences at off-target sites are difficult to detect and, therefore, more problematic than those at on-target sites. These foreign sequences can cause host immune responses (Hemmi et al. 2000; Wagner 2001), hampering the use of gene-edited primary or stem cells in cell therapy. In addition, DNA transfection is often stressful to cells. For example, plasmid DNA introduced into cells triggers cyclic GMP-AMP synthase activation (Sun et al. 2013). Furthermore, prolonged expression of RGENs from plasmid DNA, which can persist in cells for several days post-transfection, can aggravate off-target effects (Gaj et al. 2012). In line with these concerns, cells transfected with plasmids for biomedical applications or animals and plants derived from DNA-transfected cells are regarded as genetically modified by regulatory authorities (Podevin et al. 2013; Pauwels et al. 2014), requiring a costly and lengthy regulation procedure for approval in most developed countries.
In this study, we show that recombinant Cas9 protein complexed with in vitro transcribed guide RNA can be delivered into cultured human cells, including embryonic stem (ES) cells and fibroblasts, to sidestep many limitations associated with the use of plasmids. RGEN ribonucleoproteins (RNPs) induce small insertions and deletions (indels) at target sites almost immediately after delivery into cells and are degraded rapidly, reducing off-target effects.
Results
Targeted mutagenesis in human cells using RGEN RNPs
Recombinant Cas9 protein was purified from Escherichia coli (Supplemental Fig. 1) and complexed with in vitro transcribed single-chain guide RNA (sgRNA) composed of essential portions of target-specific CRISPR RNA (crRNA) and target-independent trans-activating crRNA (tracrRNA) or dualRNA that consists of crRNA and tracrRNA. The resulting RNP complex was transfected into the human leukemia K562 cell line via electroporation. At 48 h post-transfection, RGEN-induced indel frequency was measured using T7 endonuclease I (T7E1), which cleaves heteroduplexes formed by the hybridization of mutant and wild-type sequences or two different mutant sequences. The RGEN RNP designed to target the CCR5 gene, which encodes a chemokine receptor that also acts as an HIV co-receptor, was highly active, inducing mutations at frequencies of up to 57% in a dose-dependent manner, comparable to that obtained with plasmids (Fig. 1A; Supplemental Fig. 2). Small deletions or insertions, signature of error-prone DSB repair via NHEJ, were observed at the target site (Fig. 1B), reminiscent of those observed with ZFNs and TALENs (Kim et al. 2013b). We found that up to sixfold molar excess of sgRNA over Cas9 protein is required to maximize mutation frequencies (Supplemental Fig. 2). To target several other loci in K562, we prepared new RGENs by simply replacing sgRNA in the RNP complex. These RGENs delivered as RNPs were highly active, inducing mutations at frequencies that ranged from 16% to 72% (Fig. 1C; Supplemental Fig. 3). The average mutation frequency obtained with RGEN RNPs at eight different sites in K562 cells was 44 ± 7% under optimal conditions. Note that T7E1 cannot cleave homoduplexes formed by the hybridization of identical mutant sequences and, therefore, often provides an underestimate of indel frequencies, especially when nucleases are highly active, producing identical mutant sequences (Kim et al. 2014).
Figure 1.

Targeted mutagenesis in human K562 cells via direct delivery of RGEN RNPs. (A) CCR5-specific RGEN RNP-mediated mutations measured by the T7E1 assay. (B) Mutant DNA sequences at the CCR5 locus. The 20-bp target sequence is underlined and shown in bold. The PAM sequence is shown in red. (C) RGEN RNP-mediated mutagenesis at several endogenous loci. A mixture of Cas9 protein (15 μg) and sgRNA (20 μg) was transfected into 2 × 105 K562 cells. PCR amplicons around RGEN target sites were subjected to the T7E1 assay. Representative data from at least three independent experiments are shown.
