

Abstract
The rate of transcription elongation plays an important role in the timing of expression of full-length transcripts as well as in the regulation of alternative splicing. In this study, we coupled Bru-seq technology with 5,6-dichlorobenzimidazole 1-β-D-ribofuranoside (DRB) to estimate the elongation rates of over 2000 individual genes in human cells. This technique, BruDRB-seq, revealed gene-specific differences in elongation rates with a median rate of around 1.5 kb/min. We found that genes with rapid elongation rates showed higher densities of H3K79me2 and H4K20me1 histone marks compared to slower elongating genes. Furthermore, high elongation rates had a positive correlation with gene length, low complexity DNA sequence, and distance from the nearest active transcription unit. Features that negatively correlated with elongation rate included the density of exons, long terminal repeats, GC content of the gene, and DNA methylation density in the bodies of genes. Our results suggest that some static gene features influence transcription elongation rates and that cells may alter elongation rates by epigenetic regulation. The BruDRB-seq technique offers new opportunities to interrogate mechanisms of regulation of transcription elongation.
Gene transcription in eukaryotes is the highly regulated process by which RNA polymerase II (RNAPII) uses DNA as a template to produce RNA. The stages of transcription include initiation, elongation, and termination, the control of which influences gene expression. Mechanisms of transcription initiation have been studied in detail, and much is known about transcription factor activation and binding, pre-initiation complex formation, and RNAPII recruitment (Shandilya and Roberts 2012). Furthermore, the critical roles of regulatory sequences such as enhancer elements for developmental and tissue-specific gene regulation (Spitz and Furlong 2012) and the three-dimensional organization of the transcription machinery have been characterized to some level (Sutherland and Bickmore 2009). However, the importance of regulation of the rate of transcription elongation is poorly understood.
Activation of specific gene programs, such as those regulating early organism development, is thought to depend on gene size to accomplish a temporal expression pattern after simultaneous transcriptional activation (Swinburne and Silver 2008). A proposed mechanism to delay the generation of mature RNA is the inclusion of introns of various sizes (Seoighe and Korir 2011; Takashima et al. 2011). To fine-tune this timing mechanism in gene expression, cells may adjust the rates of transcription elongation, splicing, nuclear export, and ribosome access. The rate of transcriptional elongation has also been tied to alternative splicing patterns, where high transcription elongation rates favor exclusion of alternative exons, while slow elongation rates correlate with their inclusion (Close et al. 2012; Shukla and Oberdoerffer 2012).
Previous studies have measured in vivo RNAPII elongation rates in mammals using a variety of techniques including RT-PCR (Singh and Padgett 2009), tiling microarrays (Wada et al. 2009), and fluorescent labeling (Darzacq et al. 2007). These studies have been limited to a single or a small number of genes and have reported a wide range of elongation rates. A recent study utilized GRO-seq to assess elongation rates of a much larger set of genes activated by estradiol or tumor necrosis factor (TNF) and demonstrated a broad range of transcriptional elongation rates among the set of activated genes, supporting the notion that elongation rates may be regulated (Danko et al. 2013)
Here we utilize BruDRB-seq to assess transcription elongation rates genome-wide. This technique involves the transient inhibition of initiated RNAPII prior to elongation using 5,6-dichlorobenzimidazole1-β-D-ribofuranoside (DRB) (Singh and Padgett 2009). Following drug removal, RNA polymerases enter the elongation phase in a synchronized manner, and nascent RNA is labeled with bromouridine (Bru), isolated with anti-BrdU antibodies, and subjected to deep sequencing. By measuring the width of the transcription “wave” generated during the labeling period, the transcription elongation rates of all expressed genes longer than 40 kb were assessed. Our study provides the largest data set so far reported of genome-wide elongation rates in multiple cell lines. We found that high transcription elongation rates correlated with specific gene features as well as with histone modifications such as dimethylation of lysine 79 of histone H3 (H3K79me2) and monomethylation of lysine 20 of histone H4 (H4K20me1). These results indicate that cells may be able to fine-tune transcription elongation rates by epigenetic regulation.
Results
Measuring elongation rates globally reveals variation among genes
To study the elongation rates of RNAPII genome-wide, we developed BruDRB-seq, a technique based on nascent RNA labeling with Bru and assaying by deep sequencing (Paulsen et al. 2013a,b). Following a 60-min treatment of the cultured cells with DRB to arrest RNAPII at promoter-proximal sites, the drug was washed out, and the cells were incubated with Bru for 10 min either directly or after a 10-min recovery period. Cells were lysed in TRIzol, and total RNA was isolated followed by specific capturing of Bru-labeled RNA using anti-BrdU antibodies conjugated to magnetic beads. The captured Bru-labeled RNA was then reverse-transcribed, and the resulting cDNA library was subjected to deep sequencing using the Illumina HiSeq 2000 platform.
In Figure 1A (control), all expressed genes of at least 50 kb in length in the diploid human fibroblast cell line HF1 are represented by median normalized expression (an aggregate view). As expected for nascent RNA, the signal was fairly evenly distributed throughout the first 50 kb of these genes. Following a 60-min DRB treatment with Bru labeling during the last 10 min of treatment, a substantially lower yield of reads was obtained, indicating that transcription was severely reduced (Fig. 1A, 0 min). Following drug removal, a synchronized wave of transcription was observed moving out from the promoter (Fig. 1A, 10 min), and this wave moved further during the next 10 min (Fig. 1A, 20 min). These results demonstrate both the reversibility of DRB and synchronicity of transcription recovery.
Figure 1.