To rule out the possibility that RGEN RNPs cleave chromosomal DNA after cell lysis and that indels are produced by endogenous proteins associated with NHEJ in the cell lysate under cell-free conditions rather than intracellularly (Budman and Chu 2005), we transfected Cas9 protein complexed with dualRNA (rather than sgRNA) into K562 cells and isolated single cell-derived clones via limiting dilution. DualRNA was less potent than sgRNA. Mutant clones were obtained at frequencies of 12% (Supplemental Fig. 4). Furthermore, no mutations were detected using the T7E1 assay when cells were incubated with RGEN RNPs without electroporation (data not shown). These results show that RGEN RNPs entered cells and cleaved chromosomal DNA, triggering the formation of indels at target sites.
Genome editing in human primary cells and embryonic stem cells
We then investigated whether RGEN RNPs enable efficient genome editing in hard-to-transfect human primary cells such as BJ fibroblasts and pluripotent stem cells such as H9 ES cells. In these cells that are refractory to DNA transfection, RNP delivery via electroporation was at least twofold more efficient than plasmid transfection. Thus, the delivery of the CCR5-specific RGEN RNP induced indels at frequencies of 19% and 23% in BJ fibroblasts and H9 ES cells, respectively, whereas plasmid transfection gave rise to indels at frequencies of 9% and 10%, respectively (Fig. 2A). We also tested three other RGEN RNPs in H9 cells and found that the four RGENs induced indels at a frequency of 20 ± 3% on average (Fig. 2B). Notably, RNP delivery was less toxic to ES cells, producing at least twofold more colonies than did plasmid transfection (Fig. 2C). No apparent changes in the morphology of ES cell colonies were observed after RGEN RNP transfection (Fig. 2D). Furthermore, all of the colonies expressed alkaline phosphatase, a marker for pluripotency (Fig. 2D). These results suggest that use of RGEN RNPs rather than plasmids could facilitate isolation of genome-modified clones of ES cells or induced pluripotent stem cells.
Figure 2.

Genome editing in BJ fibroblasts and H9 hES cell lines via direct delivery of RGEN RNPs. (A) CCR5-specific RGEN-driven mutations detected by the T7E1 assay in H9 and BJ cells. (B) RGEN-driven mutations in H9 ES cells detected by the T7E1 assay. A mixture of Cas9 protein (75 μg) and sgRNA (100 μg) was transfected into 1 × 106 H9 cells. (C) Cytotoxicity of RGEN RNPs vs. RGEN plasmid in H9 ES cells. (**) P < 0.01, (*) P < 0.05. (D) No apparent changes in the physiology of ES cells after RGEN RNP treatment. Untransfected, RNP-, and plasmid-transfected ES cell colonies were subjected to AP staining.
Oligonucleotide-directed genome editing via homology-directed repair
Next, we investigated whether RGEN RNPs can be co-transfected with homologous donor DNA to achieve genome editing via homology-directed repair. We used single-stranded oligodeoxynucleotides (ssODNs) (Chen et al. 2011) rather than gene-targeting vectors or PCR amplicons as donor DNA to avoid the possibility that vector- or amplicon-derived double-strand DNA segments could be inserted randomly in the genome or specifically at RGEN on- or off-target sites. Co-delivery of the AAVS1-specific RNP and an ssODN containing a diagnostic XbaI site gave rise to targeted genome modification in K562 cells at a frequency of 15% measured by XbaI digestion (Fig. 3). This method can be used to insert small DNA sequences such as loxP or those that encode affinity tags or antigens at pre-determined genomic sites.
Figure 3.

Homology-directed repair using ssODNs. The 86-mer ssODN includes an XbaI restriction enzyme site, which is absent at the target site, between two short homology arms. PCR amplicons were digested with XbaI in an RFLP assay to detect sequences that resulted from homology-directed repair.
Large chromosomal deletions induced by RGEN RNPs
We and others have shown that the repair of two concurrent DSBs produced by ZFNs and TALENs gives rise to targeted chromosomal rearrangements such as deletions, inversions, and translocations (Brunet et al. 2009; Lee et al. 2010, 2012; Carlson et al. 2012; Kim et al. 2013a). Likewise, transfection of plasmids that encode Cas9 and sgRNA into human cells can cause chromosomal deletions and translocations in a targeted manner. We tested whether RGEN RNPs can induce large chromosomal deletions in K562 cells. As expected, use of two sgRNAs whose target sites are separated by 10- to 100-kb pairs caused targeted deletions of corresponding chromosomal segments (Fig. 4), demonstrating that RGEN RNP delivery enables multiplex genome engineering and targeted chromosomal rearrangements.
Figure 4.

Targeted chromosomal deletions via RGEN RNPs. (A) RGEN target sites in the region of the CCR5 locus. The distances between the CCR5 site and each of the other sites are shown. Arrows indicate PCR primers. Red arrowheads indicate sgRNA target sites. (B) PCR products corresponding to deletions in K562 cells treated with RGEN RNPs; 15 μg of Cas9 protein premixed with 20 μg each of two sgRNAs was transfected into 2 × 105 K562 cells. (C) DNA sequences of deletion-specific PCR products. In cases in which a sequence was detected more than once, the number of occurrences is shown in parentheses.
Reducing off-target effects of RGENs via RNP delivery
RGENs can induce off-target mutations at sites that are highly homologous to on-target sites (Cradick et al. 2013; Fu et al. 2013; Hsu et al. 2013; Pattanayak et al. 2013; Cho et al. 2014). Off-target DNA cleavages by RGENs can cause unwanted chromosomal rearrangements such as translocations (Cho et al. 2014). We tested whether RNP delivery could reduce off-target effects of RGENs. To this end, we chose three RGENs that were previously shown to induce off-target mutations at high frequencies in human cells (Fu et al. 2013; Cho et al. 2014) and compared RNP delivery with plasmid transfection. The three RGENs delivered via RNPs were highly active, inducing site-specific mutations at frequencies that ranged from 76% to 79%, and discriminated on-target sites from off-target sites more efficiently than did those delivered via plasmid transfection (Fig. 5). Thus, no mutations were detected using the T7E1 assay at two off-target sites that differ by 2 nucleotides (nt) from the on-target site, when the RGEN specific to the AAVS1-S3 site was delivered via RNP electroporation. In contrast, the same RGEN delivered via plasmids induced off-target mutations at these sites at frequencies of 6% and 1%, respectively. Importantly, RNP delivery did not sacrifice genome-editing activities at on-target sites, while reducing off-target effects.
Figure 5.
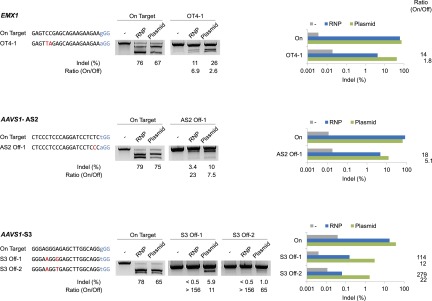
Off-target mutations caused by RGEN RNPs vs. RGEN plasmids. RGEN RNPs or plasmids that encode Cas9 and sgRNA were electroporated into K562 cells. Mutations were detected using the T7E1 assay (left) and deep sequencing (right). The PAM sequence is shown in blue. Mismatched bases are shown in red.
We then used deep sequencing to quantify off-target mutations induced by RGEN plasmids and RNPs more accurately. Again, the three RGEN RNPs discriminated on-target sites from off-target sites much more efficiently than did plasmid-driven RGENs. Thus, the RGEN RNP specific to the AAVS1-S3 site induced off-target indels at frequencies of 0.15% and 0.06%. As a result, the ratio of the indel frequency at the on-target site to that at the two off-target sites was 114 and 279, when RNPs were delivered, whereas the ratio was 12 and 22, respectively, when plasmids were transfected, demonstrating 9.5- (114/12) and 13-fold (279/22) differences, respectively, between the two methods.
Kinetics of RGEN RNP-mediated genome editing
We performed a time-course T7E1 assay and Western blotting to understand the molecular basis of the discrepancy between RNP and plasmid delivery. The T7E1 assay showed that RGEN RNPs cleaved chromosomal DNA almost immediately after delivery and that the mutation frequency reached a plateau one day after electroporation (Fig. 6). In contrast, it took three days to reach an equivalent level of mutations when RGENs were expressed via plasmid transfection. Western blotting analysis showed that Cas9 protein was rapidly degraded in cells when delivered directly: At 24 h post-transfection, Cas9 was barely detected. In contrast, Cas9 protein was expressed from plasmid for several days. Apparently, continuous expression of Cas9 and sgRNA in cells gives rise to the accumulation of off-target mutations. These results are in line with those observed with the direct delivery of ZFNs, which reduces off-target effects (Gaj et al. 2012). Unlike ZFN production, however, preparation of new RGENs does not require time-consuming, fastidious, and labor-intensive steps to de novo engineer, express, and purify sequence-specific nucleases (Liu et al. 2014).