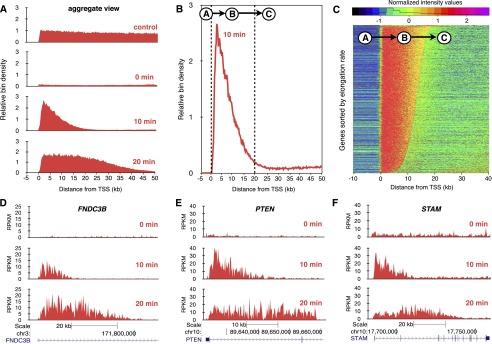
Transcription elongation rates measured genome-wide using BruDRB-seq. (A) Aggregate view of nascent RNA reads through the first 50 kb of large expressed genes in the human fibroblast cell line HF1. (Control) Bru labeling for 30 min. (0 min) Bru labeling during the last 10 min of a 60-min DRB treatment. (10 min) Appearance of a nascent transcription wave at the 5′ end of genes during a 10-min recovery after DRB removal (10-min Bru labeling during recovery period). (20 min) Advancing nascent transcription wave after a 20-min recovery time following DRB removal (Bru labeling during last 10 min of recovery). (B) Aggregate view of BruDRB-seq (10-min recovery) showing the upstream region of TSS having a low signal (A), advancing wave (B), and region downstream from the advancing wave with low signal (C). (C) A hidden Markov model was developed to identify advancing waves and measure their lengths, which are proportional to their elongation rates, having A, B, and C represent the three states of this model. Normalized signals of genes in HF1 cells ordered by elongation rate for a 10-min recovery following DRB removal are shown. Examples of transcriptional recovery in individual genes after 0-, 10-, and 20-min recovery after DRB removal in HF1 cells are shown in (D), (E), and (F).
In order to measure elongation rates of individual genes in a genome-wide fashion, we used an inference method based on a three-state hidden Markov model (HMM) (Day et al. 2007; Danko et al. 2013). The HMM was designed to identify three distinct regions of each gene: (A) the region immediately upstream of the transcription start site (TSS); (B) the advancing wave; and (C) the region of low transcription downstream from the advancing transcription wave (Fig. 1B). Quantile normalization was performed on the BruDRB-seq trace prior to HMM analysis to eliminate any effect of a gene’s expression level on the analysis (see Methods). After applying the HMM analysis to the data from the 10-min wave in HF1 cells, we ordered the genes according to their calculated elongation rates and found them to be quite variable (Fig. 1C). Examples of transcription waves moving from the promoters into the bodies of three individual genes following DRB removal are shown in Figure 1, D–F.
DRB inhibits the transition of RNAPII from the initiation/promoter paused stage into the elongation phase by blocking the phosphorylation of the C-terminal domain (CTD) of RNAPII (Dubois et al. 1994). However, DRB does not inhibit elongating RNA polymerases, and thus, DRB treatment results in a time-dependent clearing out of transcription from the promoter with a receding wave of unaffected actively transcribing polymerases. For transcribed genes longer than 200 kb in HF1 cells treated with DRB for 60 min, the receding transcription wave can be clearly observed (Fig. 2A, yellow). The elongation patterns of two large genes expressed in HF1 cells, MYO1B and TLE4, are shown in Figure 2, B and C, respectively.
Figure 2.
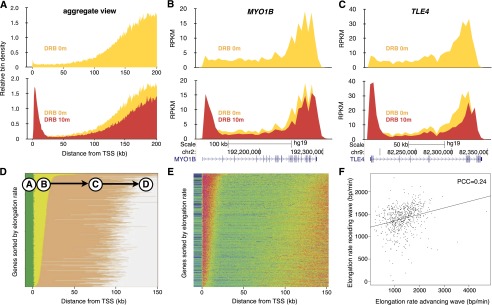
The relationship between advancing and receding transcription elongation waves. (A) Aggregate view of 189 genes longer than 200 kb in HF1 cells during DRB treatment (yellow) or following a 10-min recovery period after DRB treatment (red). Advancing and receding transcription waves in the large genes MYO1B (B) and TLE4 (C). (D) A four-state hidden Markov model was developed to take into account this receding wave in large genes. Genes in HF1 cells were ordered by the length of the advancing wave (state B) and pseudocolored by state (green, A; yellow, B; orange, C; gray, D). (E) Normalized signaling for genes ordered according to the length of state B. (F) The relationship between elongation rates calculated by the length of the advancing wave (state B) and the distance of the trough between the end of the advancing wave and the beginning of the receding wave (state C). (PCC) Pearson’s correlation coefficient.
By incorporating a fourth state in the hidden Markov model representing the receding wave of transcription, we were able to analyze the correlation between the advancing and the receding waves. A visual comparison between the states predicted by the HMM (Fig. 2D) and the normalized signal observed in those genes (Fig. 2E) indicates that predictions of the model were reasonably accurate. We compared elongation rates calculated both via the advancing (state “B”) and the receding (state “D”) wave and found that they correlate, albeit with high variability (Fig. 2F). Because the trailing edge of the receding wave is not as well defined as the advancing wave and because we can include more genes by leaving the receding wave out of the HMM, we decided to focus on the advancing wave (three-state HMM predictions) as our metric for elongation.
Elongation rates are similar in different cell lines
Transcription elongation rates have been explored in a limited number of genes and only in a few cell lines (Ardehali et al. 2009; Singh and Padgett 2009; Danko et al. 2013). In this study, we used five cell lines and BruDRB-seq to assess transcription elongation rates genome-wide. Three of these cell lines are human fibroblasts, and two cell lines, K562 and MCF-7, are cancer-derived. HF1 and TM cells are normal human fibroblasts, while Cockayne syndrome B cells (CS-B) have a genetic defect in the ERCC6 gene, which encodes the CSB protein, resulting in a defect in transcription-coupled DNA repair. It has been suggested that the CSB protein associates with the elongation transcription complex, and in vitro results suggest that CSB enhances the rate of RNAPII elongation (Selby and Sancar 1997). The median elongation rate was found to be similar across the five cell lines, with HF1, CS-B, and K562 being nearly identical (∼1.25 kb/min), and TM and MCF-7 rates slightly higher than the other cell lines (∼1.75 kb/min) (Fig. 3A). In addition, there was a positive correlation of elongation rates of individual genes between the cell lines when performing a pairwise comparison (Fig. 3B). As examples, similar transcription elongation rates for the ACTN4 and PTEN genes across the five cell lines are shown in Figure 3C. Elongation rates for the advancing transcription waves of individual genes in the five cell lines are listed in Supplemental Table 1.