Figure 6.

Time-course analyses of RGEN-mediated genome editing via RNP delivery or plasmid transfection. (A, top) Mutation frequencies were determined by the T7E1 assay. (Bottom) Western blot analysis of K562 cells transfected with the CCR5-specific RGEN via RNP or plasmid DNA delivery. (B,C) Line graphs showing the results of the T7E1 (B) and Western blot analysis (C). Note that only the relative abundance of Cas9 in each experiment is shown.
Discussion
RNA-guided genome editing with the repurposed type II CRISPR/Cas system has been transforming almost every discipline in the life sciences, biotechnology, and medicine (Kim and Kim 2014). Compared with ZFNs and TALENs, RGENs are easy to make, affordable, and scalable, lowering barriers to genome editing. However, these programmable nucleases are limited by off-target effects and unwanted integration of plasmid vectors in the genome, unless mRNA or proteins are used. Furthermore, plasmid transfection is often inefficient and stressful to cells. To sidestep these limitations, we delivered RGEN RNPs, rather than plasmids, directly into cells via electroporation and showed that RGEN RNPs enable efficient genome editing even in human primary and ES cells that are refractory to DNA transfection, while reducing off-target effects and avoiding unwanted integration of plasmid DNA in the host genome. In an accompanying paper published in this issue, Ramakrishna et al. (2014) report that a cell-penetrating peptide can be used to deliver both the Cas9 protein and sgRNA into human cells. In principle, Cas9 protein can be replaced with Cas9 mRNA. Unlike mRNA that must be translated in cells, Cas9 protein works immediately after transfection into cells. Furthermore, it is difficult to check the activity of Cas9 mRNA before transfection, whereas the activity of Cas9 protein can be tested easily in vitro before transfection. Cas9 protein can also be used for genotyping of mutations induced by the same RGENs (Kim et al. 2014).
Recently, several groups independently reported that RGENs can cause unwanted mutations at off-target sites that differ by up to 5 nt from on-target sites, raising concerns about their specificities (Cradick et al. 2013; Fu et al. 2013; Hsu et al. 2013; Pattanayak et al. 2013). Off-target effects may result in chromosomal translocations, inactivation of tumor suppressor or other essential genes, and activation of oncogenes. We and others have proposed several methods to reduce or avoid off-target effects of RGENs. First, the concentration of sgRNA and Cas9 can be titrated to improve the ratio of on-target to off-target mutation rates (Hsu et al. 2013). Unfortunately, titrating RGEN concentrations also reduces on-target mutation frequencies. Second, the use of guide RNA with two additional guanine bases at the 5′ end (that do not match with target DNA sequences), termed 5′-ggX20 sgRNA or crRNA, can discriminate on-target sites from off-target sites that differ by 2 nt efficiently without sacrificing on-target activities both in animals (Cho et al. 2013b; Sung et al. 2014) and in cell lines (Cho et al. 2013a, 2014). Interestingly, truncated sgRNAs can also decrease undesired off-target mutations, improving the specificity of RGENs (Fu et al. 2014). Third, paired Cas9 nickases (or RGENickases) that generate two adjacent single-strand breaks on opposite DNA strands, producing composite DSBs, double the specificity of RNA-guided genome engineering (Mali et al. 2013a; Ran et al. 2013; Cho et al. 2014), a strategy adopted from paired zinc finger nickases (Kim et al. 2012). Fourth, as shown in this study, RGEN RNPs can be used to reduce off-target effects. Last but not least, one should choose unique target sites that do not have homologous sequences elsewhere in the genome, a strategy we had used to avoid off-target effects of TALENs (Kim et al. 2013a). A web-based computer program that can be used for the identification of such sites is available (Bae et al. 2014). It may even be possible to combine these approaches to avoid off-target effects completely. Thus, delivery of RGENickase RNPs with two ggX20 sgRNAs to target a unique sequence in the genome may further reduce off-target effects that persist even with the use of monomeric RGEN RNPs.