Figure 3.
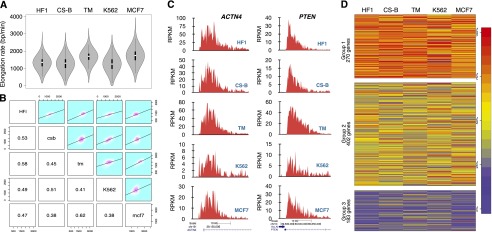
Comparisons of transcription elongation rates among five cell lines. (A) Cells were treated with DRB for 60 min followed by drug reversal and immediate incubation with 2 mM Bru for 10 min. BruDRB-seq was then performed and violin plots illustrating the distribution of elongation rates in the indicated cell lines are shown (the interquartile ranges are represented by thick vertical bars and white dots indicate the median values). Sample sizes: (HF1) 2702; (CS-B) 1932; (TM) 2469; (K562) 2270; (MCF-7) 2399. (B) Grid of pairwise comparisons of elongation rates between each of five cell lines. Each individual comparison includes those genes with measurable elongation rates that are expressed in both cell lines. Frequencies and linear regression models are plotted in the upper right panels and respective Pearson’s correlation coefficients in the lower left. (C) Examples of two individual genes showing similar elongation rates in the five cell lines. (D) Genes expressed in all five cell lines (855 genes) were clustered by normalized elongation rate into three groups using the k-medoids method. Genes in Group 1 tend to be faster-elongating in multiple cell lines, and genes in Group 3 tend to be slower in multiple cells. Genes belonging to Group 2 (47% of total genes) consist of genes with intermediate or variable elongation rates across cell lines. Genes are colored by percentile ranking within each cell line (100% is highest elongation rate [red]). (HF1) Human foreskin fibroblasts; (CS-B) Cockayne syndrome fibroblasts B; (TM) human skin fibroblasts; (K562) myelogenous leukemia; (MCF-7) breast cancer.
A clustering method was used to identify similarities in the observed elongation rates across the five cell lines. Quantile-normalized elongation rates of ∼800 genes expressed in all five cell lines were put into a k-medoids algorithm to cluster these genes into three groups based on similarities in elongation rates. The gene groups selected by the algorithm were clearly distinguished by their overall elongation rates, with a fast, slow, and variable intermediate group (Fig. 3D). These observations indicate that elongation rates of individual genes are considerably conserved among cell lines.
Gene set enrichment
To determine whether genes with similar functions or belonging to a particular pathway have similar transcription elongation rates, we assessed gene enrichment clustering among genes with similar elongation rates. We centered the average elongation rate along its own mean and provided these values and the gene symbols as preranked lists to the Gene Set Enrichment Analysis (GSEA) tool (Subramanian et al. 2005). We searched for enrichment in positional, curated (BioCarta and KEGG), gene ontology, and oncology signature gene sets obtained from the Molecular Signatures Database. We used the permissive false discovery rate (FDR) P-value suggested by the authors of GSEA (P ≤ 25%) (Subramanian et al. 2005) and focused on the gene sets that were enriched in at least three of the cell lines (Supplemental Table 2). We found that genes related to organic acid and carboxylic acid metabolism were enriched among the genes with slow elongation rates. Furthermore, genes related to regulation of the actin cytoskeleton and to leukocyte trans-endothelial migration were enriched among genes with higher elongation rates.
Gene sequence features correlate with elongation rates
Since at least half of the genes used for the clustering analysis described in Figure 3D grouped strongly by elongation rate independent of cell type, we reasoned that the rate of elongation for some genes may be correlated with specific DNA sequence features of the transcribed DNA. Certain features, such as splice site sequences and sequences with a propensity to form G-quadruplexes, have been implicated to affect RNAPII elongation rates in vitro (Belotserkovskii et al. 2013). We investigated the correlation between elongation rates and several sequence features, including GC content, exon density, regions of repetitive DNA, and sequences computationally predicted to form non-B DNA structures. For each gene, the density of each feature from the TSS to the end of the advancing wave was calculated and compared to its elongation rate (Supplemental Table 3). Because several of these features were not randomly distributed throughout the first 40 kb of the genes, a simple regression analysis was inadequate to establish a correlation. To confirm that the correlations found with these features were not solely due to the nonrandom distribution of the feature, we conducted a permutation analysis using a FDR-corrected P-value of 0.05 as the threshold for significance (Supplemental Table 4).
In all cell lines analyzed, exon density (and therefore splice site density) was negatively correlated with elongation rate (Table 1). GC content and the density of long terminal repeat sequences were also negatively correlated with elongation rates in at least three cell lines. It is possible that a higher GC content reduces elongating rates due to a higher energy requirement for breaking three hydrogen bonds between G and C versus two for A and T. The only DNA sequence feature to positively correlate with elongation rate was a high density of low complexity sequences (stretches of mono- or di-nucleotide repeats).
Table 1.
Correlations between DNA or genomic features and transcription elongation rates

Role of gene neighborhoods and genomic organization
Gene expression is often determined by whether the gene is located in an open (euchromatic) or condensed (heterochromatic) chromatin configuration. We asked whether transcription elongation rates are influenced by proximity to nearby genes, chromosomal regions, or three-dimensional organization, which we collectively refer to as “gene neighborhoods.” To examine the effects of gene neighborhoods on elongation, we compared the measured elongation rates of genes and their proximity to neighboring genes on either strand both upstream of and downstream from the gene (Table 1; Supplemental Table 5). We found a positive correlation between elongation rate and the distance to other genes. Thus, active transcription nearby has a negative impact on elongation rate. Interestingly, we also found that gene length is positively correlated with elongation rate, though it is unclear how longer genes are identified or marked for faster transcription. Next, we examined whether there was a correlation between the elongation rates of neighboring genes and found no statistically significant relationship between the elongation rates of neighboring transcribed genes (Supplemental Fig. 1A). Furthermore, inspection of the distribution of genes and their associated elongation rates along the different chromosomes suggests that genes with high or low transcription elongation rates were distributed randomly throughout the genome (Supplemental Fig. 1B).