In conclusion, RGEN RNPs enable efficient and precise genome editing in diverse human cells including recalcitrant primary and pluripotent stem cells, while avoiding unwanted integration of plasmids and reducing off-target effects. We propose that RGEN RNP delivery will broaden the utility of RNA-guided genome engineering not only in basic research but also in biomedical applications and biotechnology by circumventing regulatory requirements associated with DNA transfection.
Methods
Cell culture
K562 (ATCC, CCL-243) cells were grown in RPMI-1640 with 10% FBS and a penicillin/streptomycin mix. BJ (ATCC, CRL-2522) fibroblasts were maintained in alpha-MEM supplemented with 10% FBS, 0.1 mM nonessential amino acids, and a penicillin/streptomycin mix. H9 human ES cells were maintained in DMEM/F12 (Gibco) supplemented with 20% knockout serum replacement (Gibco), 0.1 mM 2-mercaptoethanol (Gibco), 1% nonessential amino acids (Invitrogen), 8 ng/mL fibroblast growth factor-2 (Invitrogen), 50 U/mL penicillin, and 50 μg/mL streptomycin on mitotically inactivated mouse stromal cells.
Recombinant Cas9 protein
Recombinant Cas9 protein was purchased from ToolGen, Inc. or purified from E. coli. The Cas9 DNA sequence was subcloned into pET28-b(+). Recombinant Cas9 protein containing a nuclear localization signal, the HA epitope, and the His-tag at the N terminus was expressed in BL21(DE3) strain, purified using Ni-NTA agarose beads (Qiagen), and dialyzed against 20 mM HEPES pH 7.5, 150 mM KCl, 1 mM DTT, and 10% glycerol. The purified Cas9 protein was concentrated using Ultracel 100K cellulose column (Millipore). The purity and concentration of Cas9 protein were analyzed by SDS-PAGE.
Guide RNA
RNA was in vitro transcribed through run-off reactions by T7 RNA polymerase using the MEGAshortscript T7 kit (Ambion) according to the manufacturer’s manual. Templates for sgRNA or crRNA were generated by annealing and extension of two complementary oligonuceotides (Supplemental Table 2). Transcribed RNA was purified by phenol:chloroform extraction, chloroform extraction, and ethanol precipitation. Purified RNA was quantified by spectrometry.
Transfection
To introduce DSBs in mammalian cells using an RNP complex, 2 × 105 cells were transfected with Cas9 protein (4.5–45 μg) premixed with in vitro transcribed sgRNA (6–60 μg). Cas9 protein in storage buffer (20 mM HEPES pH 7.5, 150 mM KCl, 1 mM DTT, and 10% glycerol) was mixed with sgRNA dissolved in nuclease-free water and incubated for 10 min at room temperature. No more than 4 μL of the RNP mixture was added to 20 μL of the Nucleofection solution. For plasmid-mediated expression of RGENs, 2 × 105 cells were co-transfected with 1 μg of Cas9-encoding plasmid and 1 μg of sgRNA-expressing plasmid in K562 and BJ fibroblasts or 2.4 μg of Cas9-encoding plasmid and 1.6 μg of sgRNA-expressing plasmid in H9 hES cells. K562 cells were transfected with the Amaxa SF Cell Line 4D-Nucleofector Kit using Program FF-120 (Lonza), and H9 and BJ cells were transfected with the Amaxa P3 Primary Cell 4D-Nucleofector Kit using Program CB-150 and DT-130, respectively, according to the manufacturer’s protocol. Cells were analyzed 2 d after transfection, unless indicated otherwise.
Alkaline phosphatase (AP) staining
H9 human ES cells were fixed using 100% methanol for 10 min and then washed twice with Dulbecco’s phosphate buffered saline (DPBS). AP staining was performed using the AP staining kit (Sigma) according to the manufacturer’s instructions. Briefly, staining solution was prepared by mixing sodium nitrite solution, FRV-Alkaline solution, and AS-BI alkaline solution. Cells were incubated with the staining solution for 15 min and washed three times with DPBS.