The above analyses addressed the influence of gene proximity as defined by a linear chromosome but do not consider the three-dimensional organization of genes within the nucleus. Genes that are linearly distant, even on completely different chromosomes, may interact due to long-distance DNA looping and may be transcribed by the same transcription machinery (Dekker et al. 2013). To assess whether genes associated with each other in the same transcriptional “factory” have similar elongation rates, we used publicly available data from chromatin interaction analysis by pair-end tag sequencing (ChIA-PET) for K562 and MCF-7 cells (Li et al. 2012). The elongation rates of genes that were shown by ChIA-PET to colocalize to the same transcription machinery were plotted against each other as gene 1 vs. gene 2 (Supplemental Fig. 1C). The results show that there was no significant correlation between elongation rate and chromatin interactions (Supplemental Table 6).
Elongation rates are related to specific epigenetic modifications
While we found that some DNA sequence features correlated with elongation rate (Table 1), some genes showed varied elongation rates across cells lines (Fig. 3D). Thus, the elongation rates of some genes may be regulated in a cell type-specific way, and we hypothesized that cells may regulate transcription elongation rates by specific epigenetic modifications. We first explored whether the level of DNA methylation in the body of genes correlated with transcription elongation rates. While methylation of CpG islands in promoter regions has been implicated in gene silencing, the function of CpG methylation in the body of genes is poorly understood (Jones 2012). We compared elongation rates obtained with BruDRB-seq to published genome-wide CpG methylation patterns (bisulfite sequencing) data for K562 and MCF-7 cells (The ENCODE Project Consortium 2012) and found that genes with high levels of DNA methylation tended to elongate at slower rates (Supplemental Table 4). This effect, however, was only noticeable when analyzing CpG sites with a high occurrence of methylation (at least 90%), as the correlation was not significant when including sites with a lower occurrence (e.g., at least 50% methylation) in the analysis.
To explore whether fast or slow transcription elongation rates may associate with the presence of specific histone modifications, we divided the genes into four quartiles according to elongation rates and compared them with ChIP-seq data available through ENCODE for different histone marks that have been implicated in transcription regulation (Ernst et al. 2011; The ENCODE Project Consortium 2012). Of the histone marks tested, only H3K79me2 and H4K20me1 were found to show a significant positive correlation with elongation rates (Fig. 4A,B; Supplemental Fig. 2). These histone marks have been shown to be linked to active transcription (Rao et al. 2005; Smolle and Workman 2013), but they have not previously been shown to influence the rate of elongation. We did not observe a significant correlation between transcription elongation rates and the densities of trimethylation of lysine 36 of H3 (H3K36me3) (Fig. 4C) or RNAPII (Fig. 4D). Since H3K36me3 and RNAPII densities are known to correlate with levels of gene expression, our data suggest that high elongation rates are not merely reflecting high expression levels, although a weak positive correlation was observed between transcript output measured with Bru-seq and transcript elongation rate (Kendall’s T = 0.23) (Supplemental Fig. 3). We found no significant relationship between the density of other common histone marks or transcription factors and the transcription elongation rate (Supplemental Fig. 2).
Figure 4.
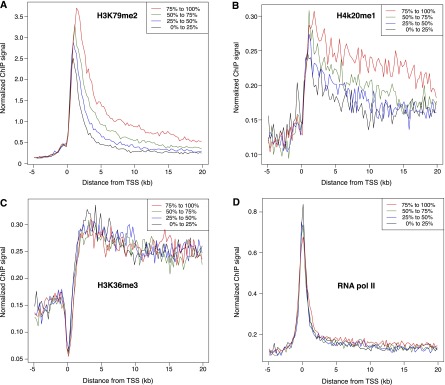
Elongation rates are associated with specific histone modifications. Genes expressed in K562 cells were ranked according to elongation rate and placed into four equal-sized groups (from fastest to slowest: red, green, blue, black). ChIP-seq data for K562 cells were obtained from ENCODE, and median binned values for each group were plotted as indicated for H3K79me2 (A), H4K20me1 (B), H3K36me3 (C), and RNA polymerase II (D). In A and B, genes with faster elongation rates have a higher density of histone modification both near the transcription start site (TSS) and within the gene bodies. In C and D, neither histone modification nor RNA polymerase II occupancy correlated to transcription elongating rates.
Discussion
Transcription elongation has recently drawn attention due to its potential role in the timing of gene expression and the regulation of alternative splicing (Mason and Struhl 2005; Darzacq et al. 2007; Singh and Padgett 2009; Wada et al. 2009; Danko et al. 2013). Here we describe a novel technique, BruDRB-seq, to measure RNAPII elongation rates genome-wide. BruDRB-seq is based on DRB-induced arrest of RNAPII at promoter sites (Singh and Padgett 2009), followed by synchronized release after drug removal. In five different human cell lines, median elongation rate estimations ranged from 1.25 to 1.75 kb/min. These transcription elongation rates, which were estimated from over 2000 genes in each cell line, are somewhat lower than previously estimated in human cells (Singh and Padgett 2009; Danko et al. 2013). It is possible that the genes analyzed by Danko and coworkers, which had been induced by estradiol or TNF, showed a higher elongation rate due to being in an “induced state” where higher transcription and RNAPII densities promoted enhanced elongation rates. Our results also differ from a previously published study that implicated a role of the CSB protein in transcription elongation (Selby and Sancar 1997). In our study, CS-B fibroblasts did not show a distinctly different elongation rate than the other cell types, suggesting that the CSB protein is not generally required for promoting rapid transcription elongation in cells, though it may be an important regulator of elongation for select genes.
Our BruDRB-seq data indicate that there is a broad range of transcription elongation rates in different genes in human cells (Fig. 3). However, the elongation rates for individual genes were reasonably conserved across the different cell lines. We speculated that this conservation may be driven by specific physical features, such as DNA sequence, gene length, or genomic position. Among the genetic features linked to elongation rate was exon density, which correlates with slow elongation. This supports the idea that RNAPII slows down at splice site junctions, which would promote exon definition and alternative splicing (Shukla and Oberdoerffer 2012). Although it has been shown that non-B DNA sequences can have a negative impact on transcription in vitro (Belotserkovskii et al. 2013), we did not find a correlation between elongation rate and potential non-B DNA sequences, though some of these sequences may not exhibit non-B DNA conformations in vivo. Furthermore, our results suggest that if a gene is located near another transcribing gene, its transcription elongation rate is decreased. It has been proposed that transcription of a downstream gene could lead to unwinding of DNA through the induction of negative supercoiling (Ljungman and Hanawalt 1992, 1995) which may affect the elongation of a proximal gene. Conversely, if neighboring genes are simultaneously transcribed in a head-to-head fashion, DNA topological barriers could emerge. Indeed, inhibition of DNA topoisomerase I, which relaxes torsional tension induced during the transcription process, has been shown to severely inhibit transcription elongation (Ljungman and Hanawalt 1996; Veloso et al. 2013). Interestingly, suppression of the rate of elongation by nearby transcription was independent of the orientation of transcription. It is possible that the slower elongation rate in gene neighborhoods is due to competition for limiting pools of ribonucleotides rather than restraints caused by transcription-induced DNA supercoiling.
Our findings that the rates of transcription elongation of nearby genes or genes interacting via DNA looping did not correlate with each other suggest that elongation rates are primarily governed by gene-specific features and epigenetic modifications. We found that H3K79me2 and H4K20me1 were enriched in genes with higher elongation rates (Fig. 4), while the density of H3K36me3 marks, which have been implicated in transcription elongation (Guenther et al. 2007), did not correlate with elongation rate in our study. Furthermore, we did not observe a strong association between high elongation rates and a high density of RNAPII (Fig. 4) or high levels of transcription in those genes (Supplemental Fig. 3). Thus, our data suggest that high elongation rates are not simply the result of high gene expression but rather are governed by specific gene features and epigenetic modifications.
The histone mark H3K79me2 is regulated by the methylase DOT1L (Min et al. 2003), an epigenetic regulator implicated in somatic cell reprogramming (Onder et al. 2012). It was found that key genes involved in the induction of a mesenchymal cell state lost dimethylation of H3K79 without a change in their expression levels during this transition. It is possible that reducing the density of H3K79me2 marks results in a reduced elongation rate of these genes and that this is allowing these cells to transition into a new cell state. DOT1L has also been found to be associated with mixed lineage leukemia (MLL) fusion proteins, resulting in aberrant methylation patterns of H3K79 and dysregulation of MLL targeted genes (Okada et al. 2005). Clinical trials are currently underway using DOT1L-targeting drugs in MLL (Anglin and Song 2013). Methylation of H4K20 is regulated by the PR-SET7 methyltransferase in a cell cycle-dependent manner (Nishioka et al. 2002), and these modifications have been shown to play a role in DNA damage responses by attracting TP53BP1 (Beck et al. 2012). Interestingly, both the H3K79me2 and H4K20me1 histone marks have been implicated in the regulation of replication origin firing (Tardat et al. 2010; Fu et al. 2013). Perhaps by regulating transcription elongation rates of long genes by H3K79 and H4K20 methylation, cells can fine-tune the firing of replication origins.
The size of a gene is a major determinant for how long it will take for transcription to be completed and for the gene to be expressed, and differences in gene lengths contribute to temporal expression patterns (Swinburne and Silver 2008). Our study shows that transcription elongation rates are associated with the histone marks H3K79me2 and H4K20me1, providing a potential mechanism by which cells can fine-tune the temporal gene expression and alternative splicing patterns despite fixed gene lengths. Future studies are needed to define the mechanisms by which cells regulate elongation rates through epigenetic modification and characterize pathological states whereby transcription elongation rates are dysregulated.
Methods
Cell culturing
HF1, hTERT immortalized foreskin-derived human fibroblasts, previously called NF (Paulsen et al. 2013a,b; Veloso et al. 2013), CS-B primary human skin fibroblasts (Coriell, GM00739) and TM, hTERT immortalized human skin fibroblasts (a gift from Dr. Tom Misteli, NCI) were grown in MEM supplemented with 10% FBS, L-glutamine, vitamin mix, and antibiotics. K562 human leukemia cells were grown in IMDM supplemented with 10% FBS and penicillin/streptomycin. MCF-7 human breast cancer cells were grown in high-glucose RPMI supplemented with 10% FBS.
Bru-seq and BruDRB-seq
The labeling of nascent RNA with bromouridine (Bru) was carried out as previously described (Paulsen et al. 2013a,b). The BruDRB-seq protocol differs from the Bru-seq protocol in that the drug 5,6-dichlorobenzimidazole 1-β-D-ribofuranoside (Sigma) is added to the media to a final concentration of 100 µM, and cells are incubated for 1 h at 37°C. After the incubation with DRB, the cells were washed with PBS twice, and nascent RNA was labeled in conditioned media containing 2 mM bromouridine (Aldrich) for 10 min at 37°C. The cells were then directly lysed in TRIzol reagent (Invitrogen). K562 cells were grown in suspension, so these cells were quickly spun down before being lysed in TRIzol. Total RNA was isolated, and the Bru-labeled RNA was isolated from the total RNA by incubation with anti-BrdU antibodies (BD Biosciences) conjugated to magnetic Dynabeads (Invitrogen) under gentle rotation for 1 h at room temperature. Finally, cDNA libraries were made from the Bru-labeled RNA using the Illumina TruSeq library kit and sequenced using Illumina HiSeq sequencers at the University of Michigan DNA Sequencing Core. The sequencing and read mapping were carried out as previously described (Paulsen et al. 2013a,b).