T7E1 assay
Genomic DNA was isolated using a genome isolation kit (Promega) according to the manufacturer’s instructions. PCR amplicons including nuclease target sites were generated using the primers listed in Supplemental Table 2. The T7E1 assay was performed as previously described (Kim et al. 2009). Briefly, the PCR amplicons were denatured by heating and annealed to form heteroduplex DNA using a thermocycler and then digested with T7 endonuclease 1 (New England Biolabs) for 20 min at 37°C and then analyzed using agarose gel electrophoresis. For sequencing analysis, PCR products corresponding to genomic modifications were purified and cloned into the T-Blunt vector using the T-Blunt PCR Cloning Kit (SolGent). Cloned products were sequenced using the M13 primer.
RFLP analysis for detection of ssODN-mediated homologous recombination
A total of 2 × 105 K562 cells were co-transfected with 15 μg of Cas9 protein mixed with 20 μg of in vitro transcribed sgRNA and 500 pmol of ssODN (Supplemental Table 2). After 48 h, genomic DNA was isolated and subjected to PCR amplification with primers that flank the target site (Supplemental Table 2). PCR amplicons were digested with XbaI.
Targeted deep sequencing
The on-target and off-target regions were amplified using Phusion polymerase (New England Biolabs) and used for library construction. Equal amounts of the PCR amplicons were subjected to paired-end read sequencing using Illumina MiSeq at Bio Medical Laboratories. Insertions or deletions located around the RGEN cleavage site (3 bp upstream of the PAM) were considered to be the mutations induced by RGENs.
Data access
The deep sequencing data are available at the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRX473144.
Competing interest statement
The authors declare competing financial interests. S.K., S.W.C., and J.-S.K. have filed a patent application based on this work.
Acknowledgments
This work was supported by the National Research Foundation of Korea (2013000718). We thank Choongil Lee and Sangsu Bae for bioinformatic analysis.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.171322.113.
Freely available online through the Genome Research Open Access option.
References
- Bae S, Park J, Kim JS 2014. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics doi: 10.1093/bioinformatics/btu048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Beumer K, Trautman JK, Carroll D 2003. Enhancing gene targeting with designed zinc finger nucleases. Science 300: 764. [DOI] [PubMed] [Google Scholar]
- Brunet E, Simsek D, Tomishima M, DeKelver R, Choi VM, Gregory P, Urnov F, Weinstock DM, Jasin M 2009. Chromosomal translocations induced at specified loci in human stem cells. Proc Natl Acad Sci 106: 10620–10625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budman J, Chu G 2005. Processing of DNA for nonhomologous end-joining by cell-free extract. EMBO J 24: 849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson DF, Tan W, Lillico SG, Stverakova D, Proudfoot C, Christian M, Voytas DF, Long CR, Whitelaw CB, Fahrenkrug SC 2012. Efficient TALEN-mediated gene knockout in livestock. Proc Natl Acad Sci 109: 17382–17387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Pruett-Miller SM, Huang Y, Gjoka M, Duda K, Taunton J, Collingwood TN, Frodin M, Davis GD 2011. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods 8: 753–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS 2013a. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230–232 [DOI] [PubMed] [Google Scholar]
- Cho SW, Lee J, Carroll D, Kim JS 2013b. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics 195: 1177–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS 2014. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 24: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick TJ, Fine EJ, Antico CJ, Bao G 2013. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res 41: 9584–9592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, Goldstein B 2013. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods 10: 1028–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland AE, Tzur YB, Esvelt KM, Colaiacovo MP, Church GM, Calarco JA 2013. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat Methods 10: 741–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD 2013. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31: 822–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK 2014. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32: 279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, et al. 2011. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29: 816–823 [DOI] [PubMed] [Google Scholar]
- Gaj T, Guo J, Kato Y, Sirk SJ, Barbas CF III 2012. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods 9: 805–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J, O’Connor-Giles KM 2013. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 194: 1029–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. 2000. A Toll-like receptor recognizes bacterial DNA. Nature 408: 740–745 [DOI] [PubMed] [Google Scholar]