Gene selection for elongation rate analysis
The Ensembl gene annotation (release 69) (Flicek et al. 2013) was used in this analysis, and the annotation data were downloaded using the biomaRt package in the R environment (Durinck et al. 2005). All transcripts from genes with biotype matching “protein coding,” “pseudogene,” “processed transcript,” or “lincRNA” were initially selected to be used in the analysis. Transcripts were selected based on their length, and the transcript’s minimum acceptable length was 40 kb in the three-state HMM analysis (see section “Hidden Markov model for elongation rate analysis”) and 150 kb in the four-state HMM analysis.
A potential source of error for the HMM analysis is the presence of additional TSSs either upstream of or downstream from a given TSS. To address this issue, we first selected genes where the value 3′ from the TSS was at least 10 times higher than the value 5′ of the TSS. Second, we rejected genes that initiated transcription from an additional TSS within the analysis range (e.g., 40 kb in the three-state HMM analysis). Third, to exclude genes with active unannotated TSSs in the analysis region, genes were rejected if the TSS-proximal signal was not more than 10 times the distal signal. Lastly, only genes with Bru-seq expression above 0.5 RPKM were used in the analysis.
Data processing and normalization for elongation rate analysis
The genomic distance analyzed for each transcript extended from 10 kb upstream of the TSS to the minimum acceptable transcript length in that analysis (40 kb in the three-state HMM analysis or 150 kb for the four-state HHM analysis). This distance was divided into 250-bp bins, and the reads along these bins were used to determine the RPKM value of each bin. To minimize the effect of any potential background contamination of unlabeled mature RNA on the elongation rate determinations, the expression signal of bins that overlapped exons was replaced by an interpolation based on the signal of the adjacent bins that did not overlap exons. In order to limit the effect of the transcript’s expression value in the elongation rate analysis, the data were quantile-normalized using the R package preprocessCore (Bolstad et al. 2003). Since most of the expression signal accumulated in the advancing and receding waves, there was a large number of bins that presented very low expression values, and the distribution of binned expression values in the analysis region was similar to a Gamma distribution. In order to improve the presentation of data to downstream analyses, a Z-score Gamma-equivalent normalization was carried out using the R package limma (Smyth et al. 2005).
Hidden Markov model for elongation rate analysis
A hidden Markov model was used to determine the elongation rate of each transcript. This analysis was carried out in two different ways. In the first analysis, the position of three states was predicted in genes that were 40 kb or longer. State 1 represented the low-signal region upstream of the TSS; State 2 represented the advancing wave with high transcription signal; State 3 represented the low-signal region downstream from the advancing elongation wave (Fig. 1B). In the second analysis, a fourth state was added representing the receding wave (Fig. 2D). This second analysis was applied to genes that were at least 150 kb long. Each gene analysis region was split into 250-bp bins, and bin RPKM values were calculated from the BruDRB-seq samples. The expression values were normalized (see section “Data processing and normalization for elongation rate analysis”) and used as the observed layer in the model. The model was trained on regions that were observed to behave as the desired states in the aggregate view of the data (Fig. 1B). The relative bin positions used to calculate the output probabilities were: (state 1) from 10 kb to 0.5 kb upstream of the TSS; (state 2) from 0.5 kb to 20 kb downstream from the TSS; (state 3) from 40 kb to 60 kb (four-state analysis) or 30 kb to 40 kb downstream (three-state analysis) from the TSS; (state 4) from 120 kb to 150 kb downstream from the TSS. The normalized expression values observed in each bin within the described ranges were pooled and used to determine the emission probabilities for each state. The model was set up so that transitions could only occur from state 1 to state 2, state 2 to state 3, and state 3 to state 4 (when analyzing long genes). The transition probabilities between these states were set to 0.00001.
The emission and transition probabilities were used to fit the multistate HMM to the data for the complete analysis region for each transcript using the R package msm (Jackson 2011). The most likely state of each bin was estimated using the Viterbi algorithm. Transcripts where the advancing wave (state 2) began more than 2 kb upstream of or downstream from the annotated TSS were removed. Transcripts where the trough (state 3) began at the annotated TSS and transcripts where a state 3 or state 4 (in the long-gene analysis) was not recognized were removed from the analysis. The calculated elongation for each transcript in the three-state analysis is given in Supplemental Table 1.
Clustering of genes according to elongation rate
In order to compare between cell lines, the measured elongation rates were quantile-normalized. A dissimilarity matrix was then calculated from the quantile-normalized elongation rates using a Euclidean distance. This metric was used for the clustering, which was carried out using the k-medoids algorithm (also known as partitioning around medoids, or PAM). The dissimilarity matrix calculation and clustering were performed using the R package cluster (Maechler et al. 2013).
Enrichment of gene sets according to elongation rate
Gene Set Enrichment Analysis (GSEA) (Subramanian et al. 2005) was used to determine if there were gene sets enriched among the genes with higher or lower elongation rates. The gene set collections used were positional, curated (BioCarta and KEGG), gene ontology (biological processes, cellular components, and molecular functions), and oncogenic signatures (downloaded from the Molecular Signatures Database [MSigDB], version 4; http://www.broadinstitute.org/gsea/msigdb/index.jsp). GSEA was run on a list of genes ranked according to elongation rate, and gene sets with at least 15 represented genes were selected for analysis. A false discovery rate adjusted P-values threshold of 0.25 was applied to determine enrichment of a gene set.
Correlation between elongation rate and gene features
To determine if the elongation rates were correlated with different physical properties of the genes such as DNA sequence, several different features were tested in a permutation test. Seven features were analyzed. (1) Transcript length (in base pairs) using the Ensembl annotation (Flicek et al. 2013). (2) Distance to nearby expressed genes (in base pairs). Genes with an expression level greater than 0.1 RPKM were considered expressed. Distances were measured to the closest upstream and downstream gene in either the sense or antisense orientation, resulting in a total of four different values. (3) Density of exons. The Ensembl’s project exon annotation was used (Flicek et al. 2013). All annotated exons were used, and exons that overlapped were merged into a single exon. (4) GC content. (5) Repetitive DNA (combined length of each class in base pairs). The RepeatMasker annotation (http://www.repeatmasker.org/) was downloaded from the UCSC Genome Browser (http://genome.ucsc.edu/). The repetitive DNA annotation was simplified to reflect only the major classes (i.e., DNA, LINE, low complexity, LTR, other, RC/Helitron, RNA, rRNA, satellite, scRNA, simple repeat, SINE, snRNA, srpRNA, tRNA, unknown). Only nonoverlapping repetitive regions were used in the analysis. (6) Non-B DNA (combined length of each class in base pairs). The Non-B DB v2.0 annotation was used in this analysis (Cer et al. 2013). The classes of non-B DNA used were: A phased repeat, direct repeat, G-quadruplex motif, inverted repeat, mirror repeat, short tandem repeat, and Z DNA motif. (7) Density of methylated CpG sites. The DNA Methylation by Reduced Representation Bisulfite-seq from the ENCODE/HudsonAlpha data set was used (http://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg19&g=wgEncodeHaibMethylRrbs). The tracks used (and their respective GEO accession ID) were as follows: K562 HudsonAlpha replicates 1 (GSM683856) and 2 (GSM68378), and MCF-7 Stanford replicates 1 (GSM720350) and 2 (GSM720353). A CpG site was only used in the analysis if it was represented in a minimum of 10 reads and if at least 90% of those reads indicated that that site was, in fact, methylated.
To assess the correlations between high and low levels of elongation and a particular DNA or chromatin feature, we divided the feature metric (e.g., exon count) by the length of elongation (providing a feature density). It was observed that certain features presented a nonrandom distribution throughout the length of the gene. For example, GC content tends to be higher near the TSS and decreases as the distance to the TSS increases, until it levels off. Therefore, if one assigned random elongation rates to genes and measured their GC content, there would be a negative correlation. Due to this limitation, a permutation test was performed by randomly distributing the observed elongation rates among the genes.
In order to limit the effect of outliers, the 5% most extreme elongation rate values (top and bottom 2.5%) were excluded from the analysis. Also, features were only analyzed if they had been measured in at least 20% of the transcripts. The feature metric was measured for all genes under the new elongation area as determined by the randomly distributed elongation rates. A regression coefficient between elongation rate and feature metric was calculated and stored. This process was repeated 2000 times. Finally, the regression coefficient observed in the original data was compared to the 2000 permutated regression coefficients. A one-tailed P-value was determined by measuring the percentage of times that a permutated regression coefficient was equal to or more extreme than the observed regression coefficient. To account for the multiple testing, the P-values were FDR-corrected. A corrected regression coefficient was calculated by subtracting the observed regression coefficient from the median value of the 2000 permutated regression coefficients. Permutation analysis results can be found in Supplemental Table 4.
Long-range promoter interaction and elongation rate
Chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) (Li et al. 2012) data was used to determine if the elongation rates of genes in contact with the same transcription machinery are correlated. A regression analysis between elongation rates of genes believed to be physically in contact with each other as assessed with ChIA-PET was carried out. The data were downloaded from the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg19&g=wgEncodeGisChiaPet). The tracks used (and their respective GEO accession ID) were: K562 POL2 replicates 1 and 2 (GSM970213), and MCF-7 POL2 replicates 3 and 4 (GSM970209). The segments that were considered to be connected by ChIA-PET analysis were intersected with the annotation of genes for which elongation rates were measured. Two genes were considered to be physically connected if two connected segments overlapped the TSS of the two genes.
Aggregate signal of ChIP-seq data for the elongation rate quartiles
To determine if transcription elongation rates were correlated to the density of specific histone modifications or proteins, we downloaded the ChIP-seq processed signal files (in the bigWig file format) from the UCSC Genome Browser. Data were obtained from http://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg19&g=wgEncodeBroadHistone and http://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg19&g=wgEncodeSydhTfbs data sets. The tracks used (and their respective GEO accession ID) were: ChIP-seq input control (GSM733780), H3K4me1 (GSM733692), H3K4me3 (GSM733680), H3K27ac (GSM733656), H3K9me1 (GSM733777), H3K9me3 (GSM733776), H3K9ac (GSM733778), H3K27me3 (GSM733658), H3K36me3 (GSM733714), H3K79me2 (GSM733653), H4K20me1 (GSM733675), CTCF (GSM733719), Pol2 (GSM733643), CCNT2 (GSM935547), GTF21 (GSM935501), NELFE (GSM935392), and MYC (GSM935516). These data were normalized according to Ram et al. (2011).
A genomic region encompassing 5 kb upstream of to 20 kb downstream from the TSS of all genes for which an elongation rate was recorded was used in the analysis. This analysis region was split into bins of 250 bp in length. For each bin, the average ChIP-seq signal of a given data set was calculated and plotted according to each quadrant of elongation rates.
Data access
All the primary sequencing data files from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE55534.
Acknowledgments
We thank Manhong Dai and Fan Meng for administration and maintenance of the University of Michigan Molecular and Behavioral Neuroscience Institute (MBNI) computing cluster and the personnel at the University of Michigan Sequencing Core for technical assistance. We also thank all members of the Ljungman laboratory for valuable discussions. This work was supported by funds from the National Institute of Environmental Health Sciences (1R21ES020946), National Human Genome Research Institute (1R01HG006786), and the National Institutes of Health (P50CA130810).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.171405.113.