- Horvath P, Barrangou R 2010. CRISPR/Cas, the immune system of bacteria and archaea. Science 327: 167–170 [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31: 827–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK 2013. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31: 227–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol 31: 233–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J 2013. RNA-programmed genome editing in human cells. eLife 2: e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kim JS 201 4. A guide to genome engineering with programmable nucleases. Nat Rev Genet 15: 321–334 [DOI] [PubMed] [Google Scholar]
- Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS 2009. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19: 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Lee HJ, Carroll D 2010. Genome editing with modularly assembled zinc-finger nucleases. Nat Methods 7: 91 (author reply 91–92) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee MJ, Kim H, Kang M, Kim JS 2011. Preassembled zinc-finger arrays for rapid construction of ZFNs. Nat Methods 8: 7. [DOI] [PubMed] [Google Scholar]
- Kim E, Kim S, Kim DH, Choi BS, Choi IY, Kim JS 2012. Precision genome engineering with programmable DNA-nicking enzymes. Genome Res 22: 1327–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kweon J, Kim A, Chon JK, Yoo JY, Kim HJ, Kim S, Lee C, Jeong E, Chung E, et al. 2013a. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol 31: 251–258 [DOI] [PubMed] [Google Scholar]
- Kim Y, Kweon J, Kim JS 2013b. TALENs and ZFNs are associated with different mutation signatures. Nat Methods 10: 185. [DOI] [PubMed] [Google Scholar]
- Kim JM, Kim D, Kim S, Kim JS 2014. Genotyping with CRISPR-Cas-derived RNA-guided endonucleases. Nat Commun 5: 3157. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Kim E, Kim JS 2010. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res 20: 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Kweon J, Kim E, Kim S, Kim JS 2012. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res 22: 539–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, et al. 2013a. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol 31: 681–683 [DOI] [PubMed] [Google Scholar]
- Li JF, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM, Sheen J 2013b. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat Biotechnol 31: 688–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Teng F, Li T, Zhou Q 2013c. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol 31: 684–686 [DOI] [PubMed] [Google Scholar]
- Liu J, Gaj T, Patterson JT, Sirk SJ, Barbas Iii CF 2014. Cell-penetrating peptide-mediated delivery of TALEN proteins via bioconjugation for genome engineering. PLoS ONE 9: e85755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM 2013a. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31: 833–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM 2013b. RNA-guided human genome engineering via Cas9. Science 339: 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. 2011. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29: 143–148 [DOI] [PubMed] [Google Scholar]
- Nekrasov V, Staskawicz B, Weigel D, Jones JD, Kamoun S 2013. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 691–693 [DOI] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR 2013. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31: 839–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels K, Podevin N, Breyer D, Carroll D, Herman P 2014. Engineering nucleases for gene targeting: safety and regulatory considerations. New Biotechnol 31: 18–27 [DOI] [PubMed] [Google Scholar]
- Podevin N, Davies HV, Hartung F, Nogue F, Casacuberta JM 2013. Site-directed nucleases: a paradigm shift in predictable, knowledge-based plant breeding. Trends Biotechnol 31: 375–383 [DOI] [PubMed] [Google Scholar]
- Porteus MH, Baltimore D 2003. Chimeric nucleases stimulate gene targeting in human cells. Science 300: 763. [DOI] [PubMed] [Google Scholar]
- Ramakrishna S, Kwaku Dad AB, Beloor J, Gopalappa R, Lee SK, Kim H 2014. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res (this issue). doi: 10.1101/gr.171264.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. 2013. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, et al. 2013. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat Biotechnol 31: 686–688 [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung YH, Baek IJ, Kim DH, Jeon J, Lee J, Lee K, Jeong D, Kim JS, Lee HW 2013. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol 31: 23–24 [DOI] [PubMed] [Google Scholar]
- Sung YH, Kim JM, Kim HT, Lee J, Jeon J, Jin Y, Choi JH, Ban YH, Ha SJ, Kim CH, et al. 2014. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res 24: 125–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner H 2001. Toll meets bacterial CpG-DNA. Immunity 14: 499–502 [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153: 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedenheft B, Sternberg SH, Doudna JA 2012. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482: 331–338 [DOI] [PubMed] [Google Scholar]