References
- Anglin JL, Song Y 2013. A medicinal chemistry perspective for targeting histone H3 lysine-79 methyltransferase DOT1L. J Med Chem 56: 8972–8983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardehali MB, Yao J, Adelman K, Fuda NJ, Petesch SJ, Webb WW, Lis JT 2009. Spt6 enhances the elongation rate of RNA polymerase II in vivo. EMBO J 28: 1067–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck DB, Oda H, Shen SS, Reinberg D 2012. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev 26: 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskii BP, Mirkin SM, Hanawalt PC 2013. DNA sequences that interfere with transcription: implications for genome function and stability. Chem Rev 113: 8620–8637 [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193 [DOI] [PubMed] [Google Scholar]
- Cer RZ, Donohue DE, Mudunuri US, Temiz NA, Loss MA, Starner NJ, Halusa GN, Volfovsky N, Yi M, Luke BT, et al. 2013. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res 41: D94–D100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close P, East P, Dirac-Svejstrup AB, Hartmann H, Heron M, Maslen S, Chariot A, Soding J, Skehel M, Svejstrup JQ 2012. DBIRD complex integrates alternative mRNA splicing with RNA polymerase II transcript elongation. Nature 484: 386–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, Siepel A, Kraus WL 2013. Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Mol Cell 50: 212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH 2007. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 14: 796–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day N, Hemmaplardh A, Thurman RE, Stamatoyannopoulos JA, Noble WS 2007. Unsupervised segmentation of continuous genomic data. Bioinformatics 23: 1424–1426 [DOI] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, Mirny LA 2013. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet 14: 390–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois MF, Nguyen VT, Bellier S, Bensaude O 1994. Inhibitors of transcription such as 5,6-dichloro-1-β-D- ribofuranosylbenzimidazole and isoquinoline sulfonamide derivatives (H-8 and H-7*) promote dephosphorylation of the carboxyl-terminal domain of RNA polymerase II largest subunit. J Biol Chem 269: 13331–13336 [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W 2005. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21: 3439–3440 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. 2013. Ensembl 2013. Nucleic Acids Res 41: D48–D55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Maunakea AK, Martin MM, Huang L, Zhang Y, Ryan M, Kim R, Lin CM, Zhao K, Aladjem MI 2013. Methylation of histone H3 on lysine 79 associates with a group of replication origins and helps limit DNA replication once per cell cycle. PLoS Genet 9: e1003542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA 2007. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130: 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson C 2011. Multi-state models for panel data: the msm package for R. J Stat Softw 38: 1–29 [Google Scholar]
- Jones PA 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492 [DOI] [PubMed] [Google Scholar]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. 2012. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148: 84–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungman M, Hanawalt PC 1992. Localized torsional tension in the DNA of human cells. Proc Natl Acad Sci 89: 6055–6059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungman M, Hanawalt P 1995. Presence of negative torsional tension in the promoter region of the transcriptionally poised dihydrofolate reductase gene in vivo. Nucleic Acids Res 23: 1782–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungman M, Hanawalt PC 1996. The anti-cancer drug camptothecin inhibits elongation but stimulates initiation of RNA polymerase II transcription. Carcinogenesis 17: 31–35 [DOI] [PubMed] [Google Scholar]
- Maechler M, Rousseeuw P, Struyf A, Hubert M, Hornik K. 2013. cluster: cluster analysis basics and extensions. R package version 1.14.4
- Mason PB, Struhl K 2005. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol Cell 17: 831–840 [DOI] [PubMed] [Google Scholar]
- Min J, Feng Q, Li Z, Zhang Y, Xu RM 2003. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112: 711–723 [DOI] [PubMed] [Google Scholar]
- Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, Chuikov S, Valenzuela P, Tempst P, Steward R, et al. 2002. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell 9: 1201–1213 [DOI] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y 2005. hDOT1L links histone methylation to leukemogenesis. Cell 121: 167–178 [DOI] [PubMed] [Google Scholar]
- Onder TT, Kara N, Cherry A, Sinha AU, Zhu N, Bernt KM, Cahan P, Marcarci BO, Unternaehrer J, Gupta PB, et al. 2012. Chromatin-modifying enzymes as modulators of reprogramming. Nature 483: 598–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Magnuson B, Wilson TE, Ljungman M 2013a. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods doi: 10.1016/j.ymeth.2013.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Tsan YC, Chang CW, Tarrier B, Washburn JG, Lyons R, et al. 2013b. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc Natl Acad Sci 110: 2240–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, Kellis M, Gymrek M, Issner R, Coyne M, et al. 2011. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell 147: 1628–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao B, Shibata Y, Strahl BD, Lieb JD 2005. Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol Cell Biol 25: 9447–9459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby CP, Sancar A 1997. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc Natl Acad Sci 94: 11205–11209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoighe C, Korir PK 2011. Evidence for intron length conservation in a set of mammalian genes associated with embryonic development. BMC Bioinformatics (Suppl 9) 12: S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shandilya J, Roberts SG 2012. The transcription cycle in eukaryotes: from productive initiation to RNA polymerase II recycling. Biochim Biophys Acta 1819: 391–400 [DOI] [PubMed] [Google Scholar]
- Shukla S, Oberdoerffer S 2012Co-transcriptional regulation of alternative pre-mRNA splicing. Biochim Biophys Acta 1819: 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Padgett RA 2009. Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol 16: 1128–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolle M, Workman JL 2013. Transcription-associated histone modifications and cryptic transcription. Biochim Biophys Acta 1829: 84–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK, Michaud J, Scott HS 2005. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21: 2067–2075 [DOI] [PubMed] [Google Scholar]
- Spitz F, Furlong EE 2012. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13: 613–626 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland H, Bickmore WA 2009. Transcription factories: gene expression in unions? Nat Rev Genet 10: 457–466 [DOI] [PubMed] [Google Scholar]
- Swinburne IA, Silver PA 2008. Intron delays and transcriptional timing during development. Dev Cell 14: 324–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y, Ohtsuka T, Gonzalez A, Miyachi H, Kageyama R 2011. Intronic delay is essential for oscillatory expression in the segmentation clock. Proc Natl Acad Sci 108: 3300–3305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E 2010. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol 12: 1086–1093 [DOI] [PubMed] [Google Scholar]
- Veloso A, Biewen B, Paulsen MT, Berg N, Carmo de Andrade Lima L, Prasad J, Bedi K, Magnuson B, Wilson TE, Ljungman M 2013. Genome-wide transcriptional effects of the anti-cancer agent camptothecin. PLoS ONE 8: e78190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Ohta Y, Xu M, Tsutsumi S, Minami T, Inoue K, Komura D, Kitakami J, Oshida N, Papantonis A, et al. 2009. A wave of nascent transcription on activated human genes. Proc Natl Acad Sci 106: 18357–18361 [DOI] [PMC free article] [PubMed] [Google Scholar]